

Abstract
Background
Building up of pathway-/disease-relevant signatures provides a persuasive tool for understanding the functional relevance of gene alterations and gene network associations in multifactorial human diseases. Ovarian cancer is a highly complex heterogeneous malignancy in respect of tumor anatomy, tumor microenvironment including pro-/antitumor immunity and inflammation; still, it is generally treated as single disease. Thus, further approaches to investigate novel aspects of ovarian cancer pathogenesis aiming to provide a personalized strategy to clinical decision making are of high priority. Herein we assessed the contribution of the AID/APOBEC family and their associated genes given the remarkable ability of AID and APOBECs to edit DNA/RNA, and as such, providing tools for genetic and epigenetic alterations potentially leading to reprogramming of tumor cells, stroma and immune cells.
Results
We structured the study by three consecutive analytical modules, which include the multigene-based expression profiling in a cohort of patients with primary serous ovarian cancer using a self-created AID/APOBEC-associated gene signature, building up of multivariable survival models with high predictive accuracy and nomination of top-ranked candidate/target genes according to their prognostic impact, and systems biology-based reconstruction of the AID/APOBEC-driven disease-relevant mechanisms using transcriptomics data from ovarian cancer samples. We demonstrated that inclusion of the AID/APOBEC signature-based variables significantly improves the clinicopathological variables-based survival prognostication allowing significant patient stratification. Furthermore, several of the profiling-derived variables such as ID3, PTPRC/CD45, AID, APOBEC3G, and ID2 exceed the prognostic impact of some clinicopathological variables. We next extended the signature-/modeling-based knowledge by extracting top genes co-regulated with target molecules in ovarian cancer tissues and dissected potential networks/pathways/regulators contributing to pathomechanisms. We thereby revealed that the AID/APOBEC-related network in ovarian cancer is particularly associated with remodeling/fibrotic pathways, altered immune response, and autoimmune disorders with inflammatory background.
Conclusions
The herein study is, to our knowledge, the first one linking expression of entire AID/APOBECs and interacting genes with clinical outcome with respect to survival of cancer patients. Overall, data propose a novel AID/APOBEC-derived survival model for patient risk assessment and reconstitute mapping to molecular pathways. The established study algorithm can be applied further for any biologically relevant signature and any type of diseased tissue.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-3001-y) contains supplementary material, which is available to authorized users.
Keywords: The AID/APOBEC family, Multigene signature, Primary serous ovarian carcinoma, Multivariable survival models, Prognostic effect, Integrated analysis of disease-relevant pathways
Background
Accumulated knowledge on dysregulated cellular checkpoints associated with cancer development and systematic studies using genomic analysis tools have suggested many new classes of cancer-causing and/or cancer-promoting genes. The discovery of AID/APOBEC gene family members with their potential multifaceted contribution to malignant transformation gave a fundamental impact [1, 2]. In humans, the AID/APOBEC family consists of eleven molecules including AID (activation-induced cytidine deaminase, gene name: AICDA) and APOBECs (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like) with the remarkable ability to edit DNA or RNA through cytosine deamination and thus providing tools to introduce DNA or RNA alterations/damages [3–5]. Under physiological conditions, AID is expressed in activated B cells within germinal centers and responsible for the diversification processes of the immunoglobulin genes by triggering both somatic hypermutation and class switch recombination events [6]. Beside genetic modifications it has been shown that AID may also contribute to epigenetic reprogramming by deaminating methylated cytosine [7]; in conjunction with T:G mismatch repair, this leads to DNA demethylation. The APOBEC3 subfamily (containing seven members) has been implicated in the innate immune defense against endogenous transposable genetic elements, endogenous retroviruses as well as exogenous viruses [8–10] based on the ability to induce DNA damage. In contrast to other family members, APOBEC1 was characterized as RNA-editing enzyme by targeting the ApoB pre-mRNA [1, 11]; later additional mRNA targets have been described [12].
Given that under pathological circumstances AID and APOBECs’ aberrant expression/activity and/or aberrant mechanisms of recruitment to target(s) and/or aberrant processing of the resulting mismatches might take place, their oncogenicity and contribution to the development and/or progression of cancer have been proposed [2, 13–23]. In B-cell malignancies, AID is responsible for DNA damage leading to double-strand DNA breaks followed by translocation of oncogenes [24–28]. In respect of solid tumors, the importance of AID for oncogenesis was strengthened since it became evident that under pathophysiological circumstances including chronic inflammation the AID expression and activity is not restricted to B cells and Ig locus; AID can also mutate non-Ig genes including among others TP53 and the CDKN2b-CDKN2a locus as targets [20, 28–32]. Among the organs of cancerous or inflamed tissues in which ectopic expression of AID was thus far detected in cells of non-B-cell origin are liver, esophagus, lung, stomach, and colon [20, 29, 31, 33–35]. Beside AID, APOBEC2 was recently identified as risk factor in liver and lung tumorigenesis [19]. Importantly, two independent meta-analyses-based studies identified a link between deleterious somatic mutations with cytosine mutation bias in several cancer types and APOBEC expression/enzymatic activities, with one member of the APOBEC3 subfamily, APOBEC3B, being responsible for the majority of cytosine mutations [13, 36]. It was proposed that for breast cancer APOBEC3B may represent a new marker and target [13, 37].
Here we tested the hypothesis that AID and/or other members of the AID/APOBEC family could be part of mechanism(s) contributing to the pathophysiology of ovarian cancer. The rationale behind is enhanced by additional puzzling evidence. Ovarian cancer shows a high degree of genomic instability; practically all classes of mutations, including point mutations and large genomic deletions and insertions, were demonstrated in high-grade serous ovarian cancer in several genes including BRCA1/2 and mutational inactivation of TP53 [38]. AID mRNA expression was shown to be induced by estrogen in an ovarian cancer cell line in vitro [39]. A recent study showed that APOBEC3B overexpression in ovarian cancer correlated with elevated levels of transversion mutations [40]; however, the clinical relevance of these findings still needs to be demonstrated including the potential prognostic relevance. Generally, the overview picture covering the mutual interrelation of all family members and their association with the clinical outcome of ovarian cancer patients is not yet available. Further aspect to consider is that the ovarian cancer cells may express several AID/APOBEC family members acting in a patient-specific manner; yet, the tumor-infiltrating immune cells of various subsets may as well express more than one molecule, each contributing to diverse, not-yet-known pathomechanisms. Thus, the systems-level overview is required. Although ovarian cancer is a heterogeneous malignancy, it is generally treated as a single disease with the use of standard chemotherapy with platinum derivatives and taxanes after surgery. The treatment strategies might undergo a substantial transformation based on the promising novel treatment options under clinical trials [41]. While high response rates to the initial regimen are observed, a relapse is seen in most of the patients due to the rapid development of drug resistance contributing to the overall poor survival characterized by a 5 year overall survival rate of < 40 % [42, 43]. Therefore, algorithms to investigate novel aspects of ovarian cancer pathophysiology aiming to identify novel molecules/pathways suitable to be used as prognostic or predictive biomarkers and/or drug targets and, thus, to provide a personalized approach to clinical decision making are of high priority.
We and others recently showed (examples in [44–47]) that building up of pathway-/disease-relevant signatures provides a persuasive tool for understanding the functional relevance of gene alterations and gene network associations in human diseases and might be taken as basis for prognostic models assessing patient risk/survival. Evidently, interpretation of a single gene expression pattern under diseased conditions might not be sufficient to understand its role in disease pathogenesis; yet, particular genes composing a multigene signature might be reciprocally interconnected within canonical or not-yet-defined disease-relevant pathway(s). We herein aimed to build up a multigene-based model that is eligible as prognostic for patients with advanced stage of serous ovarian carcinoma and to define novel key AID/APOBEC-associated aspects of ovarian cancer. A comprehensive analysis was applied linking multigene signature-based expression profiling of ovarian cancer specimens with statistical modeling followed by systems biology-based data mining and analysis of disease-relevant biological mechanisms. An overview of the analysis steps is outlined in Fig. 1.
Fig. 1.

Overview of the study design: from gene expression profiling-based data sets to prognostic models for clinical outcome and biologically meaningful, disease-associated pathways. The proposed algorithm includes three major blocks. (1) The composition of the AID/APOBEC-associated multigene signature (n = 24) is assembled based on a knowledge-driven approach and applied for the real-time PCR-based gene expression profiling of a clinically well-characterized patient cohort with primary ovarian carcinoma (n = 186). (2) Twenty one profiling-derived variables are correlated with survival data. Univariate Cox regression analysis is applied to assess the prognostic effect of each individual gene and clinical variable. Multivariable Cox regression analysis is applied to build up the survival prognostic models accounting for mutual interconnections between the genes from the signature. Two different multivariable modeling algorithms are used. As outcome, three types of models are created: (i) Clinics – the model is based on the clinicopathological parameters only; (ii) AID/APOBEC – the model is based on the multigene profiling-derived data sets; and (iii) Combined – the model is based on the clinicopathological and gene profiling-derived variables in combination. In both algorithms the standardized coefficients (STDBETA) are used for ranking the individual variables in a model by their importance. The top-ranked genes are defined as target genes for the follow-up analyses. Important to note, parameters such as proportion of explained variation (PEV), c-index and p-value are calculated and used to compare the predictive accuracy and discriminative ability of the individual models. Alignment with patients’ survival data is illustrated by Kaplan-Meier estimates showing patient stratification into low, intermediate, and high risk groups. (3) Systems biology approach is used to assign the defined target genes with prognostic impact to disease-relevant biological pathways. Firstly, the web-based analysis platform for publically available microarray datasets (GENEVESTIGATOR) is used to extract the top genes co-regulated with the target genes in ovarian cancer tissues based on inclusion criteria specified in Methods. Secondly, the obtained gene lists are subjected to the Ingenuity-based core analysis. As input, in addition to the individual lists of co-regulated genes, the combined list (“mixed”) is used to mimic the mutual interconnections within the multigene signature. The core analysis includes alignment with Canonical Pathways, Functional Annotations & Diseases and Upstream Regulators. Thirdly, Spotfire, a data discovery and visualization software, is used for large-scale IPA-derived data processing and data mining. As final outcome, the 10-top Pathways/Functions/Regulators are defined
Methods
Profile of study patients
Tumor samples of epithelial ovarian cancer (EOC) were collected in the course of the European Commission’s sixth framework program project OVCAD from five European university hospitals (Ovarian Cancer: Diagnosis of a silent killer; grant agreement no. 018698) [48]. Information on clinicopathological characteristics was documented by experienced clinicians. The clinicopathological characteristics of the 186 patients with primary EOC are summarized in Table 1; the patient group is a part of the patient cohort under study of the OVCAD consortium [49, 50]. Patient inclusion criterion comprises the epithelial ovarian cancer with advanced disease (FIGO II – IV); the majority of patients had advanced-stage ovarian cancer (FIGO III and IV, 95 %), G3 tumors (74 %), and the majority of tumors was of serous histology (88 %). 71 % of patients could be optimally cytoreduced with no residual disease after initial surgery; absence of residual disease was defined as macroscopically complete resection of tumor material. All patients received standard adjuvant chemotherapy including platinum-based anti-cancer agents. Eight percent of patients received neoadjuvant chemotherapy. Patients with recurrence or progressive disease until 6 months after the end of chemotherapy were defined as chemotherapy resistant. The median age at diagnosis was 57 years (range, 26 to 85 years); the median follow-up time was 30.0 months (95 % CI: 27.4-32.6). There were 54 cases (29 %) of death related to EOC reported during the follow-up period, designated as events below.
Table 1.
Clinicopathological characteristics of study patients
| Patients (%) | 186 | (100.0) |
|---|---|---|
| Age at diagnosis [years] | ||
| Median (range) | 57 | (26–85) |
| Progression-free survival [months] | ||
| Median (range) | 16 | (1–48) |
| Number of recurrencies (%) | 106 | (57.0) |
| Overall survival [months] | ||
| Median (range) | 24 | (1–49) |
| Number of deaths (%) | 54 | (29.0) |
| Histology (%) | ||
| Serous | 164 | (88.2) |
| Non-serous | 22 | (11.8) |
| FIGO (%) | ||
| II | 9 | (4.8) |
| III | 148 | (79.6) |
| IV | 29 | (15.6) |
| Grading (%) | ||
| 1 | 6 | (3.2) |
| 2 | 43 | (23.1) |
| 3 | 137 | (73.7) |
| Residual disease after initial surgery (%) | ||
| None | 132 | (71.0) |
| ≤ 1 cm | 34 | (18.3) |
| > 1 cm | 20 | (10.8) |
| Type of chemotherapy (%, 30 missing [16.1 %]) | ||
| Adjuvant | 124 | (66.7) |
| Neoadjuvant | 15 | (8.1) |
| Intraperitoneal | 17 | (9.1) |
| Response to first-line chemotherapy (%, 1 missing) | ||
| Responder | 138 | (74.2) |
| Non-Responder | 47 | (25.3) |
Cell lines
The human ovarian carcinoma cell lines A2780 and A2780ADR were obtained from the European Collection of Cell Cultures (Salisbury, Wiltshire, United Kingdom). A2780 is the parent line to the adriamycin resistant A2780ADR. Although adriamycin is not a therapy regimen for ovarian cancer, but considering that A2780 cell model is featured by high chemosensitivity to cisplatin, while A2780ADR cell line exhibits a collateral resistance to cisplatin, both cell lines are often used as in vitro models to study the acquisition of drug resistance [51, 52]. The human ovarian carcinoma cell lines OVCAR-3 and SK-OV-3 were obtained from the ATCC (Manassas, VA). The cell lines were maintained in phenol-red-free RPMI-1640 medium supplemented with L-glutamine (PAN-Biotech GmbH, Aidenbach, Germany), 10 % Foetal Calf Serum (FCS) (Invitrogen, Carlsbad, CA) and 1 % penicillin (10,000 U/ml)/streptomycin (10 mg/ml) solution (Invitrogen) in a humidified atmosphere at 37 °C with 5 % CO2.
RNA isolation from tumor tissues and ovarian cancer cell lines
Total RNA from tissues was isolated using the ABI 1600 nucleic acid prepstation (Applied Biosystems, Foster City, CA, USA) following the instructions of the manufacturer as described previously [49]. Total RNA from A2780, A2780ADR, OVCAR-3 and SK-OV-3 cell lines was isolated using the RNeasy Mini kit (Qiagen, Hilden, Germany) including DNase I treatment. The concentration, purity and integrity of RNA samples were determined on a Nanodrop ND-1000 (Kisker-Biotech, Steinfurt, Germany) and agarose gel electrophoresis.
Real-time PCR analysis
0.5 μg of total RNA from tissue specimens and 1 μg of total RNA from the cell lines was reverse transcribed using the High Capacity cDNA RT kit (Applied Biosystems) according to the instructions of the manufacturer. Given the patient-specific composition of various cell types within ovarian cancer tissues, for accurate normalization of mRNA between ovarian cancer tissue specimens we selected ACTB, TOP1, UBC, and YWHAZ out of a panel of 12 housekeeping genes (HKGs) as appropriate reference genes using a geNorm kit (PrimerDesign Ltd., Southampton, UK) and the geNorm software [53]. For the cancer cell lines, EEF1A1 and UBC were used as appropriate reference HKGs as estimated by DataAssist software (Applied Biosystems). Primers for genes of interest composing the AID/APOBEC-associated multigene signature were designed using Primer Express 3.0 software (Applied Biosystems) and validated using a normal tissue panel (Takara, Clontech Laboratories Inc., Mountain View, USA) as previously described [45]. Primer sequences are displayed in Additional file 1: Table S1. The assay for ACTB was from Applied Biosystems; assays for the reference HKGs TOP1, UBC, and YWHAZ were purchased from PrimerDesign Ltd; primers for ESR1 and ESR2 were purchased from Applied Biosystems (Additional file 1: Table S2).
Real-time PCR analysis was performed on ABI 7900HT instrument equipped with SDS 2.3 software (Applied Biosystems) in the 384-well plate format using POWER SYBR Green Master Mix (Applied Biosystems) or, in case hydrolysis probe assays were used, Gene Expression Master Mix (Applied Biosystems). The qPCR Human Reference Total RNA (Clontech Laboratories Inc.) was assigned as calibrator sample to which gene expression levels of all other samples are compared. Subsequently, raw Ct values were exported into Microsoft Excel and results were calculated using the ΔΔCt method [54] as relative quantities (RQ) normalized to the geometric mean of the four or of the two HKGs specified above for ovarian cancer tissues and cells lines, respectively, and shown relative to the calibrator sample.
The composition of the AID/APOBEC-associated multigene signature used for profiling of the patient cohort and the cell lines is specified in Results.
