

Abstract
Oxidative damage to mitochondria (MT) is a major mechanism for aging and neurodegeneration. We have developed a novel synthetic antioxidant, XJB-5-131, which directly targets MT, the primary site and primary target of oxidative damage. XJB-5-131 prevents the onset of motor decline in an HdhQ(150/150) mouse model for Huntington's disease (HD) if treatment starts early. Here, we report that XJB-5-131 attenuates or reverses disease progression if treatment occurs after disease onset. In animals with well-developed pathology, XJB-5-131 promotes weight gain, prevents neuronal death, reduces oxidative damage in neurons, suppresses the decline of motor performance or improves it, and reduces a graying phenotype in treated HdhQ(150/150) animals relative to matched littermate controls. XJB-5-131 holds promise as a clinical candidate for the treatment of HD.
Introduction
Mitochondrial dysfunction in Alzheimer's (AD) (1), Parkinson's (PD) (2) and Huntington's disease (HD) (3) is implied by the defects in bioenergetics (4,5), mitochondrial fusion/fission (6), mitochondrial movement (7) and transcription(8,9) that are associated with these diseases. Indeed, elevation of oxidative damage and diminished capacity to produce adenosine triphosphate (ATP) characterize neuronal toxicity (10). Treatment with antioxidants and/or supplementation with ATP producing molecules have been logical therapeutic strategies to offset at least some pathology that develops in patients. However, these attempts have met with marginal success (3,11–13).
Phosphocreatine serves as a spatial and temporal ATP buffer in tissues with high-energy requirements, such as the skeletal muscle and brain. In HD animal models, creatine improves motor performance, reduces brain atrophy, and reduces striatal Htt-aggregate burden (14,15). However, in both HD and PD patients, creatine, although well tolerated in patients, has not been successful in ameliorating disease symptoms. In PD, creatine supplementation improves mood, but without significant effects on the Unified Parkinson's Disease Rating Scale (13,16). The beneficial effects of creatine in HD patients were modest even at exceptionally high creatine levels (1–10 g/day), and the CREST-E trial at even higher levels (40 g/day) was stopped early after a futility analysis (https://nccih.nih.gov/research/extramural/crest-e).
Dietary supplementation with naturally occurring Vitamin E and Coenzyme Q10 (3,17) has been a significant focus of antioxidants therapeutics in PD and HD. CoQ10 (ubiquinone), an essential biological cofactor of the electron transport chain, functions as an important antioxidant in mitochondrial and lipid membranes. However, when administered as a dietary supplement, CoQ10 tends to be retained in cell membranes, is inefficient in entering the mitochondria (MT) (18,19) and has low permeability to the blood–brain barrier (BBB) (18,19). Nonetheless, CoQ10 has had some efficacy in both PD and HD mouse models. For example, CoQ10 treatment improves early motor deficits in several HD mouse models (20). CoQ10 treatment protects dopaminergic neurons and inhibits α-synuclein aggregation in a chemically induced 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of PD (19,21). Despite these positive preclinical outcomes, clinical trials for CoQ10 were stopped for both PD and HD patients due to the lack of efficacy (13,22).
The sources of the failure of HD therapeutics in clinical trials are unclear. The poor outcome of CoQ10 in humans is thought, at least in part, to occur due to feedback control, i.e. CoQ10 is the expressed product of an endogenous gene, and the cell compensates for a dietary increase of CoQ10 by down-regulating CoQ10 synthesis (11). Even if plasma concentrations rise during treatment, it remains controversial whether dietary supplementation of CoQ10 will significantly enhance its steady-state level or reach a pharmacological effective in vivo concentration in the brain (11,23,24). Indeed, in vivo, the effects of CoQ10 treatment have been marginal, variable or tissue specific (11,23,25). For example, MitoQ, another mitochondrial-targeted CoQ10 derivative, failed to offset neurodegeneration in PD clinical trials (26), but has unexpected promise in the periphery, as a therapeutic in treating non-alcoholic fatty liver disease (27).
The charge of CoQ10 and its derivatives can also diminish their efficacy. The loss of the charge gradient is a major feature of MT in dying neurons, yet mitochondrial entry of cationic antioxidants such as MitoQ requires the charge gradient (17). Thus, uptake of these potential-driven antioxidants is self-limiting (28) as there is inevitable depolarization of MT with disease progression. Indeed, MitoQ and MitoVit E protect cultured fibroblasts from Friedrich's ataxia (FRDA) patients; yet, their enhanced potency is abolished in cells pretreated with carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone, an agent that destroys the charge gradient (29). The diminution of efficacy in FRDA cells raises the issue as to whether MitoQ will have any benefit if treatment starts after disease onset. Many compounds have protective effects in offsetting toxicity in animals if treatment begins early. However, for most compounds, efficacy in counteracting toxicity after disease onset is typically untested, but may be a better predictor of clinical success.
Despite the limitations of antioxidant therapy, the tolerability and/or marginal success in PD and HD patients continues to fuel interest in redox-active therapeutics for neurodegeneration. However, inefficient BBB membrane penetration and the need to sustain pharmacologically effective concentrations in the brain remain significant barriers. To address these issues, we have developed XJB-5-131, a synthetic radical and electron scavenger which directly targets MT, the primary source and the primary target of reactive oxygen species (ROS). XJB's synthetic nature and neutral chemical structure allows it to permeate membranes. In preliminary characterization, we reported that pretreatment of young animals with XJB-5-131 prevents the decline of Rota-Rod performance in the HdhQ(150/150) mouse model for HD. Here, we asked the more difficult, but clinically relevant question: can XJB-5-131 reverse or attenuate toxicity in older animals that have developed disease phenotypes before the start of treatment? Our results demonstrate that XJB-5-131 is a remarkably potent inhibitor of disease progression in animals with well-developed pathology.
Results
XJB-5-131 is a bi-functional antioxidant comprising a delivery component conjugated to an antioxidant moiety (Fig. 1A) (30). The delivery portion of the molecule is an alkene peptide isostere modification of the Leu-D-Phe-Pro-Val-Orn segment of the antibiotic gramicidin S (31,32) (Fig. 1A, red). This peptide mimetic directly targets the mitochondrial membrane and delivers the antioxidant nitroxide, 2,2,6,6-tetramethyl piperidine-1-oxyl (TEMPO) (Fig. 1A, blue), to neutralize reactive radical species. We evaluated the efficacy of XJB-5-131 in the HdhQ(150/150) HD mouse model, which harbored the disease-length 150 CAG tract knocked into both endogenous alleles (33). These animals are referred to as HdhQ(150/150) while the littermate controls are referred to as HdhQ(wt/wt). The HdhQ(150/150) knock-in model more closely mimics disease progression relative to fragment models (33), and the slow onset provides a large window to observe phenotypic changes with XJB-5-131 treatment. Homozygous HdhQ(150/150) animals were used in the analysis to produce the most robust phenotypes. In the following figures, HdhQ(150/150) (HD) and HdhQ(wt/wt) [wild-type (WT)] animals were treated either with XJB-5-131 (tr) or the saline vehicle (vh), and the treatment groups are abbreviated in the figures as HD-tr, HD-vh, WT-tr and WT-vh, respectively.
Figure 1.
