

Abstract
Riboflavin, also known as vitamin B2, is essential for cellular reduction-oxidation reactions, but is not readily synthesized by mammalian cells. It has been proposed that riboflavin absorption occurs through solute carrier family 52 members (SLC52) A1, A2 and A3. These transporters are also candidate genes for the childhood onset-neural degenerative syndrome Brown–Vialetto–Van Laere (BVVL). Although riboflavin is an essential nutrient, why mutations in its transporters result in a neural cell-specific disorder remains unclear. Here, we provide evidence that Slc52a3 is the mouse ortholog of SLC52A3 and show that Slc52a3 deficiency results in early embryonic lethality. Loss of mutant embryos was associated with both defects in placental formation and increased rates of apoptosis in embryonic cells. In contrast, Slc52a3 −/− embryonic stem cell lines could be readily established and differentiated into motor neurons, suggesting that this transporter is dispensable for neural differentiation and short-term maintenance. Consistent with this finding, examination of Slc52a3 gene products in adult tissues revealed expression in the testis and intestine but little or none in the brain and spinal cord. Our results suggest that BVVL patients with SCL52A3 mutations may be good candidates for riboflavin replacement therapy and suggests that either the mutations these individuals carry are hypomorphic, or that in these cases alternative transporters act during human embryogenesis to allow full-term development.
Introduction
Riboflavin and its derivatives flavin adenine dinucleotide (FAD) and flavin mononucleotide act as essential cofactors for a variety of metabolic processes, including the electron transport chain. Because riboflavin cannot be readily synthesized in mammalian cells, it must be absorbed from the diet via riboflavin transporters, which have recently been identified in human and rat (1–3). As a result, riboflavin deficient diets have been associated with a variety of developmental and growth disorders (4).
Brown–Vialetto–Van Laere (BVVL) syndrome is a rare neurodegenerative disease characterized by progressive ponto-bulbar palsy, bilateral sensorineural hearing loss, muscle weakness and amyotrophy (5–8). Respiratory failure as a result of diaphragm denervation is a main cause of death. The origins of this disorder are poorly understood and discovery of genetic variants contributing to this disorder has been slowed by the death of patients prior to reproductive age and its low incidence (9). However, exome sequencing of BVVL patients has recently shown that a subset of patients may be homozygous for rare coding variants in C20ORF54 (9–16). C20ORF54 encodes SLC52A3, one of the three recently identified candidate riboflavin transporters, which share considerable homology. Subsequent exome and candidate gene sequencing studies have further implicated this class of transporters in BVVL, showing that distinct patients can harbor rare variants in another riboflavin transporter gene, SLC52A2 (10,16–18). Thus far, SLC52A1 mutations have yet to be seen in BVVL patients. These discoveries have led to reported attempts to administer riboflavin to children with BVVL, with some reports of anecdotal improvement in their condition (19).
However, it remains untested whether there is a causal relationship between mutations in the class of putative riboflavin transporters and neural degeneration in BVVL. Furthermore, it is unknown in which cell types each of these putative transporters acts to allow the movement of riboflavin or other substrates. Addressing these questions experimentally might allow the validity of riboflavin replacement therapy in BVVL to be modeled and provide insight into the optimal route of administration.
Here, we show that Slc52a3 is the mouse ortholog of C20ORF54 (SLC52A3) and that a homozygous disruption of this locus results in early embryonic lethality. Although mutant embryos did not survive beyond mid-gestation, we were able to generate homozygous mutant embryonic stem (ES) cells and found that they could be readily differentiated into motor neurons. Consistent with this observation, we found that Slc52a3 was most strongly expressed in the intestine, placenta as well as the testis, but was largely absent from the adult brain, an expression pattern mirrored by its human ortholog, C20ORF54 (SLC52A3). In total, our studies in the mouse suggest Slc52a3 functions during early development and in the intestine based on the protein expression profile, making BVVL patients with SLC52A3 mutations interesting candidates for riboflavin replacement therapy.
Results
SLC52A2 and 3 but not 1 conserved in mouse
Three putative riboflavin transporters have been identified in human and are encoded by three independent but homologous genes (SLC52A1, SLC52A2 and SLC52A3) (2,3,20,21). At the protein level, SLC52A1 and SLC52A2 are the most conserved displaying >87% sequence similarity, while SLC52A3 shows more divergence with 44% overlap to SLC52A1, and 43% conservation with SLC52A2 (Fig. 1A). In contrast, the rat and mouse genomes only appear to carry two genes with similarity to this family of transporters, with the mouse SLC52A2 protein showing 80% identity to human SLC52A2 and mouse SLC52A3 showing >74% identity to human SLC52A3 (Fig. 1A and B and Supplementary Material, Fig. S1A). In contrast, we could find no gene with conserved homology to SLC52A1 in the syntenic portion of the mouse genome: orthologs of human genes surrounding SLC52A1 on human chromosome 17 were found on mouse chromosome 11 in similar positions and orientations (Supplementary Material, Fig. S1B). At the site where the mouse ortholog of SLC52A1 might be expected to be found there were instead an apparent lincRNA-encoding gene (Gm12320) and an unstudied protein-coding gene (Gm12318), with no homology to SLC52A family members. Further searches for genes with homology to SLC52A members in the mouse genome also failed to locate an SLC52A1 mouse ortholog. We, therefore, conclude that while humans have three SLC52A family members, mice appear to contain only two.