Statistical analysis
Profiling-derived values were log2 transformed for Cox regression models to avoid disproportional impact of outliers. Missing values were imputed using the R package mice [55]. Correlation coefficients were calculated by Pearson's correlation for log2 transformed values using SPSS. Hazard ratios and corresponding 95 % confidence intervals were estimated by univariate Cox regression analysis for both the clinicopathological variables and the gene profiling-based variables using the IBM SPSS statistical package (version 20.0; SPSS Inc., an IBM company, Chicago, USA). Regularized multivariable Cox regression was applied to develop prognostic models using two types of regularization as specified below. Calculations were performed with the R (R Foundation for Statistical Computing, Vienna, Austria) package glmnet [56]. In the first approach, Cox regression with a ridge penalty (ridge) was used for estimating multivariable models. In the second approach, models were generated by simultaneous parameter shrinkage and variable selection using the LASSO (L1-norm penalization). Both approaches introduce a penalty to the likelihood function in order to reduce the inflation of variance of the predictions (overfit) caused by a critical ratio of number of outcome events and number of variables. While by the ridge penalty all variables will enter the final model but with severely shrunken regression coefficients, the LASSO penalty selects only some of the variables for the final model, and assigns regression coefficients of 0 to all other variables. The tuning parameter lambda of the ridge and the LASSO penalties were optimized by minimizing the Cox model’s partial deviance in a leave-one-out cross-validation procedure. An additional leave-one-out cross-validation loop was wrapped around the model development process to obtain cross-validated predictors for each patient. Here, the model was re-estimated N times each time omitting one patient in turn, and the cross-validated predictor for that patient was computed as the vector product of the re-estimated regression coefficients and the patient’s corresponding gene expression values. Global p-values for each model were calculated in SPSS using univariate Cox proportional hazards analysis with the corresponding cross-validated predictors of the model as single covariate. Overall survival (OS) and progression-free survival (PFS) were shown by Kaplan-Meier graphs, stratified by quantiles of the cross-validated linear predictors, and accompanied by corresponding log-rank test p-values. Using the cross-validated predictors, we also assessed the discriminative ability of the model by determining the concordance index (c-index) [57] and its proportion of explained variation (PEV) [58]. The c-index is a discrimination measure and describes, as an average measure over all possible pairs of patients, the concordance of survival times and linear predictors derived from the model. The measure is adjusted by inverse probability weighting techniques to accommodate censored survival times. PEV describes the relative gains in predictive accuracy of the survival status at any time point during follow-up when prediction based on covariates replaces unconditional prediction. Absolute values of standardized regression coefficients (STDBETA or ,) were used for comparing and ranking the variables by their importance in prediction. The standardized regression coefficient of a variable Xj is the natural logarithm of the hazard ratio between two patients who differ in Xj by 1 SD (ceteris paribus), and can be calculated as SD(Xj). Standardized coefficients were then visually compared by depicting Ŝ36 and , which are the average 36 months overall survival rate and the estimated 36 months overall survival probability in a subject whose value of Xj differs from the mean of Xj by 1 SD, respectively.
To further estimate the predictive accuracy of the above-described modeling algorithm, we performed the same analyses on the basis of 21 pseudo genes which were obtained by permuting the full block of the original AID/APOBEC-associated 21-gene data set, preserving the distributions and correlation structure within those genes. As outcome, no models could be built for pseudo AID/APOBEC using both ridge and LASSO penalties with respect to OS and PFS; when combined with clinicopathological variables, pseudo AID/APOBEC did not improve the predictive accuracy and discrimination ability of clinicopathological variables. This provides evidence that the applied modeling algorithm is robust against falsely identifying any relevance of randomly selected gene sets.
Correlation coefficients were calculated by Pearson's correlation for log2 transformed values using SPSS; Bonferroni-Holm method was used as multiple-testing correction. P-values ≤ 0.05 were considered as indicating statistical significance.
Group differences were assessed by two-way analysis of variance (ANOVA) and Tukey's post hoc test.
Expression profiling of signature-associated genes in ovarian cancer cell lines using the published microarray-based data sets
We examined the expression profiles of genes comprising the AID/APOBEC signature across previously published microarray data sets using the GENEVESTIGATOR platform. GENEVESTIGATOR is a manually curated web-based analysis platform for publicly available transcriptomic data sets [59, 60]. For analysis, we selected data sets from the Affymetrix Human Genome U133 Plus 2.0 Array platform; out of a total of 54037 arrays, we selected data attributed to ovarian cancer cell lines applying the filter “Cell Lines_Pathological Cell Lines_Neoplastic Cell Lines_Ovary_All”; this selection included 149 arrays. The expression values (log2 transformed) were exported from GENEVESTIGATOR for follow-up clustering and statistical analyses. Clustering analysis and follow-up graphical representation was performed using Cluster 3.0 and Java TreeView programs.
Analysis of signature-associated, co-expressed genes using the published microarray-based data sets
For the in silico identification of genes showing co-regulation with the top candidate genes ranked within the combined model (ridge) by maximal impact to the prognostic effect (our heuristic solution is to use the cutpoint at STDBETA ≥ |0.15|), we used the GENEVESTIGATOR search engine. The top candidate genes subjected to GENEVESTIGATOR-based analysis were designated in the text below as target genes. Members of the APOBEC3 subfamily exhibit high sequence homologies. We checked the specificity of Affymetrix probes covering the APOBEC3 members using BLAST and ensured that the eligible Affymetrix probe set (ID 204205_at) for APOBEC3G is highly specific, whereas the probe set 214995_s_at is cross-reactive with APOBEC3F and the probe set 215579_at does not recognize APOBEC3G. This is in line with previously made conclusions [13]. The specific APOBEC3G probe set was used in GENEVESTIGATOR-based analysis. For the GENEVESTIGATOR-based analysis the following inclusion criteria were applied: (i) we selected data from the Affymetrix Human Genome U133 Plus 2.0 Array platform; (ii) from a total of 709 arrays of EOC, only those with annotated FIGO stage (I-IV) were selected (n = 538); (iii) only those target genes were subjected for analysis which showed detectable microarray expression based on the normalized signal intensity in ovarian carcinoma tissues; (iv) analysis was restricted to samples with lowest (10th percentile as threshold) and highest (90th percentile as threshold) target gene expression levels (n = 106 selected, Additional file 1: Figure S1); the applied sample selection strategy leads to the exclusion of those genes which show co-expression with the target gene purely based on the non-modulated expression patterns. Additionally, to ensure that changes in target gene expression across the pre-selected tumor samples within both groups are caused by intrinsic gene regulation and not by potential differences in sample quality, a correlation analysis was done between 45 HKGs demonstrating high homogeneity with correlation coefficients > 0.99 (Additional file 1: Figure S2). Next, the lists of the top 50 co-expressed probe sets for each target gene, ranked according to the Pearson correlation coefficient, were exported for further analysis; a combined gene list has been created covering the co-expressed genes of all individual target probe sets (named as mixed list below). The content of the mixed list thus reflects the combined input of the multigene signature accounting for/mimicking the mutual interconnections between the individual genes. The Ingenuity Pathway Analysis (IPA) tool was used to assign the co-regulated genes to common biological pathways, biological functions and/or diseases as well as upstream regulating molecules [61]. The IPA Core analysis included the following categories: (i) Canonical Pathways, (ii) Functional Annotations, and (iii) Upstream Regulators. The significance of the association between each gene list and a canonical pathway was measured by right-tailed Fisher’s exact test. As a result, a p-value was obtained, determining the probability that the association between the genes from our data set and a Canonical Pathway/Functional Category/Upstream Regulator can be explained by chance alone. The top ranking was based on the p-value. Only significant outcomes (p < 0.05) were taken for follow-up analyses. For alignment of the IPA-derived large-scale data sets, data mining and data visualization the Spotfire software was used [62]. Given the complexity of the follow-up analyses, various approaches were applied. We present herein two algorithms. (I) The top 10 output results (herein designated as the 10-top-output_mixed) were ranked by the corresponding IPA-derived p-values using the mixed list as input data. Subsequently, the position of each 10-top-output_mixed candidate was assessed within the individual target-associated gene list (output_individual). Of particular interest were those which appeared in both the 10-top-output_mixed and at least one of the 10-top-output_individual. (II) The output_mixed results were aligned with the output_individual results searching for the strongest overlap and meaning the mandatory presence in output_mixed and the maximal number of the output_individual (e.g. 5 out of 5 > 4 out of 5 > 3 out of 5; named as output_overlap). Subsequently, the 10-top-output_overlap was ranked by the IPA-derived p-values of the output_mixed results. The unweighted pair group method with arithmetic mean was used as clustering method with Euclidean distance measure and average value as ordering weight.
Study approval
The study was approved in accordance to the requirements of the ethical committees of the individual institutions participating in OVCAD (EK207/2003, ML2524, HEK190504, EK366, EK260). Informed consent for the scientific use of biological material was obtained from all patients in accordance with the requirements of the ethics committees of the institutions involved; the herein participating OVCAD partners include Department of Gynecology, European Competence Center for Ovarian Cancer at Campus Virchow Klinikum, Charité – Medical University Berlin (Berlin, Germany), Division of Gynecological Oncology, Department of Obstetrics and Gynecology, Universitaire Ziekenhuizen Leuven, Katholieke Universiteit Leuven (Leuven, Belgium), Department of Obstetrics and Gynecology, Medical University of Vienna (Vienna, Austria), and Department of Gynecology, University Medical Center Hamburg - Eppendorf (Hamburg, Germany).
Results
A multigene signature approach to assess the patient-specific transcriptional profiles in the context of the clinical relevance
The selection of genes composing the multigene signature is knowledge- and biology-driven. Our expert-designed gene signature is thereby influenced by previous published work and disease relevance, and in this sense is biased towards previous knowledge; importantly, this selection is not based on pre-tested prognostic impact in our study cohort and thereby does not lead to a real bias. This approach represents relatively new way of addressing the pathophysiological relevance of transcriptional profiles and methodologically has indisputable advantage of the real-time PCR-based analysis that ultimately provides results which do not need further methodological validation. It is an appropriate strategy of choice for low-level expressed genes and for genes with high sequence similarity which is truly relevant for AID and APOBEC3 subfamily, respectively. Furthermore, considering the complex cellular composition of ovarian cancer tissue and the current limited knowledge linking the cell type-specific expression patterns of AID and APOBECs with their functionality under diseased conditions, we included all members of the AID/APOBEC family, regardless of other potential ways of their regulation besides those on the transcriptional levels.
The applied gene signature includes the entire AID/APOBEC family consisting of AID (AICDA), APOBEC1, APOBEC2, APOBEC3A, APOBEC3B, APOBEC3C, APOBEC3D, APOBEC3F, APOBEC3G, APOBEC3H, and APOBEC4; PTPRC (also known as CD45), PAX5 (also known as BSAP), CD23, NUGGC (also known as SLIP-GC), and PRDM1 (also known as BLIMP1), ID2 and ID3 were added accounting for ovarian cancer tissue infiltrating immune cells, B-cell biology and transcriptional control of AID, respectively [17, 63, 64]; the estrogen receptors ESR1 and ESR2 were included given the hormone-dependent nature of the analyzed tumor type and the potential involvement of estrogen in AID regulation [39]; DPPA3 (also known as STELLA) and NANOG are pluripotency-associated genes whose expression was shown to be linked to the AID functional activity [7, 65]; XRCC5 (also known as KU80) and XRCC6 (also known as KU70) are involved in DNA repair mechanism downstream of AID [66]. In sum, the gene panel includes B-cell identity markers, AID/APOBEC family members, genes involved in their regulation, and their functional co-factors or target genes (n = 24). Gene names, Gene ID, short functional description from NCBI, synonyms and accession numbers are provided in Additional file 1: Table S3.
Univariate associations of the individual gene expression-derived variables and the clinicopathological parameters with overall and progression free survival
To determine the clinical relevance of the gene expression data sets of each individual gene of the signature and of the clinicopathological parameters, we first used the classical Cox regression analysis strategy whereby the values were aligned with OS and PFS. Two genes showed statistically significant associations with OS, namely AID (HR = 1.18, 95 % CI: 1.04–1.33, p = 0.008) and ID3 (HR = 1.36, 95 % CI: 1.12–1.64, p = 0.002), as estimated by univariate Cox regression analysis and summarized in Additional file 1: Table S4. Among clinical risk factors, five variables were associated with OS, namely age (HR = 1.03, 95 % CI: 1.01–1.06, p = 0.011), FIGO stage (HR = 1.83, 95 % CI: 1.02–3.29, p = 0.044), grading (HR = 2.10, 95 % CI: 1.03–4.31, p = 0.042), peritoneal carcinomatosis (HR = 4.17, 95 % CI: 1.78–9.78, p = 0.001), and residual disease (HR = 1.98, 95 % CI: 1.15–3.44, p = 0.014; Additional file 1: Table S4). With respect to PFS, no significant associations with the gene expression-derived variables were observed (Additional file 1: Table S4). Three clinical variables showed a strong association with PFS: FIGO stage (HR = 2.33, 95 % CI: 1.53–3.54, p < 0.001), peritoneal carcinomatosis (HR = 3.88, 95 % CI: 2.27–6.61, p < 0.001) and residual disease (HR = 1.95, 95 % CI: 1.29–2.94, p = 0.002). APOBEC1, APOBEC2 and DPPA3 mRNAs were not expressed or expressed at the detection limit in the ovarian cancer tissues of the herein investigated cohort of patients and therefore those variables were excluded from the univariate Cox regression and all subsequent analyses. Thus, the final number of the gene profiling-derived variables included to the follow-up analyses was equal to n = 21.
Of note, the follow-up statistical models using Cox regression with ridge and LASSO penalties are not based on the pre-selection of variables according to their significance estimated by univariate Cox regression analysis.
We further performed a correlation analysis for profiling-derived variables. As summarized in Additional file 1: Table S5 a strong correlation (r > 0.6; p < 0.001) was found between PTPRC/CD45 and APOBEC3 family members such as A3C, A3D, A3G, and A3H as well as NUGGC and PRDM1; between APOBEC3D and both APOBEC3G and APOBEC3H, XRCC6/Ku70, NUGGC, and PRDM1; between ID2 and ID3; between XRCC6/Ku70 and XRCC5/Ku80 as well as APOBEC3D, ID2 and ID3; between PAX5 and FCER2; between NANOG and XRCC6/Ku70 and XRCC5/Ku80; between PRDM1 and ID3, XRCC6/Ku70, XRCC5/Ku80, and NUGGC. In respect of AID, the strongest correlation was found with PTPRC/CD45 (r = 0.521; p < 0.001). ESR1 and ESR2 did not show significant correlation with any other gene of the multigene signature.
Prognostic models for OS and PFS
Using Cox regression with ridge and LASSO penalties, we developed multivariable models for evaluating patient prognosis and for stratifying patients into risk groups. Calculations were done for three sets of explanatory variables: (i) using the six clinicopathological variables (Clinics), (ii) using the gene profiling-derived AID/APOBEC variables (AID/APOBEC), and (iii) combining clinical and multigene-derived variables (Combined). The results for the multivariable ridge models are summarized in Tables 2 and 3 and Additional file 1: Table S6. The clinicopathological variables-based model predicts both OS (PEV = 8.8 %, c-index = 0.69, p < 0.001) and PFS (PEV = 17.0 %, c-index = 0.68, p < 0.001). The AID/APOBEC model showed moderate predictive accuracy and discrimination for OS (PEV = 2.5 %, c-index = 0.59, p = 0.025). The combined model had the highest predictive accuracy with respect to OS (PEV = 11.1 %, c-index = 0.7, p < 0.001). According to their standardized regression coefficients, the following five genes proved most important for prediction within the AID/APOBEC model: ID3, AID, APOBE3G, PTPRC/CD45, and ESR1 (Table 3). Among the covariates within the combined model, ID3 emerged as the prognostically most important variable for OS, exceeding the clinical risk factors such as peritoneal carcinomatosis or age. Together with ID3, PTPRC/CD45, AID, and APOBEC3G showed high importance for survival prediction standing ahead of the clinical risk factors such as grading and histology (Table 3, Fig. 2).
Table 2.
Comparative analysis of multivariable models (ridge) for prognostication of OS and PFS
| OS | PFS | |||||
|---|---|---|---|---|---|---|
| PEV % | c-index | P | PEV % | c-index | P | |
| Clinics | 8.77 | 0.69 | <0.001 | 17.0 | 0.68 | <0.001 |
| AID/APOBEC | 2.53 | 0.59 | 0.025 | n.a. | n.a. | n.a. |
| Combined | 11.13 | 0.70 | <0.001; 0.021* | 13.6 | 0.65 | <0.001; 0.320* |
Cross-validated performance assessment of Cox regression models (ridge) by proportion of explained variation (PEV), concordance index (c-index) and p-value. Models were developed using (i) clinicopathological variables designated as Clinics; (ii) AID/APOBEC multigene-based variables designated as AID/APOBEC; (iii) their combination designated as Combined
* p-value for added value of AID/APOBEC on top of Clinics in bivariable models with cross-validated predictors
Table 3.