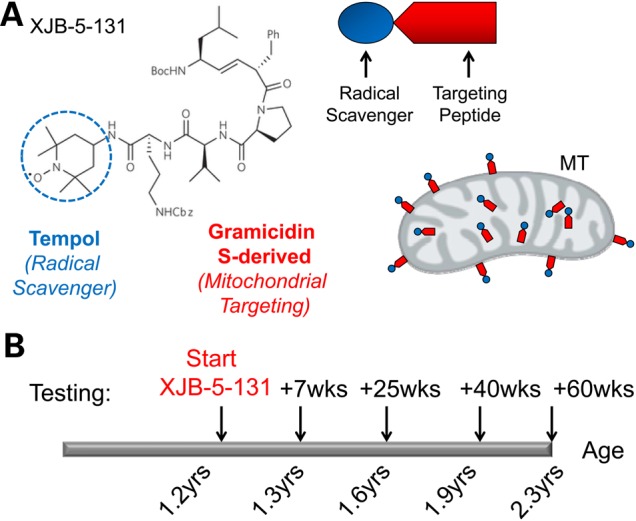
The structure of XJB-5-131 and treatment strategy. (A) XJB-5-131 is a bi-functional antioxidant comprising a mitochondrial targeting alkene peptide isostere (blue) attached to an antioxidant component (red). The targeting portion of the molecule is derived from the Leu-d-Phe-Pro-Val-Orn segment of the antibiotic gramicidin S. This peptide mimetic directly targets the mitochondrial membrane and delivers the antioxidant, TEMPO (blue box) that is able to prevent or neutralize oxidative damage. MT targeting is depicted as black lines in the red oblong structure of the MT. (B) A schematic diagram of treatment regimen. Animals were aged to 1.2 years (60 weeks) to develop features of HD pathology. XJB-5-131 treatment was initiated at 1.2 years and continued through age 2.3 years. Animals were tested at 7, 10, 25, 40 and 60 weeks after the start of treatment for changes in CNS and motor activity.
HdhQ(150/150) and littermate controls develop pathology by 60 weeks
HD is characterized by early striatal degeneration, cognitive deficits, involuntary choreiform movements and wasting (34,35), which progress with age. In the experimental paradigm, HdhQ(150/150) animals were allowed to age for roughly 1.2 years (60 weeks) to develop features of pathology, at which point treatment began and continued for an additional 1.2 years (60 weeks of treatment) (Fig. 1B). Animals were tested at various times of treatment for physical appearance, central nervous system (CNS) pathology, motor function and the impact of XJB-5-131 on oxidative DNA damage. Collectively, changes in these parameters tested whether the features of disease could be pharmacologically reversed after disease onset (Fig. 1B). Indeed, Hdh(Q150/Q150) animals before the start of treatment (60 weeks) had developed measureable pathology (Supplementary Material, Fig. S1). Similar to our previous report, Hdh(Q150/Q150) animals weighed less (Supplementary Material, Fig. S1A and B), and performed poorly on the Rota-Rod relative to their HdhQ(wt/wt) littermate controls (Supplementary Material, Fig. S1E). At 60 weeks of age, animals of both genotypes had similar neuronal counts (see Fig. 3). However, the level of ubiquitin-containing inclusions were significantly higher in Hdh(Q150/Q150) animals (Supplementary Material, Fig. S1C and D). Thus, untreated Hdh(Q150/150) animals at 60 weeks were characterized by well-developed phenotypes associated with HD progression.
Figure 3.

XJB-5-131 treatment of Hdh(Q150/150) prevents loss of striatal neurons in animals with age. (A) Coronal sections of the forebrain were chosen at interaural 3.8 mm as defined by the Brain Atlas (dotted line). At this position, the dorsal portion (yellow outline) of the caudate putamen of the STR (red region) was assayed for neuronal counts. The left side image is Nissl stained (image credit: Allen Institute for Brain Science) with the STR indicated in red. The right side image is IF stained for neurons using NeuN (green). (B) Images of anti-NeuN IF staining of neurons (green) in the dorsal part of the caudate putamen of the STR of animals at various times of treatment (as indicated), which started at 60 weeks of age. Vehicle-treated control animals is (WT-vh), vehicle-treated HdhQ(150/150) is (HD-vh) and XJB-5-131-treated HdhQ(150/150) animals is (HD-tr). Neuronal counts within the STR indicate HD-vh animals lose neurons by 85 weeks of age, whereas XJB-5-131 treatment from the age of 60 weeks significantly prevents neuronal loss and is similar to WT-vh. [n = 3 mice per sample, 1 × 10 µm brain slice per animal was tested, 10 imaging fields (>900 cells total) per slice] (*P < 0.05, **P < 0.005, Student's t-test, one-tailed homoscedastic) (yellow bar indicates 50 µm). (C) Quantification of the results from all animals (represented in B). (D) Comparison of striatal neuron loss with the neuron numbers of the HIP [in the pyramidal layer (sp) of Ammon's horn field (CA2)]. At the 60 + 60 weeks of treatment, the control CA3 region of the more resistant HIP displayed no neuronal loss. The color key indicates vehicle-treated control animals is (WT-vh), vehicle-treated HdhQ(150/150) is (HD-vh). The images (E) and quantification (F) of anti-Iba1 staining (green) of the striatum; images (G) and quantification (H) of glial projection stained with an anti-GFAP antibody (green). Images are 3D renderings of 10 µm Z-stacks, 0.5 µm spacing per optical slice (yellow bar indicate 50 µm). Quantifications were of n = 3 mice per sample, five imaging fields per mouse.
Treatment with XJB-5-131 alters physical appearance of aging HdhQ(150/150) animals relative to controls
As HdhQ(150/150) animals aged, they developed striking physical changes that were blocked by XJB-5-131 treatment. At 1.2 years (60 weeks), animals of both genotypes were well groomed and maintained healthy black coats (Fig. 2A, Panels 1 and 2). However, in HdhQ(150/150) animals, 8% developed salt and pepper coats at advanced age (2.3 years), which was prominent around the temples, muzzle and underside (Fig. 2B and C). Furthermore, vehicle-treated animals of both genotypes developed some degree of hair loss at advanced age, but the loss was particularly prominent in older HdhQ(150/150) animals (16%) at 2.3 years (Fig. 2A, Panel 3; Fig. 2C). This phenotype was consistent with the excessive grooming activity that has been previously reported in HD animal models (36). XJB-5-131 prevented coat graying (salt and pepper colored coats) (Fig. 2B and C), and significantly reduced hair loss in HdhQ(150/150) animals (Fig. 2A and C). The sleek coat appearance of XJB-5-131-treated HdhQ(150/150) animals, irrespective of gender, was largely indistinguishable from that of controls at all times tested (Fig. 2A and C).
Figure 2.
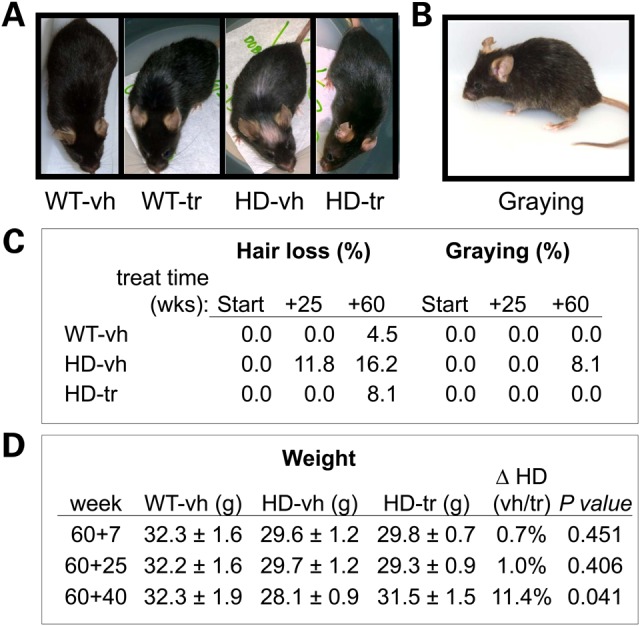
XJB-5-131 treatment prevents weight reduction and decreases hair loss and graying phenotypes in HdhQ(150/150) and controls with age. (A) A representative example of the hair loss on the forehead and dorsal side (A, Panel 3) that developed in a HdhQ(150/150)-vh (HD-vh) mouse at 120 weeks of age. (B) A representative example of ‘salt and pepper’ coats developed in HD-vh animals at 120 weeks, particularly around the temples, muzzle and underside at old ages (in this case 120 weeks of age). The coat appearance of HD-tr (60 + 60weeks treatment) animals was indistinguishable from that of age-matched WT-vh or WT-tr animals of 120 weeks (A, Panels 1, 2 and 4). (C) Quantification of the hair loss and graying phenotypes (in A and B) among mice at 120 weeks, expressed as a percentage of all animals in their treatment group, WT-vh (n = 37), HD-vh (n = 46), HD-tr (n = 34). (D) The weight of male HD-vh mice with age relative to control littermates. XLB-5-131 promotes weight gain at 40 weeks of treatment (P < 0.05, Student's t-test, one-tailed homoscedastic). WT-vh (n = 7), HD-vh (n = 12), HD-tr (n = 9).
