

Abstract
Neurofibromatosis type 1 (NF1) is a common neurogenetic condition characterized by significant clinical heterogeneity. A major barrier to developing precision medicine approaches for NF1 is an incomplete understanding of the factors that underlie its inherent variability. To determine the impact of the germline NF1 gene mutation on the optic gliomas frequently encountered in children with NF1, we developed genetically engineered mice harboring two representative NF1-patient-derived Nf1 gene mutations (c.2542G>C;p.G848R and c.2041C>T;p.R681X). We found that each germline Nf1 gene mutation resulted in different levels of neurofibromin expression. Importantly, only R681XCKO but not G848RCKO, mice develop optic gliomas with increased optic nerve volumes, glial fibrillary acid protein immunoreactivity, proliferation and retinal ganglion cell death, similar to Nf1 conditional knockout mice harboring a neomycin insertion (neo) as the germline Nf1 gene mutation. These differences in optic glioma phenotypes reflect both cell-autonomous and stromal effects of the germline Nf1 gene mutation. In this regard, primary astrocytes harboring the R681X germline Nf1 gene mutation exhibit increased basal astrocyte proliferation (BrdU incorporation) indistinguishable from neoCKO astrocytes, whereas astrocytes with the G848R mutation have lower levels of proliferation. Evidence for paracrine effects from the tumor microenvironment were revealed when R681XCKO mice were compared with conventional neoCKO mice. Relative to neoCKO mice, the optic gliomas from R681XCKO mice had more microglia infiltration and JNKThr183/Tyr185 activation, microglia-produced Ccl5, and glial AKTThr308 activation. Collectively, these studies establish that the germline Nf1 gene mutation is a major determinant of optic glioma development and growth through by both tumor cell-intrinsic and stromal effects.
Introduction
Neurofibromatosis type 1 (NF1) is a common genetic disorder, affecting 1 in 3000 people worldwide (1). While NF1 is a classic autosomal dominant inherited monogenic condition with complete penetrance, expression of the clinical features of this disorder is extremely variable (2,3). In this regard, children and adults with NF1 are at risk for developing a wide range of medical problems, including benign and malignant peripheral nerve sheath tumors, bone defects, cognitive and attentional deficits, heart defects, breast cancer, autism and brain tumors. Unfortunately, it is currently not possible to predict which of these various clinical abnormalities will manifest in any given individual. The lack of prognostic risk factors to guide patient management represents one of the most significant barriers to actualizing personalized (precision) medicine strategies for children and adults affected with NF1.
Converging evidence from epidemiologic and human NF1-patient-induced pluripotent stem cell (iPSC) studies has raised the possibility that the germline NF1 gene mutation may be one such predictive risk factor. As such, several groups have reported genotype–phenotype correlations, including clustering of 5′-tertile NF1 gene mutations in individuals with NF1 optic pathway glioma (OPG) (4,5) and two specific germline NF1 gene mutations in people who do not develop cutaneous neurofibromas [c.2970-2972_delAAT; (6); c.5425C>T; (7)]. In addition, patients with large 1.4 Mb genomic microdeletions, including the entire NF1 gene locus, have an increased number of neurofibromas and are at an elevated risk for cardiac malfunction, skeletal anomalies, facial dysmorphism, and malignant tumor development (8). Moreover, studies employing NF1-patient-derived iPSCs and derivative neural progenitor (NP) cells have recently revealed that different NF1 germline mutations have distinct effects on NF1 protein (neurofibromin) expression and function (9). While these early-phase genotype–phenotype studies are intriguing, the potential mechanistic impact of the germline NF1 gene mutation on disease pathogenesis has not been formally investigated.
One of the most common tumors affecting children with NF1 is the OPG (10). In this regard, 15–20% of children with NF1 develop World Health Organization grade I pilocytic astrocytomas, a low-grade brain tumor characterized by low proliferative indices and infiltration of microglia (11). While seldom fatal, these OPGs are often associated with visual decline (12,13). Over the past two decades, the cellular and molecular mechanisms responsible for NF1-associated optic glioma (14,15) have been investigated using genetically engineered mouse (GEM) models, resulting in the identification of several promising therapeutic drug targets (16,17). However, these Nf1 mutant mice harbor a germline inactivating disruption of the murine Nf1 gene created by introducing a neomycin targeting cassette into exon 31, a mutation not found in people with NF1 (18,19). In this regard, the impact of this knockout allele could either under- or over-represent the effects of actual NF1-patient germline NF1 gene mutations, and as such, fails to capture the full spectrum of clinical heterogeneity that characterizes the NF1 patient population.
To formally evaluate the potential impact of the germline NF1 gene mutation on optic glioma formation and growth, we performed proof-of-principle studies using Nf1 GEM strains harboring two distinct NF1 patient-derived germline Nf1 gene mutations. Herein, we demonstrate that mice harboring the R681X, but not the G848R, mutation develop optic gliomas. In addition, we show that the R681X mutation results in gliomas with greater microglia infiltration, tumor proliferation, and associated retinal ganglion cell loss relative to conventional knockout mice, revealing both cell-autonomous and stromal effects for the germline Nf1 gene mutation on disease pathogenesis. Collectively, these data provide the first demonstration that the germline NF1 gene mutation is one important factor that underlies clinical heterogeneity relevant to risk assessment in this common neurogenetic condition.
Results
Neurofibromin expression is dictated by germline Nf1 gene mutation
To establish the impact of the germline Nf1 gene mutation on optic glioma formation, we leveraged GEM technology to engineer two distinct patient-derived NF1 gene mutations into C57BL/6 ES cells and generate mice harboring these germline mutations (Li K and Kesterson R, manuscript in preparation). The first line harbors a c.2542G>C missense mutation in exon 21 [Gly848Arg; G848R; (20)], while the second harbors a c.2041C>T nonsense mutation [Arg681*; R681X; (21)], causing a premature stop codon in exon 18 (Fig. 1A). Since murine Nf1 optic glioma formation requires the coupling of somatic Nf1 inactivation in neuroglial progenitors with a germline Nf1 gene mutation (14,22), we employed the glial fibrillary acid protein (GFAP)-Cre strain to generate conditional Nf1 knockout (CKO) mice (23) bearing each NF1-patient germline Nf1 gene mutation: Nf1flox/G484R; GFAP-Cre (G848RCKO) and Nf1flox/R681X; GFAP-Cre (R681XCKO) mice. All mice were maintained on a pure C57BL/6 genetic background. To determine whether neurofibromin expression was differentially impacted by these NF1 patient-derived germline Nf1 gene mutations, G848RCKO, neoCKO and R681XCKO mouse brainstem samples were analyzed by western blot. G848RCKO, neoCKO and R681XCKO mice exhibited 38, 47 and 77% reductions in brainstem neurofibromin expression, respectively, relative to control mice (Fig.1B). A similar pattern of neurofibromin expression was observed in the optic nerves from G848RCKO, neoCKO and R681XCKO mice by immunohistochemistry (Fig.1C).