Relative importance of individual variables in multivariable models (ridge) for OS
| Clinics | Combined | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Pos. | Variables | beta | HR | STDBETA | Pos. | Variables | beta | HR | STDBETA |
| 1. | Peritoneal carcinomatosis | 0.83 | 2.30 | 0.38 | 1. | ID3 | 0.30 | 1.35 | 0.52 |
| 2. | Age | 0.02 | 1.02 | 0.27 | 2. | Peritoneal carcinomatosis | 1.05 | 2.86 | 0.48 |
| 3. | Histology | 0.58 | 1.79 | 0.19 | 3. | Age | 0.03 | 1.03 | 0.40 |
| 4. | FIGO stage | 0.48 | 1.62 | 0.18 | 4. | PTPRC (CD45) | 0.19 | 1.21 | 0.34 |
| 5. | Residual disease | 0.37 | 1.45 | 0.17 | 5. | AICDA | 0.14 | 1.15 | 0.33 |
| 6. | Grading | 0.34 | 1.41 | 0.15 | 6. | APOBEC3G | −0.19 | 0.83 | −0.31 |
| AID/APOBEC | 7. | Grading | 0.53 | 1.70 | 0.23 | ||||
| Pos. | Variables | beta | HR | STDBETA | 8. | Histology | 0.67 | 1.96 | 0.22 |
| 1. | ID3 | 0.18 | 1.19 | 0.30 | 9. | ID2 | −0.12 | 0.89 | −0.22 |
| 2. | AICDA | 0.09 | 1.09 | 0.21 | 10. | FIGO stage | 0.50 | 1.64 | 0.18 |
| 3. | APOBEC3G | −0.11 | 0.89 | −0.19 | 11. | Residual disease | 0.36 | 1.44 | 0.17 |
| 4. | PTPRC (CD45) | 0.08 | 1.08 | 0.14 | 12. | NUGGC | −0.08 | 0.92 | −0.16 |
| 5. | ESR1 | −0.06 | 0.94 | −0.11 | 13. | PAX5 | −0.06 | 0.94 | −0.16 |
| 6. | PRDM1 | 0.06 | 1.06 | 0.10 | 14. | ESR1 | −0.08 | 0.92 | −0.16 |
| 7. | APOBEC3B | 0.05 | 1.05 | 0.09 | 15. | ESR2 | 0.04 | 1.04 | 0.14 |
| 8. | XRCC5 (Ku80) | −0.06 | 0.95 | −0.07 | 16. | APOBEC3A | −0.07 | 0.93 | −0.14 |
| 9. | APOBEC3A | −0.03 | 0.97 | −0.07 | 17. | APOBEC3C | −0.10 | 0.91 | −0.11 |
| 10. | APOBEC3C | −0.06 | 0.95 | −0.06 | 18. | APOBEC3D | 0.06 | 1.06 | 0.09 |
| 11. | PAX5 | −0.02 | 0.98 | −0.06 | 19. | APOBEC3F | 0.06 | 1.06 | 0.07 |
| 12. | ID2 | −0.03 | 0.97 | −0.05 | 20. | XRCC6 (Ku70) | 0.05 | 1.05 | 0.06 |
| 13. | XRCC6 (Ku70) | 0.04 | 1.04 | 0.05 | 21. | NANOG | 0.04 | 1.04 | 0.06 |
| 14. | FCER2 | −0.02 | 0.98 | −0.05 | 22. | APOBEC3H | 0.03 | 1.03 | 0.06 |
| 15. | APOBEC3F | 0.03 | 1.03 | 0.03 | 23. | PRDM1 | 0.03 | 1.03 | 0.04 |
| 16. | ESR2 | 0.01 | 1.01 | 0.03 | 24. | APOBEC3B | 0.02 | 1.02 | 0.03 |
| 17. | APOBEC3H | −0.01 | 0.99 | −0.01 | 25. | APOBEC4 | 0.01 | 1.01 | 0.02 |
| 18. | APOBEC4 | 0.00 | 1.00 | 0.01 | 26. | XRCC5 (Ku80) | −0.02 | 0.98 | −0.02 |
| 19. | APOBEC3D | 0.00 | 1.00 | −0.01 | 27. | FCER2 | 0.00 | 1.00 | 0.00 |
| 20. | NUGGC | 0.00 | 1.00 | −0.01 | |||||
| 21. | NANOG | 0.00 | 1.00 | −0.01 | |||||
Within each model, variables are ranked by descending importance as expressed by their absolute standardized regression coefficients. Histology was encoded as “0” for serous and “1” for non-serous; Peritoneal carcinomatosis was encoded as “0” for no and “1” for yes; Grading was encoded as “0” for Grade 1 and 2 and “1” for Grade 3; Residual disease was encoded as “0” for no and “1” for yes; beta, regression coefficient (log hazard ratio); HR, hazard ratio; STDBETA, standardized regression coefficients. By multivariate modeling with penalized likelihood, the multivariate-adjusted HR shows the direction and magnitude of prognostic effect if adjusted for other variables. Ridge regression shifts the HR towards the value of 1.0 to avoid overestimation bias and to decrease variance in models with many variables
Fig. 2.
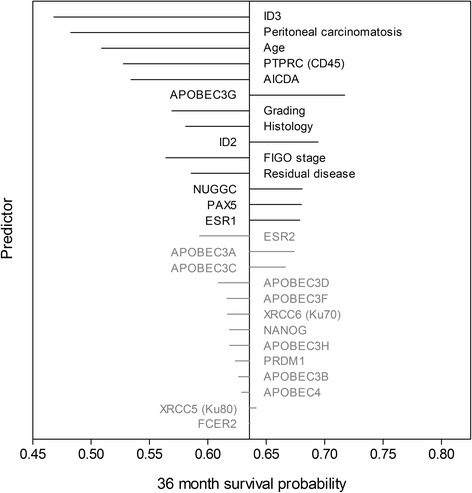
Impact of individual variables on survival prediction of the multivariable model (ridge) for OS if predictors are changed by +1 SD. Survival probabilities are estimated at 36 months of follow-up time. The length of the lines is proportional to the change in prediction in case the value of the indicated variable changes by +1 SD (left: negative effect; right: positive effect). Variables are ranked according to the absolute changes in prediction which also corresponds to the order within the combined model (Table 3). Gene profiling variables not used for follow-up analyses are displayed in grey color
Patients were sub-divided into low, intermediate, and high risk groups according to their cross-validated predictors for OS based on the ridge model. Figure 3 shows the corresponding Kaplan-Meier graphs. The combined model showed statistically significant differences between the risk groups (log-rank test: p < 0.001) giving major improvement of patient stratification. With respect to PFS, the combination of gene expression data sets with the clinical variables did not result in improved patient stratification in comparison to the clinicopathological variables-based model (Fig. 3). Importantly, the results of the second regularization method (LASSO) were in line with those described above and indicated similar predictive abilities with respect to OS by the clinicopathological variables-based model (PEV = 7.54 %, c-index = 0.67, p < 0.001), by the AID/APOBEC multigene-based model (PEV = 5.76 %, c-index = 0.64, p < 0.001), and superior performance of the combined model (PEV = 10.80 %, c-index = 0.70, p < 0.001) (Additional file 1: Tables S7-S9, Additional file 1: Figure S3). The corresponding Kaplan-Meier graphs for OS and PFS using the LASSO penalization are shown in Additional file 1: Figure S4.
Fig. 3.
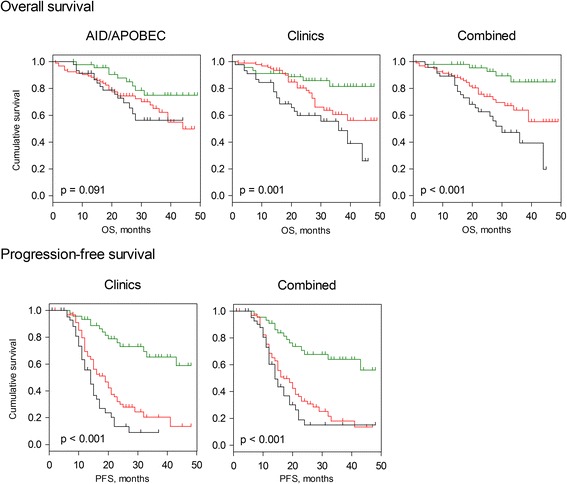
Kaplan-Meier estimates for patient stratification based on the AID/APOBEC model, the clinical model, and the combined one (ridge-based). Kaplan-Meier curves for OS and PFS are shown giving patients’ stratification into low risk (n = 46, green), intermediate risk (n = 94, red), and high risk (n = 46, black) groups with the 25th and 75th percentiles serving as thresholds (lower than the 25th percentile indicates low risk). No stable and well calibrated model for AID/APOBEC was found in respect of PFS, thus only models for Clinics and Combined are shown. P-value of the log-rank test is indicated
Expression of genes from the AID/APOBEC multigene signature in ovarian cancer cell lines
Given the complex cellular composition of the ovarian cancer tissues we assessed the mRNA expression of genes composing the AID/APOBEC multigene signature by real-time PCR in four ovarian cancer cell lines such as A2780, A2780ADR, OVCAR-3, and SK-OV-3. Expression profiling revealed that the majority of genes were found to be expressed in at least one of the four examined cell lines (Additional file 1: Figure S5); of note, among those are the top genes, which showed the highest impact to the prognostic power of the multivariable model.
We next expanded the scope of expression analysis to a wider range of ovarian cancer cell lines (n = 55) across previously published microarray data sets using the GENEVESTIGATOR platform (Additional file 1: Figure S6). The analyzed set of cell lines included, among others, the ovarian cancer cell lines, which had been previously ranked and sub-grouped by their suitability as high-grade serous ovarian cancer tumors on the basis of genomic profiling [67]. With the exception of genes exhibiting low expression and/or expression at the microarray detection limit (AICDA, APOBEC1, APOBEC3A, PAX5, and PTPRC), the genes composing the AID/APOBEC multigene signature were expressed in various cell lines representing the above mentioned sub-groups. We next applied a hierarchical cluster analysis on the basis of expression values of the signature genes across the analyzed cell lines. The resulting two main clusters for ovarian cancer cell lines accentuate the expression differences for APOBEC3B, APOBEC3C, and APOBEC3G as well as of ID2 and ID3 (Additional file 1: Figure S7).
Systems biology approach linking the gene expression data sets with the disease-relevant biological pathways and functions
To define the AID/APOBEC-attributed biological pathways potentially associated with the pathogenesis of ovarian cancer, a systems biology approach was applied (described in detail in Fig. 1 of study design and in Methods). The eligible genes from the profiling-derived candidate genes with the highest impact to the combined prognostic model include AID, APOBEC3G, ESR1, ID2, ID3, NUGGC, PAX5, and PTPRC/CD45. However, 3 genes were excluded such as NUGGC, since no corresponding probesets do exist on the U133 Plus 2.0 Array, and AID and PAX5 due to the low mRNA expression levels in ovarian cancer tissue when detected by microarray. Thus, APOBEC3G, ESR1, ID2, ID3, and PTPRC/CD45 were used as target genes for follow-up analyses. Next, for each target gene we assessed the co-expressed genes in ovarian cancer tissues by GENEVESTIGATOR. The exported gene lists are summarized in Additional file 1: Tables S10-S14. To assign the co-regulated genes to common biological pathways, biological functions and/or diseases as well as upstream regulating molecules the Ingenuity Pathway Analysis (IPA) tool was used.
Data-driven signature-associated Canonical pathways
Results of the follow-up IPA-based core analysis in respect of the Canonical Pathways are listed in Table 4 (based on the algorithm I, where the priority is given to the outcome_mixed as specified in Methods). The Canonical Pathway designated as Hepatic Fibrosis/Hepatic Stellate Cell Activation was ranked to position 1. Of note, 8 out of the top 10 canonical pathways derived from the output_mixed were found in at least one output_individual within the 10-top positions (with p-values all < 0.05; Table 4). The strongest overlap between the 10-top-output pathways of mixed and individual was observed for APOBEC3G (5/10) and PTPRC/CD45 (5/10) indicating the strongest contribution from those two genes; in contrast, no overlap was found for ESR1. Similarly to the algorithm I-derived results, Hepatic Fibrosis/Hepatic Stellate Cell Activation pathway was as well ranked to position 1 when the algorithm II was applied, where the priority is given to the overlap between the output_mixed and output_individual (Additional file 1: Table S15). The results of overall alignment illustrated by the heat map (Additional file 1: Figure S8, A) and by pie chart (Additional file 1: Figure S9, A) indicate that the Canonical Pathways attributed to the individual target genes are mostly diverse among each other but show a strong overlap with the mixed-attributed Canonical Pathways. To align the results of herein pathway analysis with the related studies, we additionally included the references to the published studies in the area of ovarian carcinoma and others (Table 4 and Additional file 1: Table S15).
Table 4.
The 10-top-AID/APOBEC signature-linked Canonical Pathways identified by systems biology approach (algorithm I)
| Canonical 10-top-output_mixeda | Canonical output_individualb | Related studiesc | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| APOBEC3G | ESR1 | ID2 | ID3 | PTPRC (CD45) | |||||||||||
| Pos. | Canonical pathways | P-value | Molecules | Pos. | Molecules | Pos. | Molecules | Pos. | Molecules | Pos. | Molecules | Pos. | Molecules | Ovarian Cancer | Others |
| 1 | Hepatic Fibrosis / Hepatic Stellate Cell Activation | 8.32E-08 | COL1A1, IGFBP4, CCR5, FN1, CTGF, TIMP1, ACTA2, IL10RA, CCL5, FAS, EGFR | 1 | COL1A1, IGFBP4, CTGF, TIMP1, ACTA2, FAS, EGFR | 5 | IGFBP4, FN1 | 14 | CCR5, IL10RA, CCL5 | [79, 80, 98] | [81] | ||||
| 2 | Altered T Cell and B Cell Signaling in Rheumatoid Arthritis | 1.51E-06 | HLA-DOA, IL15, TLR8, FCER1G, CD86, TLR3, TNFSF13B, FAS | 3 | HLA-DOA, IL15, TLR3, TNFSF13B | 6 | TLR8, FCER1G, CD86 | [83] | |||||||
| 3 | Antigen Presentation Pathway | 2.40E-05 | PSMB9, HLA-DOA, CIITA, HLA-DPB1, HLA-DPA1 | 1 | PSMB9, HLA-DOA, CIITA, HLA-DPB1, HLA-DPA1 | [82, 99] | |||||||||
| 4 | Communication between Innate and Adaptive Immune Cells | 2.45E-05 | IL15, TLR8, FCER1G, CD86, CCL5, TLR3, TNFSF13B | 4 | IL15, CCL5, TLR3, TNFSF13B | 1 | TLR8, FCER1G, CD86, CCL5 | [100] | |||||||
| 5 | Role of Pattern Recognition Receptors in Recognition of Bacteria and Viruses | 3.47E-05 | IFIH1, CLEC7A, TLR8, CASP1, CCL5, TLR3, C3AR1 | 2 | IFIH1, CLEC7A, CASP1, CCL5, TLR3 | 9 | TLR8, CCL5, C3AR1 | [101–103] | |||||||
| 6 | Allograft Rejection Signaling | 1.35E-04 | HLA-DOA, FCER1G, CD86, HLA-DPB1, HLA-DPA1, FAS | 6 | HLA-DOA, HLA-DPB1, HLA-DPA1 | 25 | FCER1G, CD86 | [104] | |||||||
| 7 | Crosstalk between Dendritic Cells and Natural Killer Cells | 2.09E-04 | CSF2RB, ACTA2, IL15, CD86, TLR3, FAS | 21 | IL15, TLR3 | 40 | ACTA2,FAS | 27 | CSF2RB, CD86 | [105, 106] | |||||
| 8 | CCR5 Signaling in Macrophages | 5.25E-04 | CCR5, FCER1G, CCL5, GNG12, FAS | 27 | GNG12, FAS | 4 | CCR5, FCER1G, CCL5 | [107, 108] | [109] | ||||||
| 9 | CD28 Signaling in T Helper Cells | 8.13E-04 | PTPRC, HLA-DOA, FCER1G, CD86, ITPR1, LCP2 | 2 | PTPRC, FCER1G, CD86, LCP2 | [110, 111] | |||||||||
| 10 | Graft-versus-Host Disease Signaling | 9.12E-04 | HLA-DOA, FCER1G, CD86, FAS | 16 | FCER1G, CD86 | [112] | |||||||||
aIPA nomenclature is used for canonical pathways. The 10-top-output_mixed results are shown; the ranking is based on the corresponding IPA-based p-value; the molecules mapped to the pathway are listed
bPosition of the canonical pathway within the output_individual for each target gene, the corresponding p-value and the molecules mapped to the pathway are indicated. Bold, within the 10-top-output_individual; Italic, outside the 10-top-output_individual. Empty cell: the corresponding pathway was not significant in output_individual
cLiterature search-based results annotating those canonical pathways in ovarian cancer or other related studies
Data-driven signature-associated functional annotations and/or diseases
IPA was further used to align the output gene lists with the biological functions and/or diseases named as Functional Annotations. Due to affiliation of functional annotations into several functional categories, the broader IPA classification system, only algorithm II was applied. Results of the 10-top are shown in Tables 5, 6 and 7 and include the results of the output_mixed overlaid with 5, 4, or 3 output_individual. We observed the strong overlap between mixed and ID2 > PTPRC/CD45 > APOBEC3G > ID3 and the minor overlap with ESR1. Within the top outcomes, the over-representation of the basic cellular functions with the accents to the cell movement linked to the immune system and inflammation-driven diseases was documented. Furthermore, at this categorical level the cancer-related processes appeared (metastasis, triple-negative breast cancer). Additionally, the total alignment patterns are illustrated by heat map (Additional file 1: Figure S8, B) and by pie chart (Additional file 1: Figure S9, B).