XJB-5-131 treatment remediated other features of the disease. Weight loss is associated with HD pathology in humans. Before treatment started, both male and female mice were lighter relative to their littermate controls (9% lighter and 13.8%, respectively). Untreated male HdhQ(150/150) animals lost an average of 6% of their body weight as they aged from 1.2 to 2.3 years (Fig. 2D). Remarkably, in these animals, XJB-5-131 not only prevented further weight loss during the first 25 weeks of treatment (Fig. 2D, 60 + 25 weeks), but also promoted an average 11.4% weight gain by 60 weeks of treatment (Fig. 2D, 60 + 60 weeks). Surprisingly, there was no effect of XJB-5-131 on the weights of female HdhQ(150/150) animals (not shown) and they remained smaller than their HdhQ(wt/wt) littermates throughout the experiments. For the most part, XJB-5-131 had obvious beneficial effects on the physiology of HdhQ(150/150) animals.
Treatment with XJB-5-131 reduces neuronal loss and decreases brain lesions in HdhQ(150/150) animals
We examined the impact of XJB-5-131 on brain pathology in HdhQ(150/150) animals and littermate controls at equivalent ages. The brains of HdhQ(150/150) and HdhQ(wt/wt) littermates were sectioned and the features compared at the same position in each sample (interaural 3.8 mm, Mouse Brain Atlas) (Fig. 3A). The sensitive striatum (STR) (Fig. 3B) and the more resistant hippocampus (HIP) (Fig. 3D) were stained with anti-NeuN and 4′,6-diamidino-2-phenylindole (DAPI) to quantify the neuronal number and density (Supplementary Material, Fig. S2). At the start of treatment (60 weeks), vehicle-treated HdhQ(150/150) animals, and their HdhQ(wt/wt) littermate controls had similar numbers of neurons in the STR (Fig. 3B and C). However, there was a substantial loss of ∼40–50% in aging vehicle-treated HdhQ(150/150) animals by 85 weeks (60 + 25 weeks) of treatment that was not observed in the age-matched littermate controls (Fig. 3B and C) (Supplementary Material, Table S1). As judged by histological examination (Fig. 3E–G), there was no measureable gliosis (Fig. 3E and F, anti-Iba1 staining of activated microglia) or changes in the number of glial projections [Fig. 3G and H, anti-glial fibrillary acidic protein (GFAP) staining of astrocytes] in either genotype at any age tested.
The decline in tissue integrity within the affected brain regions was also obvious during tissue handling (Supplementary Material, Fig. S3). The STR developed a distinctly spongy consistency with age (Supplementary Material, Fig. S3A and B), and frozen sections became increasingly difficult to slice without tearing the tissue (Supplementary Material, Fig. S3B). The number and volume of the striatal gaps (Supplementary Material, Fig. S3B–D, red) increased with age. Tearing was specific to the affected brain regions, i.e. gaps were present around the STR, but in the same slice were absent in more resistant regions of the brain such as the HIP (Supplementary Material, Fig. S3B and C, blue).
Remarkably, XJB-5-131 treatment attenuated the neuronal loss in the STR of HdhQ(150/150) and improved tissue quality in these animals as they aged (Fig. 3B and C). The neuronal number and density (Supplementary Material, Fig. S2) (Fig. 3B and C) in the STR of XJB-5-131-treated HdhQ(150/150) animals was similar to control littermates up to 2.3 years (Fig. 3B and C). The decline in neuronal number was region-specific as the neuronal number in the more resistant HIP was indistinguishable among treated groups of both genotypes throughout the experiment (Fig. 3D). The consistency of the STR improved with treatment. Tissue tearing and gaps were infrequent in XJB-5-131-treated HdhQ(150/150) animals (Supplementary Material, Fig. S3D), whose brain tissue densities and integrity were similar to those of their control littermates (Supplementary Material, Fig. S3B and D).
HD pathology is marked by the progressive appearance of inclusions. Indeed, we detected few inclusions in control animals, but aggregates were prominent in age-matched vehicle-treated HdhQ(150/150) mice by 60 weeks (Fig. 4A and B). XJB-5-131 treatment suppressed the rise in inclusions compared with vehicle-treated HdhQ(150/150) littermates (Fig. 4A and B). Although vehicle-treated HdhQ(150/150) animals had developed significant pathology, XJB-5-131 treatment beginning after onset prevented additional loss of neurons and further inclusion formation in animals that had developed disease symptoms before the start of treatment. Taken together, XJB-5-131 had clear benefits in offsetting pathological benchmarks in the CNS of HdhQ(150/150) animals.
Figure 4.
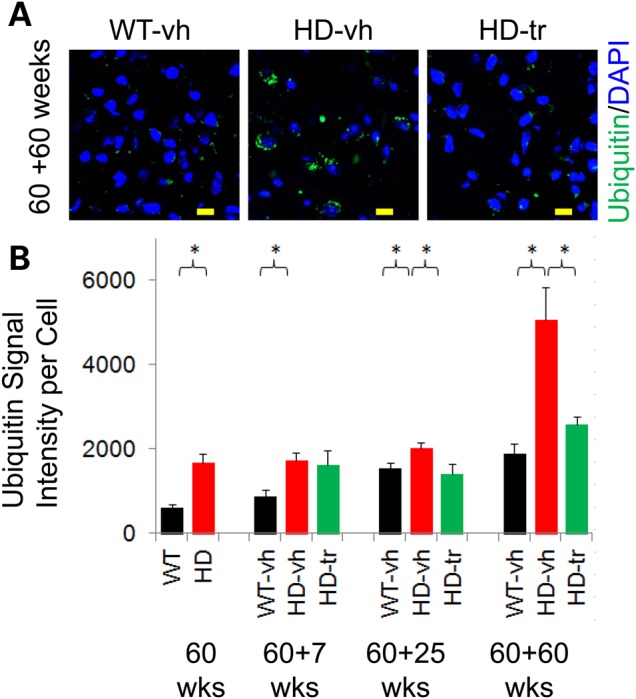
XJB-5-131 treatment of HdhQ(150/150) animals reduces inclusions in the brain. (A) The signal staining intensity for anti-ubiquitin antibody (green) after 60 weeks of XJB-5-131 treatment (60 + 60 weeks). Inclusions are elevated in the striatal cells of HD-vh mice relative to striatal cells WT-vh, and are suppressed in HD-tr by XJB-5-131 treatment. Scale bars indicate 10 µm. Blue is DAPI staining of DNA in the nucleus. (B) Quantified results of (A) for WT-vh (black) HD-vh (red), and HD-tr (green), as indicated. The start of treatment is 60 weeks; the time of treatment is n, as in (60 + n). There are significantly fewer inclusion bodies (measured by IF signal of ubiquitin) after XJB-5-131 treatment in the striatal cells of HD-tr relative to WT-vh and HD-vh and at all measured time points (n = 3 animals per genotype, >250 cells each) (*P < 0.05, Student's t-test, one-tailed homoscedastic).