Figure 1.
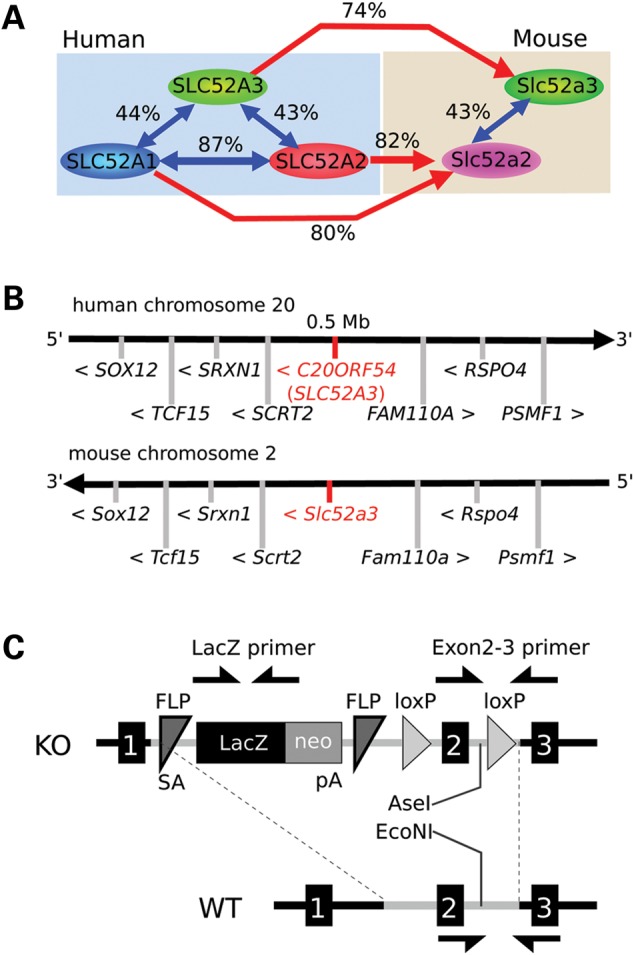
Gene information of riboflavin transporters, the target genes in BVVL syndromes. (A) Riboflavin transporter members in human and mouse. Human and the other primates have three members; SLC52A1, SLC52A2 and SLC52A3, though rodents have two; Slc52a2 and Slc53a3. Each transporter has high homologies between human and mouse. Similarity of SLC52A3 with SLC52A1 and 2 is lower (<50%). No homolog of SLC52A1 exists in mouse, but human SLC52A1 has high similarity with human SLC52A2, suggesting SLC52A1 is a duplicated gene from SLC52A2. Similarity values between these proteins were calculated by NCBI BLAST. (B) Comparing human and mouse riboflavin transporter gene: SLC52A3 and the surrounding genes. The genes surrounding human and mouse SLC52A3 are highly conserved between human chromosome 20 and mouse chromosome 2, suggesting those genes are definitively homolog genes. (C) Gene schema of Slc52a3 knock-out mouse. Genotyping was performed with primers against LacZ and Exons 2 and 3 (arrows). After amplifying Exons 2 and 3 region by PCR, restriction enzyme digests with AseI and EcoNI was performed. In homozygous mutant mice, PCR products with Exons 1 and 2 primer sets are digested with AseI, but undigested with EcoNI. PCR products from WT are digested with EcoNI only. DNA fragments from PCR products of heterozygous mutant mice are found in both AseI and EcoNI digestion.
The human SLC52A3 protein is well conserved between vertebrates and has 11 predicted integral membrane regions, which likely function to transport riboflavin across biological membranes (Fig. 1A, Supplementary Material, Fig. S1C). Alignment comparison of presumptive SLC52A3 protein sequences between all vertebrates in NCBI BLAST showed that BVVL patient mutations were biased to highly conserved regions of the protein (Supplementary Material, Figs S1D and S2). These mutant residues were also highly conserved in the distinct human transporters SLC52A1 and SLC52A2, suggesting these regions may be critical for correct protein folding or riboflavin transport activity (Supplementary Material, Fig. S2).
Slc52a3 knock-out is embryonic lethal
In order to investigate the function of Slc52a3 in a mouse model, we obtained mutant embryos harboring a targeted insertion between Exons 1 and 2 from the knock-out mouse project (KOMP) consortium (17). This insertion contained a LacZ reporter and transcriptional terminator downstream of a strong splice-acceptor. Thus, this allele would be predicted to serve as both a transcriptional reporter of Slc52a3 activity and a loss of function mutation (Fig. 1C). After transfer of frozen embryos to recipient females, we obtained candidate Slc52a3 +/− heterozygous mutant mice. We observed no genotype dependent change in weight over the first 24 weeks of their life (n = 11 mice for each genotype) or other overt phenotypes in these animals over 2 years of maintenance (n = 6 mice of each genotype) (Supplementary Material, Fig. S3A). As children diagnosed with BVVL were found to carry independent rare variants in each of their two alleles of SLC52a3, we next attempted to produce homozygous mutant animals. However, extensive intercrossing between heterozygotes failed to generate homozygous Slc52a3 mutant animals and instead produced only +/− and WT animals in a 2:1 ratio (n = 174 pups) (Table 1). Although our genotyping strategy could in principal clearly distinguish between heterozygous and homozygous mutants (Supplementary Material, Fig. S3B), the substantial number of crosses we performed also allowed us to statistically demonstrate that it was unlikely homozygous mutants were hidden in our heterozygous class of animals (P < 0.05, Table 1). Thus, we concluded that in the mouse, Slc52a3 loss of function results in embryonic lethality. To determine when Slc52a3 became essential, we further analyzed embryos from Slc52a3 +/− crosses at several early developmental stages.