Table 5.
The top-AID/APOBEC signature-linked Functional Annotations identified by systems biology approach (algorithm II; overlap with 5 individual target genes)
| Functional Annotation top-output_mixed_and_5_individual | ||||||||
|---|---|---|---|---|---|---|---|---|
| Pos. | Functional Annotations | Mixed | APOBEC3G | ESR1 | ID2 | ID3 | PTPRC (CD45) | |
| P-value | Molecules | Molecules | Molecules | Molecules | Molecules | Molecules | ||
| 1 | proliferation of cells | 1.16E-16 | AEBP1, AIF1, ALOX5, AR, ARMC10, BGN, C3AR1, CAMK2N1, CASP1, CCL5, CCR5, CD2, CD47, CD48, CD84, CD86, CDKN1A, CIITA, CLEC7A, COL1A1, COL6A1, COL6A2, CSF2RB, CTGF, CTSS, CYBB, DKK3, DLC1, DOCK2, EGFR, EMILIN1, ENG, EPS8, ESR1, FAS, FBN1, FCER1G, FN1, FOXP1, FYB, GBP2, HCK, HEXB, HLA-DPB1, ID1, ID2, ID3, IGFBP4, IGFBP7, IL10RA, IL15, IRF1, ITGAM, ITPR1, LAIR1, LAMB1, LCP2, LIMA1, LPAR3, LRIG1, LST1, MEIS1, MNDA, MTUS1, MUC16, NID2, NPAS3, NPDC1, PMEPA1, PMP22, PPAP2A, PRRX1, PSMB10, PTPRC, RARRES3, RHBDF1, RHOB, ROCK2, SAMSN1, SASH3, SDC2, SNAI2, SOX17, SPOCK1, SULF2, TIMP1, TLR3, TNFRSF14, TNFSF13B, VCAN, ZFP36 | ALOX5, CASP1, CCL5, CIITA, CLEC7A, CTSS, GBP2, HLA-DPB1, IL15, IRF1, ITGAM, PSMB10, RARRES3, TLR3, TNFRSF14, TNFSF13B | AR, ARMC10, CD47, ESR1, LPAR3, LRIG1, MEIS1, MUC16, NPAS3, SOX17 | CAMK2N1, CDKN1A, COL1A1, COL6A1, COL6A2, CTGF, DLC1, EGFR, ENG, EPS8, FAS, FOXP1, HEXB, ID1, ID2, ID3, IGFBP4, IGFBP7, LIMA1, MTUS1, NPDC1, PPAP2A, RHBDF1, RHOB, SDC2, SNAI2, SULF2, TIMP1, ZFP36 | AEBP1, COL6A2, DKK3, DLC1, EMILIN1, FBN1, FN1, ID1, ID2, ID3, IGFBP4, ITPR1, LAMB1, LIMA1, PMEPA1, PMP22, PPAP2A, PRRX1, RHOB, SDC2, SNAI2, SULF2, VCAN | AIF1, C3AR1, CCL5, CCR5, CD2, CD48, CD84, CD86, CSF2RB, CYBB, DOCK2, FCER1G, FYB, HCK, IL10RA, LAIR1, LCP2, LST1, MNDA, PTPRC, SAMSN1, SASH3 |
IPA nomenclature was used for Functional Annotations. The top-output_mixed_and_5 individual results are shown; the ranking is based on the corresponding IPA-based p-value of the output_mixed; the molecules mapped to the Functional Annotation are listed
Table 6.
The top-AID/APOBEC signature-linked Functional Annotations identified by systems biology approach (algorithm II; overlap with 4 individual target genes)
| Functional Annotations-top-output_mixed_and_4_individual | ||||||||
|---|---|---|---|---|---|---|---|---|
| Pos. | Functional Annotations | Mixed | APOBEC3G | ESR1 | ID2 | ID3 | PTPRC (CD45) | |
| P-value | Molecules | Molecules | Molecules | Molecules | Molecules | Molecules | ||
| 1 | arthritis | 1.53E-17 | AIF1, ALOX5, AR, BGN, C3AR1, CASP1, CCL5, CCR5, CD86, CDH11, CDKN1A, CIITA, COL6A1, CSF2RB, CTSS, FAS, FCER1G, FCGRT, FN1, FSTL1, GBP2, GZMA, HCK, HLA-DOA, HLA-DPA1, HLA-DPB1, IGFBP4, IGFBP7, IL10RA, IL15, IRF1, ITGAM, ITPR1, JAM3, LST1, MS4A6A, PSMB9, PTPRC, SAMD9L, SDC2, SNAI2, SPOCK1, TAGAP, TIMP1, TLR3, TNFSF13B, ZFP36 | ALOX5, CASP1, CCL5, CIITA, CTSS, GBP2, HLA-DOA, HLA-DPA1, HLA-DPB1, IL15, IRF1, ITGAM, MS4A6A, PSMB9, SAMD9L, TAGAP, TLR3, TNFSF13B | CDH11, CDKN1A, COL6A1, FAS, FCGRT, IGFBP4, IGFBP7, SDC2, SNAI2, TIMP1, ZFP36 | BGN, CDH11, FN1, IGFBP4, ITPR1, JAM3, SDC2, SNAI2, SPOCK1 | AIF1, C3AR1, CCL5, CCR5, CD86, CSF2RB, FCER1G, GZMA, HCK, IL10RA, LST1, MS4A6A, PTPRC | |
| 2 | cell death of immune cells | 8.52E-15 | CASP1, CCL5, CCR5, CD2, CD47, CD86, CDKN1A, CIITA, COL1A1, CSF2RB, CYBB, EGFR, ESR1, EYA2, FAS, FCER1G, FN1, GIMAP4, GZMA, HCK, ID2, ID3, IFIH1, IL15, IRF1, ITGAM, ITPR1, LAIR1, LRIG1, MEIS1, PTPRC, SNAI2, TIMP1, TLR3, TNFSF13B | CASP1, CCL5, CIITA, IFIH1, IL15, IRF1, ITGAM, TLR3, TNFSF13B | CD47, ESR1, EYA2, LRIG1, MEIS1 | CDKN1A, COL1A1, EGFR, FAS, ID2, ID3, SNAI2 | CCL5, CCR5, CD2, CD86, CSF2RB, CYBB, FCER1G, GIMAP4, GZMA, LAIR1, PTPRC | |
| 3 | rheumatoid arthritis | 2.2E-13 | AIF1, ALOX5, BGN, C3AR1, CCL5, CCR5, CD86, CDH11, CIITA, CSF2RB, FCGRT, GBP2, GZMA, HCK, HLA-DOA, HLA-DPA1, HLA-DPB1, IGFBP4, IGFBP7, IL10RA, ITPR1, JAM3, LST1, MS4A6A, PSMB9, PTPRC, SAMD9L, SDC2, SNAI2, SPOCK1, TAGAP, TIMP1, TLR3, TNFSF13B, ZFP36 | ALOX5, CCL5, CIITA, GBP2, HLA-DOA, HLA-DPA1, HLA-DPB1, MS4A6A, PSMB9, SAMD9L, TAGAP, TLR3, TNFSF13B | CDH11, FCGRT, IGFBP4, IGFBP7, SDC2, SNAI2, TIMP1, ZFP36 | BGN, CDH11, IGFBP4, ITPR1, JAM3, SDC2, SNAI2, SPOCK1 | AIF1, C3AR1, CCL5, CCR5, CD86, CSF2RB, GZMA, HCK, IL10RA, LST1, MS4A6A, PTPRC | |
| 4 | apoptosis | 2.9E-12 | AIF1, ALOX5, ANTXR2, APOL6, AR, ARMC10, BGN, CASP1, CCL5, CCR5, CD2, CD47, CD48, CD53, CDH11, CDKN1A, CIITA, COL1A1, CSF2RB, CTGF, CTSS, CYBB, DKK3, DLC1, EGFR, ENG, ESR1, EVA1C, EYA2, FAS, FBN1, FCER1G, FN1, FOXP1, FSTL1, GIMAP4, GZMA, HCK, HEXB, ID1, ID2, ID3, IFIH1, IGFBP4, IGFBP7, IL15, IRF1, ITGAM, ITPR1, LAIR1, LRIG1, MNDA, MUC16, PMEPA1, PMP22, PTPRC, RHOB, SDC2, SGPP1, SNAI2, SOX17, SPOCK1, SULF2, TIMP1, TLR3, TNFRSF14, TNFSF13B, VCAN, XAF1, ZFP36 | ALOX5, APOL6, CASP1, CCL5, CIITA, CTSS, EVA1C, IFIH1, IL15, IRF1, ITGAM, TLR3, TNFSF13B, XAF1 | ANTXR2, CDKN1A, COL1A1, CTGF, DLC1, EGFR, ENG, FAS, FOXP1, HEXB, ID1, ID2, ID3, IGFBP4, IGFBP7, RHOB, SDC2, SGPP1, SNAI2, SULF2, TIMP1, ZFP36 | BGN, DKK3, DLC1, FBN1, FN1, FSTL1, ID1, ID2, ID3, IGFBP4, ITPR1, PMEPA1, PMP22, RHOB, SDC2, SNAI2, SPOCK1, SULF2 | CCL5, CCR5, CD2, CD48, CD53, CSF2RB, CYBB, FCER1G, GZMA, HCK, LAIR1, MNDA, PTPRC | |
| 5 | necrosis | 1.36E-11 | ALOX5, ANTXR2, AR, ARMC10, BGN, CASP1, CCL5, CCR5, CD2, CD47, CD48, CD86, CDH11, CDKN1A, CIITA, COL1A1, COL6A1, CSF2RB, CTGF, CTSS, CYBB, DKK3, DPYD, EGFR, ENG, ESR1, EVA1C, EYA2, FAS, FCER1G, FN1, FSTL1, GIMAP4, GZMA, HCK, =ID1, ID2, ID3, IFIH1, IGFBP4, IGFBP7, IL15, IRF1, ITGAM, ITPR1, LAIR1, LRIG1, MEIS1, MUC16, PMEPA1, PMP22, PTPRC, RHOB, ROCK2, SDC2, SGPP1, SNAI2, SOX17, SPOCK1, SULF2, TIMP1, TLR3, TNFRSF14, TNFSF13B, VCAN, XAF1 | ALOX5, CASP1, CCL5, CIITA, CTSS, DPYD, EVA1C, IFIH1, IL15, IRF1, ITGAM, TLR3, TNFRSF14, TNFSF13B, XAF1 | ANTXR2, CDKN1A, COL1A1, COL6A1, CTGF, EGFR, ENG, FAS, ID1, ID2, ID3, IGFBP4, IGFBP7, RHOB, ROCK2, SDC2, SGPP1, SNAI2, SULF2, TIMP1 | BGN, DKK3, FN1, FSTL1, ID1, ID2, ID3, IGFBP4, ITPR1, PMEPA1, PMP22, RHOB, SDC2, SNAI2, SPOCK1, SULF2 | CCL5, CCR5, CD2, CD48, CD86, CSF2RB, CYBB, FCER1G, GIMAP4, GZMA, HCK, LAIR1, PTPRC | |
IPA nomenclature was used for Functional Annotations. Since analysis for the 10-top-output_mixed_and_5 individual revealed only one functional annotation, the results of the top-output_mixed_and_4 individual are additionally shown. The ranking is based on the corresponding IPA-based p-value of the output_mixed; the molecules mapped to the Functional Annotation are listed
Table 7.
The 10-top-AID/APOBEC signature-linked Functional Annotations identified by systems biology approach (algorithm II; overlap with 3 individual target genes)
| Functional Annotations-10-top-output_mixed_and_3_individual | ||||||||
|---|---|---|---|---|---|---|---|---|
| Pos. | Functional Annotations | Mixed | APOBEC3G | ESR1 | ID2 | ID3 | PTPRC (CD45) | |
| P-value | Molecules | Molecules | Molecules | Molecules | Molecules | Molecules | ||
| 1 | Rheumatic Disease | 1.66E-18 | ACTA2, AIF1, ALOX5, AR, BGN, C3AR1, CASP1, CCL5, CCR5, CD86, CDH11, CDKN1A, CIITA, COL1A1, COL6A1, CSF2RB, CTSS, FAS, FBN1, FCER1G, FCGRT, FN1, FSTL1, GBP2, GZMA, HCK, HLA-DOA, HLA-DPA1, HLA-DPB1, IGFBP4, IGFBP7, IL10RA, IL15, IRF1, ITGAM, ITPR1, JAM3, LST1, MS4A6A, PSMB9, PTPRC, SAMD9L, SDC2, SNAI2, SPOCK1, TAGAP, TIMP1, TLR3, TLR8, TNFSF13B, ZFP36 | ACTA2, CDH11, CDKN1A, COL1A1, COL6A1, FAS, FCGRT, IGFBP4, IGFBP7, SDC2, SNAI2, TIMP1, ZFP36 | BGN, CDH11, FBN1, FN1, IGFBP4, ITPR1, JAM3, SDC2, SNAI2, SPOCK1 | AIF1, C3AR1, CCL5, CCR5, CD86, CSF2RB, FCER1G, GZMA, HCK, IL10RA, LST1, MS4A6A, PTPRC, TLR8 | ||
| 2 | cell movement of leukocytes | 8.68E-17 | AIF1, ALOX5, AR, BGN, C3AR1, CASP1, CCL5, CCR5, CD2, CD47, CD48, CD86, CDKN1A, CIITA, COL1A1, CTGF, CTSS, CYBB, DOCK2, EGFR, ENG, EPS8, ESR1, FAS, FCER1G, FN1, FYB, HCK, IL10RA, IL15, ITGAM, JAM3, LCP2, PTPRC, RHOB, ROCK2, TIMP1, TLR3, TNFRSF14, TNFSF13B | ALOX5, CASP1, CCL5, CIITA, CTSS, IL15, ITGAM, TLR3, TNFSF13B | CDKN1A, COL1A1, EGFR, ENG, EPS8, FAS, RHOB, ROCK2, TIMP1 | AIF1, C3AR1, CCL5, CCR5, CD2, CD48, CD86, CYBB, DOCK2, FCER1G, FYB, HCK, IL10RA, LCP2 | ||
| 3 | quantity of leukocytes | 1.51E-16 | ALOX5, C3AR1, CCL5, CCR5, CD47, CD48, CD84, CD86, CDKN1A, CIITA, CLEC7A, CSF2RB, CTSS, CYBB, DKK3, DOCK2, ESR1, FAS, FCER1G, FOXP1, FYB, HCK, HEXB, ID1, ID2, IL10RA, IL15, IRF1, ITGAM, JAM3, LAIR1, LCP2, MEIS1, PSMB10, PSMB9, PTPRC, SAMSN1, SASH3, SLA, SNAI2, TIMP1, TLR3, TNFSF13B, ZFP36 | ALOX5, CCL5, CIITA, CLEC7A, CTSS, IL15, IRF1, ITGAM, PSMB10, PSMB9, TLR3, TNFSF13B | CDKN1A, FAS, FOXP1, HEXB, ID1, ID2, SNAI2, TIMP1, ZFP36 | C3AR1, CCL5, CCR5, CD48, CD84, CD86, CSF2RB, CYBB, DOCK2, FCER1G, FYB, HCK, IL10RA, LAIR1, LCP2, PTPRC, SAMSN1, SASH3, SLA | ||
| 4 | cell movement of myeloid cells | 4.14E-13 | AIF1, ALOX5, AR, C3AR1, CASP1, CCL5, CCR5, CD2, CD47, CD48, CDKN1A, COL1A1, CTGF, CTSS, CYBB, DOCK2, EGFR, ENG, FAS, FCER1G, FN1, HCK, IL15, ITGAM, JAM3, PTPRC, RHOB, ROCK2, TLR3 | ALOX5, CASP1, CCL5, CTSS, IL15, ITGAM, TLR3 | CDKN1A, COL1A1, EGFR, ENG, FAS, RHOB, ROCK2 | AIF1, C3AR1, CCL5, CCR5, CD2, CD48, CYBB, DOCK2, FCER1G, HCK | ||
| 5 | cell movement of phagocytes | 9.33E-13 | AIF1, ALOX5, AR, C3AR1, CASP1, CCL5, CCR5, CD47, CD86, CDKN1A, COL1A1, CTGF, CTSS, CYBB, DOCK2, EGFR, ENG, EPS8, FAS, FCER1G, FN1, HCK, IL10RA, IL15, ITGAM, JAM3, RHOB, TIMP1, TLR3 | ALOX5, CASP1, CCL5, CTSS, ITGAM, TLR3 | CDKN1A, COL1A1, EGFR, ENG, EPS8, FAS, RHOB, TIMP1 | AIF1, C3AR1, CCL5, CCR5, CD86, CYBB, DOCK2, FCER1G, HCK, IL10RA | ||
| 6 | cell death | 7.48E-11 | AIF1, ALOX5, ANTXR2, APOL6, AR, ARMC10, BGN, CASP1, CCL5, CCR5, CD2, CD47, CD48, CD53, CD86, CDH11, CDKN1A, CIITA, COL1A1, COL6A1, CSF2RB, CTGF, CTSS, CYBB, DKK3, DLC1, DPYD, EGFR, ENG, ESR1, EVA1C, EYA2, FAS, FBN1, FCER1G, FN1, FOXP1, FSTL1, GIMAP4, GZMA, HCK, HEXB, ID1, ID2, ID3, IFIH1, IGFBP4, IGFBP7, IL15, IRF1, ITGAM, ITPR1, LAIR1, LCP2, LRIG1, MEIS1, MNDA, MUC16, PMEPA1, PMP22, PTPRC, RHOB, ROCK2, SDC2, SGPP1, SNAI2, SOX17, SPOCK1, SULF2, TFEC, TIMP1, TLR3, TNFRSF14, TNFSF13B, VCAN, XAF1, ZFP36 | ALOX5, APOL6, CASP1, CCL5, CIITA, CTSS, DPYD, EVA1C, IFIH1, IL15, IRF1, ITGAM, TLR3, TNFRSF14, TNFSF13B, XAF1 | ANTXR2, CDKN1A, COL1A1, COL6A1, CTGF, DLC1, EGFR, ENG, FAS, FOXP1, HEXB, ID1, ID2, ID3, IGFBP4, IGFBP7, RHOB, ROCK2, SDC2, SGPP1, SNAI2, SULF2, TIMP1, ZFP36 | AIF1, CCL5, CCR5, CD2, CD48, CD53, CD86, CSF2RB, CYBB, FCER1G, GIMAP4, GZMA, HCK, LAIR1, LCP2, MNDA, PTPRC | ||
| 7 | metastasis | 1.05E-10 | ACTA2, AR, CASP1, CD47, CD86, CDKN1A, CTGF, CYBB, EGFR, ESR1, FN1, FOXP1, ID1, ID2, ID3, IGFBP7, IL15, IRF1, JAM3, MEIS1, NID2, PAPSS2, PRRX1, RAB31, RHOB, TIMP1, TLR8, VCAN | AR, CD47, ESR1, MEIS1 | ACTA2, CDKN1A, CTGF, EGFR, FOXP1, ID1, ID2, ID3, IGFBP7, PAPSS2, RAB31, TIMP1 | FN1, ID1, ID2, ID3, JAM3, PRRX1 | ||
| 8 | triple-negative breast cancer | 1.15E-10 | AR, CDH11, CDKN1A, CTGF, DLC1, EGFR, EPS8, ESR1, FAS, HCK, IRF1, LAMB1, TIMP1, WFDC2 | AR, ESR1, WFDC2 | CDH11, CDKN1A, CTGF, DLC1, EGFR, EPS8, FAS, TIMP1 | CDH11, DLC1, LAMB1 | ||
| 9 | binding of cells | 1.52E-10 | AR, BGN, CCL5, CCR5, CD2, CD47, CD48, CD84, CD86, CLEC7A, CSF2RB, ENG, FAS, FN1, FYB, HCK, HS3ST1, ITGAM, JAM3, LCP2, MUC16, PTPRC, RHOB, SDC2, SNX9 | ENG, FAS, HS3ST1, RHOB, SDC2, SNX9 | BGN, FN1, JAM3, RHOB, SDC2 | CCL5, CCR5, CD2, CD48, CD86, CSF2RB, FYB, HCK, LCP2, PTPRC | ||