Treatment with XJB-5-131 prevents motor decline
Early signs of HD pathology manifest as motor abnormalities which progress with time. To test whether XJB-5-131 was able to improve motor function in aging animals with well-developed pathology, we measured the Rota-Rod performance, grip strength-endurance and open-field activity of 60-week HdhQ(150/150) animals at various points following the start of treatment (Fig. 5, from 7 to 60 weeks).
Figure 5.

XJB-5-131 treatment of HdhQ(150/150) improves motor function with age. (A) Representative examples of the grip test for three treatment groups at three ages, as indicated. The HdhQ(150/150)-vh animals lose the ability to grip the bar with age, while treatment with XJB-5-131 prevents this deterioration, as HD-tr animals appear equivalent to vehicle (vh) or treated (tr) WT animals (see Supplementary Material, Video S1). (B) Quantification of (A). The results for the change in performance of each animal were normalized for their performance relative to the start of treatment, which was denoted as one for each animal. Treated diseased animals performed significantly better with age over their untreated counterparts (P < 0.05) (P = Student's t-test, one-tailed homoscedastic). (C) The quantified results of the Rota-Rod assay also indicate that XJB-5-131 improved performance of HdhQ(150/150) relative to untreated animals (vehicle, vh). Vh and treated (tr) HdhQ(150/150) animals were normalized to the performance of their WT littermates for each time point. The color key identifies the genotypes and treatment groups. (D) XJB-5-131 treatment in HdhQ(150/150) animals reduces rear feet dragging phenotypes. A sample video is provided (Supplementary Material, Video S3). The HdhQ(150/150) untreated or vehicle-treated mice occasionally dragged their back feet during forward motion at 20–40 weeks of treatment (80–100 weeks of age). Treated HdhQ(150/150) had a significantly reduced occurrence of this phenotype (P = Student's t-test, one-tailed homoscedastic) (n = 3 mice per sample type, five videos of 5 min each were scored per mouse).
XJB-5-131 administration led to a striking improvement in grip strength-endurance (Fig. 5A and B). Each animal was normalized for its performance at the start of treatment (at 60 weeks) (Fig. 5B) and followed with age. Aging HdhQ(wt/wt) littermate maintained good performance within the 25 weeks treatment period (total age was 85 weeks) (Fig. 5C), while vehicle-treated HdhQ(150/150) animals substantially declined in the same period (Supplementary Material, Video S1). In contrast, there was no significant decline in grip strength in XJB-5-131-treated HdhQ(150/150) animal during the same 25-week treatment period (Fig. 5B, green) (see Supplementary Material, Video S1). XJB-5-131-treated animals were similar to vehicle-treated HdhQ(wt/wt) littermates (black), and indistinguishable from XJB-5-131-treated HdhQ(wt/wt) (green) controls at advanced age (Fig. 5B).
XJB-5-131 improvement on Rota-Rod performance of HdhQ(150/150) animals was equally striking (Fig. 5C). The vehicle-treated HdhQ(150/150) animals were characterized by reduced motor performance on the Rota-Rod, which progressed with age. XJB-5-131 treatment of HdhQ(150/150) animals significantly improved performance that was well developed at the beginning of treatment (Fig. 5C). Even at advanced age, the Rota-Rod performance of XJB-treated HdhQ(150/150) animals was better than that at the beginning of treatment (Fig. 5C, compare 0 and 25 weeks), and was similar to their age-matched HdhQ(wt/wt) littermates at all ages tested.
We also tested the open-field activity in vehicle- and XJB-5-131-treated HdhQ(150/150) animals using video imaging. Quantified for each animal was the speed of motion, acceleration speed, body area, body axes ratio and the orientation of movement from individual animals, measured at the same time each day (see the ‘Materials and Methods’ section) (Supplementary Material, Fig. S4A). The activity of animals in the open field was highly variable and sensitive to handling conditions (examples in Supplementary Material, Video S2). Some HdhQ(150/150) animals did not move (Supplementary Material, Fig. S4B), but others were active and did not appear distinct from control littermates. Similarly, most control animals had strong exploratory activity (Supplementary Material, Fig. S4C), but others had poor motility. Consequently, the average activity among treated and untreated animals for these parameters was not statistically different.
In contrast, quantitative video analysis revealed a significant frequency of ‘foot dragging’ in vehicle-treated HdhQ(150/150) animals that was not observed in control or in XJB-5-131-treated HdhQ(150/150) littermates (Fig. 5D and Supplementary Material, Fig. S5) (see Supplementary Material, Video S3). As they pushed themselves forward to initiate movement, these animals were often slow to fully engage their hind legs (Supplementary Material, Fig. S5) (see Supplementary Material, Video S3). These instances were manually counted and corroborated by video analysis as arcs when centroid speed is plotted versus body area (see Supplementary Material, Video S3). An average of 10 occurrences of ‘foot dragging’ were detected in HdhQ(150/150) animals during a 5 min period, while no instances were observed in XJB-5-131-treated littermates or in age-matched HdhQ(wt/wt) littermates (Fig. 5D). Collectively, XJB-5-131 significantly reduced the motor deficits in HdhQ(150/150) mice.
Treatment with XJB-5-131 reduces oxidative damage to MT and nuclear DNA in neurons
XJB-5-131 is designed to target MT and reduce oxidative damage. Indeed, we previously published that boron-dipyrromethene (BODIPY)-fluorescent (FL)-XJB-5-131, a FL-BODIPY-labeled derivative of XJB-5-131, crosses the plasma membrane of MT in primary cultured striatal neurons, and co-stained with MitoTracker Deep Red (37). If the beneficial effects of XJB-5-131 occurred by its expected mechanisms, then XJB-5-131 should reduce the level of oxidative damage in brain mtDNA. In the first experiments, we sliced brains sections, used an antibody to 8-oxo-7,8-dihydroguanine (8-oxo-G) on each slice to determine whether XJB-5-131 reduced staining intensity (Fig. 6).
Figure 6.

XJB-5-131 reduces oxidative damage in MT and somatic DNA. (A) At the start of treatment there are = low levels of oxidative DNA lesions in untreated HdhQ(wt/wt) (WT-vh) animals as visualized by anti-8-oxo-G IF staining. There is little discernible difference between WT and HdhQ(150/150) (HD) littermates at these early ages (up to 85 weeks of age) (image not shown). At late ages (120 weeks), there is a multi-fold increase in DNA lesions in HD animals, which is suppressed by XJB-5-131 treatment, as seen both by IF staining for 8-oxo-G (A,B) and by (C) q-PCR analysis (n = 3 mice per group) (*P < 0.05) (P = Student's t-test, one-tailed homoscedastic). (B) 8-oxo-dG (red) is present both in MT and in the nucleus of striatal cells of old HD mice [in this case an HdhQ(150/150)-vh mouse 120 weeks of age]. (B, Panel 2) The localization of the oxidative damage is primarily perinuclear foci, along with a fainter and diffuse nuclear pattern. (B, Panel 3 and insets) The foci co-localize with the MT [anti-VDAC (green)] and also with DAPI (viewable when DAPI imaging exposure is increased). Yellow bars represent 10 µm (images ∼21 µm confocal Z-stack 3D reconstruction, frame: 84.9 µm2).