Table 1.
Genotyping frequency in each embryo stage and after wean (3 weeks old).
| Stage | −/− | +/− | WT | Total | P | c2 |
|---|---|---|---|---|---|---|
| After wean | 0 | 117 | 57 | 174 | 2.50019E-13 | – |
| E10.5 | 2 | 22 | 11 | 35 | 0.0310 | 6.943 |
| E9.0 | 7 | 35 | 19 | 61 | 0.0486 | 6.049 |
| ESC | 6 | 10 | 4 | 20 | 0.8187 | 0.4 |
P: possibilities calculated by χ2 tests (C2).
Slc52a3 mutants lost at mid-gestation
We found that we could readily recover homozygous mutant embryos at 9.5 dpc and that although they were recovered at slightly less than expected frequency, they seemed to have no overt phenotypes (Fig. 2A and B). By Day 10.5, the frequency of mutants became even further skewed from expected ratios and those embryos that were recovered displayed abnormal morphologies (Table 1 and Fig. 2C and D). Staining with antibodies specific to the apoptotic indicator terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) also demonstrated a significant increase in the number of apoptotic cells in E10.5 mutants relative to controls (Fig. 2E and F).
Figure 2.

Slc52a3 KO mouse is embryonic lethal. (A and B) Day 9.5 embryos resulting from heterozygous mutant mice crossing. (A) Genotyping PCR results. (B) Recovered embryos of each genotype. Bar; 300 μm. (C and D) Day 10.5 embryos resulting from heterozygous mutant mouse crossing. (C) Genotyping PCR results. (D) Recovered embryos of each genotype. Homozygous KO embryos have severe developmental abnormality. Bar; 1 mm. (E) TUNEL assay in E10.5 embryos of each genotype. Bars: 100 μm. (F) Ratio of TUNEL positive cells to DAPI positive cells. **P < 0.01.
Establishment of Slc52a3 knock-out ES cells
To attempt to create homozygous mutant cells for validation of the loss-of-function effects of the Slc52a3 allele we studied in embryos, we attempted to produce Slc52a3 mutant ES cell lines. After intercrossing heterozygous animals, we harvested blastocysts, then cultivated inner cell mass outgrowths under standard (2i) growth condition (22,23) (Fig. 3A). We found that we could generate ES cell lines of each of the expected three genotypes at the expected Mendelian ratios (Table 1 and Fig. 3B). These cells expressed pluripotency markers such as Oct-3/4 and Sox2 (Fig. 3C). Western blotting analysis demonstrated the expression of Slc52a3 in heterozygous and WT ES cells. However, the levels of Slc52a3 in −/− mutant ES cells was significantly reduced with <3% of the normal protein level remaining in homozygous mutant cells (Fig. 3C–E). Interestingly, we noted that Slc52a3 −/− ES cells exhibited a modest but significant decrease in the expression of Oct3/4 (Fig. 5B). However, we did not note any change in the morphology and growth characteristics of mutant ES cells, nor did we note any overt change in the expression of indicators of pluripotency as assessed by immuno-staining (Fig. 3C). Thus, we conclude that while embryonic development seemed highly sensitive to depletion of Slc52a3, normal level of this protein were not essential for development to the blastocyst stage or normal ES cell growth and maintenance.
Figure 3.

Establishment of Slc52a3 KO ES cells. (A) Day 4.5 embryos (blastocysts) from heterozygous crossing were harvested and cultured in ES media with 2i/LIF (left panels). Five-day-culture of blastocysts led the outgrowth of the inner cell mass (middle panels). Outgrowing cell colonies in 2i/LIF condition on gelatin-coated dish were picked and cell lines were established (right panels). The total number of the established cell lines and the genotype ratio are shown shown in Table 1. (B) Genotyping results of the established ES cells. (C) Established cell lines stained with anti-pluripotent markers; Sox2 and Oct-3/4 and anti-Slc52a3 in each genotype. Slc52a3 −/− cell lines have limited expression of the target gene even though expression of the pluripotent markers seems to be normal. (D and E) Western blotting of pluripotent ES cell lysates. Relative expression levels were normalized by α-tubulin expression.
Figure 5.