| 10 | Lymphocyte migration | 2.14E-10 | ALOX5, CCL5, CCR5, CD2, CD47, CD48, CD86, COL1A1, CTSS, DOCK2, EGFR, ENG, FAS, FN1, FYB, IL15, JAM3, TIMP1, TNFRSF14, TNFSF13B | ALOX5, CCL5, IL15, TNFSF13B | COL1A1, EGFR, ENG, FAS, TIMP1 | CCL5, CCR5, CD2, CD48, CD86, DOCK2, FYB | ||
IPA nomenclature was used for Functional Annotations. Since analysis for the 10-top-output_mixed_and_4 individual revealed only 5 functional annotations, the results of the 10-top-output_mixed_and_3 individual are additionally shown. The ranking is based on the corresponding IPA-based p-value of the output_mixed; the molecules mapped to the Functional Annotation are listed
Data-driven signature-associated upstream regulators
To include the biological information for the higher-level overview, the IPA-based analysis for Upstream Regulators was performed. In this case, both algorithms I and II were used. The 10-top most significantly over-represented regulators, when applying algorithm I, were LPS, IFNalpha, IFNgamma, TGFbeta1, TNF, IL10, STAT3, IL6, IL13, and tretinoin (Table 8). Of note, 6 out of 10 regulators derived from the output_mixed were found in at least one output_individual within the 10-top positions (with p-values all < 0.05). The strongest overlap between the 10-top-output_mixed and _individual was observed for APOBEC3G (4/10) and PTPRC/CD45 (2/10); in contrast, no overlap was found for ESR1, thus the observed distribution is similar to the one described for Canonical Pathways analysis (Table 4). Complementary, the heat map and pie chart illustrate the total overlap patterns between output_mixed and output_individuals (Figures S8, C and S9, C). When applying the algorithm II, the following additional molecules were identified such as APOE, CD44, TECAM1, Ifi204, alpha Catenin, INFbeta1, TGFBR1, SPI1, and CD40 (Additional file 1: Tables S16 and S17).
Table 8.
The 10-top-AID/APOBEC signature-linked Upstream Regulators identified by systems biology approach (algorithm I)
| Upstream Regulators-10-top-output_mixeda | Upstream Regulators output_individualb | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| APOBEC3G | ESR1 | ID2 | ID3 | PTPRC (CD45) | |||||||||
| Pos. | Upstream Regulator | P-value | Molecules | Pos. | Molecules | Pos. | Molecules | Pos. | Molecules | Pos. | Molecules | Pos. | Molecules |
| 1 | lipopolysaccharide | 2.52E-27 | ACTA2, ALOX5, APOBEC3F, APOBEC3G, APOL6, AR, BGN, C3AR1, CARD6, CASP1, CCL5, CCR5, CD48, CD53, CD86, CDH11, CDKN1A, CIITA, CLEC7A, COL1A1, COL5A1, COL6A1, CSF2RB, CTGF, CYBB, DKK3, FAS, FBN1, FCER1G, FCGRT, FN1, FYB, GBP2, GZMA, HCK, ID2, IFIH1, IGFBP4, IL10RA, IL15, IRF1, ITGAM, KIAA1551, LAMB1, LCP2, LST1, NID1, NID2, PAPSS2, PLEK, PSMB10, PSMB9, RARRES3, RHOB, SAMSN1, TFEC, TIMP1, TLR3, TLR8, TNFSF13B, TRIM22, VCAN, XAF1, ZFP36 | ||||||||||
| 2 | Interferon alpha | 3.48E-19 | APOBEC3F, APOBEC3G, APOL3/APOL4, C3AR1, CASP1, CCL5, CCR5, CD86, CDKN1A, CIITA, CSF2RB, EGFR, FAS, GBP2, IFIH1, IGFBP4, IL10RA, IL15, IRF1, ITGAM, MNDA, PSMB9, RARRES3, TLR3, TLR8, TNFSF13B, TRIM22 | 4 | APOBEC3F, APOBEC3G, APOL3/APOL4, CASP1, CIITA, IL15, IRF1, TLR3, TNFSF13B | 591 | CDKN1A, IGFBP4 | 146 | CSF2RB, TLR8 | ||||
| 3 | IFNG | 7.59E-18 | AIF1, APOBEC3G, APOL6, CARD6, CASP1, CCL5, CCR5, CD2, CD86, CDKN1A, CIITA, COL1A1, CSF2RB, CTGF, CTSS, CYBB, FAS, FCER1G, FN1, GBP2, GMPR, HLA-DOA, ID1, IFIH1, IGFBP4, IL10RA, IL15, IRF1, ITGAM, LCP2, LST1, MNDA, PLEK, PSMB10, PSMB9, RARRES3, RHOB, SNAI2, TIMP1, TLR3, TLR8, TNFRSF14, TNFSF13B, TRIM22, ZFP36 | 1 | APOL6, CARD6, CASP1, CCL5, CIITA, CTSS, GBP2, IFIH1, IL15, IRF1, ITGAM, PSMB10, PSMB9, RARRES3, TLR3, TNFSF13B, TRIM22 | 7 | AIF1, CCL5, CCR5, CD2, CD86, CYBB, FCER1G | ||||||
| 4 | TGFB1 | 8.38E-18 | ACTA2, ALOX5, BGN, CASP1, CCL5, CCR5, CD86, CDH11, CDKN1A, CIITA, COL1A1, COL5A1, COL6A1, COL6A2, CTGF, CTSS, CYBB, DKK3, DOCK2, EMILIN1, ENG, FAS, FBN1, FCER1G, FN1, GMPR, GZMA, HEXB, ID1, ID2, ID3, IFIH1, IGFBP4, IGFBP7, IL10RA, IL15, IRF1, ITGAM, ITPR1, PDLIM5, PLOD1, PMEPA1, PTPRC, RAB31, RHOB, SNAI2, SPOCK1, TIMP1, TNFRSF14, TNFSF13B, VCAN, ZFP36 | 161 | ALOX5, CCL5, IRF1, ITGAM | 3 | ACTA2, CDH11, CDKN1A, COL1A1, CTGF, ENG, FAS, HEXB, ID1, RAB31, RHOB, SNAI2, TIMP1, ZFP36 | 3 | BGN, CDH11, COL5A1, FBN1, FN1, ID1, PMEPA1, RHOB, SNAI2, SPOCK1 | ||||
| 5 | TNF | 1.25E-16 | ABR, AEBP1, ALOX5, AR, BGN, CARD16, CARD6, CASP1, CCL5, CCR5, CD47, CD86, CDH11, CDKN1A, CIITA, COL1A1, CSF2RB, CTGF, CTSS, CYBB, EGFR, ENG, ESR1, FAS, FCER1G, FCGRT, FN1, GBP2, HEXB, ID1, ID3, IFIH1, IGFBP4, IL10RA, IL15, IRF1, ITGAM, ITPR1, NID1, PPAP2A, PSMB10, PSMB9, RARRES3, SDC2, TIMP1, TLR3, TLR8, TNFSF13B, ZFP36 | 7 | CASP1, CCL5, CTSS, GBP2, IL15, IRF1, ITGAM, PSMB10, PSMB9, RARRES3, TLR3, TNFSF13B | 69 | CDH11, CDKN1A, EGFR, FAS, HEXB, NID1, PPAP2A, TIMP1 | 52 | AEBP1, CDH11, FN1, ITPR1, NID1, PPAP2A | 181 | CCL5, CCR5, CD86, CYBB | ||
| 6 | IL10 | 1.91E-15 | ACTA2, CCL5, CCR5, CD2, CD86, CDKN1A, CIITA, CLEC7A, COL1A1, CSF2RB, CTSS, FAS, FCER1G, GZMA, IGFBP4, IL10RA, IRF1, PSMB9, TIMP1, TLR3, TLR8, TNFSF13B, VCAN, ZFP36 | 87 | CCL5, CIITA, TLR3 | 97 | ACTA2, CDKN1A, TIMP1, ZFP36 | 45 | CCL5, CD2, CD86 | ||||
| 7 | STAT3 | 5.98E-15 | CASP1, CCL5, CCR5, CD86, CDKN1A, CIITA, COL5A1, ESR1, FAS, FCER1G, FN1, GBP2, ID2, IFIH1, IRF1, ITGAM, PSMB9, SMAD9, TIMP1, TLR3, TRIM14, TRIM22, VCAN, XAF1, ZFP36 | 9 | CASP1, CIITA, GBP2, IRF1, ITGAM, PSMB9, TLR3, XAF1 | 461 | CDKN1A, TIMP1, ZFP36 | 106 | FN1, SMAD9, VCAN | ||||
| 8 | IL6 | 2.87E-14 | BGN, CASP1, CCL5, CCR5, CD48, CD53, CD86, CDKN1A, CIITA, CSF2RB, CTGF, CYBB, EGFR, FAS, FCGRT, FN1, GBP2, ID1, ID2, IGFBP4, IL15, IRF1, ITGAM, PSMB10, PSMB9, PTPRC, RNASE6, TIMP1, TLR3, TLR8 | 13 | CASP1, CIITA, GBP2, IRF1, ITGAM, PSMB9, TLR3 | 211 | CDKN1A, ID1, ID2, TIMP1 | ||||||
| 9 | IL13 | 7.28E-14 | C3AR1, CASP1, CCL5, CCR5, CD48, CD86, CLEC7A, COL1A1, COL6A2, CTGF, CTSS, CYBB, EGFR, FAS, FGD2, HOMER2, NID1, SAMSN1, SLA, SNAI2, TFEC, TIMP1, TNFSF13B | 70 | CASP1, CCL5, CTSS, FGD2 | 512 | FAS, NID1, TIMP1 | 1 | C3AR1, CCL5, CCR5, CD48, CD86, CYBB, SAMSN1, SLA | ||||
| 10 | tretinoin | 1.15E-13 | C3AR1, CASP1, CCR5, CD53, CD86, CDKN1A, COL1A1, CTGF, CYBB, DDX60L, DLC1, EGFR, EYA2, FCER1G, FN1, GZMA, HS3ST1, ID1, ID2, ID3, IFIH1, IGFBP4, IGFBP7, IL10RA, IRF1, ITGAM, LAMB1, MEIS1, MNDA, NID2, PLEK, PSMB9, RARRES3, SAMD9L, SLA, SMAD9, SOX17, THSD4, TIMP1, TLR3, TRIM22, XAF1 | ||||||||||
aThe 10-top-output_mixed results are shown; the ranking is based on the corresponding IPA-based p-value; the molecules associated with the corresponding Upstream Regulator are listed
bPosition of the Upstream Regulator within the output_individual for each target gene, the molecules associated with the Upstream Regulator are indicated. Color code: grey, within the 10-top-output_individual; white, outside the 10-top-output_individual. Empty cell: the corresponding Upstream Regulator was not significant in output_individual
Discussion
Multigene profiling and survival models
The herein presented study is, to our knowledge, the first one linking expression of the entire AID/APOBEC family and interacting genes with clinical outcome with respect to survival of cancer patients. High efforts are invested in the field of cancer research to evaluate the applicability of gene expression for the use in risk prediction; nevertheless, no established standard is available for the implemented methodology to study gene expression profiles in the pathophysiology of disease. Different approaches have certain advantages and disadvantages. Data-driven approaches using curated microarray expression data from various studies offer the advantage of a transcriptome-wide screening, but face a lack of sensitivity for very low-level expressed genes and/or the specificity for genes with high sequence similarity; the latter two aspects are fully relevant for AID and APOBEC3 subfamily, respectively, as discussed herein and by others [13]. A knowledge-driven approach, where the composition of a gene signature characterizing certain biological aspects is assembled based on data mining followed by real-time PCR-based gene expression profiling of clinical specimens has by definition the advantage of high sensitivity and reproducibility as real-time PCR methodology is the gold standard in expression profiling. To maximize the outcome, we applied herein a rational integration of both approaches.
We structured the study by three consecutive analytical modules: (i) multigene-based expression profiling, (ii) statistical modeling for survival prediction and (iii) delineation of AID/APOBEC-associated gene network(s) and pathways by applying bioinformatics tools (as summarized in Fig. 1). Each module resulted in novel outcomes with respect to the pathophysiology of ovarian cancer; their combination in turn provided an advantageous comprehensive overview. The herein established study algorithm, named by us as MuSiCO (from Multigene Signature to the Patient-Orientated Clinical Outcome), can be further applied for any gene-/pathway-/disease-related signature and any type of tumor under investigation.
One objective here is the development of proper multivariable-based models for evaluating patient prognosis on the basis of the patient-specific gene expression data sets. Although proceedings in whole genome analysis have triggered the developments of statistical methods for survival models [68], multivariable Cox regression analysis of multigene-based prognostic models is still a challenging task based on the following aspects. Classical statistical regression methods based on maximum likelihood have been widely used when the number of outcome events highly exceeds the number of variables. In practice, however, we frequently face restricted availability of well-characterized high quality clinical samples. Thus, in case of a multigene approach, a relatively large number of profiling-derived variables is often accompanied by relatively few outcome events (with respect to OS this is the number of cancer-related deaths and not the number of patients in the examined cohort). Given that, herein we established and applied state-of-the-art algorithms for multivariable modeling allowing (i) to reduce the risk of getting an overoptimistic or overfitted survival model and, thus, to increase predictive accuracy; (ii) to be wrapped in a cross-validation loop in order to estimate the performance of the model and, importantly, to compare the prognostic power and accuracy between different models; and (iii) to be able to rank the contributions of individual variables to predictions by their importance. We explicitly applied the leave-one-out strategy of cross-validation instead of dividing the data set in to a training and test set. We believe that the latter approach is inferior for several reasons: (i) in a situation of a relatively small number of events, it would waste a lot of data which would be needed to obtain more stable estimates; (ii) it can easily be manipulated by selecting a split that yields optimal results. To characterize the performances of our prognostic models, we used PEV, c-index, and p-value, and these parameters could be used to compare models within one study but also to make comparisons between independent studies and laboratories. Thereby, by usage of those appropriate measures, the truly independent validation can be performed by an independent group of researchers and using an independent cohort of patients.
