

Abstract
The symptoms of Clostridium difficile infection are caused by two closely related toxins, TcdA and TcdB, which are encoded by the 19.6 kb Pathogenicity Locus (PaLoc). The PaLoc is variably present among strains, and in this respect it resembles a mobile genetic element. The C. difficile population structure consists mainly of five phylogenetic clades designated 1–5. Certain genotypes of clade 5 are associated with recently emergent highly pathogenic strains causing human disease and animal infections. The aim of this study was to explore the evolutionary history of the PaLoc in C. difficile clade 5. Phylogenetic analyses and annotation of clade 5 PaLoc variants and adjoining genomic regions were undertaken using a representative collection of toxigenic and nontoxigenic strains. Comparison of the core genome and PaLoc phylogenies obtained for clade 5 and representatives of the other clades identified two distinct PaLoc acquisition events, one involving a toxin A+B+ PaLoc variant and the other an A−B+ variant. Although the exact mechanism of each PaLoc acquisition is unclear, evidence of possible homologous recombination with other clades and between clade 5 lineages was found within the PaLoc and adjacent regions. The generation of nontoxigenic variants by PaLoc loss via homologous recombination with PaLoc-negative members of other clades was suggested by analysis of cdu2, although none is likely to have occurred recently. A variant of the putative holin gene present in the clade 5 A−B+ PaLoc was likely acquired via allelic exchange with an unknown element. Fine-scale phylogenetic analysis of C. difficile clade 5 revealed the extent of its genetic diversity, consistent with ancient evolutionary origins and a complex evolutionary history for the PaLoc.
Keywords: evolution, phylogeny, toxin locus
Introduction
The gram-positive, spore-forming bacterium Clostridium difficile is a major cause of healthcare-associated diarrhea, and the most commonly reported nosocomial pathogen in the United States (Magill et al. 2014). Clostridium difficile infection (CDI) encompasses a wide clinical spectrum ranging from asymptomatic carriage to mild self-limiting diarrhea, and ultimately fulminant colitis and toxic megacolon. The C. difficile population structure consists of six distinct phylogenetic clades designated 1, 2, 3, 4, 5, and C-I (Dingle et al. 2011, 2014; Stabler et al. 2012). Toxin production varies by clade; the most divergent clade, C-I, produces neither binary toxin, nor the two main toxins, toxin A and toxin B. Strains belonging to clades 2, 3, and 5 produce binary toxin, whereas most genotypes within clades 1–3 produce both toxins A and B. Clade 4 is largely nontoxigenic except for multilocus sequence type (MLST) ST37 (polymerase chain reaction [PCR]-ribotype 017) toxinotype VIII strains which only produce toxin B.
Clade 5 is noteworthy because it contains the clinically important PCR-ribotype 078 or ST11. This genotype causes illness which is more severe and associated with higher mortality rates than other clinically important strains (Walker et al. 2013). Clade 5 is also notable for its divergence from the first four clades, separating an estimated 85 Ma (He et al. 2010). Ribotype 078 has a strong association with animal infections (Rodriguez-Palacios et al. 2007; Pirs et al. 2008; Rupnik et al. 2008; Pelaez et al. 2013), and its incidence in cases of symptomatic human infection is increasing (Goorhuis et al. 2008; Bauer et al. 2011; Eckert et al. 2013). Less abundant clade 5 genotypes have also been identified in animals, including ribotypes 033 (toxinotype XIa/b), 045 (toxinotype XIV), 126 (toxinotype XXVIII), and 127 and 237 (toxinotype XXXI) (Schneeberg et al. 2012, 2013; Squire et al. 2013; Knight and Riley 2013; Knight et al. 2013).
The symptoms of CDI are caused by two closely related toxins, designated A and B, and encoded by the pathogenicity locus (PaLoc), a 19 kb chromosomally integrated DNA sequence (Braun et al. 1996). The PaLoc’s single genomic location (Braun et al. 1996; Dingle et al. 2011), clade-specificity of its genetic variants (Dingle et al. 2011), and a lack of genes conferring mobility (Braun and von Eichel-Streiber 1999) suggest that it is stably integrated. However, nontoxigenic strains exist which intermingle phylogenetically with toxigenic strains of clades 1, 4, and 5, suggesting multiple gain and loss events (Elliott et al. 2009; Dingle et al. 2011, 2014). Transfer of the PaLoc to nontoxigenic strains and resultant toxigenic conversion has been demonstrated experimentally (Brouwer et al. 2013).