Motor neuron differentiation of Slc52a3 KO ES cells. (A–C) qPCR assay of Slc52a3, Oct-3/4 and Isl1 were performed through the differentiation into motor neurons at Day 0, 7 and 14. *P < 0.05; **P < 0.01, N.S.: no significance, ND: not detected. All expression levels were normalized to GAPDH. (D) Motor neurons derived from ES cells from each genotype were stained with anti-Isl1/2 and Tuj1. (E) Percentage of motor neurons from each culture was determined by quantification of Isl1/2 / Tuj1 positive cells. Bars: 200 μm.
Slc52a3 knock-out causes placental deficiency
To further investigate potential causes for the embryonic loss of −/− mutants we examined the expression pattern of Slc52a3 in the developing embryo. X-gal staining of +/− embryos suggested that Slc52a3 was expressed in the placenta at E10.5 (Fig. 4A). Western blotting confirmed that the Slc52a3 protein was expressed in the placenta of control embryos (Fig. 4B). Immunofluorescent staining of dissected embryos suggested minimal expression of Slc52a3 in fetal tissues at E8.5 and E10.5. In contrast, we observed strong staining in extraembryonic tissues at both Days 8.5 (Fig. 4C) and 10.5 (Fig. 4D). Based on our finding that Slc52a3 seemed to be predominantly expressed in extraembryonic tissues, we isolated E10.5 conceptuses and histologically examined their developing placentas. We found Slc52a3 −/− embryos lost most of their placental tissues by Day 10.5 (Fig. 4E). We could confirm the presence of decidual, giant cell, and placental labyrinth tissues in the extra embryonic field of WT embryos. However, homozygous mutant embryos maintained only the giant cell layer between the decidua and yolk sac, and had lost the placental labyrinth.
Figure 4.

Slc52a3 KO embryo has placental loss at Day10.5 extraembryonic tissues. (A) X-gal staining of Slc52a3 +/− and WT embryos with extraembryonic tissues at Day 10.5. (B) Western blotting of WT adult tissues and placenta at Day 10.5. (C) Days 8.5 and (D) 10.5 embryos stained with antibodies of Slc52a3. (E) Sequencial dissections of Slc52a3 −/− and WT embryos at Day 10.5 were stained with DAPI. Broken lines show the placental regions from giant cell layers to amniotic membranes. Bars: 500 μm.
Motor neuron differentiation of Slc52a3 mutant ES cells
Because BVVL syndrome is a neurodegenerative disease with upper and lower motor neuropathy (5–8), the loss of homozygous mutant embryos early in development precluded us from addressing whether or not Slc52a3 has an important function in the normal development or short-term survival of motor neurons in the spinal cord. To address this issue, which has important implications for the role mutations in this gene might play in BVVL patients, we differentiated mouse ES cell lines with +/+, +/− and −/− genotypes into motor neurons using a well-established method (24). To quantify the efficiency of motor neuron differentiation and persistence in culture we measured the expression of ISL transcription factors, which are expressed in many motor neuron subtypes (24). Examination of the relative abundance of Isl1transcripts by reverse transcriptase–polymerase chain reaction (RT-PCR) suggested that there was no statistically significant difference between mutant and control cultures at either 7 or 14 days of motor neuron differentiation (Fig. 5A–C). Quantification of the number of presumptive motor neurons expressing both Tuj1 and Isl1/2 after 14 days of differentiation of the three genotypes of stem cell lines also indicated there were no overt changes in the survival of this neuronal sub-type imposed by Slc52a3 −/− loss of function (Fig. 5D and E). To provide further insight into whether Slc52a3 plays an important role in motor neuron biology, we also monitored its expression during directed differentiation into this cell type. Strikingly, we found that Slc52a3 expression was significantly and dramatically reduced after 7 days of differentiation and essentially absent at 14 days when compared with ES cell levels (Fig. 5A). We, therefore conclude Slc52a3 is unlikely to play an important role in the initial development of spinal motor neurons.
Slc52a3 expression in adult tissues
Although we could not readily assess whether Slc52a3 serves an important postnatal function due to the loss of mutant embryos, we examined its expression pattern in adult animals to determine whether the resulting information might provide further insight into how mutations in this gene could contribute to the phenotype of individuals with BVVL. A broad survey of the relative expression of Slc52a3 by quantitative RT-PCR indicated that transcripts were present in intestine and testis, but absent or rare in the brain, spinal cord, heart, lung, liver and musculature (Fig. 6A). Western blotting demonstrated the presence of Slc52a3 protein in the intestine, seminal vesicles and testes as well as its apparent absence in the cerebrum (Fig. 6B). Immuno-staining and X-gal staining confirmed expression in intestine and the male reproductive organs (Fig. 6C–E). We found Slc52a3 was expressed in the intestinal villus (Fig. 6C), suggesting Slc52a3 may play a role in the absorption of riboflavin from the diet. Examination of the testis indicated strong expression of Slc52a3 in the stromal Leydig cells, but not in seminiferous tubules themselves, where sperm maturation occurs (Fig. 6D). In the seminal vesicle, where mature sperm accumulate, we observed Slc52a3 expression in epithelial cells (Fig. 6E).
Figure 6.

Slc52a3 expression in adult tissues. (A) qPCR assay of Slc52a3 expression in adult tissues. Expression levels were normalized by GAPDH. (B) Western blotting of adult tissues. (C–E) Immunofluorescent staining and X-gal staining of anti-Slc52a3 in intestine (C), testis (D) and seminal vesicle (E). V, intestinal villus; IC: intestinal crypt; S, seminiferous tubule; L, Leydig cells; CT, connective tissue; MM, mucous membrane. Bars: 200 μm.