Figure 1.

The germline Nf1 gene mutation determines the level of neurofibromin expression. (A) Schematic illustration of the mouse Nf1 gene, with the alternate exons and RAS-GAP domain indicated. The positions of the three germline Nf1 gene mutations are depicted above exons 18, 21 and 31. (B) Neurofibromin expression was reduced in the brainstem of mice with Cre-mediated Nf1flox inactivation and G848RCKO and R681XCKO or null KO (neoCKO) alleles by 38, 77 and 47%, respectively, relative to control mice following normalization to α-tubulin. (C) The effect of these germline Nf1 gene mutations on neurofibromin expression was examined in the optic nerve by immunohistochemistry. At least three mice were used per genotype.
Optic glioma formation is dependent on the germline Nf1 gene mutation
Based on prior studies demonstrating that Nf1flox/neo; GFAP-Cre CKO (neoCKO) mice (harboring the neomycin knockout allele) develop optic gliomas with >95% penetrance by 3 months of age (16,24), G848RCKO and R681XCKO mice were aged to 3 months prior to optic nerve analysis. We analyzed a minimum of 12 mice per genotype in total. Whereas G848RCKO mice had optic nerve volumes indistinguishable from controls, there was a 43% increase in optic nerve volumes from R681XCKO mice (>95% penetrance by 3 months), similar to neoCKO mice (48% increase; Fig. 2A). In addition, R681XCKO mouse optic nerves had increased cellularity and atypical cells, histopathologic features similar to neoCKO mouse optic gliomas. These features were not found in G848RCKO mouse optic nerves (Fig. 2B). Immunostaining for GFAP, a marker of glial cells, revealed the highest level of immunoreactivity in the optic nerves of R681XCKO mice, slightly greater than observed in neoCKO mouse optic gliomas. Similarly, R681XCKO optic nerves exhibited increased proliferation (%Ki67+ cells) compared with control and G848RCKO mice (∼14-fold increase) as well as to neoCKO optic nerves (2.2-fold increase) (Fig. 2D).
Figure 2.

The germline Nf1 gene mutation dictates optic glioma formation and proliferation. (A) Increased optic nerve volumes were observed in conditional knockout (neoCKO) (1.49-fold) and R681XCKO (1.43-fold) optic nerves relative to Nf1flox/flox control littermates (CTL), while no significant changes were detected between G848RCKO mice (mean = 0.07 ± 0.005 mm3) and controls (mean = 0.062 ± 0.005 mm3). (n = 6 mice per genotype). (B) Haematoxylin and eosin staining of the optic nerves revealed hypercellularity and nuclear atypia in neoCKO and R681XCKO optic nerves (n = 6 mice per genotype). (C) Increased GFAP immunoreactivity is observed in neoCKO and R681XCKO compared with no increase seen between G868RCKO and controls. (D) R681XCKO mice exhibited increased Ki67 immunoreactivity (14-fold; 2.2-fold) compared with control and neoCKO mice, respectively (n = 5 mice per genotype). Scale bars: 100 µm. All data are represented as means ± s.e.m. (*P < 0.05; one-way ANOVA with Bonferroni post-test).
One of the common morbidities seen in children with NF1-associated optic gliomas is reduced visual acuity (12,13) due to retinal ganglion cell (RGC) loss and retinal nerve fiber layer (RNFL) thinning (25). We used phospho-neurofilament heavy (pNF-H) chain immunohistochemistry as a surrogate marker of axonal injury in the optic nerve, based on previous studies in other models of nervous system injury (26,27). Increased pNF-H immunoreactivity was detected in the optic nerves from neoCKO mice, which was further elevated with punctate staining in R681XCKO mice (Fig. 3A) Consistent with the differential impact of the germline Nf1 gene mutation on optic glioma formation and growth (proliferation), the retinae from R681XCKO mice exhibited a 5.3-fold increase in TUNEL+ cells, a 3.2-fold reduction of Brn3a+ RGCs, and a 3.3-fold decrease in RNFL thickness relative to G848RCKO and control mice (Fig. 3B). Importantly, there was a 1.5-fold increase in TUNEL+ cells, 27% reduction of Brn3a+ RGCs, and 1.78-fold decrease in RNFL thickness in R681XCKO mice compared with neoCKO mice (Fig. 3C). Together with the results described above, these data firmly demonstrate that neither the R681X nor the G848R mutation is equivalent to the conventional neomycin knockout allele, thus establishing differential effects of the germline Nf1 gene mutation on optic glioma formation, growth, and associated RGC survival.
Figure 3.

The germline Nf1 gene mutation dictates the degree of optic glioma-induced retinal dysfunction. (A) pNF-H immunostaining in the optic nerves of control, G848RCKO, neoCKO and R681XCKO mice revealed increased immunoreactivity in neoCKO and R681XCKO mice, with R681XCKO mice exhibiting additional punctate staining (arrowheads). (B) The retinae of R681XCKO and neoCKO mice had increased percentages of TUNEL+ cells (5.9- and 4.8-fold increase, respectively) compared with control mice (n = 5 mice per genotype). (C) The retinae of R681XCKO and neoCKO mice had lower percentages of Brn3a+ cells (50 and 41% decrease, respectively) compared with control mice (n = 5 mice per genotype). (D) The retinae of R681XCKO and neoCKO mice had RNFL thinning as revealed by SMI-32 immunostaining (3.4-and 3.2-fold decrease, respectively) compared with control mice (n = 5 mice per genotype). GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; RNFL, retinal nerve fiber layer. Scale bars: 100 µm. All data are represented as means ± s.e.m. (**P < 0.01; *P < 0.05; one-way ANOVA with Bonferroni post-test). n.s., not significant.