The C. difficile toxins belong to the large clostridial toxin family, members of which have also been identified in Clostridium sordellii, Clostridium novyi, and Clostridium perfringens (Amimoto et al. 2007). The large clostridial toxins comprise four domains (Davies et al. 2011): A glucosylating enzymatic domain, an autocatalytic processing/protease domain, a membrane-translocating domain and a repetitive, and receptor-binding domain. Because of the large size of the toxin-encoding genes, analysis of genetic polymorphism within the C. difficile PaLoc has frequently been confined to toxinotyping, a method based on PCR and restriction endonuclease digests which infers nucleotide sequence variation indirectly (Rupnik et al. 1998). Within clade 5, toxinotypes are largely congruent with genotype (Stabler et al. 2012); for example, the 078 PaLoc is designated toxinotype V. Deletions which can abrogate toxin production (Rupnik 2008) are common within the repetitive sequences of the toxin A receptor-binding domain and have been described in clades 1, 2, 4, and 5 (Rupnik 2008; Zaiß et al. 2009). The PaLoc encodes a further three genes: tcdR (an alternative sigma factor), tcdE (a bacteriophage-like putative holin), and tcdC (a negative regulator), and contains a phage-like endolysin fragment (Monot et al. 2011). Changes to PaLoc genes, most notably tcdB (Lanis et al. 2013) and more controversially, tcdC (Carter et al. 2011; Bakker et al. 2012; Cartman et al. 2012), have been proposed to impact on the severity of clinical disease.
The purpose of this study was to explore the evolutionary history of the PaLoc in C. difficile clade 5. To do so, the phylogeny of clade 5 was reconstructed based on whole-genome sequences (WGS) from a representative collection of isolates comprising both toxigenic and nontoxigenic strains. The toxigenic strains within clade 5 exhibited significant PaLoc variation consistent with the previously assigned toxinotypes XIa/b, XIV, XXVIII, and XXXI (Schneeberg et al. 2012, 2013; Squire et al. 2013; Knight and Riley 2013; Knight et al. 2013). The extent of clade 5 PaLoc variation was studied and compared with this locus in members of other clades. By examining the whole genome and PaLoc phylogenies, we aimed to understand the evolutionary events leading to PaLoc acquisition and/or loss by clade 5.
Materials and Methods
Isolates and Genome Sequencing
A total of 23 isolates from clade 5 were included in the study. The reference isolate L033 was kindly supplied by Prof. Mark Wilcox, Leeds Institute of Molecular Medicine, University of Leeds, United Kingdom cultured originally from a patient in the Netherlands. The WGS of this isolate was reported previously (Dingle et al. 2013). All other isolates were from Australia and included 16 human clinical isolates from: Western Australia (n = 9); New South Wales (n = 1); Victoria (n = 1); and Queensland (n = 1). These were isolated between December 11, 2005 and August 7, 2010, except for HCD52 which was first cultured in 1998. The Western Australian human isolates were cultured at the state pathology laboratory, PathWest, while the others were referred to PathWest from diagnostic laboratories in other states for PCR ribotyping. Animal isolates (n = 6) of bovine (n = 1), equine (n = 1), and porcine (n = 4) origin, cultured between March 26, 2007 and November 23, 2009 in New South Wales and Western Australia, were also included. The presence of PaLoc sequences within the isolates was determined previously using published PCR assays for tcdA (including both the repetitive and nonrepetitive regions; Kato et al. 1998), tcdB (Kato et al. 1991), and for both binary toxin component genes cdtA and cdtB (Stubbs et al. 2000).
Genomes were sequenced using Illumina Technology (Bentley et al. 2008) as described previously (Dingle et al. 2013). De novo assemblies were constructed using Velvet (Zerbino and Birney 2008) and A5 (Tritt et al. 2012). All the isolates had previously been toxin genotyped by PCR to detect the presence of the toxin A and toxin B genes, tcdA and tcdB, respectively, with regions in both the repetitive and nonrepetitive domain of tcdA targeted (Kato et al. 1991, 1998). The presence of both binary toxin genes was also tested for by PCR (Stubbs et al. 2000). Previously sequenced genomes for strain CD630 (UK 012, ST54, A+B+CDT−) (Sebaihia et al. 2006), Ox160 (UK 027, ST1, A+B+CDT+), Ox1485 (UK 023, ST5, A+B+CDT+), and Ox920 (UK 017, ST37, A−B+CDT−) were used as references for clades 1–4, respectively (Dingle et al. 2014).