Expression of riboflavin transporter family in human and mouse tissues
We next sought to reconcile our observations in Slc52a3 mutant mice with the pathology observed in BVVL patients harboring SLC52A3 mutations by mining and examining the expression pattern of the human gene in existing expression databases (Supplementary Material, Fig. S4). By examining data from BioGPS (http://biogps.gnf.org/), we found that similar to the situation in mouse, human SLC52A3 transcript was present in the intestine, testis and placenta. However, we also noted that SLC52A1, the candidate transporter not present in the mouse genome was also expressed in the human placenta as well the digestive and reproductive tracks (Supplementary Material, Fig. S4). Examination of these data sets independently confirmed our observation that Slc52A3 expression was largely or completely absent in the nervous system. Instead, SLC52A2, which exhibited a broad expression profile, seemed to be the primary candidate riboflavin transporter expressed in the brain.
Discussion
Here, we have shown that a loss-of-function mutation in Slc52a3 leads to embryo lethality around Day 10.5, potentially due to a failure in placental development (Fig. 4). In the placental labyrinth, a complex vascular structure allows for the exchange of nutrients and oxygen between maternal and fetal blood (Fig. 7A). Our studies indicate that in the absence of Slc52A3, the placental labyrinth either does not form properly, or cannot function normally and is not maintained. As a result, it would appear that the mouse embryo is deprived of not only riboflavin, but also other key maternal factors necessary for survival. Although it is not clear precisely how loss of this transporter impacts extraembryonic development, recent report have shown that one riboflavin derivative, FAD, activates lysine-specific demethylation 1, which can contribute to histone H3-K4 demethylation (25) and therefore changes in cellular behavior.
Figure 7.

Scheme of the SLC52A3 function in embryo development and BVVL patients. (A) Mechanism of riboflavin absorption in mouse placenta. Embryo vein absorbs riboflavin from the intervillus blood pool in which maternal blood supplies essential nutrients. Riboflavin is suggested to be absorbed through Slc52a3 to the fetal vein and supplied to fetus through umbilical cord. (B) A hypothesis of pathogenesis in BVVL syndrome. SLC52A3 in the intestine and spinal cord. Riboflavin is absorbed in the intestine through SLC52A3 transport. Riboflavin is expected to be absorbed from the blood stream to neural tissues via SLC52A2.
Our findings provide further insight into the role that candidate causative mutations in SLC52A gene family members may play in BVVL (9–16). Although the early loss of mutant embryos precluded a functional test of the hypothesis, our gene expression studies support the notion that Slc52A3 plays an important role in absorbing riboflavin in the gut, either to support specific aspects of gut function, or to facilitate uptake of riboflavin into the blood (1,26). In contrast, these data support the view that patient mutations in SLC52A2, may influence the function of a much more widely expressed transporter, which is also expressed in the brain (10,16–18).
SLC52A1 was first riboflavin transporter identified in humans and it seems to exist only within the primate lineage (2). No obvious orthologs have been identified in other vertebrates. SLC52A1 is highly similar to SLC52A2, suggesting that SLC52A1 may have arisen through gene duplication of an ancestral form SLC52A2 (Fig. 1A and Supplementary Material, Fig. S2). Interestingly, SLC52A1 is a candidate gene for multiple acyl-CoA dehydrogenation deficiency, which results from riboflavin deficiency in pregnancy (27). Although we found that the loss of Slc52a3 function resulted in early termination, compound heterozygous carriers of mutations in human SLC52A3 development, apparently normally, until well into childhood. Our studies combined with patient observations suggest that SLC52A1 expression may compensate for reduced SLC52A3 function in those individuals (27).
Although SLC52A1 and SLC52A3 are both expressed in the human intestine, BVVL patients with SLC52A3 mutations have a significant decrease in riboflavin plasma levels (11). Therefore, it may be that failure of riboflavin absorption in the intestine and the resulting short supply in neural tissues might lead to BVVL in such patients (Fig. 7B). Our analysis of the expression pattern of Slc52a3 in mice suggests it may also play an important role in intestinal riboflavin transport. Given the early embryonic lethality of Slc52a3 mutations in the mouse, generation of a conditional mutant allele will be required to test this hypothesis. Nevertheless, the expression pattern of SLC52A3 and its mouse ortholog support the proposal that supplying riboflavin intravenously to BVVL patients harboring mutations in this gene may bypass the primary site of its activity and provide some therapeutic value.
Materials and Methods
Genomic DNA and total RNA preparation
Genomic DNA was harvested with the Tail Direct PCR kit (Allele Biotechnology) according to the manufacturer's protocol. Total RNA samples were isolated with Trizol (Invitrogen). cDNA samples were reverse transcribed using the iScriptc DNA Synthesis Kit (Bio-Rad) for subsequent qPCR assay with SYBR green qPCR Supermix (Bio-Rad).