Oxidation occurs continually throughout life. As expected, the level of 8-oxo-G staining was obvious in the histology section of both genotypes, even in the brain sections of HdHQ(wt/wt) animals at early treatment times (Fig. 6B, Wt-vh. 60 + 7 weeks, shown is the STR). However, the level of 8-oxo-G increases significantly in the aging brain, and some of the 8-oxo-G converts to other forms of oxidative damage, which are no longer detected by the 8-oxo-G antibodies (38,39). Thus, antibody staining intensity poorly discriminated between the lesion level in brain tissue from aging HdHQ(wt/wt) and HdhQ(150/150) animals (Supplementary Material, Table S2), but served as a robust probe of signal reduction by XJB-5-131. Indeed, XJB-5-131 treatment dramatically reduced the level of 8-oxo-G staining in the brains of both HdhQ(150/150) and HdHQ(wt/wt) littermates (Fig. 6B, compare Panels 2 and 3) (Supplementary Material, Table S2).
In the histology sections, oxidation was present in the nuclear DNA, as judged by the diffuse staining intensity throughout the neuronal nucleus (Fig. 6A, overlay the blue DAPI in Panel 1 to the co-incident diffuse, red 8-oxoG antibody staining in Panels 2 and 3). However, the majority of 8-oxo-G staining was confined to organelles (Fig. 6A, Panels 1 and 2), which were identified as MT by co-staining with the mitochondrial voltage-dependent anion channel (VDAC) (Fig. 6A, compare the red 8-oxo-G staining in Panel 2 with green VDAC antibody staining in Panel 3). Indeed, the individual fluorophore patterns (Fig 6A insets) confirmed that 8-oxo-G co-stained with mtDNA and VDAC in the same organelles. The oxidative damage in MT occurred in both neurons and in glial cells, but the lesion level was roughly 3-fold higher in neurons (67% neurons versus 22% of glia) (Supplementary Material, Table S2).
To more accurately compare the overall damage, we isolated the DNA in the MT of the striatum, and used quantitative polymerase chain reaction (qPCR) to measure the level of lesions directly in HdHQ(wt/wt) and HdhQ(150/150) animals. qPCR intensity increases linearly with the lesion load, and detects a wide range of DNA oxidative lesion based on their ability to slow or to block polymerase progression in DNA (38). Importantly, 8-oxo-G has little impact in polymerase progression, and the detection of this common lesion does not dominate the PCR products. Thus, qPCR better reflected differences in oxidative damage between the HdHQ(wt/wt) and HdhQ(150/150) animals by quantifying lesions that are typically present at the lower level.
Indeed, qPCR provided evidence that the level of oxidative DNA damage in mtDNA was higher in the brains of vehicle-treated HdhQ(150/150) animals relative to their HdhQ(wt/wt) littermates (Fig. 6C), and confirmed that XJB-5-131 reduced the level of oxidative damage in HdhQ(150/150) animals. Thus, XJB-5-131 operated by its expected mechanism in brain MT. Collectively, the reduced load of oxidative damage after XJB-5-131 treatment was accompanied by rescue of neurons in XJB-5-131-treated HdhQ(150/150) animals and improvement in their motor performance.
Discussion
There are no effective therapeutics available that offset the devastating progressive effects of HD or other neurodegenerative diseases. Previously, we have reported that the synthetic radical and electron scavenger, XJB-5-131, attenuates the decline in Rota-Rod performance if treatment begins at a young age before phenotypes develop (37). Here, we report that XJB-5-131 treatment attenuates or reverses the effects of disease if treatment begins after disease onset. In aging animals with well-developed pathology, XJB-5-131 treatment promotes weight gain, attenuates neuronal loss, reduces inclusion formation, prevents the performance decline in multiple motor tests and decreases oxidative damage in the brains of aging HdhQ(150/150) animals relative to age-matched vehicle-treated HdhQ(150/150) animals. The treated HdhQ(150/150) animals were often indistinguishable from age-matched HdhQ(wt/wt) control animals. XJB-5-131 also reduces the somatic expansion of the CAG repeat tracts in these animals (40), and this feature has been confirmed in R6/2 animals (A. Polyzos and J. Morton, Univ. of Cambridge, submitted for publication). To our knowledge, this is the first demonstration of a sustained reversal/attenuation of pathophysiology by an antioxidant therapy.
The key to its efficacy is the mitochondrial targeting function of XJB-5-131. A peptide mimetic moiety in this compound targets the antioxidant directly to the mitochondrial membrane (Fig. 1A) where the compound is enriched ∼600-fold relative to the cytosol. Neither untargeted TEMPO (antioxidant component) nor the peptide mimetic (targeting component) alone are effective in offsetting toxicity (30). We show here that XJB-5-131 effectively reduces ROS in brain MT, but also reduces 8-oxo-G staining in the mtDNA, consistent with its functions in mitochondrial targeting and antioxidant efficacy. Thus, the peptide mimetic portion of XJB-5-131 delivers the antioxidant moiety directly to the site where reactive species are formed (Fig. 1A), and there the drug exerts a sustained homeostatic effect.
TEMPO, the active nitroxide moiety in XJB-5-131, can undergo one- or two-electron transfer processes to hydroxylamines or oxoammoniumcations (41,42) thereby supporting redox homeostasis. Due to these redox cycling abilities, XJB-5-131 prevents the leakage of electrons during the reduction of O2 to H2O at cytochrome c-oxidase and avoids the formation of superoxide radical anion (31). The nitroxide moiety is able to return the electrons to the electron transport chain, and therefore XJB-5-131 can exert antioxidant properties without significantly diminishing ATP generation.
XJB-5-131 overcomes some potential sources of past clinical failures of antioxidant therapies. Since XJB-5-131 is a synthetic antioxidant it is not modulated by gene expression, and is capable of achieving and maintaining pharmacologically effective concentrations in cells. XJB-5-131 crosses the BBB and is readily visualized in the brains of rats by in vivo EPR imaging (43). Thus, the synthetic compound has proven direct access to the neurons. Furthermore, XJB-5-131 is charge neutral and does not influence the mitochondrial membrane potential nor does it depend on the electrostatic gradient to enter MT. This property of XJB-5-131 provides a mechanism for improving mitochondrial function even after membrane depolarization.
Taken together, direct targeting of a synthetic antioxidant to MT is poised to provide a level of specificity and efficacy that enables a viable therapeutic strategy. Because mitochondrial decline is a common and central feature of toxicity in the brain, targeted nitroxides such as XJB-5-131 are expected to be useful in a wide spectrum of neurodegenerative diseases, or in other deleterious ROS-induced conditions and mitochondrial malfunctions. Indeed, we have previously shown that XJB-5-131 and the related JP4-039 have efficacy in a rat model of hemorrhagic shock (44), a mouse model of radiation mitigation (45), a rat model of traumatic brain injury (43) and on cardiac resistance to ischemia-reperfusion-induced oxidative stress in aged rats (46).
A therapeutic compound is far less useful if it cannot reverse the disease phenotypes that have developed in a patient before treatment. The lack of such efficacy in animal testing may account, at least in part, for the current disconnect between preclinical and clinical outcomes. Delivery of XJB-5-131 to neuronal protects MT against damage before or after onset in models of advanced HD. XJB-5-131 has not yet been tested in humans for efficacy, however, based on the preclinical results reported here, a compound with these or similar properties is desirable. Individuals with HD are typically aware that the disease gene is passed down in their family line, but not all individuals in a family will choose to be genetically tested. Thus, subjects who have a confirmed diagnosis may opt to start treatment with an effective therapeutic at a young age. At the same time, a compound that is effective after the development of phenotypes would bring enormous benefits to patients who choose not to be tested or are unaware of the disease but develop symptoms later in life.