For the examined cohort of patients we herein confirmed strong prognostic relevance of clinical risk factors and showed that six clinicopathological variables such as peritoneal carcinomatosis, age, histology, FIGO stage, residual disease, and grading can be assembled into a survival model which has prognostic power for both OS and PFS. Importantly, by inclusion of the AID/APOBEC signature-based variables, the combined model significantly improved the prognostication of OS. Furthermore, several of the gene profiling-derived variables within the combined model such as ID3, PTPRC/CD45, AID, APOBEC3G, and ID2 exceed the prognostic impact of some clinicopathological variables to the model. Remarkably, in both models (ridge and LASSO) ID3 was ranked at the 1st position overcoming the impact of all six clinical and profiling-derived variables. Given in addition the strong significance of ID3 in univariate Cox regression analysis, the data nominate ID3 as prognostic factor for OS. Moreover, since higher ID3 mRNA levels were associated with poor survival, further functional studies are needed to validate whether ID3 might in addition act as a “driver” of pathogenesis of ovarian cancer. ID molecules (ID1-4) are functional inhibitors/antagonists of the basic helix-loop-helix transcription factors and thus control the expression of multiple targets including among others AID [63, 64]. The critical implications of dysregulated IDs in multiple cancer hallmarks are highly recognized (reviewed in [69]) including contribution to pathomechanisms of ovarian cancer [38, 70–72]. Small molecule inhibitors of IDs are in development and might be considered as novel combinatorial therapeutic approach for treatment of cancer [73, 74].
For several cancer types (breast, bladder, cervical, head and neck and lung) an APOBEC mutation pattern was identified and the APOBEC-mediated mutagenesis was found to correlate with APOBEC mRNA levels, particularly with APOBEC3B [13, 36]. Furthermore, during our data mining, the study of Leonard et al. was published [40] showing elevated expression of APOBEC3B in the majority of ovarian cancer cell lines examined and in a subset of high-grade primary ovarian cancer in comparison to the normal ovarian or fallopian tube epithelial cells and non-malignant ovarian tissues, respectively. Although a direct comparison of expression levels between serous tumor samples and normal ovarian tissues is the point of debates, which has been as well discussed by the authors, the accompanied functional studies revealed a positive association between APOBEC3B expression in cancer tissues from 16 patients and elevated levels of transversion mutations, thus, suggesting a contributing role of APOBEC3B in genomic instability attributed to ovarian cancer. Against the logical expectations, our data did not reveal a prognostic relevance of APOBEC3B mRNA levels in the examined cohort of patients when assessed by univariate Cox regression analysis. In the multivariable prognostic models such as AID/APOBEC or Combined, according to standardized regression coefficients-based ranking APOBEC3B was assigned to the positions 7 and 24, respectively, and, thus, showed a moderate/minimal impact on the prognostic ability of the models. Our data, however, does not exclude any additional ways of regulation of APOBEC3B activity in a patient-specific manner with respect to disease pathobiology. Besides AID, among APOBEC3 subfamily members, APOBEC3G contributed to the prognostic models (both ridge and LASSO). This might indicate that in tumor cells during the cancer progression the interplay between individual APOBEC3 family members plays a contributing role. Additionally, one should consider the complex composition of the ovarian cancer tissues used for the gene expression profiling, which besides the tumor cells includes the tumor stroma with significant component attributed to infiltrated immune cell populations; thus, the AID/APOBEC mRNA expression values likely reflex the sum from all positive cells. Indeed, on the one side, the herein performed expression analysis of the signature genes using a wide range of ovarian cancer cell lines showed that those innate/adaptive immunity-related genes might as well be expressed in ovarian tumor cells per se; our data are generally in line with the profiling results reported recently [40]. On the other side, the correlation analysis of multigene-derived data sets across ovarian cancer tissues revealed strong positive association between PTPRC/CD45, the classical immune cell marker, and APOBEC3 subfamily members such as A3C, A3D, A3H, and the prognostically relevant A3G as well as AID. Furthermore, although weaker, a positive association was observed with PAX5, the B-cell transcription factor. These data suggest that, besides expression by tumor cells, certain contributions from immune cells, including B lymphocytes, to the total mRNA expression levels of individual APOBECs indeed might take place which thereby impacts the prognostic power of the model. Of note, no significant correlation was found between PTPRC/CD45 and APOBEC3B, thus, likely excluding the major impact of the CD45-positive immune cells to this variable.
It is important to emphasize that within the prognostic model APOBEC3G behaves as a protective factor with potential anti-tumor action since higher APOBEC3G mRNA expression levels were associated with better clinical outcome in respect of OS. Previous cell-based studies showed that APOBEC3G does not fall into the subclass of APOBECs (which among others includes APOBEC3B) grouped based on mutational specificity for TC motifs [75] suggesting somewhat different APOBEC3G-mediated biological consequences. Considering the APOBEC3 functions in virus, naked foreign DNA or retrotransposon restriction, a potential association between the APOBEC expression, the APOBEC-mediated cancer-related mutagenesis and the viral infection/viral carcinogenesis is appealing and was discussed recently, when two cancer types, cervical and head and neck cancer, which are highly associated with human papillomavirus, HPV, were found among those six types with strong enrichment of APOBEC-mediated mutagenic patterns [36, 76]; HPV in turn is one of the known APOBEC3-targeted viruses [77, 78] (besides well-studied HIV-1, the list includes HTLV, HCV, HBV, HPV, HSV-1, and EBV). Such association in ovarian cancer is currently not known.
Data-driven disease-relevant pathways
The third analytical module applied herein allowed us to extend the signature- and modeling-based knowledge and dissect potential mechanisms/pathways/factors contributing to disease pathogenesis and patient survival. The reconstructed network was created and visualized using the IPA software on the basis of the target molecules defined by prognostic modeling and the molecules from co-expressed genes derived from the 10-top Canonical Pathways and Upstream Regulators (Fig. 4). This integration and visualization of both experimental and in silico microarray-based data illustrate the existence of mutual interconnections between four target genes such as PTPRC/CD45, ID3, APOBEC3G, ID2 and point to more separate biological function(s) of the node around ESR1 in advanced stage serous ovarian cancer.
Fig. 4.
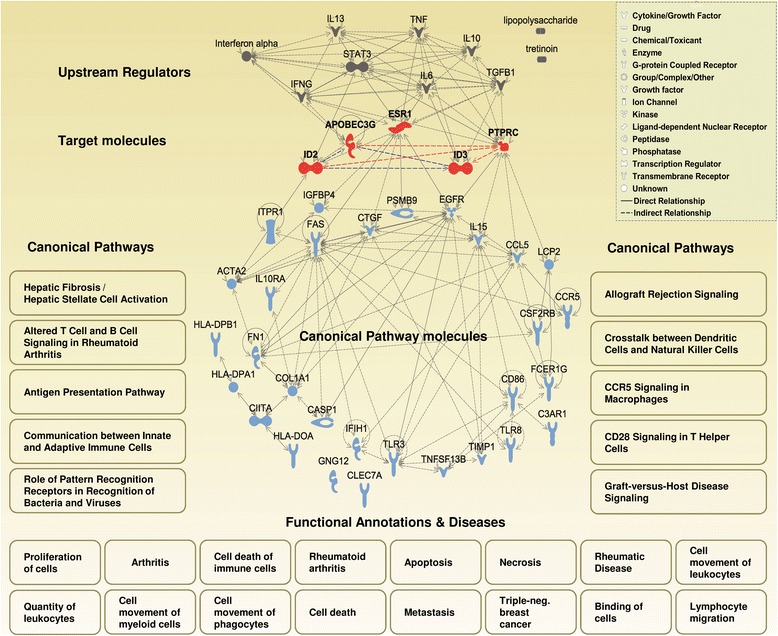
Representation of the top Canonical Pathways, Functional Annotations and Upstream Regulators detected upon analysis of target genes and their co-regulated genes. A reconstructed gene network was created using the Ingenuity Pathway Analysis Software (IPA) on the basis of the target molecules, the molecules from the 10-top Canonical Pathways (see Table 4, Molecules) and the 10-top Upstream Regulators (see Table 8). Solid lines in grey display the IPA-identified direct interactions between the molecules; dashed lines display indirect interactions. The multigene approach-based correlation analysis was used to find additional biological associations between the target genes. Statistically significant study-based associations (SPSS program, Additional file 1: Table S5) are displayed by dashed lines; red for correlation coefficient ≥ 0.6, p < 0.001; blue for correlation coefficient < 0.6, p < 0.001. The 10-top Canonical Pathways are listed according to the IPA-based ranking (from left to right). For a complete overview, also the most significant Functional Annotations & Diseases are shown (see Tables 5, 6 and 7)
Generally, the 10-top Canonical Pathways contain molecules which are characteristic for tissue remodeling/fibrotic pathway, altered immune response including antigen presentation mechanisms, and communication between various immune cell populations involved in innate and adaptive immune responses. It gives a link to autoimmune disorders with inflammatory background as rheumatoid arthritis and to transplant rejection by the recipient’s immune system and, unsuspectedly, does not highlight the cancer-related processes. Notably, among the top Canonical Pathways, the first top-ranked was Hepatic Fibrosis/Hepatic Stellate Cell Activation based on such molecules as COL1A1, IGFBP4, CCR5, FN1, CTGF, TIMP1, ACTA2, IL10RA, CCL5, FAS, EGFR. The significant association between fibrosis and the clinical outcome of ovarian cancer patients was observed recently by others, although it was identified by applying completely different approaches such as miRNA screening or histological examinations, respectively [79, 80]. Surprisingly, the same pathway was identified as one of the most relevant canonical pathways in granulosa cells from bovine ovarian follicles during atresia, which represents one of the physiological processes in healthy ovaries [81]. Thus, aberrant modulation of the pathway’s underlying molecules might turn the physiological processes to the direction of malignant transformation.
Multiple Canonical Pathways within the identified 10-top are linked to the antigen processing and presentation machinery and antigen recognition by lymphocytes (Fig. 4 and Table 4, pos. 2, 3, 6, 9, and 10); HLA class II transcripts are among the molecules underlying these pathways. In this respect it is interesting to note that the recent study by Yoshihara et al. [82] identified the antigen presentation pathway to be significantly modulated (as estimated by downregulation of HLA class I molecules) in a high risk group compared with a low risk group of patients with high-grade serous ovarian cancer.
The top Functional Annotations & Diseases and Upstream Regulators of the AID/APOBEC-associated network reconstruction further indicate the particular significance of immunity, aberrant immunity/autoimmunity and inflammation. Rather unexpectedly, the categories such as rheumatic diseases, arthritis, rheumatoid arthritis were top-ranked together with more broad functions such as proliferation of cells, binding of cells, cell movement including movement of various immune cell subsets (lymphocytes, myeloid cells, phagocytes), and quantity of leukocytes. The identified association with rheumatoid arthritis – the progressive inflammatory autoimmune disorder – further points out to the importance/relevance of inflammation and suggests an autoimmune phenomenon as potential novel aspect in pathophysiology of ovarian cancer. It is important to note that the cytokines most directly implicated in the pathophysiology of rheumatoid arthritis are proinflammatory TNFalpha and IL-6 [83]; herein these molecules are ranked within the 10-top most significantly over-represented regulators. Intriguingly, both TNFalpha and in particular IL-6 have been previously shown to promote epithelial ovarian tumorigenesis and cancer progression (reviewed in [84]). Numerous preclinical and translational studies emphasize the rational of targeting the IL-6/IL-6-signaling pathways in cancer, considering among others ovarian carcinoma, either as single treatment or in combination with other chemotherapeutic drugs [85, 86]; reviewed in [87]. The present study supports this notion. Furthermore, it proposes for consideration/reconsideration the assessment of IL-6 and other markers of arthritis including systemic autoantibodies and, as proposed previously, C-reactive protein [88] for monitoring the disease and therapy response in ovarian cancer. Noteworthy, data of recent epidemiological studies suggest an increased risk of developing ovarian cancer for patients with rheumatoid arthritis at advanced stage of disease; for entire, unstratified patient group the association was reported to be inverse [89].
Besides that, the data unexpectedly suggest that the therapeutic regiments considered for treatment of HIV by mechanism(s) of enhancing the APOBEC3G expression/activity might be added for consideration for the stratified group of high risk patients with serous ovarian cancer. Among those are IFNalpha and novel IFN-related mimetics preserving beneficial antiviral roles while minimizing negative effects [90]. It is important to emphasize that based on different argumentations and accenting the immunomodulatory and antiproliferative activities of IFNs family, the attempts have been already made to establish IFN as a standard in the treatment of ovarian cancer [91, 92]. However, the results were not monosemantic among the various clinical trials. This stresses the complexity of the disease and strongly indicates the necessity to stratify the patient population prior to drug application. Small molecules as agonists or antagonists [93, 94], which are able to modulate specifically the APOBEC3G and APOBEC3B levels and activities, respectively, can be as well considered as starting points for further development of combinatorial drug applications in ovarian cancer. Still, there is much to clarify regarding the expression patterns of APOBEC3G (as well as APOBEC3B) on the protein transcript levels in respect of the immune contexture and tumor anatomy applying the methodology of computerized assessment of large-scale ovarian cancer tissue sections (examples in [95, 96]).
We herein used a well-characterized patient cohort at advanced stage of ovarian cancer. This cohort reflects the current clinical situation in the medical care of ovarian cancer patients as most cases of ovarian cancer are diagnosed at advanced stages of disease due to inconspicuous symptoms and lack of reliable biomarkers [97]. Based on that, the data-driven conclusion likely suggests the contribution of AID/APOBEC-triggered mechanisms to the disease progression. However, their cancer-causing role cannot be as well excluded.
Conclusions
The herein defined analysis algorithm, MuSiCO, allows to establish a link between AID/APOBEC-associated gene expression profiles and patient survival, and to further delineate novel disease-associated pathways/networks. Based on the results of complex multivariable modeling, we propose a novel strategy for risk assessment of patients with primary ovarian cancer by integration of AID/APOBEC signature-based data sets and clinical risk factors into a combined survival model. We evaluated the performance of various prognostic models based on PEV, c-index and p-value. We propose to use these parameters to compare models not only within one study but also to make comparisons between independent studies and laboratories. Furthermore, we reconstructed a gene regulatory network on the basis of target molecules defined by prognostic modeling and the molecules from co-expressed genes derived from curated transcriptome-based expression data from the serous ovarian cancer-based studies. These findings link the expression pattern of AID/APOBEC-associated genes with remodeling/fibrotic pathways, altered immune response, and autoimmune disorders with inflammatory background (Fig. 4), and propose for a consideration of potential novel biomarkers and/or targets and therapeutic regiments, although with a strong indication for necessity to stratify the patient population prior to drug application. Among them are APOBEC3G, AID, ID3, IL-6, IFNalpha and novel IFN-related mimetics. This study additionally suggest to consolidate the acquired knowledge and research efforts in the fields of virology and cancer research around AID/APOBECs expression and functionality as well as drug targeting and drugs in development.
Abbreviations
AID (or AICDA), activation-induced cytidine deaminase; APOBEC, apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like; C-index, concordance index; EOC, epithelial ovarian cancer; FIGO, International Federation of Obstetrics and Gynecology; HR, hazard ratio; CI, confidence interval; IPA, Ingenuity Pathway Analysis; MuSiCO, from Multigene Signature to the Patient-Orientated Clinical Outcome; OS, overall survival; PEV, proportion of explained variation; PFS, progression free survival
Acknowledgments
This work was supported by the Austrian Science Fund (FWF; http://www.fwf.ac.at/) projects P22441-B13 and P23228-B19 (to D. Mechtcheriakova); the Sixth Framework Programme (FP6) Project of the European Union (EU) called ‘Ovarian Cancer: Diagnosis of a silent killer – OVCAD’, no. 018698.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its Additional file 1.
Authors' contributions
Conception and design: DM, MS, AM, GH, TT, and RZ. Experimental part, analysis and interpretation of data: MS, DM, AM, GH, MJ, DP, and PZ. Preparation of the manuscript: DM, MS, AM, GH, MJ and PZ with support of RZ, DCCT, DP, EJJ, PB, SM, and TT. Sample collection and maintaining patient database: RZ, DCCT, DP, GH, IB, JS, SL, IV, and SM. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
The study was approved in accordance to the requirements of the ethical committees of the individual institutions participating in OVCAD (EK207/2003, ML2524, HEK190504, EK366, EK260). Informed consent for the scientific use of biological material was obtained from all patients in accordance with the requirements of the ethics committees of the institutions involved; the herein participating OVCAD partners include Department of Gynecology, European Competence Center for Ovarian Cancer at Campus Virchow Klinikum, Charité – Medical University Berlin (Berlin, Germany), Division of Gynecological Oncology, Department of Obstetrics and Gynecology, Universitaire Ziekenhuizen Leuven, Katholieke Universiteit Leuven (Leuven, Belgium), Department of Obstetrics and Gynecology, Medical University of Vienna (Vienna, Austria), and Department of Gynecology, University Medical Center Hamburg - Eppendorf (Hamburg, Germany).