The differential impact of the germline Nf1 gene mutation results from effects on both astrocytes and microglia
The differences observed between neoCKO, G848RCKO and R681XCKO optic nerves could reflect changes in the levels of astrocyte hyperproliferation following somatic Nf1 gene inactivation. To assess this potential effect, we analyzed brainstem astrocytes, since primary astrocyte cultures derived from murine optic nerves contain a heterogeneous population of macroglia (28) and the brainstem is the second most common brain region where gliomas arise in children with NF1 (29). As such, primary brainstem astrocytes were isolated from Nf1flox/G848R, Nf1flox/neo and Nf1flox/R681X mice and were infected with an adenovirus containing either Cre recombinase (Cre) to inactivate the Nf1flox allele or β-galactosidase (LacZ) to serve as controls. Importantly, the degree of neurofibromin reduction was similar between G848R, neo, and R861X astrocytes following Cre-mediated recombination of the Nf1flox allele relative to their respective controls (not shown), suggesting that any differences in proliferation observed using these three astrocyte populations reflected the contribution of the germline Nf1 gene mutation. Following loss of the Nf1flox allele (Cre infection), we observed no significant increase in G848RCKO astrocyte proliferation relative to Nf1flox/flox controls, whereas neoCKO and R681XCKO astrocytes exhibited a 3.2–4-fold increase in proliferation (Fig. 4A).
Figure 4.

Optic glioma formation and growth is partly determined by the effect of the germline Nf1 gene mutation on astrocyte RAS effector activation and proliferation. (A) Astrocyte proliferation was increased by 3.2- to 4.0-fold following Cre-mediated Nf1flox inactivation (Cre) in Nf1G681X/flox and Nf1neo/flox astrocytes, but only by 1.3-fold in Nf1G848R/flox astrocytes. Normalization is relative to control Nf1flox/flox astrocytes (LacZ infection) (n > 3 samples per genotype). Data are represented as means ± s.e.m. (*P < 0.05; one-way ANOVA with Dunn's multiple comparison test). Immunostaining with pAktSer473 (B) pERKThr202/Tyr204 (C) and pS6Ser240/244 (D) antibodies revealed a greater percentage of pAktSer473, pERKThr202/Tyr204 and pS6Ser240/244-immunoreactive cells in the optic nerves of neoCKO (pAktSer473: 9-fold increase, pERKThr202/Tyr204: 7.4-fold increase, pS6Ser240/244: 2.8-fold increase) and R681XCKO (pAktSer473: 10.4-fold increase, pERKThr202/Tyr204: 7.1-fold increase, pS6Ser240/244: 3.3-fold increase) optic nerves relative to Nf1flox/flox littermate controls (CTL). A minimum of four animals per genotype was assayed. n.s., not significant. Scale bars: 100 µm. Data are represented as means ± s.e.m. (*P < 0.05, ***P < .001; one-way ANOVA with Bonferroni post-test).
We next examined the downstream signaling pathways hyperactivated following neurofibromin loss. For these studies, immunohistochemistry was employed to determine the percentage of cells in the murine optic nerves that exhibit AKT (phospho-AktSer473; pAktSer473), ERK (phospho-ERKThr202/Tyr204, pERKThr202/Tyr204) and mTOR (phospho-S6Ser240/244; pS6Ser240/244) activation in vivo. This analysis revealed increased numbers of cells with neurofibromin downstream effector activation in neoCKO (pAktSer473, 9-fold increase, pERKThr202/Tyr204, 7.4-fold increase, pS6Ser240/244, 2.8-fold increase) and R681XCKO (pAktSer473; 10.4-fold increase, pERKThr202/Tyr204; 7.1-fold increase, pS6Ser240/244; 3.3-fold increase) mice compared with control and G848RCKO mice (Fig. 4B–D). Collectively, these findings demonstrate that one mechanism by which the germline Nf1 gene mutation dictates optic glioma formation and growth is by creating cell-autonomous differences in neurofibromin downstream effector activation and astrocyte proliferation in vitro and in vivo.
While this cell-autonomous effect of the germline Nf1 gene mutation on astrocyte proliferation in vitro explains the ability of neoCKO and R681XCKO, but not G848RCKO, mice to form optic gliomas, it does not account for the greater proliferation observed in R681XCKO mice relative to neoCKO mice in vivo. Previous studies from our laboratory have established an obligate role for non-neoplastic (stromal) cells in the formation and maintenance of murine optic gliomas (30–33). In particular, we have shown that brain microglia are required for optic glioma development and continued growth, such that silencing microglia function delays gliomagenesis and reduces optic glioma growth. Consistent with a critical role for microglia in these brain tumors, there were 2-fold more microglia in R681XCKO compared with neoCKO mouse optic nerves (Fig. 5A).
Figure 5.

Optic glioma formation and growth is partly determined by the effect of the germline Nf1 gene mutation on microglia number and function. (A) Iba1 immunostaining demonstrates increased numbers of microglia in the optic nerves of neoCKO (3.6-fold) and R681XCKO (7.1-fold) mice relative to controls (CTL). Whereas R681XCKO mice have more microglia than neoCKO mice (2-fold), no increase in microglia numbers was detected in G848RCKO mice relative to controls (n = 5 mice per genotype). (B) Increased percentages of pJNK+ cells were observed in neoCKO (4.66-fold) and R681XCKO (11.28-fold) optic nerves relative to control optic nerves (n = 4 mice per genotype). (C) Immunofluorescence revealing pJNK staining in Iba1+ cells (microglia) within the optic nerves of R681XCKO mice. White arrowheads depict two independent cells positively labeled with both pJNK and Iba1. (D) Increased percentages of CCL5+ cells were observed in neoCKO (14.56-fold) and R681XCKO (27.37-fold) optic nerves compared with control mice (n = 5 mice per genotype). (E) A greater percentage of pAktThr308-immunoreactive cells was found in R681XCKO mice relative to control (4.68-fold), and neoCKO (2.88-fold) optic nerves (n = 4 mice per genotype). Scale bars: 100 µm. All data are represented as means ± s.e.m. (***P < 0.001; **P < 0.01; *P < 0.05; one-way ANOVA with Bonferroni post-test).