Phylogenetic Analysis
All isolates were PCR ribotyped at PathWest using published oligonucleotide primers (O'Neill et al. 1996), and the products were separated by capillary electrophoresis and patterns compared against the local reference library. A subset (n = 8) that did not match any of the previously determined banding patterns available in Australia was referred to the United Kingdom Anaerobe Reference Unit (University Hospital of Wales, Heath Park, Cardiff, Wales, UK) for comparison against their library. Seven were found to be novel types and assigned new UK PCR-ribotype designations. Following the move of the UK C. difficile reference library to the Microbiology Department, Leeds General Infirmary (Leeds, England, UK), a further eight isolates were referred for typing, six of which were novel and assigned new UK designations. A subset of isolates chosen to represent closely phylogenetically related clade 5 groups on the basis of their PCR-ribotyping patterns, underwent toxinotyping using the A3 and B1 fragments, as previously described (Rupnik et al. 1998). Data were extracted bioinformatically from unclosed WGS using BIGSdb (Jolley and Maiden 2010) to determine MLST as per a published scheme (Griffiths et al. 2010). The sequence type (ST) was determined for each isolate using the database available at http://pubmlst.org/cdifficile. Newly identified alleles and STs were submitted to the database and assigned numbers in order of discovery. Fine-scale phylogenies were constructed using the core genomes for all clade 5 isolates. These were constructed using ClonalFrame (Didelot and Falush 2007), and ancestry times were calculated in years using isolate dates and previous estimate of the C. difficile molecular clock (Didelot et al. 2012). Polymorphisms between four pairs of isolates were mapped along the whole M120 genome (Sebaihia et al. 2006) by aligning their de novo assemblies against M120 using MuMMER version 3.25 (Kurtz et al. 2004). The distribution of polymorphisms in the PaLoc region was plotted using DNAplotter (Carver et al. 2009).
Gene Prediction, Annotation, and Comparison
The PaLoc region and insertion sites were studied in detail, sequences being translated and annotated using Artemis genome browser and annotation tool (Rutherford et al. 2000). Sequence comparisons were performed using the Artemis Comparison Tool (Carver et al. 2005). Analysis of the PaLoc genes tcdR, tcdB, tcdA, tcdC, and tcdE was performed using MEGA 5.2 (Tamura et al. 2011) to construct neighbor-joining trees. BLAST searches of genes and predicted translation products against GenBank were used to identity their putative functions. Secondary structure prediction of nucleic acids was performed using the RNAfold server (Gruber et al. 2008), and putative RNA genes searched against the Rfam database (Griffiths-Jones et al. 2003).
Results
Isolates and Genotypes
Clostridium difficile isolates identified during previous studies as belonging to clade 5 on the basis of MLST data or PCR ribotype were chosen (n = 23) (Elliott et al. 2009; Elliott et al. 2011; Stabler et al. 2012; Squire et al. 2013; Elliott B, Hong S, Knight DR, Riley TV, unpublished data). The presence of the PaLoc and therefore the genetic potential for toxin expression had been assessed previously by PCR assay for the presence of toxin genes. Isolates which were PCR positive for tcdA and/or tcdB are subsequently referred to as toxigenic and designated A+B+ or A−B+. Isolates for which toxin gene PCR assays were negative, and the lok1/3 PCR (Braun et al. 1996) was positive were designated either nontoxigenic or A−B−. The absence of toxin A production was confirmed by enzyme immunoassay for isolates in which tcdA deletions were detected, with the Triage C. difficile Panel (Biosite). Production of toxin B was confirmed using the cell culture cytotoxicity assay as previously described (Bowman and Riley 1986). All isolates (toxigenic [n = 18] and nontoxigenic [n = 5]) were genotyped by MLST; a total of nine STs were identified, eight of which were novel (table 1). No ST occurred in both toxigenic and nontoxigenic variants. Corresponding PCR ribotype and toxinotyping data are indicated in table 1. Four STs were represented by only one isolate and one ST by two isolates belonging to the same ribotype. The other four STs contained both multiple isolates and PCR ribotypes (table 1). ST11 was unusual in being further discriminated into four PCR-ribotypes.
Table 1.