Additional file
The following additional data are available with the online version of this paper. Figure S1. Graphical view of the expression range of target genes used in GENEVESTIGATOR-based analysis. Figure S2. Correlation analysis of reference HKGs expression values in microarray data sets. Figure S3. Figure shows the impact of individual variables on survival prediction of the multivariable model (LASSO) for OS if predictors are changed by +1 SD. Figure S4. Figure shows LASSO-based Kaplan/Meier estimates for OS and PFS. Figure S5. Figure shows expression profiles of the AID/APOBEC-based multigene signature in ovarian cancer cell lines. Figure S6. Figure shows the extended analysis of expression profiles of the AID/APOBEC signature genes in a wide range of ovarian cancer cell lines (n = 55). Figure S7. Shown is the result of hierarchical clustering for individual genes composing the AID/APOBEC signature across arrays/samples of 55 ovarian cancer cell lines. Figure S8. The heat maps show the distribution of the Canonical Pathways/Functional Annotations/Upstream Regulators between corresponding target genes. Figure S9. The pie charts indicate the overlap of Canonical Pathways/Functional Annotations/Upstream Regulators between output_mixed and output_individual. Table S1. Real-time PCR primer sequences. Table S2. Real-time PCR primers. Table S3. Genes composing the AID/APOBEC multigene signature. Table S4. Univariate Cox regression analysis of clinicopathological variables and gene profiling-derived data sets for OS and PFS. Table S5. Correlation analysis for the AID/APOBEC multigene-derived variables. Table S6. Multivariable models (ridge) for PFS. Table S7. Comparative analysis of multivariable models (LASSO) for prognostication of OS and PFS. Table S8. Multivariable models (LASSO) for OS. Table S9. Multivariable models (LASSO) for PFS. Table S10. Top 50 Affymetrix probe sets co-regulated with APOBEC3G. Table S11. Top 50 Affymetrix probe sets co-regulated with ESR1. Table S12. Top 50 Affymetrix probe sets co-regulated with ID2. Table S13. Top 50 Affymetrix probe sets co-regulated with ID3. Table S14. Top 50 Affymetrix probe sets co-regulated with PTPRC/CD45. Table S15. 10-top-AID/APOBEC signature-linked Canonical Pathways. Table S16. Top-AID/APOBEC signature-linked Upstream Regulators. Table S17. 10-top-AID/APOBEC signature-linked Upstream Regulators. (PDF 4761 kb)
Contributor Information
Martin Svoboda, Email: martin.svoboda@gmx.net.
Anastasia Meshcheryakova, Email: anastasia.meshcheryakova@meduniwien.ac.at.
Georg Heinze, Email: georg.heinze@meduniwien.ac.at.
Markus Jaritz, Email: Markus.Jaritz@imp.ac.at.
Dietmar Pils, Email: dietmar.pils@univie.ac.at.
Dan Cacsire Castillo-Tong, Email: dan.cacsire-castillo@meduniwien.ac.at.
Gudrun Hager, Email: gudrunhager@ymail.com.
Theresia Thalhammer, Email: theresia.thalhammer@meduniwien.ac.at.
Erika Jensen-Jarolim, Email: erika.jensen-jarolim@meduniwien.ac.at.
Peter Birner, Email: peter.birner@meduniwien.ac.at.
Ioana Braicu, Email: elena.braicu@charite.de.
Jalid Sehouli, Email: jalid.sehouli@charite.de.
Sandrina Lambrechts, Email: sandrina.lambrechts@med.kuleuven.be.
Ignace Vergote, Email: ignace.vergote@med.kuleuven.be.
Sven Mahner, Email: s.mahner@uke.de.
Philip Zimmermann, Email: phz@nebion.com.
Robert Zeillinger, Email: robert.zeillinger@meduniwien.ac.at.
Diana Mechtcheriakova, Email: diana.mechtcheriakova@meduniwien.ac.at.
References
- 1.Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, et al. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J Biol Chem. 1999;274(26):18470–6. doi: 10.1074/jbc.274.26.18470. [DOI] [PubMed] [Google Scholar]
- 2.Okazaki IM, Hiai H, Kakazu N, Yamada S, Muramatsu M, Kinoshita K, et al. Constitutive expression of AID leads to tumorigenesis. J Exp Med. 2003;197(9):1173–81. doi: 10.1084/jem.20030275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Conticello SG. Creative deaminases, self-inflicted damage, and genome evolution. Ann N Y Acad Sci. 2012;1267:79–85. doi: 10.1111/j.1749-6632.2012.06614.x. [DOI] [PubMed] [Google Scholar]
- 4.Smith HC, Bennett RP, Kizilyer A, McDougall WM, Prohaska KM. Functions and regulation of the APOBEC family of proteins. Semin Cell Dev Biol. 2012;23(3):258–68. doi: 10.1016/j.semcdb.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vaidyanathan B, Yen WF, Pucella JN, Chaudhuri J. AIDing Chromatin and Transcription-Coupled Orchestration of Immunoglobulin Class-Switch Recombination. Front Immunol. 2014;5:120. doi: 10.3389/fimmu.2014.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuraoka M, Holl TM, Liao D, Womble M, Cain DW, Reynolds AE, et al. Activation-induced cytidine deaminase mediates central tolerance in B cells. Proc Natl Acad Sci U S A. 2011;108(28):11560–5. doi: 10.1073/pnas.1102571108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morgan HD, Dean W, Coker HA, Reik W, Petersen-Mahrt SK. Activation-induced cytidine deaminase deaminates 5-methylcytosine in DNA and is expressed in pluripotent tissues: implications for epigenetic reprogramming. J Biol Chem. 2004;279(50):52353–60. doi: 10.1074/jbc.M407695200. [DOI] [PubMed] [Google Scholar]
- 8.Hultquist JF, Lengyel JA, Refsland EW, LaRue RS, Lackey L, Brown WL, et al. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J Virol. 2011;85(21):11220–34. doi: 10.1128/JVI.05238-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424(6944):99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- 10.Stenglein MD, Burns MB, Li M, Lengyel J, Harris RS. APOBEC3 proteins mediate the clearance of foreign DNA from human cells. Nat Struct Mol Biol. 2010;17(2):222–9. doi: 10.1038/nsmb.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Teng B, Burant CF, Davidson NO. Molecular cloning of an apolipoprotein B messenger RNA editing protein. Science. 1993;260(5115):1816–9. doi: 10.1126/science.8511591. [DOI] [PubMed] [Google Scholar]
- 12.Rosenberg BR, Hamilton CE, Mwangi MM, Dewell S, Papavasiliou FN. Transcriptome-wide sequencing reveals numerous APOBEC1 mRNA-editing targets in transcript 3' UTRs. Nat Struct Mol Biol. 2011;18(2):230–6. doi: 10.1038/nsmb.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature. 2013;494(7437):366–70. doi: 10.1038/nature11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jeon S, Han S, Lee K, Choi J, Park SK, Park AK et al. Genetic variants of AICDA/CASP14 associated with childhood brain tumor. Genetics and molecular research : GMR. 2013;12(AOP). doi:10.4238/2013.January.30.1. [DOI] [PubMed]
- 15.Klein IA, Resch W, Jankovic M, Oliveira T, Yamane A, Nakahashi H, et al. Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell. 2011;147(1):95–106. doi: 10.1016/j.cell.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lada AG, Dhar A, Boissy RJ, Hirano M, Rubel AA, Rogozin IB, et al. AID/APOBEC cytosine deaminase induces genome-wide kataegis. Biol Direct. 2012;7:47. doi: 10.1186/1745-6150-7-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mechtcheriakova D, Svoboda M, Meshcheryakova A, Jensen-Jarolim E. Activation-induced cytidine deaminase (AID) linking immunity, chronic inflammation, and cancer. Cancer Immunol Immunother. 2012;61(9):1591–8. doi: 10.1007/s00262-012-1255-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nowarski R, Wilner OI, Cheshin O, Shahar OD, Kenig E, Baraz L, et al. APOBEC3G enhances lymphoma cell radioresistance by promoting cytidine deaminase-dependent DNA repair. Blood. 2012;120(2):366–75. doi: 10.1182/blood-2012-01-402123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okuyama S, Marusawa H, Matsumoto T, Ueda Y, Matsumoto Y, Endo Y, et al. Excessive activity of apolipoprotein B mRNA editing enzyme catalytic polypeptide 2 (APOBEC2) contributes to liver and lung tumorigenesis. Int J Cancer. 2012;130(6):1294–301. doi: 10.1002/ijc.26114. [DOI] [PubMed] [Google Scholar]
- 20.Shinmura K, Igarashi H, Goto M, Tao H, Yamada H, Matsuura S, et al. Aberrant expression and mutation-inducing activity of AID in human lung cancer. Ann Surg Oncol. 2011;18(7):2084–92. doi: 10.1245/s10434-011-1568-8. [DOI] [PubMed] [Google Scholar]
- 21.Shinohara M, Io K, Shindo K, Matsui M, Sakamoto T, Tada K, et al. APOBEC3B can impair genomic stability by inducing base substitutions in genomic DNA in human cells. Sci Rep. 2012;2:806. doi: 10.1038/srep00806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamanaka S, Balestra ME, Ferrell LD, Fan J, Arnold KS, Taylor S, et al. Apolipoprotein B mRNA-editing protein induces hepatocellular carcinoma and dysplasia in transgenic animals. Proc Natl Acad Sci U S A. 1995;92(18):8483–7. doi: 10.1073/pnas.92.18.8483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamane A, Resch W, Kuo N, Kuchen S, Li Z, Sun HW, et al. Deep-sequencing identification of the genomic targets of the cytidine deaminase AID and its cofactor RPA in B lymphocytes. Nat Immunol. 2011;12(1):62–9. doi: 10.1038/ni.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dorsett Y, McBride KM, Jankovic M, Gazumyan A, Thai TH, Robbiani DF, et al. MicroRNA-155 suppresses activation-induced cytidine deaminase-mediated Myc-Igh translocation. Immunity. 2008;28(5):630–8. doi: 10.1016/j.immuni.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feldhahn N, Henke N, Melchior K, Duy C, Soh BN, Klein F, et al. Activation-induced cytidine deaminase acts as a mutator in BCR-ABL1-transformed acute lymphoblastic leukemia cells. J Exp Med. 2007;204(5):1157–66. doi: 10.1084/jem.20062662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gruber TA, Chang MS, Sposto R, Muschen M. Activation-induced cytidine deaminase accelerates clonal evolution in BCR-ABL1-driven B-cell lineage acute lymphoblastic leukemia. Cancer Res. 2010;70(19):7411–20. doi: 10.1158/0008-5472.CAN-10-1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klemm L, Duy C, Iacobucci I, Kuchen S, von Levetzow G, Feldhahn N, et al. The B cell mutator AID promotes B lymphoid blast crisis and drug resistance in chronic myeloid leukemia. Cancer Cell. 2009;16(3):232–45. doi: 10.1016/j.ccr.2009.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robbiani DF, Bothmer A, Callen E, Reina-San-Martin B, Dorsett Y, Difilippantonio S, et al. AID is required for the chromosomal breaks in c-myc that lead to c-myc/IgH translocations. Cell. 2008;135(6):1028–38. doi: 10.1016/j.cell.2008.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kou T, Marusawa H, Kinoshita K, Endo Y, Okazaki IM, Ueda Y, et al. Expression of activation-induced cytidine deaminase in human hepatocytes during hepatocarcinogenesis. Int J Cancer. 2007;120(3):469–76. doi: 10.1002/ijc.22292. [DOI] [PubMed] [Google Scholar]
- 30.Matsumoto Y, Marusawa H, Kinoshita K, Niwa Y, Sakai Y, Chiba T. Up-regulation of activation-induced cytidine deaminase causes genetic aberrations at the CDKN2b-CDKN2a in gastric cancer. Gastroenterology. 2010;139(6):1984–94. doi: 10.1053/j.gastro.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 31.Morita S, Matsumoto Y, Okuyama S, Ono K, Kitamura Y, Tomori A, et al. Bile acid-induced expression of activation-induced cytidine deaminase during the development of Barrett's oesophageal adenocarcinoma. Carcinogenesis. 2011;32(11):1706–12. doi: 10.1093/carcin/bgr194. [DOI] [PubMed] [Google Scholar]
- 32.Ramiro AR, Jankovic M, Eisenreich T, Difilippantonio S, Chen-Kiang S, Muramatsu M, et al. AID is required for c-myc/IgH chromosome translocations in vivo. Cell. 2004;118(4):431–8. doi: 10.1016/j.cell.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 33.Endo Y, Marusawa H, Kou T, Nakase H, Fujii S, Fujimori T, et al. Activation-induced cytidine deaminase links between inflammation and the development of colitis-associated colorectal cancers. Gastroenterology. 2008;135(3):889–98. doi: 10.1053/j.gastro.2008.06.091. [DOI] [PubMed] [Google Scholar]
- 34.Matsumoto Y, Marusawa H, Kinoshita K, Endo Y, Kou T, Morisawa T, et al. Helicobacter pylori infection triggers aberrant expression of activation-induced cytidine deaminase in gastric epithelium. Nat Med. 2007;13(4):470–6. doi: 10.1038/nm1566. [DOI] [PubMed] [Google Scholar]
- 35.Svoboda M, Kaufmann T, Meshcheryakova A, Bajna E, Jensen-Jarolim E, Mechtcheriakova D. Multigene Signature Approach to Assess the Role of AID/APOBEC Family Members During the Epithelial-to-mesenchymal Transition Program. Eur J Cancer. 2012;48:S111–S. doi: 10.1016/S0959-8049(12)71137-1. [DOI] [Google Scholar]
- 36.Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet. 2013;45(9):970–6. doi: 10.1038/ng.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sieuwerts AM, Willis S, Burns MB, Look MP, Meijer-Van Gelder ME, Schlicker A, et al. Elevated APOBEC3B correlates with poor outcomes for estrogen-receptor-positive breast cancers. Hormones Cancer. 2014;5(6):405–13. doi: 10.1007/s12672-014-0196-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–15. doi:10.1038/nature10166. [DOI] [PMC free article] [PubMed]
- 39.Pauklin S, Sernandez IV, Bachmann G, Ramiro AR, Petersen-Mahrt SK. Estrogen directly activates AID transcription and function. J Exp Med. 2009;206(1):99–111. doi: 10.1084/jem.20080521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leonard B, Hart SN, Burns MB, Carpenter MA, Temiz NA, Rathore A, et al. APOBEC3B upregulation and genomic mutation patterns in serous ovarian carcinoma. Cancer Res. 2013;73(24):7222–31. doi: 10.1158/0008-5472.CAN-13-1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu JF, Konstantinopoulos PA, Matulonis UA. PARP inhibitors in ovarian cancer: current status and future promise. Gynecol Oncol. 2014;133(2):362–9. doi: 10.1016/j.ygyno.2014.02.039. [DOI] [PubMed] [Google Scholar]
- 42.Agarwal R, Kaye SB. Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat Rev Cancer. 2003;3(7):502–16. doi: 10.1038/nrc1123. [DOI] [PubMed] [Google Scholar]
- 43.Markman M. Antineoplastic agents in the management of ovarian cancer: current status and emerging therapeutic strategies. Trends Pharmacol Sci. 2008;29(10):515–9. doi: 10.1016/j.tips.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 44.Gillet JP, Calcagno AM, Varma S, Davidson B, Bunkholt Elstrand M, Ganapathi R, et al. Multidrug resistance-linked gene signature predicts overall survival of patients with primary ovarian serous carcinoma. Clin Cancer Res. 2012;18(11):3197–206. doi: 10.1158/1078-0432.CCR-12-0056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mechtcheriakova D, Sobanov Y, Holtappels G, Bajna E, Svoboda M, Jaritz M, et al. Activation-induced cytidine deaminase (AID)-associated multigene signature to assess impact of AID in etiology of diseases with inflammatory component. PLoS One. 2011;6(10):e25611. doi: 10.1371/journal.pone.0025611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yoshihara K, Tajima A, Komata D, Yamamoto T, Kodama S, Fujiwara H, et al. Gene expression profiling of advanced-stage serous ovarian cancers distinguishes novel subclasses and implicates ZEB2 in tumor progression and prognosis. Cancer Sci. 2009;100(8):1421–8. doi: 10.1111/j.1349-7006.2009.01204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meshcheryakova A, Svoboda M, Tahir A, Kofeler HC, Triebl A, Mungenast F et al. Exploring the role of sphingolipid machinery during the epithelial to mesenchymal transition program using an integrative approach. Oncotarget. 2016. doi:10.18632/oncotarget.7947. [DOI] [PMC free article] [PubMed]
- 48.Chekerov R, Braicu I, Castillo-Tong DC, Richter R, Cadron I, Mahner S, et al. Outcome and clinical management of 275 patients with advanced ovarian cancer International Federation of Obstetrics and Gynecology II to IV inside the European Ovarian Cancer Translational Research Consortium-OVCAD. Int J Gynecol Cancer. 2013;23(2):268–75. doi: 10.1097/IGC.0b013e31827de6b9. [DOI] [PubMed] [Google Scholar]
- 49.Pils D, Bachmayr-Heyda A, Auer K, Svoboda M, Auner V, Hager G, et al. Cyclin E1 (CCNE1) as independent positive prognostic factor in advanced stage serous ovarian cancer patients - a study of the OVCAD consortium. Eur J Cancer. 2014;50(1):99–110. doi: 10.1016/j.ejca.2013.09.011. [DOI] [PubMed] [Google Scholar]
- 50.Pils D, Hager G, Tong D, Aust S, Heinze G, Kohl M, et al. Validating the impact of a molecular subtype in ovarian cancer on outcomes: a study of the OVCAD Consortium. Cancer Sci. 2012;103(7):1334–41. doi: 10.1111/j.1349-7006.2012.02306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pepe A, Sun L, Zanardi I, Wu X, Ferlini C, Fontana G, et al. Novel C-seco-taxoids possessing high potency against paclitaxel-resistant cancer cell lines overexpressing class III beta-tubulin. Bioorg Med Chem Lett. 2009;19(12):3300–4. doi: 10.1016/j.bmcl.2009.04.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Prislei S, Mariani M, Raspaglio G, Mozzetti S, Filippetti F, Ferrandina G, et al. RON and cisplatin resistance in ovarian cancer cell lines. Oncol Res. 2010;19(1):13–22. doi: 10.3727/096504010X12828372551713. [DOI] [PubMed] [Google Scholar]
- 53.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3(7):RESEARCH0034. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25(4):402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 55.Van Buuren S, Groothuis-Oudshoorn K. mice: Multivariate Imputation by Chained Equations in R. J Stat Softw. 2011;45(3):1–67. doi: 10.18637/jss.v045.i03. [DOI] [Google Scholar]
- 56.Simon N, Friedman J, Hastie T, Tibshirani R. Regularization Paths for Cox's Proportional Hazards Model via Coordinate Descent. J Stat Softw. 2011;39(5):1–13. doi: 10.18637/jss.v039.i05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Uno H, Cai T, Pencina MJ, D'Agostino RB, Wei LJ. On the C-statistics for evaluating overall adequacy of risk prediction procedures with censored survival data. Stat Med. 2011;30(10):1105–17. doi: 10.1002/sim.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schemper M, Henderson R. Predictive accuracy and explained variation in Cox regression. Biometrics. 2000;56(1):249–55. doi: 10.1111/j.0006-341X.2000.00249.x. [DOI] [PubMed] [Google Scholar]
- 59.GENEVESTIGATOR - Shaping biological discovery. https://www.genevestigator.com/gv/. Accessed 26 July 2013 and 29 April 2016.