The need for new therapeutics for neurodegeneration is extraordinary. Due to rapid increases in the elderly populations, age-related diseases are expected to grow exponentially in the coming years (47). Among these, neurodegenerative diseases will be the most devastating in terms of their emotional and economic impacts. Because the number of affected individuals will grow significantly, the gap between the size of the problem and our capabilities for treatment will widen. Recently, a reduction in the level of expanded mhtt protein by anti-sense RNA to human Huntington has led to reversible efficacy in an animal model (48). XJB-5-131 alone is able to produce the same outcome based on its distinct mechanism of action. These features support XJB-5-131's role as a promising clinical candidate, and warrant further investigation of this class of compounds for their efficacy either as a stand-along therapeutic or in combination with other treatments.
Materials and Methods
Animals, breeding and weights
The Institutional Animal Care and Use Committee approved all procedures. Animals were treated under guidelines for the ethical treatment of animals, and approved by IACUC protocol #274005 at Lawrence Berkeley Laboratory. All animal work was conducted according to national and international guidelines. The following mouse models were used: homozygous HdhQ(150/150), which have been previously described and characterized (33). The original HdhQ(150/wt) line (33) was used in these experiments, and not a derived line that is commercially available, but was generated later (strain B6.129P2-Htttm2Detl/150J) (Jackson Labs, Bar Harbor, ME, USA). The HdhQ(150/wt) were generated on the C57Bl6 background. The homozygous mice are referred to as HdhQ(150/150) and their WT littermates are referred to as HdhQ(wt/wt).
Treatments and groups
Mice were divided into three treatment groups: untreated, vehicle treated (vh) and XJB-5-131 treated (tr) (at 2 mg/kg) for each genotype [HdhQ(150/150) and their WT littermates]. There are 30 mice in each group. XJB-5-131 was administered by intra-peritoneal injection three times per week for the duration of the study. Vehicle treatments were identical except that XJB-5-131 was replaced by filtered phosphate-buffered saline (PBS). Mice were weighed, and tested for behavior beginning at 60 weeks, and after 7, 10, 25 and 40 or 60 weeks of treatment. Treatment began ∼60 weeks, followed by treatment period of up to 60 weeks. The animal age at the end of the study was ∼120 weeks.
XJB-5-131
XJB-5-131 was synthesized as previously reported (3). Lyophilized, powdered XJB-5-131 was reconstituted in dimethyl sulfoxide at a concentration of 1 mg/µl, as previously described (49). These samples were aliquoted and kept at −80°C. On the day of injection, the XJB-5-131 solution was mixed with 0.2 µm filtered and pre-warmed PBS (100°C) and heated for 10 s to reach a final concentration of 2 mg/kg mouse body weight in 200 µl solution. The solution (200 µl) was injected within 30 min of preparation.
Rota-Rod
Motor activity was tested with a Rota-Rod (UgoBasile, USA/Italy), as previously described (7,37,40). Weights of each animal were recorded once per week. Animals in each group were evaluated for Rota-Rod performance and grip strength at the indicated ages. Mice were lowered on to the already spinning Rota-Rod at the required speed (20 rpm was used in this study). The amount of time the animals stayed on the Rota-Rod was determined by a built-in magnetic trip-switch, which was stopped when the animal fell-off. Mice were measured for Rota-Rod activity for a maximum of 120 s, with three attempts given for each mouse. Animals were trained for one session each day for 5 consecutive trials, and tested for 3 consecutive days at each time point (0, 7, 10 and 25 weeks of treatment). The best (longest) performance was recorded and used for the comparative analysis.
Grip test
For the grip strength-endurance test, mice were lowered onto a parallel rod (D < 0.25 cm) placed 50 cm above a padded surface. The mice were allowed to grab the rod with their forelimbs, after which they were released and scored for the length of time they could hold onto the bar (maximum 30 s). Mice were tested consecutively three times for each time point (at 0, 7, 10 and 25 weeks of treatment). The maximum length of time they were able to hold on was recorded for analysis.
Antibodies, immunofluorescence and quantification
Primary antibodies used were mouse anti-NeuN Alexa488 conjugate (Millipore #MAB377X) (used at 1:400), mouse anti-GFAP Cy3 conjugate (Abcam #ab49874) (used at 1:400), rabbit anti-Iba1 (Wako #019-19741) (used at 1:400), rabbit anti-Ubiquitin (DAKO #Z0458) (used at 1:400), mouse anti-8-oxo-dG (Trevigen #4354-mc-050) (used at 1:400) and rabbit anti-VDAC (Thermo Fisher #PA1-954A) (used at 1:400). Secondary antibodies used were goat anti-rabbit Alexa-555 conjugated (Invitrogen #A31630) (used at 1:400), goat anti-mouse Alexa-488 conjugated (Invitrogen #A31620) (used at 1:400). Brain sections cryoembedded in optimal cutting temperature (OCT) were sliced (10 µm thick using a Leica Cryostat set at: −14°C for the sample and −12°C for the blade) and placed onto Histobond microscope slides (VWR). They were immediately fixed and OCT removed in 100% methanol (10 min). Samples were rehydrated in 75, 50, 25 and 0% ethanol in PBS (2 min each). Tissue was treated with Image-iT FX signal enhancer (Thermo Fisher #I36933) to reduce autofluorescence (30 min) and blocked (2–18 h) in blocking solution (PBS, 3% bovine serum albumin, 5% goat serum, 0.7% donkey serum, 0.03% triton X-100). Antibody staining was performed overnight, followed by three washes with PBS (5 min ea.). Secondary antibody was applied later as required along with 0.5 µm DAPI (1–2 h) followed by three washes in PBS. Slides were coated with Vectashield + DAPI, sealed with a coverslip and stored (−20°C) until they were imaged. Slides were imaged using a Zeiss 710 confocal microscope using either of 20 × (0.8N/A)/air, 40 × (1.2N/A)/water or 100 × (1.4N/A)/oil lenses. Image analysis was done using ImageJ: Fiji (50).
Video imaging and foot dragging quantification
Mouse behavior in the open-field arena was tracked at high-resolution from raw videos (30 Hz) processed with Matlab scripts (The MathWorks) as a custom-made adaptation of freely available animal tracking software (51). Quantitative analyses of low-level kinematic quantities were based on the animal centroid coordinates, its instantaneous speed and acceleration, and morphological features such as the animal's body area, and its orientation via an ellipsoidal fit on its posture. Foot dragging effects were suggested via phase plots combining centroid speed as a function of body area.
Lesion counts by quantitative PCR
The relative level of mtDNA lesions in the mouse brain was performed as previously described (52,53). The determination of mtDNA lesion number consisted of amplifying a 10 kp mtDNA fragment by performing an initial denaturation for 45 s at 94°C, followed by 22 cycles of denaturation for 15 sat 94°C, annealing/extension at 64°C for 12 min, and a final extension for 10 min at 72°C. We used the following primer nucleotide sequences: 5′-CCAGTCCATGCAGGAGCATC-3′ (forward) and 5′-CGAGAAGAGGGGCATTGGTG-3′ (reverse). Levels of damage were calculated as the relative amplification of the HdhQ(150/150) STR compared wiith the WT Hdh(wt/wt) controls. The data were corrected for possible changes in mtDNA replication rates. The results were derived from three qPCR assays in triplicate on each animal. n = 7 WT-vh mice; n = 9 HD-vh mice and n = 5 HD-tr mice.
Supplementary Material
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
Funding
This work was supported by National Institutes of Health (grants ES020766-01, NS060115, CA092584 to C.T.M.; SC1NS095380 to S.A.P.; GM061838) and the University of Puerto Rico Infrastructural (grant MD007600 to S.A.P.; AG04337601 to P.W.) and Portuguese Foundation for Science and Technology (grant no SFRH/BPD/97544/2013 to A.G.-M.).