To determine whether this increase in microglia explains the higher levels of proliferation observed in R681XCKO mouse optic gliomas, we focused on microglia-mediated paracrine effects. First, we examined JNK activation (JNKThr183/Tyr185; pJNK), based on our previous finding that Nf1+/− microglia have increased levels of JNK activation in vitro and that pharmacological JNK inhibition reduces Nf1 mouse optic glioma proliferation in vivo (33). Consistent with increased microglia functional activity, optic gliomas from R681XCKO mice had a 2.4-fold greater percentage of pJNK+ cells relative to neoCKO mice (Fig. 5B). It is important to note that the JNK activity (pJNK) was localized to Iba1+ microglia (Fig. 5C). Second, since microglia promote glioma growth through the elaboration of specific chemokines, like Ccl5 (34), we next quantified the percentage of Ccl5+ cells in these optic nerves, and observed a 1.9-fold greater percentage of Ccl5+ cells in R681XCKO relative to neoCKO optic gliomas (Fig. 5D). Third, Ccl5 can activate phosphoinositol-3-kinase (35–37), leading to AktThr308 phosphorylation (AktThr308) (38). As such, increased AKTThr308 phosphorylation (2.9-fold) was only observed in the optic nerves from R681XCKO mice relative to controls (Fig. 5E). Moreover, AktThr308 activation was localized to astrocytes, as revealed by GFAP/pAKTThr308 double labeling (data not shown). Together, these data reveal both cell-intrinsic (astrocyte) and non-cell-intrinsic (microglia) mechanisms by which the germline Nf1 gene mutation differentially dictates optic glioma formation and growth (Fig. 6).
Figure 6.
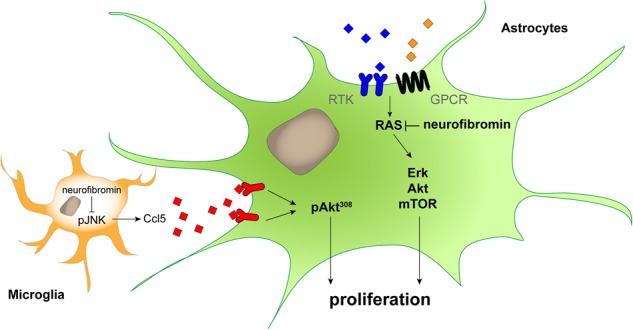
The germline Nf1 gene mutation independently determines astrocyte and microglia dysfunction relative to optic glioma formation and growth. Proposed model of the cell-autonomous (astrocytes) and non-cell-autonomous (microglia) effects of the germline Nf1 gene mutation on murine optic glioma growth. In microglia, the germline Nf1 gene mutation differentially impacts on JNK activation and Ccl5 expression, which results in varying levels of chemokine (Ccl5)-mediated AktThr308 activation in neoplastic astrocytes to increase optic glioma growth in vivo. Similarly, the germline Nf1 gene mutation differentially determines neurofibromin-regulated downstream signaling pathway activation and glial cell proliferation, leading to differing levels of cell-intrinsic astrocyte growth in the tumor.
Discussion
Little is known about the factors that underlie the significant clinical heterogeneity that characterizes many human monogenic diseases. Using NF1 as an instructive model of a common monogenic disorder, we sought to critically evaluate the differential impact of the germline NF1 gene mutation on optic glioma formation. With over 1300 different germline NF1 gene mutations documented and few recurrent mutations (39), robust correlations between germline mutations and clinical features have been largely unsuccessful (2,40). Nonetheless, several intriguing genotype–phenotype correlations have been reported (4–6,41). In addition, recent work from our laboratory employing NF1-patient-derived iPSCs with different germline NF1 gene mutations has revealed differences in neurofibromin expression and downstream signaling (9). However, since these iPSCs harbor different genomic backgrounds, which could also influence neurofibromin levels and function, we sought to formally evaluate the impact of the germline NF1 gene mutation engineered in mice on the identical genetic background.
For these proof-of-principle studies, two GEM strains were engineered to harbor germline Nf1 gene mutations most representative of the types of mutations found in individuals with NF1: nonsense (R681X) and missense (G848R) mutations (9,42). For the first time, we demonstrate distinct effects of two different germline NF1 gene mutations on murine optic glioma formation, growth, and associated retinal pathology. Herein, we show that Nf1 mutant mice harboring the R681X, but not the G848R, germline Nf1 gene mutation develop optic gliomas characterized by higher proliferative indices, increased GFAP immunoreactivity, and greater microglial infiltration. In addition, we demonstrate that these mutations are different from the conventional Nf1 knockout allele in terms of disease severity, revealing both cell-autonomous and non-cell-autonomous (stromal) mechanisms. Collectively, these novel findings argue that the germline NF1 gene mutation is one important factor in dictating disease pathogenesis.
It is interesting to note that the two germline Nf1 gene mutations have distinct effects on neurofibromin expression in vivo. The G848R missense mutation results in <38% reduction of neurofibromin levels, whereas the R681X nonsense mutation confers >77% reduction. These findings are similar to the neurofibromin expression profiles observed in NF1 patient-derived fibroblasts and iPSC-NPs, where patients clustered into two subgroups based on neurofibromin expression: Group 1 with minor reductions (<25%) and Group 2 with >70% reduced expression (9). Importantly, neurofibromin expression in the neomycin KO (neoCKO) mouse is reduced by ∼50%. Further underscoring the inability of current GEM models to fully capture the impact of patient mutation-driven phenotypes, optic nerve gliomas developing in mice harboring the nonsense R681X mutation (R681XCKO) were more aggressive than those developing in neoCKO mice (more microglia and increased proliferative indices).
These findings echo similar experiences in modeling other genetic conditions. For example, mice with a targeted disruption of the N-acetylglucosamine-1-phosphotransferase (GNPTAB) gene lack some of the characteristic features seen in patients with mucolipidosis II (MLII) (43), whereas mice engineered with a patient MLII GNPTAB mutation show facial and skeletal anomalies, lysosomal malfunction and reduced lifespan (44). In addition, mice with constitutively active oncogenic KRASG12D mutations fail to develop cardiomyopathy, a common feature of Noonan Syndrome (45), whereas GEM models harboring the recurrent patient-derived KRAS+/V14I mutation exhibit the characteristic growth, craniofacial, and cardiac defects associated with the human disease (46).