- 60.Hruz T, Laule O, Szabo G, Wessendorp F, Bleuler S, Oertle L, et al. Genevestigator v3: a reference expression database for the meta-analysis of transcriptomes. Adv Bioinforma. 2008;2008:420747. doi: 10.1155/2008/420747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ingenuity. http://www.ingenuity.com/. Accessed 11 August 2014.
- 62.TIBCO Spotfire. http://spotfire.tibco.com/. Accessed 18 July 2014.
- 63.Sayegh CE, Quong MW, Agata Y, Murre C. E-proteins directly regulate expression of activation-induced deaminase in mature B cells. Nat Immunol. 2003;4(6):586–93. doi: 10.1038/ni923. [DOI] [PubMed] [Google Scholar]
- 64.Xu Z, Pone EJ, Al-Qahtani A, Park SR, Zan H, Casali P. Regulation of aicda expression and AID activity: relevance to somatic hypermutation and class switch DNA recombination. Crit Rev Immunol. 2007;27(4):367–97. doi: 10.1615/CritRevImmunol.v27.i4.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bhutani N, Brady JJ, Damian M, Sacco A, Corbel SY, Blau HM. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature. 2010;463(7284):1042–7. doi: 10.1038/nature08752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kotnis A, Du L, Liu C, Popov SW, Pan-Hammarstrom Q. Non-homologous end joining in class switch recombination: the beginning of the end. Philos Trans R Soc Lond Ser B Biol Sci. 2009;364(1517):653–65. doi: 10.1098/rstb.2008.0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun. 2013;4:2126. doi: 10.1038/ncomms3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Simon RM, Subramanian J, Li MC, Menezes S. Using cross-validation to evaluate predictive accuracy of survival risk classifiers based on high-dimensional data. Brief Bioinform. 2011;12(3):203–14. doi: 10.1093/bib/bbr001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lasorella A, Benezra R, Iavarone A. The ID proteins: master regulators of cancer stem cells and tumour aggressiveness. Nat Rev Cancer. 2014;14(2):77–91. doi: 10.1038/nrc3638. [DOI] [PubMed] [Google Scholar]
- 70.Ren Y, Cheung HW, von Maltzhan G, Agrawal A, Cowley GS, Weir BA, et al. Targeted tumor-penetrating siRNA nanocomplexes for credentialing the ovarian cancer oncogene ID4. Sci Transl Med. 2012;4(147):147ra12. doi: 10.1126/scitranslmed.3003778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schindl M, Schoppmann SF, Strobel T, Heinzl H, Leisser C, Horvat R, et al. Level of Id-1 protein expression correlates with poor differentiation, enhanced malignant potential, and more aggressive clinical behavior of epithelial ovarian tumors. Clin Cancer Res. 2003;9(2):779–85. [PubMed] [Google Scholar]
- 72.Shepherd TG, Theriault BL, Nachtigal MW. Autocrine BMP4 signalling regulates ID3 proto-oncogene expression in human ovarian cancer cells. Gene. 2008;414(1–2):95–105. doi: 10.1016/j.gene.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 73.Chaudhary J, Garland W, Salvador R. A novel small molecule inhibitor of Id proteins (AGX-51) blocks cell survival in vitro and diminishes angiogenesis and tumor growth in vivo. Faseb J. 2009;23:761.
- 74.Garland W, Salvador R, Chaudhary J. Abstract 758: Studies in cancer models with AGX51, a Direct Transcriptional RegulatorTM. Cancer Res. 2013;73(8). doi:10.1158/1538-7445.AM2013-758.
- 75.Beale RC, Petersen-Mahrt SK, Watt IN, Harris RS, Rada C, Neuberger MS. Comparison of the differential context-dependence of DNA deamination by APOBEC enzymes: correlation with mutation spectra in vivo. J Mol Biol. 2004;337(3):585–96. doi: 10.1016/j.jmb.2004.01.046. [DOI] [PubMed] [Google Scholar]
- 76.Henderson S, Chakravarthy A, Su X, Boshoff C, Fenton TR. APOBEC-mediated cytosine deamination links PIK3CA helical domain mutations to human papillomavirus-driven tumor development. Cell Rep. 2014;7(6):1833–41. doi: 10.1016/j.celrep.2014.05.012. [DOI] [PubMed] [Google Scholar]
- 77.Vieira VC, Soares MA. The role of cytidine deaminases on innate immune responses against human viral infections. BioMed Res Int. 2013;2013:683095. doi: 10.1155/2013/683095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vartanian JP, Guetard D, Henry M, Wain-Hobson S. Evidence for editing of human papillomavirus DNA by APOBEC3 in benign and precancerous lesions. Science. 2008;320(5873):230–3. doi: 10.1126/science.1153201. [DOI] [PubMed] [Google Scholar]
- 79.Batista L, Gruosso T, Mechta-Grigoriou F. Ovarian cancer emerging subtypes: role of oxidative stress and fibrosis in tumour development and response to treatment. Int J Biochem Cell Biol. 2013;45(6):1092–8. doi: 10.1016/j.biocel.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 80.Samrao D, Wang D, Ough F, Lin YG, Liu S, Menesses T, et al. Histologic parameters predictive of disease outcome in women with advanced stage ovarian carcinoma treated with neoadjuvant chemotherapy. Transl Oncol. 2012;5(6):469–74. doi: 10.1593/tlo.12265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hatzirodos N, Hummitzsch K, Irving-Rodgers HF, Harland ML, Morris SE, Rodgers RJ. Transcriptome profiling of granulosa cells from bovine ovarian follicles during atresia. BMC Genomics. 2014;15:40. doi: 10.1186/1471-2164-15-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yoshihara K, Tsunoda T, Shigemizu D, Fujiwara H, Hatae M, Masuzaki H, et al. High-risk ovarian cancer based on 126-gene expression signature is uniquely characterized by downregulation of antigen presentation pathway. Clin Cancer Res. 2012;18(5):1374–85. doi: 10.1158/1078-0432.CCR-11-2725. [DOI] [PubMed] [Google Scholar]
- 83.Choy E. Understanding the dynamics: pathways involved in the pathogenesis of rheumatoid arthritis. Rheumatology (Oxford) 2012;51(Suppl 5):v3–11. doi: 10.1093/rheumatology/kes113. [DOI] [PubMed] [Google Scholar]
- 84.Maccio A, Madeddu C. Inflammation and ovarian cancer. Cytokine. 2012;58(2):133–47. doi: 10.1016/j.cyto.2012.01.015. [DOI] [PubMed] [Google Scholar]
- 85.Dijkgraaf EM, Welters MJ, Nortier JW, van der Burg SH, Kroep JR. Interleukin-6/interleukin-6 receptor pathway as a new therapy target in epithelial ovarian cancer. Curr Pharm Des. 2012;18(25):3816–27. doi: 10.2174/138161212802002797. [DOI] [PubMed] [Google Scholar]
- 86.Guo Y, Nemeth J, O'Brien C, Susa M, Liu X, Zhang Z, et al. Effects of siltuximab on the IL-6-induced signaling pathway in ovarian cancer. Clin Cancer Res. 2010;16(23):5759–69. doi: 10.1158/1078-0432.CCR-10-1095. [DOI] [PubMed] [Google Scholar]
- 87.Yao X, Huang J, Zhong H, Shen N, Faggioni R, Fung M, et al. Targeting interleukin-6 in inflammatory autoimmune diseases and cancers. Pharmacol Ther. 2014;141(2):125–39. doi: 10.1016/j.pharmthera.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 88.Hefler LA, Concin N, Hofstetter G, Marth C, Mustea A, Sehouli J, et al. Serum C-reactive protein as independent prognostic variable in patients with ovarian cancer. Clin Cancer Res. 2008;14(3):710–4. doi: 10.1158/1078-0432.CCR-07-1044. [DOI] [PubMed] [Google Scholar]
- 89.Hemminki K, Liu X, Ji J, Forsti A, Sundquist J, Sundquist K. Effect of autoimmune diseases on risk and survival in female cancers. Gynecol Oncol. 2012;127(1):180–5. doi: 10.1016/j.ygyno.2012.07.100. [DOI] [PubMed] [Google Scholar]
- 90.Vazquez N, Schmeisser H, Dolan MA, Bekisz J, Zoon KC, Wahl SM. Structural variants of IFNalpha preferentially promote antiviral functions. Blood. 2011;118(9):2567–77. doi: 10.1182/blood-2010-12-325027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gavalas NG, Karadimou A, Dimopoulos MA, Bamias A. Immune response in ovarian cancer: how is the immune system involved in prognosis and therapy: potential for treatment utilization. Clin Dev Immunol. 2010;2010:791603. doi: 10.1155/2010/791603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lawal AO, Musekiwa A, Grobler L. Interferon after surgery for women with advanced (Stage II-IV) epithelial ovarian cancer. Cochrane Database Syst Rev. 2013;6:CD009620. doi: 10.1002/14651858.CD009620.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li M, Shandilya SM, Carpenter MA, Rathore A, Brown WL, Perkins AL, et al. First-in-class small molecule inhibitors of the single-strand DNA cytosine deaminase APOBEC3G. ACS Chem Biol. 2012;7(3):506–17. doi: 10.1021/cb200440y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Matsui M, Shindo K, Izumi T, Io K, Shinohara M, Komano J, et al. Small molecules that inhibit Vif-induced degradation of APOBEC3G. Virol J. 2014;11:122. doi: 10.1186/1743-422X-11-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Meshcheryakova A, Tamandl D, Bajna E, Stift J, Mittlboeck M, Svoboda M, et al. B cells and ectopic follicular structures: novel players in anti-tumor programming with prognostic power for patients with metastatic colorectal cancer. PLoS One. 2014;9(6):e99008. doi: 10.1371/journal.pone.0099008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39(4):782–95. doi: 10.1016/j.immuni.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 97.Permuth-Wey J, Sellers TA. Epidemiology of ovarian cancer. Methods Mol Biol. 2009;472:413–37. doi: 10.1007/978-1-60327-492-0_20. [DOI] [PubMed] [Google Scholar]
- 98.Mateescu B, Batista L, Cardon M, Gruosso T, de Feraudy Y, Mariani O, et al. miR-141 and miR-200a act on ovarian tumorigenesis by controlling oxidative stress response. Nat Med. 2011;17(12):1627–35. doi: 10.1038/nm.2512. [DOI] [PubMed] [Google Scholar]
- 99.Leffers N, Fehrmann RS, Gooden MJ, Schulze UR, Ten Hoor KA, Hollema H, et al. Identification of genes and pathways associated with cytotoxic T lymphocyte infiltration of serous ovarian cancer. Br J Cancer. 2010;103(5):685–92. doi: 10.1038/sj.bjc.6605820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Scarlett UK, Cubillos-Ruiz JR, Nesbeth YC, Martinez DG, Engle X, Gewirtz AT, et al. In situ stimulation of CD40 and Toll-like receptor 3 transforms ovarian cancer-infiltrating dendritic cells from immunosuppressive to immunostimulatory cells. Cancer Res. 2009;69(18):7329–37. doi: 10.1158/0008-5472.CAN-09-0835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Casartelli N, Guivel-Benhassine F, Bouziat R, Brandler S, Schwartz O, Moris A. The antiviral factor APOBEC3G improves CTL recognition of cultured HIV-infected T cells. J Exp Med. 2010;207(1):39–49. doi: 10.1084/jem.20091933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Peng G, Lei KJ, Jin W, Greenwell-Wild T, Wahl SM. Induction of APOBEC3 family proteins, a defensive maneuver underlying interferon-induced anti-HIV-1 activity. J Exp Med. 2006;203(1):41–6. doi: 10.1084/jem.20051512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhou L, Wang X, Wang YJ, Zhou Y, Hu S, Ye L, et al. Activation of toll-like receptor-3 induces interferon-lambda expression in human neuronal cells. Neuroscience. 2009;159(2):629–37. doi: 10.1016/j.neuroscience.2008.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wasiuk A, Dalton DK, Schpero WL, Stan RV, Conejo-Garcia JR, Noelle RJ. Mast cells impair the development of protective anti-tumor immunity. Cancer Immunol Immunother. 2012;61(12):2273–82. doi: 10.1007/s00262-012-1276-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Harizi H. Reciprocal crosstalk between dendritic cells and natural killer cells under the effects of PGE2 in immunity and immunopathology. Cell Mol Immunol. 2013;10(3):213–21. doi: 10.1038/cmi.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wong JL, Berk E, Edwards RP, Kalinski P. IL-18-primed helper NK cells collaborate with dendritic cells to promote recruitment of effector CD8+ T cells to the tumor microenvironment. Cancer Res. 2013;73(15):4653–62. doi: 10.1158/0008-5472.CAN-12-4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Milliken D, Scotton C, Raju S, Balkwill F, Wilson J. Analysis of chemokines and chemokine receptor expression in ovarian cancer ascites. Clin Cancer Res. 2002;8(4):1108–14. [PubMed] [Google Scholar]
- 108.Tsukishiro S, Suzumori N, Nishikawa H, Arakawa A, Suzumori K. Elevated serum RANTES levels in patients with ovarian cancer correlate with the extent of the disorder. Gynecol Oncol. 2006;102(3):542–5. doi: 10.1016/j.ygyno.2006.01.029. [DOI] [PubMed] [Google Scholar]
- 109.Lee C, Liu QH, Tomkowicz B, Yi Y, Freedman BD, Collman RG. Macrophage activation through CCR5- and CXCR4-mediated gp120-elicited signaling pathways. J Leukoc Biol. 2003;74(5):676–82. doi: 10.1189/jlb.0503206. [DOI] [PubMed] [Google Scholar]
- 110.Kormendy D, Hoff H, Hoff P, Broker BM, Burmester GR, Brunner-Weinzierl MC. Impact of the CTLA-4/CD28 axis on the processes of joint inflammation in rheumatoid arthritis. Arthritis Rheum. 2013;65(1):81–7. doi: 10.1002/art.37714. [DOI] [PubMed] [Google Scholar]
- 111.Michel F, Acuto O. CD28 costimulation: a source of Vav-1 for TCR signaling with the help of SLP-76? Sci STKE. 2002;2002(144):e35. doi: 10.1126/stke.2002.144.pe35. [DOI] [PubMed] [Google Scholar]
- 112.Choi SW, Levine JE, Ferrara JL. Pathogenesis and management of graft-versus-host disease. Immunol Allergy Clin N Am. 2010;30(1):75–101. doi: 10.1016/j.iac.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analysed during this study are included in this published article and its Additional file 1.