Isolate Details
| Isolate Code | Isolate No. | Collection Date | Host | State | Toxin Profile | Ribotype | Toxinotype | ST |
|---|---|---|---|---|---|---|---|---|
| ES 153 | 06-4555666 | July 19, 006 | Human | NSW | A + B + CDT+ | UK 126 | ND | 11 |
| ES 210 | JH09M67811 | July 29, 2009 | Human | NSW | A + B + CDT+ | UK 126 | ND | 11 |
| AI 155 | H.WA2 | August 2009 | Equine | NSW | A − B + CDT+ | UK 605 | ND | 163 |
| ES 223 | JH10M3970 | January 12, 2010 | Human | NSW | A − B + CDT+ | UK 606 | ND | 163 |
| WA 16 | P06-4500199M | January 5, 2006 | Human | WA | A − B + CDT+ | UK 584 | ND | 163 |
| HCD 52 | Cdiff 642 | 1998 | Human | WA | A − B − CDT+ | UK 585 | NT | 164 |
| Q 6 | 324060196 | May 3, 2007 | Human | QLD | A − B − CDT+ | UK 586 | NT | 167 |
| AI 6 | 07-321 | March 26, 2007 | Porcine | WA | A − B − CDT+ | UK 238 | NT | 169 |
| WA 12 | Q05-3910805C | December 11, 2005 | Human | WA | A − B − CDT+ | UK 239 | NT | 168 |
| WA 94 | P06-9511410S | January 29, 2006 | Human | WA | A + B + CDT+ | UK 078 | V | 11 |
| ES 280 | 98700735 | August 7, 2010 | Human | VIC | A + B + CDT+ | UK 078 | V | 11 |
| RPH 123 | P07-9572883U | June 28, 2007 | Human | WA | A + B + CDT+ | UK 078 | V | 11 |
| WA 27 | P06-1321417R | January 31, 2006 | Human | WA | A + B + CDT+ | UK 127 | VI | 11 |
| L033 | Unknown | Human | UK | A − B − CDT+ | UK 033 | XI | 11 | |
| WA 13 | Q05-3033322T | December 31, 2005 | Human | WA | A − B + CDT+ | UK 291 | XXX | 164 |
| ES 30 | 06-081-2908 | March 22, 2006 | Human | NSW | A − B + CDT+ | UK 280 | XXX | 166 |
| PMH 25 | P07-7325065W | August 19, 2007 | Human | WA | A + B + CDT+ | UK 280 | XXX | 166 |
| AI 27 | 0048 | October 9, 2008 | Bovine | NSW | A − B + CDT+ | UK 583 | XXX | 173 |
| ES 166 | 06-5509002 | September 20, 2006 | Human | NSW | A − B + CDT+ | UK 281 | XXX | 174 |
| AI 15 | P06-2941A | April 18, 2007 | Porcine | WA | A − B + CDT+ | UK 237 | XXXI | 167 |
| AI 152 | R11-GG1 | November 23, 2009 | Porcine | WA | A − B + CDT+ | UK 285 | XXXI | 167 |
| AI 178 | GG1-FS(1) | October 15, 2009 | Porcine | WA | A − B + CDT+ | UK 237 | XXXI | 167 |
| WA 151 | P06-4574276A | July 11, 2006 | Human | WA | A − B + CDT+ | UK 237 | XXXI | 167 |
Note.— ND, not done; NT, nontoxigenic (i.e., PaLoc-negative); NSW, New South Wales; WA, Western Australia; QLD, Queensland; VIC, Victoria; UK, United Kingdom.
Genetic Organization of the Clade 5 PaLocs
Unclosed WGS were determined for the 23 isolates. The closed genomes of clade 5 strain M120 (ST11) (He et al. 2010) and clade 1 reference strain 630 (ST54) (Sebaihia et al. 2006; Monot et al. 2011) were used for comparative purposes (GenBank accession numbers FN665653 and AM180355, respectively). They provided scaffolds against which the de novo assembled clade 5 PaLoc sequences could be aligned for the toxigenic strains. The unclosed genome sequence of isolates Ox160, Ox1485, and Ox920 were used as references for clades 2, 3, and 4, respectively (Dingle et al. 2011, 2014).
The region of the chromosome containing the PaLoc was annotated in toxigenic strains and the PaLoc insertion site in nontoxigenic strains. All 18 isolates previously designated toxigenic by PCR contained PaLoc sequences of 10,211–19,742 kb located within the single chromosomal integration site described previously (Braun et al. 1996). Of these, 12 isolates were negative for tcdA and designated A−B+, whereas the remainder was A+B+. These data suggested the possibility that clade 5 contained two phylogenetically distinct PaLoc variants.