Genotyping PCR
Primer sequences for genotyping are as follows: LacZ forward: 5′- ATCACGACGCGCTGTATC, reverse: 5′- ACATCGGGCAAATAATATCG; Exons 1 and 2 forward: 5′-CAGGGACCACCTTGAACACT, reverse: 5′-AGAAGAGCAGTGGCGAGAAG. Genomic DNA and primers (50 nm each) were mixed with AmpliTaq Gold 360 Master Mix (Thermo Fisher Scientific). Thermal cycling conditions were 5 min at 95°C followed by 35 cycles of 30 s at 95°C, 30 s at 55°C and 1 min at 72°C. After enzymatic amplification for 35 cycles with Exons 1 and 2 primers, the PCR products were digested with the restriction enzyme AseI (New England Biolabs, R0526S) and EcoNI (New England Biolabs, R0521S) for 3 h at 37°C followed by 10 min at 80°C, and resolved on a 1% agarose gel in Tris acetate-ethylenediaminetetraacetic acid (EDTA) buffer.
Primers for quantitative PCR
Primer sequences for qPCR are as follows; mSlc52a3 forward: 5’-CCTGGTACCTGCCTTCTTCTTACC, reverse: 5′-TGTATCCAGGAGGTCACGTTC, mOct-3/4 forward: 5′-TCTTTCCACCAGGCCCCCGGCTC, reverse: 5′-TGCGGGCGGACATGGGGAGATCC; mIsl1/2 forward: 5′-TATCCAGGGGATGACAGGAAC, reverse: 5′-GCTGTTGGGTGTATCTGGGAG, mGAPDH forward: 5′-TTCACCACCATGGAGAAGGC, reverse: 5′-GGCATGGACTGTGGTCATGA.
Antibodies
Antibodies used in this study included anti-C20ORF54 for mouse Slc52a3 staining (rabbit polyclonal, Santa Cruz, sc85426), anti-mouse Oct-3/4 (mouse monoclonal, Santa Cruz, sc5279), anti-mouse Sox2 (goat polyclonal, Santa Cruz, sc17320), anti-mouse Isl-1/2 (mouse monoclonal IgG1, DSHB, 40.2D6), anti-mouse Tuj1 (mouse monoclonal IgG2a, Covance Antibody Products, MMS-435P) and anti-α-Tubulin (mouse monoclonal, Sigma-Aldrich, T6199). Cell nuclei were stained with the VECTASHIELD Mounting medium with 4’,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, H-1200).
Quantitative polymerase chain reaction
qPCR with SYBR green was performed in triplicate in three independent experiments using CFX96™ Real-Time System (Bio-Rad). The housekeeping gene glyceraldehyde-3-phosphate dehydrogenase, GAPDH, was used as an internal control to normalize the expression levels using the ΔΔCt method for quantification. The thermal cycling conditions were 5 min at 95°C followed by 35 cycles of 30 s at 95°C, 30 s at 55°C and 1 min at 72°C.
Slc52a3 mutant mice
All of the experimental protocols and procedures were approved by the Institutional Animal Care and Use Committee at Harvard University. The Slc52a3tm2a(KOMP)Wtsi frozen embryos were purchased from the KOMP, and transferred into foster mothers. Heterozygous mutants were confirmed by genotyping PCR of those pups. Those mutants were crossed and the F1 generations were obtained. The genotype ratio of a total of 174 mice bred by crossing heterozygous mutants was quantified. Using a χ2 test, statistical analysis was performed by the following equation.
O: observed frequency count, E: expected frequency count (Table 1).
Harvesting mouse embryos
Harvested embryos in each stage were genotyped and fixed in 4% paraformaldehyde (PFA) solution at 4°C overnight for subsequent cryodissection into 20 μm sections with OCT compound (Tissue Tek). The samples were blocked with 5% Donkey serum (Jackson ImmunoResearch Laboratories) in PBS followed by antibody staining. TUNEL assay was performed on Day 10.5 embryos from heterozygous mutant crossings. TUNEL staining was performed according to the manufacturer's instructions (Click-iT® TUNEL Alexa Fluor® 488 Imaging Assay, Invitrogen). The genotype ratio of a total of 45 embryos at Days 10.5 and 61 embryos at Day 9.5 resulting from crossing heterozygous mutants was determined. Statistical significance was calculated using a χ2 test (Table 1).
Establishment of mouse Slc52a3 KO ES cells
Three-month-old female mice injected with pregnant mare's serum gonadotrophin followed 48 h later by human chorionic gonadotropin were mated. Four days following the crossing date, the pre-implantation embryos were flushed directly from the uterus, and cultured the harvested blastocysts in mouse ES media containing 20% fetal bovine serum, 1% glutamax (GIBCO), 2-mercaptoethanol and 1% penicillin/streptomycin (GIBCO) in the Dulbecco's modified Eagle's medium (DMEM) (GIBCO) with 1 μm PD0325901, 3 μm CHIR99021 and 1000 units/ml leukocyte inhibiting factor (LIF) on a gelatin-coated dish. After 5 days, the inner cell masses were picked and cultured in ES media with 2i/LIF to establish ES cell lines. The protein samples in the pluripotent state were prepared in extraction buffer containing 7 m urea, 2 m thiourea, 0.2% sodium dodecyl sulphate, 20 mm 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (pH7.4), 10 mm NaF, 1 mm EDTA, 1 mm dithiothreitol, and proteinase inhibitor cocktail (Complete, Roche). A statistical calculation was performed with genotyped ratio of established cell lines.