Moreover, we demonstrate that the specific germline NF1 gene mutation differentially impacts on optic glioma severity in at least two distinct ways. In this regard, we report both cell-autonomous and stromal (non-cell-autonomous) effects of the germline NF1 gene mutation on disease pathogenesis in vitro and in vivo. The cell-autonomous effect on optic glioma results from the effect of reduced neurofibromin expression on the downstream effector pathways most critical for mediating tumor growth. Previous studies from our laboratory and others have shown that loss of neurofibromin function results in increased ERK and AKT activation, which each converge on mTOR to drive Nf1-deficient astrocyte proliferation in vitro and Nf1 optic glioma growth in vivo (16,47–49). In the current report, loss of the Nf1flox allele by Cre-mediated excision had more significant effects on astrocyte proliferation when coupled with the neo or R681X germline Nf1 gene mutation than the G848R mutation in vitro. Similarly, in the optic nerves in vivo, greater numbers of cells with increased ERK (phospho-ERKThr202/Tyr204), AKT (phospho-AktSer473), and mTOR (phospho-S6Ser240/244) activation were observed in neoCKO and R681XCKO mice relative to G848RCKO and control mice, which were indistinguishable from each other. Studies are underway to more precisely define how the different germline Nf1 gene mutations impact on neurofibromin-regulated effector pathway activation in response to specific growth factors present in these optic gliomas.
In addition, there was a non-cell-autonomous (stromal) effect that reflected the impact of the germline NF1 gene mutation on non-neoplastic cells in the tumor microenvironment (microglia). Microglia are brain tissue macrophages that promote optic glioma development and growth in Nf1 GEM strains. These immune system-like cells alter their function in response to changes in Nf1 gene expression, such that Nf1+/neo microglia exhibit increased JNK activation (33). Consistent with the importance of microglial JNK hyperactivation in stroma-mediated glioma growth, pharmacologic JNK inhibition reduces Nf1 optic glioma proliferation in vivo. As such, optic gliomas in R681XCKO mice had greater numbers of phospho-JNK-immunoreactive microglia and increased microglial expression of Ccl5, a chemokine recently shown to maintain murine optic glioma growth in vivo (34). In these latter studies, Ccl5 was sufficient to increase Nf1-deficient optic nerve astrocyte growth in vitro and treatment with Ccl5 neutralizing antibodies reduced optic glioma growth in vivo. While the precise mechanism responsible for microglial Ccl5-mediated astrocyte hyperproliferation is still under investigation, increased Ccl5 expression in the tumor is most likely to drive astroglial cell proliferation through AktThr308 hyperactivation, as observed in other cancers (35–37). Current work is focused on determining how growth factors, like Ccl5, elaborated by cells in the tumor microenvironment regulate Nf1 optic glioma growth relevant to the development and evaluation of future stroma-directed brain tumor therapies.
In summary, the proof-of-concept studies presented in this report provide the first evidence for a clear effect of the germline Nf1 gene mutation on disease pathogenesis in NF1. Combined with other risk factors, such as genomic single nucleotide polymorphisms (50) and patient sex (51,52), further investigations using similar strains with different germline Nf1 gene mutations found in people with NF1, specifically those with particular clinical phenotypes (e.g. p1809) may yield critical new insights into the impact of the germline mutation on other medical issues affecting children and adults with NF1. Together with preliminary findings using NF1 patient-derived iPSCs and NPs, these murine studies raise the intriguing possibility that identifying the germline NF1 gene mutation may have some predictive utility in this at-risk patient population.
Materials and Methods
Mice
All animals were maintained on an inbred C57BL/6 background using a 12 h light–dark cycle with ad libitum access to food and water. Heterozygous Nf1 mice were generated with one wild-type copy of the Nf1 gene and one copy containing either a missense mutation in exon 21 (corresponding to the human c.2542G>C NF1 gene mutation; Gly848Arg, p.G848R) (Li and Kesterson, manuscript in preparation; draft readily available to reviewers upon request), a nonsense mutation in exon 18 (corresponding to the human c.2041C>T NF1 gene mutation; Arg681*, p.R681) (Li and Kesterson, manuscript in preparation; draft readily available to reviewers upon request) or a null inactivating allele created by the insertion of a neomycin cassette into exon 31 (Nf1+/neo) (19). G848R and R681X mice were generated using C57BL/6 ES cells and backcrossed a minimum of 10 times to wild-type C57BL/6 mice, while neo31 mice have been extensively backcrossed onto the C57BL/6 genetic background for over 15 years in the laboratory. Conditional knockout mice were generated with the G848R, neo31 or R681X mutation as the germline Nf1 allele, with somatic Nf1 gene inactivation resulting from Cre-mediated excision of the Nf1flox allele in neuroglial progenitor cells (22). The resulting strains included Nf1flox/G848R; GFAP-Cre (G848RCKO), Nf1flox/neo; GFAP-Cre (neoCKO) (14) and Nf1flox/R681X; GFAP-Cre (R681XCKO). Littermate Nf1+/+ and Nf1flox/flox (FF) mice were used as controls. All experiments were performed on 3-month-old animals, unless otherwise stated, under active Animal Studies Committee protocols at Washington University. Mice were randomly assigned to all experimental groups without bias, and the investigators were blinded to sample group allocation and subsequent analysis during all of the experiments.
Optic nerve volume measurements
Optic nerves with an intact chiasm were microdissected following transcardial perfusion. Optic nerves were photographed and diameters were measured at the chiasm (150, 300 and 450 μm anterior to the chiasm) to generate optic nerve volumes (16). A minimum of six animals per genotype were employed for these experiments.
Retinal nerve fiber layer measurements
RNFL thickness was quantitated using the average of 15 measurements of SMI-32-stained axons 0–250 µm proximal to the optic nerve head (ImageJ software) (53). A minimum of five animals per genotype were used for these experiments.