The genetic organization of the PaLoc in clade 5 was investigated. Within the six clade 5 A+B+ isolates the five PaLoc open-reading frames (ORFs) were intact, resembling typical PaLocs as exemplified by the reference genomes used comparatively (fig. 1A; Sebaihia et al. 2006). Sequence analysis of the A−B+ clade 5 isolates (toxinotypes XXX and XXXI) identified their common genetic organization and a large deletion within the PaLoc (10,366 bp relative to reference M120) encompassing tcdA and tcdC (fig. 1B).
Fig. 1.—

Distinct variants of the PaLoc found in clade 5. The genetic organization of the clade 5 PaLoc and flanking genes: (A) A+B+, toxinotypes XXX and XXXI, (B) A−B+, (C) PaLoc-negative strains, (D) A−B− toxinotype XI strains, and (E) WA12 containing nontoxigenic clade C-1 related (non-PaLoc) sequence at the PaLoc integration site and consequently A−B−.
The PaLoc Integration Site in Nontoxigenic Clade 5
Investigation of the empty clade 5 PaLoc integration site in the five nontoxigenic isolates revealed that the majority (3/5) lacked the entire PaLoc, containing the 115 bp sequence described previously in its place (Braun et al. 1996; fig. 1C), among nontoxigenic isolates of clades 1 and 4 (Dingle et al. 2014). The first exception was an ST11(033) isolate that contained a PaLoc fragment consistent with toxinotype XI (Geric and Rupnik 2010; fig. 1D): only tcdC and the 3′-terminal 2,456 bp of tcdA containing a 681 bp deletion in the repetitive sequences were present, together representing 6,521 bp of the PaLoc. This reflected a 51,379 bp deletion relative to strain M120 (He et al. 2010), removing tcdR, tcdB, tcdE, and part of tcdA as well as the region immediately upstream of the PaLoc. A variant of the mobile element Tn6218 (Dingle et al. 2014) was present immediately upstream of this element in ST11(033) (fig. 1D). The second exception was the previously described isolate WA12 (fig. 1E) containing a 7,183 kb sequence at the PaLoc integration site (Elliott et al. 2009). A closely related sequence (87% nt sequence identity) is also present in nontoxigenic clade C-I strains (Dingle et al. 2014).
Inter and Intraclade Phylogenetic Relationships of the Clade 5 PaLoc
The tcdB gene was present and intact in all 18 toxigenic clade 5 PaLoc variants, and was therefore used for focused investigation of the PaLoc phylogenetic relationships with one another and with representatives from other clades (fig. 2A–D). This analysis identified two distinct clade 5 PaLoc variants corresponding to A+B+ and A−B+, each of which was genetically homogeneous. The genetic distance between the two clade 5 variants changed across tcdB (fig. 2A–D). They were clearly distinct from one another within the glucosyltransferase and protease domains (fig. 2A–B), but more closely related within the translocation and receptor-binding domains (fig. 2C–D). The only other intact gene to be present in all toxigenic clade 5 PaLoc variants, tcdR, also supported this separation between A+B+ and A−B+ (fig. 3A). The tcdR gene of isolate AI27, which was intermediate between the two groups, was a hybrid of the two. The glucosyltransferase domain of the A−B+ clade 5 strains was notable for its homology to clade 4 ST37(017), which is also A−B+ (fig. 2A). These data suggest an evolutionary history involving recombination of PaLoc sequences involving clade 5 and members of other clades.
Fig. 2.—

Independent clade 5 PaLoc acquisitions are supported by the crosspopulation phylogeny of tcdB functional domains. Phylogenies constructed from the four domains of tcdB: (A) The glucosyltransferase domain, (B) the cysteine protease domain, (C) the translocation domain, and (D) the receptor-binding domain. Colored labels indicate clade (see Key). Strain labels are shown in supplementary figure S1, Supplementary Material online.
Fig. 3.—
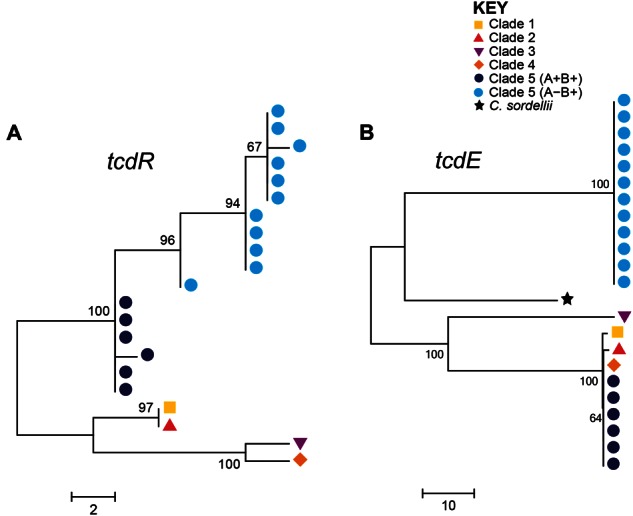
Phylogenetic relationship of PaLoc accessory genes. Phylogenies of the PaLoc accessory genes (A) tcdR and (B) tcdE, which encode a positive regulator and a putative holin gene, respectively. The A−B+ tcdR gene (from strain AI27, marked with asterisk) clustered closest to the A+B+ lineage (A) was a hybrid of the two sequences suggesting it derived from a recombination event (data not shown). The putative holin gene present in the PaLoc of clade 5 A−B+ strains is distantly related to that found in the other clades (B), suggesting it was gained via allelic exchange with another source. Strain labels are shown in supplementary figure S2, Supplementary Material online.