Supplementary Material
References
- 1.Huang Y., Mucke L. (2012) Alzheimer mechanisms and therapeutic strategies. Cell, 148, 1204–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Exner N., Lutz A.K., Haass C., Winklhofer K.F. (2012) Mitochondrial dysfunction in Parkinson's disease: molecular mechanisms and pathophysiological consequences. EMBO J., 31, 3038–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johri A., Beal M.F. (2012) Antioxidants in Huntington's disease. Biochim. Biophys. Acta, 1822, 664–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Naseri N.N., Xu H., Bonica J., Vonsattel J.P., Cortes E.P., Park L.C., Arjomand J., Gibson G.E. (2015) Abnormalities in the tricarboxylic Acid cycle in Huntington disease and in a Huntington disease mouse model. J. Neuropathol. Exp. Neurol., 74, 527–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Desler C., Hansen T.L., Frederiksen J.B., Marcker M.L., Singh K.K., Juel Rasmussen L. (2012) Is there a link between mitochondrial reserve respiratory capacity and aging? J. Aging.Res., 2012, 192503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Song W., Chen J., Petrilli A., Liot G., Klinglmayr E., Zhou Y., Poquiz P., Tjong J., Pouladi M.A., Hayden M.R. et al. (2011) Mutant huntingtin binds the mitochondrial fission GTPasedynamin-related protein-1 and increases its enzymatic activity. Nat. Med., 17, 377–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trushina E., Dyer R.B., Badger J.D. II, Ure D., Eide L., Tran D.D., Vrieze B.T., Legendre-Guillemin V., McPherson P.S., Mandavilli B.S. et al. (2004) Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol. Cell Biol., 24, 8195–8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weydt P., Pineda V.V., Torrence A.E., Libby R.T., Satterfield T.F., Lazarowski E.R., Gilbert M.L., Morton G.J., Bammler T.K., Strand A.D. et al. (2006) Thermoregulatory and metabolic defects in Huntington's disease transgenic mice implicate PGC-1alpha in Huntington's disease neurodegeneration. Cell. Metab., 4, 349–362. [DOI] [PubMed] [Google Scholar]
- 9.Valor L.M. (2015) Transcription, epigenetics and ameliorative strategies in Huntington's disease: a genome-wide perspective. Mol. Neurobiol., 51, 406–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaturvedi R.K., Flint Beal M. (2013) Mitochondrial diseases of the brain. Free Radic. Biol. Med., 63, 1–29. [DOI] [PubMed] [Google Scholar]
- 11.Kwong L.K., Kamzalov S., Rebrin I., Bayne A.C., Jana C.K., Morris P., Forster M.J., Sohal R.S. (2002) Effects of coenzyme Q(10) administration on its tissue concentrations, mitochondrial oxidant generation, and oxidative stress in the rat. Free Radic. Biol. Med., 33, 627–638. [DOI] [PubMed] [Google Scholar]
- 12.Littarru G.P., Tiano L. (2010) Clinical aspects of coenzyme Q10: an update. Nutrition, 26, 250–254. [DOI] [PubMed] [Google Scholar]
- 13.Beal M.F., Oakes D., Shoulson I., Henchcliffe C., Galpern W.R., Haas R., Juncos J.L., Nutt J.G., Voss T.S., Ravina B. et al. (2014) A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: no evidence of benefit. JAMA Neurol., 71, 543–552. [DOI] [PubMed] [Google Scholar]
- 14.Andreassen O.A., Dedeoglu A., Ferrante R.J., Jenkins B.G., Ferrante K.L., Thomas M., Friedlich A., Browne S.E., Schilling G., Borchelt D.R. et al. (2001) Creatine increase survival and delays motor symptoms in a transgenic animal model of Huntington's disease. Neurobiol. Dis., 8, 479–491. [DOI] [PubMed] [Google Scholar]
- 15.Dedeoglu A., Kubilus J.K., Yang L., Ferrante K.L., Hersch S.M., Beal M.F., Ferrante R.J. (2003) Creatine therapy provides neuroprotection after onset of clinical symptoms in Huntington's disease transgenic mice. J. Neurochem., 85, 1359–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bender A., Koch W., Elstner M., Schombacher Y., Bender J., Moeschl M., Gekeler F., Muller-Myhsok B., Gasser T., Tatsch K. et al. (2006) Creatine supplementation in Parkinson disease: a placebo-controlled randomized pilot trial. Neurology, 67, 1262–1264. [DOI] [PubMed] [Google Scholar]
- 17.Oyewole A.O., Birch-Machin M.A. (2015) Mitochondrial-targeted antioxidants. FASEB J., 12, 4766–4771. [DOI] [PubMed] [Google Scholar]
- 18.Palamakula A., Khan M.A. (2004) Evaluation of cytotoxicity of oils used in coenzyme Q10 self-emulsifying drug delivery systems (SEDDS). Int. J. Pharm., 273, 63–73. [DOI] [PubMed] [Google Scholar]
- 19.Orsucci D., Mancuso M., Ienco E.C., LoGerfo A., Siciliano G. (2011) Targeting mitochondrial dysfunction and neurodegeneration by means of coenzyme Q10 and its analogues. Curr. Med. Chem., 18, 4053–4064. [DOI] [PubMed] [Google Scholar]
- 20.Schilling G., Savonenko A.V., Coonfield M.L., Morton J.L., Vorovich E., Gale A., Neslon C., Chan N., Eaton M., Fromholt D. et al. (2004) Environmental, pharmacological, and genetic modulation of the HD phenotype in transgenic mice. Exp. Neurol., 187, 137–149. [DOI] [PubMed] [Google Scholar]
- 21.Cleren C., Yang L., Lorenzo B., Calingasan N.Y., Schomer A., Sireci A., Wille E.J., Beal M.F. (2008) Therapeutic effects of coenzyme Q10 (CoQ10) and reduced CoQ10 in the MPTP model of Parkinsonism. J. Neurochem., 104, 1613–1621. [DOI] [PubMed] [Google Scholar]
- 22.Shannon K.M., Fraint A. (2015) Therapeutic advances in Huntington's disease. Mov. Disord., 30, 1539–1546 [DOI] [PubMed] [Google Scholar]
- 23.Sohal R.S., Forster M.J. (2007) Coenzyme Q, oxidative stress and aging. Mitochondrion, 7 (Suppl.), S103–S111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salama M., Yuan T.F., Machado S., Murillo-Rodriguez E., Vega J.A., Menendez-Gonzalez M., Nardi A.E., Arias-Carrion O. (2013) Co-enzyme Q10 to treat neurological disorders: basic mechanisms, clinical outcomes, and future research direction. CNS Neurol. Disord. Drug. Targets, 12, 641–664. [DOI] [PubMed] [Google Scholar]
- 25.Sumien N., Heinrich K.R., Shetty R.A., Sohal R.S., Forster M.J. (2009) Prolonged intake of coenzyme Q10 impairs cognitive functions in mice. J. Nutr., 139, 1926–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Snow B.J., Rolfe F.L., Lockhart M.M., Frampton C.M., O'Sullivan J.D., Fung V., Smith R.A., Murphy M.P., Taylor K.M. (2010) A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson's disease. Mov. Disord., 25, 1670–1674. [DOI] [PubMed] [Google Scholar]
- 27.Farhangi M.A., Alipour B., Jafarvand E., Khoshbaten M. (2014) Oral coenzyme Q10 supplementation in patients with nonalcoholic fatty liver disease: effects on serum vaspin, chemerin, pentraxin 3, insulin resistance and oxidative stress. Arch. Med. Res., 45, 589–595. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y., Aberg F., Appelkvist E.L., Dallner G., Ernster L. (1995) Uptake of dietary coenzyme Q supplement is limited in rats. J. Nutr., 125, 446–453. [DOI] [PubMed] [Google Scholar]