Western blotting, immunohistochemistry and immunofluorescence
Western blotting was performed on snap-frozen hippocampi or WBC pellets, lysed in RIPA buffer supplemented with protease inhibitors using appropriate primary antibodies (Supplementary Material, Table S1), secondary horseradish peroxidase-conjugated antibodies (Sigma) and ECL (Fisher) chemiluminescence (24). Western signal band intensity was quantified using ImageJ Software (National Institutes of Health, USA, http://imagej.nih.gov/ij Java 1.7.0_67). The western blot images illustrated are representative of separate sets of blots performed on a minimum of 10 independent animals per genotype. Quantitative data were obtained from analysis of all individual western blots. Immunohistochemistry and immunofluorescence were performed as previously described (54), on mice transcardially perfused with 4% PFA (Sigma) in 0.1 m sodium phosphate buffer (pH 7.4) and post-fixed in 4% PFA prior to paraffin embedding. For some immunofluorescence experiments (Brn3a, Ccl5), antibody signal amplification was performed using the TSA Cy3 Plus system (Perkin Elmer) (53). Four to 6 animals per genotype were used for these experiments.
Primary astrocyte culture and proliferation assay
Primary astrocytes were generated from the brainstems of postnatal Days 0–1 mouse pups and maintained in astrocyte growth medium (Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum and 1% penicillin/streptomycin) (53). To inactivate the conditional Nf1flox allele, astrocytes (passage 1) were infected with adenovirus type 5 (Ad5) containing Cre recombinase (Ad5-Cre) (University of Iowa Gene Transfer Vector Core, Iowa City, IA, USA). Control infections employed Ad5 containing β-galactosidase (Ad5-LacZ). 4 days post-infection, astrocytes were passaged and serum starved for 48 h prior to western blotting and proliferation analysis. Astrocyte proliferation was assessed using the BrdU Cell Proliferation ELISA kit (Roche) following manufacturer's instructions. Briefly, 6000 serum-starved astrocytes were labeled with BrdU for 18 h followed by 2-h incubation in peroxidase-conjugated anti-BrdU antibody. Proliferating astrocytes were identified using a colorimetric substrate reaction measured at 450 nm on a spectrophotometer (Bio-Rad). A minimum of six animals per genotype were used for each experiment, and these studies were replicated at least four independent times.
Statistical analyses
All statistical analyses were performed using GraphPad Prism 5 software (GraphPad Software). One- or two-way ANOVA with Bonferroni post-test correction analyses were employed for multiple comparisons.
Supplementary Material
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
Funding
J.A.T. was supported on the Vision Sciences (5-T32-EY13360) and Neuroscience (5-T32-NS007205-33) T32 training grants. These studies were funded by grants from the National Cancer Institute (1R01-CA195692-01) and Alex's Lemonade Stand Foundation to D.H.G.
Supplementary Material
References
- 1.Jett K., Friedman J.M. (2010) Clinical and genetic aspects of neurofibromatosis 1. Genet. Med., 12, 1–11. [DOI] [PubMed] [Google Scholar]
- 2.Szudek J., Joe H., Friedman J.M. (2002) Analysis of intrafamilial phenotypic variation in neurofibromatosis 1 (NF1). Genet. Epidemiol., 23, 150–164. [DOI] [PubMed] [Google Scholar]
- 3.Friedman J.M. (1999) Epidemiology of neurofibromatosis type 1. Am. J. Med. Genet., 89, 1–6. [PubMed] [Google Scholar]
- 4.Bolcekova A., Nemethova M., Zatkova A., Hlinkova K., Pozgayova S., Hlavata A., Kadasi L., Durovcikova D., Gerinec A., Husakova K. et al. (2013) Clustering of mutations in the 5′ tertile of the NF1 gene in Slovakia patients with optic pathway glioma. Neoplasma, 60, 655–665. [DOI] [PubMed] [Google Scholar]
- 5.Sharif S., Upadhyaya M., Ferner R., Majounie E., Shenton A., Baser M., Thakker N., Evans D.G. (2011) A molecular analysis of individuals with neurofibromatosis type 1 (NF1) and optic pathway gliomas (OPGs), and an assessment of genotype-phenotype correlations. J. Med. Genet., 48, 256–260. [DOI] [PubMed] [Google Scholar]
- 6.Upadhyaya M., Huson S.M., Davies M., Thomas N., Chuzhanova N., Giovannini S., Evans D.G., Howard E., Kerr B., Griffiths S. et al. (2007) An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): evidence of a clinically significant NF1 genotype-phenotype correlation. Am. J. Hum. Genet., 80, 140–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pinna V., Lanari V., Daniele P., Consoli F., Agolini E., Margiotti K., Bottillo I., Torrente I., Bruselles A., Fusilli C. et al. (2015) p.Arg1809Cys substitution in neurofibromin is associated with a distinctive NF1 phenotype without neurofibromas. Eur. J. Hum. Genet., 23, 1068–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Raedt T., Brems H., Wolkenstein P., Vidaud D., Pilotti S., Perrone F., Mautner V., Frahm S., Sciot R., Legius E. (2003) Elevated risk for MPNST in NF1 microdeletion patients. Am. J. Hum. Genet., 72, 1288–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anastasaki C., Woo A.S., Messiaen L.M., Gutmann D.H. (2015) Elucidating the impact of neurofibromatosis-1 germline mutations on neurofibromin function and dopamine-based learning. Hum. Mol. Genet., 24, 3518–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Listernick R., Charrow J., Greenwald M.J., Esterly N.B. (1989) Optic gliomas in children with neurofibromatosis type 1. J. Pediatr., 114, 788–792. [DOI] [PubMed] [Google Scholar]