Clade 5 PaLoc Acquisition
A ClonalFrame tree was constructed using the core genomes of all clade 5 isolates (fig. 4). This tree was time scaled to allow the possibility of approximately dating the evolutionary events which led to the current distribution of the PaLoc within clade 5. The most parsimonious scenario involves separate gains of the A−B+ and A+B+ PaLoc variants and several subsequent losses. The acquisition of the A−B+ variant was dated at approximately 1,300 years ago whereas the gain of the A+B+ variant could either be ancient, or as recent as approximately 100 years ago due to its occurrence on a very long branch.
Fig. 4.—

The two PaLoc variants were each acquired independently approximately 1,300 years ago. A time-scaled ClonalFrame tree of the core genome dating (approximately) the events of acquisition and loss of the PaLoc within clade 5. The different lineages are color coded according to toxin profiles: A+B+ (red), A−B+ (orange), and A−B− (blue). The A−B+ isolates within clade 5 can be divided into two separate branches based on toxinotype (shown in black text on the right).
Construction of phylogenies at four loci directly flanking the PaLoc integration site revealed different relationships between toxigenic clade 5 strains, nontoxigenic clade 5 strains, and representatives of other clades (fig. 5). The cdu2, cdu1, and cdd2 genes in A+B+ and A−B+ clade 5 strains were more closely related to one another than with other clades, but gene cdd1 showed a strikingly different pattern (fig. 5C). The same cdd1 allele was present in all six A+B+ strains, as well as in the M120 reference strain and the closely related L033 strain, and this allele was more closely related to those found in clades 1–4, than in the A−B+ strains of clade 5 (fig. 5C). This suggests that cdd1 may have been acquired along with the PaLoc in the A+B+ strains. This further supports the two independent PaLoc acquisitions by clade 5, possibly also involving homologous recombination of flanking sequences within the A+B+ strains. The deletion of most of the PaLoc in ST11(033) was estimated to be quite recent, occurring approximately 50 years ago.
Fig. 5.—

Acquisition and loss of the clade 5 PaLoc via homologous recombination. In A−B− clade 5 strains, the phylogenies of cdu2 (A) and cdu1 (B) genes upstream of the PaLoc are distinct from other clade 5 strains, with the exception of WA12. The genes seen in these strains are more closely related to those of clades 1–4, suggesting that the PaLoc was lost via homologous recombination with a nontoxigenic strain from one of these clades. In the A+B+ clade 5 strains, the same is seen with the cdd1 gene (C), suggesting these strains gained cdd1 along with the PaLoc via homologous recombination with a toxigenic strain from another clade, whereas the phylogeny of cdd2 (D) remains relatively consistent throughout clade 5. L033 is not included in the trees for cdu2 and cdu1 as these genes are not present in this strain. Strain labels are shown in supplementary figure S3, Supplementary Material online.
Clade 5 PaLoc Loss by Homologous Recombination
The phylogeny of the tree constructed using the core genomes of clade 5 isolates suggested that nontoxigenic strains have been generated by PaLoc loss from clade 5 genomes on multiple occasions, possibly by homologous recombination with nontoxigenic C. difficile strains (fig. 4). To investigate these losses in more detail, the genetic organization and phylogeny of loci flanking the PaLoc insertion site (Braun et al. 1996) in all available clade 5 genomes was compared with isolates representing the other four clades.