Motor neuron differentiation
Mouse ES cells from each genotype were differentiated into motor neurons as described previously (24,28). Briefly, trypsinized ES cells were cultured via EB formation in DMEM-F12 (GIBCO) with 10% knock-out serum (GIBCO), 1% glutamax, 2-mercaptoethanol and 1% penicillin/streptomycin. The EBs were treated with 100 nm retinoic acid and 1 μm smoothened antagonist from Days 3–7. On Day 7, the EBs were digested into single-cells with papain containing 1000 units/ml DNaseI (Worthington Biochemical). Resuspended cells were plated on Matrigel (FISHER) -coated dish with a concentration of 10 000 cells/ml in the F12 medium (GIBCO) containing 5% horse serum (GIBCO), B-27 supplement and N2 supplement (GIBCO) with 10 ng/ml glial derived neurotrophic factor, ciliary neurotrophic factor and brain derived neurotrophic factor (R&D Systems). Total RNA samples were prepared at Days 7 and 14. For the motor neuron survival experiments, differentiated cells were stained using immunofluorescence at Day 14.
Preparing adult tissues for experiments
Four-month-old mice were anesthetized with avertin and perfusion-fixed with 4% PFA. Tissues were harvested and post-fixed in 4% PFA overnight at 4°C, washed with PBS, immersed in 30% sucrose overnight at 4°C and frozen in OCT compound for sectioning using cryostat. Tissues were also harvested from anesthetized mice without 4% PFA treatment, digested in protein extraction buffer described above, and Trizol to prepare RNA samples, respectively.
X-gal staining
X-gal activity was assessed by incubating 20 µm thick sections with freshly prepared LacZ staining solution (1.0 mg/ml of X-gal, 4 mm potassium ferrocyanide, 4 mm potassium ferricyanide and 2 mm MgCl2 in phosphate buffer) overnight at 37°C. The sections were examined and photographed with a Zeiss AX10.
Supplementary Material
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
Funding
This work was supported by the Thisbe and Noah Scott Foundation and the Howard Hughes Medical Institute.
Supplementary Material
References
- 1.Fujimura M., Yamamoto S., Murata T., Yasujima T., Inoue K., Ohta K.Y., Yuasa H. (2010) Functional characteristics of the human ortholog of riboflavin transporter 2 and riboflavin-responsive expression of its rat ortholog in the small intestine indicate its involvement in riboflavin absorption. J. Nutr., 140, 1722–1727. [DOI] [PubMed] [Google Scholar]
- 2.Yonezawa A., Masuda S., Katsura T., Inui K. (2008) Identification and functional characterization of a novel human and rat riboflavin transporter, RFT1. Am. J. Physiol. Cell. Physiol., 295, C632–C641. [DOI] [PubMed] [Google Scholar]
- 3.Yao Y., Yonezawa A., Yoshimatsu H., Masuda S., Katsura T., Inui K. (2010) Identification and comparative functional characterization of a new human riboflavin transporter hRFT3 expressed in the brain. J. Nutr., 140, 1220–1226. [DOI] [PubMed] [Google Scholar]
- 4.Powers H.J. (2003) Riboflavin (vitamin B-2) and health. Am. J. Clin. Nutr., 77, 1352–1360. [DOI] [PubMed] [Google Scholar]
- 5.BROWN C.H. (1894) Infantile amyotrophic lateral sclerosis of the family type. J. Nerv. Ment. Dis., 19, 707–716. [Google Scholar]
- 6.Vialetto E. (1936) Contributo alla forma ereditaria della paralisi bulbare progressive. Riv. Sper. Freniat., 40, 1–24. [Google Scholar]
- 7.Van Laere J. (1977) Un nouveau cas de paralysie bulbo-pontine chronique progressive avec surdité. Rev. Neurol., 133, 119–124. [PubMed] [Google Scholar]
- 8.Sathasivam S. (2008) Brown-Vialetto-Van Laere syndrome. Orphanet. J. Rare. Dis., 3, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Green P., Wiseman M., Crow Y.J., Houlden H., Riphagen S., Lin J.-P., Raymond F.L., Childs A.-M., Sheridan E., Edwards S. (2010) Brown-Vialetto-Van Laere syndrome, a Ponto-Bulbar Palsy with deafness, is caused by mutations in C20orf54. Am. J. Hum. Genet., 86, 485–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson J.O., Gibbs J.R., Van Maldergem L., Houlden H., Singleton A.B. (2010) Exome sequencing in Brown-Vialetto-van Laere syndrome. Am. J. Hum. Genet., 87, 567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bosch A.M., Abeling N.G., IJlst L., Knoester H., van der Pol W.L., Stroomer A.E., Wanders R.J., Visser G., Wijburg F.A., Duran M. (2011) Brown-Vialetto-Van Laere and Fazio Londe syndrome is associated with a riboflavin transporter defect mimicking mild MADD, a new inborn error of metabolism with potential treatment. J. Inherit. Metab. Dis., 34, 159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dezfouli M.A., Yadegari S., Nafissi S., Elahi E. (2012) Four novel C20orf54 mutations identified in Brown–Vialetto–Van Laere syndrome patients. J. Hum. Genet., 57, 613–617. [DOI] [PubMed] [Google Scholar]