- 11.Louis D.N., Ohgaki H., Wiestler O.D., Cavenee W.K., Burger P.C., Jouvet A., Scheithauer B.W., Kleihues P. (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol., 114, 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fisher M.J., Loguidice M., Gutmann D.H., Listernick R., Ferner R.E., Ullrich N.J., Packer R.J., Tabori U., Hoffman R.O., Ardern-Holmes S.L. et al. (2012) Visual outcomes in children with neurofibromatosis type 1-associated optic pathway glioma following chemotherapy: a multicenter retrospective analysis. Neuro. Oncol., 14, 790–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Listernick R., Ferner R.E., Liu G.T., Gutmann D.H. (2007) Optic pathway gliomas in neurofibromatosis-1: controversies and recommendations. Ann. Neurol., 61, 189–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bajenaru M.L., Hernandez M.R., Perry A., Zhu Y., Parada L.F., Garbow J.R., Gutmann D.H. (2003) Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer Res., 63, 8573–8577. [PubMed] [Google Scholar]
- 15.Hegedus B., Hughes F.W., Garbow J.R., Gianino S., Banerjee D., Kim K., Ellisman M.H., Brantley M.A. Jr., Gutmann D.H. (2009) Optic nerve dysfunction in a mouse model of neurofibromatosis-1 optic glioma. J. Neuropathol. Exp. Neurol., 68, 542–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hegedus B., Banerjee D., Yeh T.H., Rothermich S., Perry A., Rubin J.B., Garbow J.R., Gutmann D.H. (2008) Preclinical cancer therapy in a mouse model of neurofibromatosis-1 optic glioma. Cancer Res., 68, 1520–1528. [DOI] [PubMed] [Google Scholar]
- 17.Kaul A., Toonen J.A., Cimino P.J., Gianino S.M., Gutmann D.H. (2015) Akt- or MEK-mediated mTOR inhibition suppresses Nf1 optic glioma growth. Neuro. Oncol., 17, 843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacks T., Shih T.S., Schmitt E.M., Bronson R.T., Bernards A., Weinberg R.A. (1994) Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat. Genet., 7, 353–361. [DOI] [PubMed] [Google Scholar]
- 19.Brannan C.I., Perkins A.S., Vogel K.S., Ratner N., Nordlund M.L., Reid S.W., Buchberg A.M., Jenkins N.A., Parada L.F., Copeland N.G. (1994) Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev., 8, 1019–1029. [DOI] [PubMed] [Google Scholar]
- 20.Pascual-Castroviejo I., Pascual-Pascual S.I., Velazquez-Fraqua R., Botella P., Viano J. (2007) Familial spinal neurofibromatosis. Neuropediatrics, 38, 105–108. [DOI] [PubMed] [Google Scholar]
- 21.Kocova M., Kochova E., Sukarova-Angelovska E. (2015) Optic glioma and precocious puberty in a girl with neurofibromatosis type 1 carrying an R681X mutation of NF1: case report and review of the literature. BMC Endocr. Disord., 15, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bajenaru M.L., Zhu Y., Hedrick N.M., Donahoe J., Parada L.F., Gutmann D.H. (2002) Astrocyte-specific inactivation of the neurofibromatosis 1 gene (NF1) is insufficient for astrocytoma formation. Mol. Cell. Biol., 22, 5100–5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu Y., Romero M.I., Ghosh P., Ye Z., Charnay P., Rushing E.J., Marth J.D., Parada L.F. (2001) Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev., 15, 859–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bajenaru M.L., Garbow J.R., Perry A., Hernandez M.R., Gutmann D.H. (2005) Natural history of neurofibromatosis 1-associated optic nerve glioma in mice. Ann. Neurol., 57, 119–127. [DOI] [PubMed] [Google Scholar]
- 25.Avery R.A., Liu G.T., Fisher M.J., Quinn G.E., Belasco J.B., Phillips P.C., Maguire M.G., Balcer L.J. (2011) Retinal nerve fiber layer thickness in children with optic pathway gliomas. Am. J. Ophthalmol., 151, 542–549 e542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosenfeld J., Dorman M.E., Griffin J.W., Gold B.G., Sternberger L.A., Sternberger N.H., Price D.L. (1987) Distribution of neurofilament antigens after axonal injury. J. Neuropathol. Exp. Neurol., 46, 269–282. [DOI] [PubMed] [Google Scholar]
- 27.Parrilla-Reverter G., Agudo M., Nadal-Nicolas F., Alarcon-Martinez L., Jimenez-Lopez M., Salinas-Navarro M., Sobrado-Calvo P., Bernal-Garro J.M., Villegas-Perez M.P., Vidal-Sanz M. (2009) Time-course of the retinal nerve fibre layer degeneration after complete intra-orbital optic nerve transection or crush: a comparative study. Vis. Res., 49, 2808–2825. [DOI] [PubMed] [Google Scholar]
- 28.Solga A.C., Gianino S.M., Gutmann D.H. (2014) NG2-cells are not the cell of origin for murine neurofibromatosis-1 (Nf1) optic glioma. Oncogene, 33, 289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guillamo J.S., Creange A., Kalifa C., Grill J., Rodriguez D., Doz F., Barbarot S., Zerah M., Sanson M., Bastuji-Garin S. et al. (2003) Prognostic factors of CNS tumours in Neurofibromatosis 1 (NF1): a retrospective study of 104 patients. Brain, 126, 152–160. [DOI] [PubMed] [Google Scholar]
- 30.Daginakatte G.C., Gutmann D.H. (2007) Neurofibromatosis-1 (Nf1) heterozygous brain microglia elaborate paracrine factors that promote Nf1-deficient astrocyte and glioma growth. Hum. Mol. Genet., 16, 1098–1112. [DOI] [PubMed] [Google Scholar]
- 31.Pong W.W., Higer S.B., Gianino S.M., Emnett R.J., Gutmann D.H. (2013) Reduced microglial CX3CR1 expression delays neurofibromatosis-1 glioma formation. Ann. Neurol., 73, 303–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simmons G.W., Pong W.W., Emnett R.J., White C.R., Gianino S.M., Rodriguez F.J., Gutmann D.H. (2011) Neurofibromatosis-1 heterozygosity increases microglia in a spatially and temporally restricted pattern relevant to mouse optic glioma formation and growth. J. Neuropathol. Exp. Neurol., 70, 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Daginakatte G.C., Gianino S.M., Zhao N.W., Parsadanian A.S., Gutmann D.H. (2008) Increased c-Jun-NH2-kinase signaling in neurofibromatosis-1 heterozygous microglia drives microglia activation and promotes optic glioma proliferation. Cancer Res., 68, 10358–10366. [DOI] [PubMed] [Google Scholar]