All toxigenic clade 5 strains (n = 18) and the atypical nontoxigenic WA12 contained a distinctive cdu2 locus, lacking the 5’ terminal 600 bp of the gene and containing two frameshift mutations, a 228 bp deletion, and a 31 bp insertion with weak homology to C. difficile IStrons relative to CD630 (Sebaihia et al. 2006). A deletion coupled with an insertion also occurred in the cdu3−cdu2 intergenic region (fig. 1A, B, and E). In contrast, the remaining nontoxigenic clade 5 isolates contained an intact cdu2 gene, with the single exception of L033, due to its upstream deletion (fig. 1D). A neighbor-joining tree was built for cdu2 using the 396 bp of the gene common to all isolates (fig. 5A). Although the number of polymorphic sites was low (65 variant sites), the cdu2 fragment of toxigenic isolates was clearly distinct from all the nontoxigenic isolates except WA12 which clustered with the toxigenic-like clade 5. The remaining nontoxigenic clade 5 cdu2 fragment clustered with the other four clades, away from toxigenic clade 5. Gene cdu1 showed a very similar phylogenetic pattern to cdu2 (fig. 5B). These data suggested that nontoxigenic clade 5 strains were generated (and whole cdu2 genes acquired) during an interclade homologous recombination originating from nontoxigenic members of other clades.
To investigate whether any of the PaLoc losses had occurred recently, four pairs of closely related genomes were chosen from the whole-genome tree (fig. 4), with one nontoxigenic and one toxigenic strain within each pair. The lack of any clear increase or decrease in single nucleotide polymorphism (SNP) density between these pairs prevented the identification of recombination boundaries, probably because the PaLoc losses in these clade 5 isolates were not recent (fig. 6).
Fig. 6.—

Investigation of mechanisms of PaLoc acquisition and modification post acquisition using SNP plots. The distribution of polymorphisms between five different pairs of strains is shown for the genomic region containing the PaLoc relative to the reference strain M120. The PaLoc region is indicated by the blue dashed box. Each row represents a pairwise comparison, and individual polymorphisms are shown in red.
Changes in the A−B+ Lineage PaLoc Postacquisition
In all of the isolates belonging to the clade 5 A−B+ lineage, there is an ORF at the 3′-end of the PaLoc (fig. 1B) closely related (79% identity at the amino acid level) to that seen at the PaLoc integration site of WA 12 (fig. 1E) This suggests that a mobile element may have been involved in the loss of tcdA and tcdC after the PaLoc was acquired.
Investigation of the phylogeny of the PaLoc tcdE gene across the C. difficile population structure revealed that the putative holin gene present in the A−B+ clade 5 PaLoc was only distantly related to that found in clades 1–4 (65–66% at the nucleotide level). It had slightly greater identity to tcsE from C. sordellii than the M120 tcdE (75% at the amino acid level vs. 71%) (fig. 5). Although BLAST searches of viral and bacterial databases were not able to identify a more closely related sequence, the A−B+ clade 5 tcdE was probably gained via allelic exchange from an unknown source.
Discussion
Phylogenetic analysis of C. difficile clade 5 revealed its genetic diversity and highlighted the complex evolutionary history of its PaLoc. Clade 5 contained at least eight STs in addition to the well-characterized “hypervirulent” ST11. Two distinct phylogenetic branches were identified, suggesting the clade itself is not recently emergent as a clinical problem, in contrast to the emergent ST11(078) clone (Goorhuis et al. 2008; Walker et al. 2013). Clostridium difficile is estimated to have diverged from Clostridium tetani between 1.1 and 85 Ma, (He et al. 2010), although these two species are thought not to belong to the same family, let alone genus (Collins et al. 1994; Ludwig et al. 2009), making it difficult to date the origin of C. difficile as a species. Nevertheless, clade 5 is known to have diverged from the rest of the species very early on, making it potentially several million years old (He et al. 2010).
Fine-scale phylogenetic analysis suggested that clade 5 has acquired the PaLoc on two occasions. Clade 5 is relatively unusual in this respect, since clade 2 is the only other lineage in which multiple PaLoc variants have been identified to date (Dingle et al. 2011).
The exact mechanisms involved in PaLoc acquisition remain unclear, although the presence of an incongruent cdd1 in the clade 5 A+B+ lineage implicates homologous recombination involving PaLoc flanking sequences in this case. An alternative mechanism that has been proposed is PaLoc acquisition by site-specific recombination, catalysed by an integrase supplied in trans (Dingle et al. 2014). This would be consistent with the single PaLoc integration site observed in clades 1–5, and the congruence of PaLoc flanking genes with clade (Dingle et al. 2014). Horizontal transfer of the PaLoc from strain CD630 to a nontoxigenic strain and consequent toxigenic conversion has been demonstrated (Brouwer et al. 2013).The PaLoc was transferred on variably sized DNA fragments, at a frequency similar to conjugative transposons seen in C. difficile (Brouwer et al. 2013). The first C. difficile genome to be sequenced contained an estimated approximately 10% mobile elements, hence there are a large number of candidates which could act as a helper element (Sebaihia et al. 2006).