- 29.Jauslin M.L., Meier T., Smith R.A., Murphy M.P. (2003) Mitochondria-targeted antioxidants protect Friedreich Ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. FASEB J., 17, 1972–1974. [DOI] [PubMed] [Google Scholar]
- 30.Fink M.P., Macias C.A., Xiao J., Tyurina Y.Y., Delude R.L., Greenberger J.S., Kagan V.E., Wipf P. (2007) Hemigramicidin-TEMPO conjugates: novel mitochondria-targeted antioxidants. Crit. Care Med., 35, S461–S467. [DOI] [PubMed] [Google Scholar]
- 31.Wipf P., Xiao J., Jiang J., Belikova N.A., Tyurin V.A., Fink M.P., Kagan V.E. (2005) Mitochondrial targeting of selective electron scavengers: synthesis and biological analysis of hemigramicidin-TEMPO conjugates. J. Am. Chem. Soc., 127, 12460–12461. [DOI] [PubMed] [Google Scholar]
- 32.Hoye A.T., Davoren J.E., Wipf P., Fink M.P., Kagan V.E. (2008) Targeting mitochondria. Acc. Chem. Res., 41, 87–97. [DOI] [PubMed] [Google Scholar]
- 33.Lin C.H., Tallaksen-Greene S., Chien W.M., Cearley J.A., Jackson W.S., Crouse A.B., Ren S., Li X.J., Albin R.L., Detloff P.J. (2001) Neurological abnormalities in a knock-in mouse model of Huntington's disease. Hum. Mol. Genet., 10, 137–144. [DOI] [PubMed] [Google Scholar]
- 34.Vonsattel J.P., DiFiglia M. (1998) Huntington disease. J. Neuropathol. Exp. Neurol., 57, 369–384. [DOI] [PubMed] [Google Scholar]
- 35.Vonsattel J.P., Myers R.H., Stevens T.J., Ferrante R.J., Bird E.D., Richardson E.P. Jr. (1985) Neuropathological classification of Huntington's disease. J. Neuropathol. Exp. Neurol., 44, 559–577. [DOI] [PubMed] [Google Scholar]
- 36.Orvoen S., Pla P., Gardier A.M., Saudou F., David D.J. (2012) Huntington's disease knock-in male mice show specific anxiety-like behaviour and altered neuronal maturation. Neurosci. Lett., 507, 127–132. [DOI] [PubMed] [Google Scholar]
- 37.Xun Z., Rivera-Sanchez S., Ayala-Pena S., Lim J., Budworth H., Skoda E.M., Robbins P.D., Niedernhofer L.J., Wipf P., McMurray C.T. (2012) Targeting of XJB-5-131 to mitochondria suppresses oxidative DNA damage and motor decline in a mouse model of Huntington's disease. Cell. Rep., 2, 1137–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luo W., Muller J.G., Rachlin E.M., Burrows C.J. (2001) Characterization of hydantoin products from one-electron oxidation of 8-oxo-7,8-dihydroguanosine in a nucleoside model. Chem. Res. Toxicol., 14, 927–938. [DOI] [PubMed] [Google Scholar]
- 39.Muftuoglu M., Mori M.P., de Souza-Pinto N.C. (2014) Formation and repair of oxidative damage in the mitochondrial DNA. Mitochondrion, 17, 164–181. [DOI] [PubMed] [Google Scholar]
- 40.Budworth H., Harris F.R., Williams P., Lee do Y., Holt A., Pahnke J., Szczesny B., Acevedo-Torres K., Ayala-Pena S., McMurray C.T. (2015) Suppression of somatic expansion delays the onset of pathophysiology in a mouse model of Huntington's disease. PLoS Genet., 11, e1005267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parry J.D., Pointon A.V., Lutz U., Teichert F., Charlwood J.K., Chan P.H., Athersuch T.J., Taylor E.L., Singh R., Luo J. et al. (2009) Pivotal role for two electron reduction in 2,3-dimethoxy-1,4-naphthoquinone and 2-methyl-1,4-naphthoquinone metabolism and kinetics in vivo that prevents liver redox stress. Chem. Res. Toxicol., 22, 717–725. [DOI] [PubMed] [Google Scholar]
- 42.Sheng Y., Abreu I.A., Cabelli D.E., Maroney M.J., Miller A.F., Teixeira M., Valentine J.S. (2014) Superoxide dismutases and superoxide reductases. Chem. Rev., 114, 3854–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ji J., Kline A.E., Amoscato A., Samhan-Arias A.K., Sparvero L.J., Tyurin V.A., Tyurina Y.Y., Fink B., Manole M.D., Puccio A.M. et al. (2012) Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury. Nat. Neurosci., 15, 1407–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Macias C.A., Chiao J.W., Xiao J., Arora D.S., Tyurina Y.Y., Delude R.L., Wipf P., Kagan V.E., Fink M.P. (2007) Treatment with a novel hemigramicidin-TEMPO conjugate prolongs survival in a rat model of lethal hemorrhagic shock. Ann. Surg., 245, 305–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gokhale A., Rwigema J.C., Epperly M.W., Glowacki J., Wang H., Wipf P., Goff J.P., Dixon T., Patrene K., Greenberger J.S. (2010) Small molecule GS-nitroxide ameliorates ionizing irradiation-induced delay in bone wound healing in a novel murine model. In Vivo, 24, 377–385. [PMC free article] [PubMed] [Google Scholar]
- 46.Escobales N., Nunez R.E., Jang S., Parodi-Rullan R., Ayala-Pena S., Sacher J.R., Skoda E.M., Wipf P., Frontera W., Javadov S. (2014) Mitochondria-targeted ROS scavenger improves post-ischemic recovery of cardiac function and attenuates mitochondrial abnormalities in aged rats. J. Mol. Cell Cardiol., 77, 136–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koplan J.P., Fleming D.W. (2000) Current and future public health challenges. JAMA, 284, 1696–1698. [DOI] [PubMed] [Google Scholar]
- 48.Kordasiewicz H.B., Stanek L.M., Wancewicz E.V., Mazur C., McAlonis M.M., Pytel K.A., Artates J.W., Weiss A., Cheng S.H., Shihabuddin L.S. et al. (2012) Sustained therapeutic reversal of Huntington's disease by transient repression of huntingtin synthesis. Neuron, 74, 1031–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Skoda E.M., Davis G.C., Wipf P. (2012) Allylic amines as key building blocks in the synthesis of (E)-alkene peptide isosteres. Org. Process Res. Dev., 16, 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schneider C.A., Rasband W.S., Eliceriri K.W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods, 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gomez-Marin A., Partoune N., Stephens G.J., Louis M., Brembs B. (2012) Automated tracking of animal posture and movement during exploration and sensory orientation behaviors. PLoS One, 7, e41642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ayala-Torres S., Chen Y., Svoboda T., Rosenblatt J., Van Houten B. (2000) Analysis of gene-specific DNA damage and repair using quantitative polymerase chain reaction. Methods, 22, 135–147. [DOI] [PubMed] [Google Scholar]
- 53.Siddiqui A., Rivera-Sanchez S., Castro Mdel R., Acevedo-Torres K., Rane A., Torres-Ramos C.A., Nicholls D.G., Andersen J.K., Ayala-Torres S. (2012) Mitochondrial DNA damage is associated with reduced mitochondrial bioenergetics in Huntington's disease. Free Radic. Biol. Med., 53, 1478–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.