- 34.Solga A.C., Pong W.W., Kim K.Y., Cimino P.J., Toonen J.A., Walker J., Wylie T., Magrini V., Griffith M., Griffith O.L. et al. (2015) RNA sequencing of tumor-associated microglia reveals Ccl5 as a stromal chemokine critical for neurofibromatosis-1 glioma growth. Neoplasia, 17, 776–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang C.Y., Fong Y.C., Lee C.Y., Chen M.Y., Tsai H.C., Hsu H.C., Tang C.H. (2009) CCL5 increases lung cancer migration via PI3K, Akt and NF-kappaB pathways. Biochem. Pharmacol., 77, 794–803. [DOI] [PubMed] [Google Scholar]
- 36.Tyner J.W., Uchida O., Kajiwara N., Kim E.Y., Patel A.C., O'Sullivan M.P., Walter M.J., Schwendener R.A., Cook D.N., Danoff T.M. et al. (2005) CCL5-CCR5 interaction provides antiapoptotic signals for macrophage survival during viral infection. Nat. Med., 11, 1180–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu G.T., Chen H.T., Tsou H.K., Tan T.W., Fong Y.C., Chen P.C., Yang W.H., Wang S.W., Chen J.C., Tang C.H. (2014) CCL5 promotes VEGF-dependent angiogenesis by down-regulating miR-200b through PI3K/Akt signaling pathway in human chondrosarcoma cells. Oncotarget, 5, 10718–10731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vincent E.E., Elder D.J., Thomas E.C., Phillips L., Morgan C., Pawade J., Sohail M., May M.T., Hetzel M.R., Tavare J.M. (2011) Akt phosphorylation on Thr308 but not on Ser473 correlates with Akt protein kinase activity in human non-small cell lung cancer. Br. J. Cancer, 104, 1755–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Messiaen L., Yao S., Brems H., Callens T., Sathienkijkanchai A., Denayer E., Spencer E., Arn P., Babovic-Vuksanovic D., Bay C. et al. (2009) Clinical and mutational spectrum of neurofibromatosis type 1-like syndrome . JAMA, 302, 2111–2118. [DOI] [PubMed] [Google Scholar]
- 40.Sabbagh A., Pasmant E., Laurendeau I., Parfait B., Barbarot S., Guillot B., Combemale P., Ferkal S., Vidaud M., Aubourg P. et al. (2009) Unravelling the genetic basis of variable clinical expression in neurofibromatosis 1. Hum. Mol. Genet., 18, 2768–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rojnueangnit K., Xie J., Gomes A., Sharp A., Callens T., Chen Y., Liu Y., Cochran M., Abbott M.A., Atkin J. et al. (2015) High incidence of noonan syndrome features including short stature and pulmonic stenosis in patients carrying NF1 missense mutations affecting p.Arg1809: genotype-phenotype correlation. Hum. Mutat, 36, 1052–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alkindy A., Chuzhanova N., Kini U., Cooper D.N., Upadhyaya M. (2012) Genotype-phenotype associations in neurofibromatosis type 1 (NF1): an increased risk of tumor complications in patients with NF1 splice-site mutations? Hum. Genomics, 6, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gelfman C.M., Vogel P., Issa T.M., Turner C.A., Lee W.S., Kornfeld S., Rice D.S. (2007) Mice lacking alpha/beta subunits of GlcNAc-1-phosphotransferase exhibit growth retardation, retinal degeneration, and secretory cell lesions. Invest. Ophthalmol. Vis. Sci., 48, 5221–5228. [DOI] [PubMed] [Google Scholar]
- 44.Paton L., Bitoun E., Kenyon J., Priestman D.A., Oliver P.L., Edwards B., Platt F.M., Davies K.E. (2014) A novel mouse model of a patient mucolipidosis II mutation recapitulates disease pathology. J. Biol. Chem., 289, 26709–26721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dalin M.G., Zou Z., Scharin-Tang M., Safari R., Karlsson C., Bergo M.O. (2014) Myocardial KRAS(G12D) expression does not cause cardiomyopathy in mice. Cardiovasc. Res., 101, 229–235. [DOI] [PubMed] [Google Scholar]
- 46.Hernandez-Porras I., Fabbiano S., Schuhmacher A.J., Aicher A., Canamero M., Camara J.A., Cusso L., Desco M., Heeschen C., Mulero F. et al. (2014) K-RasV14I recapitulates Noonan syndrome in mice. Proc. Natl. Acad. Sci. USA, 111, 16395–16400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dasgupta B., Yi Y., Chen D.Y., Weber J.D., Gutmann D.H. (2005) Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1-associated human and mouse brain tumors. Cancer Res., 65, 2755–2760. [DOI] [PubMed] [Google Scholar]
- 48.Lee D.Y., Yeh T.H., Emnett R.J., White C.R., Gutmann D.H. (2010) Neurofibromatosis-1 regulates neuroglial progenitor proliferation and glial differentiation in a brain region-specific manner. Genes Dev., 24, 2317–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dasgupta B., Li W., Perry A., Gutmann D.H. (2005) Glioma formation in neurofibromatosis 1 reflects preferential activation of K-RAS in astrocytes. Cancer Res., 65, 236–245. [PubMed] [Google Scholar]
- 50.Roth J.J., Santi M., Rorke-Adams L.B., Harding B.N., Busse T.M., Tooke L.S., Biegel J.A. (2014) Diagnostic application of high resolution single nucleotide polymorphism array analysis for children with brain tumors. Cancer Genet., 207, 111–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Diggs-Andrews K.A., Brown J.A., Gianino S.M., Rubin J.B., Wozniak D.F., Gutmann D.H. (2014) Sex is a major determinant of neuronal dysfunction in neurofibromatosis type 1. Ann. Neurol., 75, 309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Warrington N.M., Sun T., Luo J., McKinstry R.C., Parkin P.C., Ganzhorn S., Spoljaric D., Albers A.C., Merkelson A., Stewart D.R. et al. (2015) The cyclic AMP pathway is a sex-specific modifier of glioma risk in type I neurofibromatosis patients. Cancer Res., 75, 16–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kaul A., Toonen J.A., Gianino S.M., Gutmann D.H. (2015) The impact of coexisting genetic mutations on murine optic glioma biology. Neuro. Oncol, 17, 670–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brown J.A., Gianino S.M., Gutmann D.H. (2010) Defective cAMP generation underlies the sensitivity of CNS neurons to neurofibromatosis-1 heterozygosity. J. Neurosci., 30, 5579–5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.