- 13.Nabokina S.M., Subramanian V.S., Said H.M. (2012) Effect of clinical mutations on functionality of the human riboflavin transporter-2 (hRFT-2). Mol. Genet. Metab., 105, 652–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bandettinidi Poggio M., Gagliardi S., Pardini M., Marchioni E., Monti Bragadin M., Reni L., Doria-Lamba L., Roccatagliata L., Ceroni M., Schenone A. et al. (2013) A novel compound heterozygous mutation of C20orf54 gene associated with Brown-Vialetto-Van Laere syndrome in an Italian family. Eur. J. Neurol., 20, e94–e95. [DOI] [PubMed] [Google Scholar]
- 15.Koy A., Pillekamp F., Hoehn T., Waterham H., Klee D., Mayatepek E., Assmann B. (2012) Brown-Vialetto-Van Laere syndrome, a riboflavin-unresponsive patient with a novel mutation in the C20orf54 gene. Pediatr. Neurol., 46, 407–409. [DOI] [PubMed] [Google Scholar]
- 16.Ciccolella M., Corti S., Catteruccia M., Petrini S., Tozzi G., Rizza T., Carrozzo R., Nizzardo M., Bordoni A., Ronchi D. et al. (2013) Riboflavin transporter 3 involvement in infantile Brown-Vialetto-Van Laere disease, two novel mutations. J. Med. Genet., 50, 104–107. [DOI] [PubMed] [Google Scholar]
- 17.Haack T.B., Makowski C., Yao Y., Graf E., Hempel M., Wieland T., Tauer U., Ahting U., Mayr J.A., Freisinger P. (2012) Impaired riboflavin transport due to missense mutations in SLC52A2 causes Brown-Vialetto-Van Laere syndrome. J. Inherit. Metab. Dis., 35, 943–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Foley A.R., Menezes M.P., Pandraud A., Gonzalez M.A., Al-Odaib A., Abrams A.J., Sugano K., Yonezawa A., Manzur A.Y., Burns J. et al. (2014) Treatable childhood neuronopathy caused by mutations in riboflavin transporter RFVT2. Brain, 137, 44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anand G., Hasan N., Jayapal S., Huma Z., Ali T., Hull J., Blair E., Mcshane T., Jayawant S. (2012) Early use of high-dose riboflavin in a case of Brown–Vialetto–Van Laere syndrome. Dev. Med. Child. Neurol., 54, 187–189. [DOI] [PubMed] [Google Scholar]
- 20.Yamamoto S., Inoue K., Ohta K.-Y., Fukatsu R., Maeda J.-Y., Yoshida Y., Yuasa H. (2009) Identification and functional characterization of rat riboflavin transporter 2. J. Biochem., 145, 437–443. [DOI] [PubMed] [Google Scholar]
- 21.Yonezawa A., Inui K. (2013) Novel riboflavin transporter family RFVT/SLC52, identification, nomenclature, functional characterization and genetic diseases of RFVT/SLC52. Mol. Aspects Med., 34, 693–701. [DOI] [PubMed] [Google Scholar]
- 22.Ying Q.-L., Nichols J., Chambers I., Smith A. (2003) BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell, 115, 281–292. [DOI] [PubMed] [Google Scholar]
- 23.Ying Q.-L., Wray J., Nichols J., Batlle-Morera L., Doble B., Woodgett J., Cohen P., Smith A. (2008) The ground state of embryonic stem cell self-renewal. Nature, 453, 519–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wichterle H., Lieberam I., Porter J.A., Jessell T.M. (2002) Directed differentiation of embryonic stem cells into motor neurons. Cell, 110, 385–397. [DOI] [PubMed] [Google Scholar]
- 25.Hino S., Sakamoto A., Nagaoka K., Anan K., Wang Y., Mimasu S., Umehara T., Yokoyama S., Kosai K., Nakao M. (2012) FAD-dependent lysine-specific demethylase-1 regulates cellular energy expenditure. Nat. Commun., 3, 758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Subramanian V.S., Rapp L., Marchant J.S., Said H.M. (2011) Role of cysteine residues in cell surface expression of the human riboflavin transporter-2 (hRFT2) in intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol., 301, G100–G109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ho G., Yonezawa A., Masuda S., Inui K., Sim K.G., Carpenter K., Olsen R.K., Mitchell J.J., Rhead W.J., Peters G. (2011) Maternal riboflavin deficiency, resulting in transient neonatal-onset glutaric aciduria Type 2, is caused by a microdeletion in the riboflavin transporter gene GPR172B. Hum. Mutat., 32, E1976–E1984. [DOI] [PubMed] [Google Scholar]
- 28.Di Giorgio F.P., Carrasco M.A., Siao M.C., Maniatis T., Eggan K. (2007) Non–cell autonomous effect of glia on motor neurons in an embryonic stem cell–based ALS model. Nat. Neurosci., 10, 608–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.