It has been proposed that the ancestral C. difficile population may have been nontoxigenic, and that different PaLoc variants were acquired independently by five different clades after they diverged (Dingle et al. 2014). This was supported by the distribution of nontoxigenic strains throughout clades 1, 4 (Dingle et al. 2014), and 5, the distinct phylogeny of the PaLoc, and the existence of a divergent clade designated C-I, members of which have all been found to be nontoxigenic to date. Among the clade 5 isolates available to the present study, little evidence was found of a putative nontoxigenic ancestor, with analysis of cdu2 suggesting all but one (WA12) of the nontoxigenic genotypes within this clade were originally toxigenic, but have lost their PaLoc via interclade homologous recombination with nontoxigenic isolates. The only nontoxigenic genotype to still have the signature clade 5 cdu2, isolate WA12, has lost part of its 115 bp PaLoc integration site upon insertion of the C-I clade-like sequence. SNP plots and fine-scale phylogeny suggested that none of these events was recent.
Previously reported data have shown that intraclade homologous recombination can involve sequences up to 234 kb (Dingle et al. 2014). Although the PaLoc appears to be stably integrated, like other regions of the C. difficile chromosome, it is susceptible to the activity of mobile elements, often resulting in large-scale deletions. The PaLoc seen in the toxinotype XXX/XXXI branch of clade 5 (fig. 1B) has a particularly complex history, with evidence of homologous recombination with other clades and potentially other species. A remnant of the C-I sequence may have been acquired during the same event which caused the large deletion at the 3′-end of the PaLoc. Fragments of putative mobile elements have been identified in the region of the deletion in the toxinotype X (A−B+) isolate 8864 (Song et al. 1999). The most common A−B+ type, UK 017, toxinotype VIII, which belongs to clade 4 has a approximately 1,200 bp deletion at the 3′-end of the tcdA gene which appears to have resulted from homologous recombination of the repeats within the cell-binding domain, in addition to a nonsense mutation further upstream (von Eichel-Streiber et al. 1999).
The most obvious evidence linking a partial deletion of the PaLoc to a mobile element occurs in L033, where Tn6218 is present at the deletion site (fig. 7). Such deletions adjacent to mobile elements may be linked to failed attempts at transposition (Roberts et al. 1991). Also within the toxinotype XXX/XXXI lineage is a putative holin that appears to derive from an unknown source. The toxinotype XXX/XXXI PaLoc therefore appears to be the product of multiple evolutionary events including interspecies homologous recombination, and a possible insertion/deletion event involving a mobile element. However, not all mobile elements associated with the PaLoc have resulted in major deletions to it sequence. Tn6218 is found within the clade 3 PaLoc near tcdE (Dingle et al. 2014) without the large deletions seen in the toxinotype XI PaLoc. A novel type of element, termed an IStron, has been found within tcdA (Braun et al. 2000). The C. sordellii PaLoc also seems to be susceptible to changes due to the activity of mobile elements. The fragment of an ORF near the large deletion of tcsH in C. sordellii ATCC 9714 (fig. 8) suggests that mobile elements are responsible for the generation of variant phenotypes (TcsH−TcsL+) in this species as well. It was hoped that the availability of the toxin-encoding locus of the reference C. sordellii isolate would further enhance our understanding of the C. difficile PaLoc. The C. sordellii toxins are more closely related to those of C. difficile than any of the other LCT (large clostridial toxin) family, and the presence of two homologs accompanied by a putative holin hinted at a similar arrangement (fig. 8). However, data available to this study indicate that the C. sordellii toxin locus is apparently part of a much larger DNA element (possibly a plasmid), but its borders could not be defined (Elliott B, Hong S, Knight DR, Riley TV, unpublished data). The genome sequences from C. sordellii ATCC 9714 and VPI 9018 (TcsH+TcsL+) have been published, and the authors were also unable to properly define the element (Sirigi Reddy et al. 2013). The origins of Pathogenicity Islands are particularly enigmatic in gram-positive organisms (Hacker et al. 2004). The presence of a putative holin and lysin within the PaLoc is suggestive of a bacteriophage origin, and alpha toxin (TcnA) of C. novyi is still carried by a bacteriophage (Eklund et al. 1976).
Fig. 7.—

Modification of clade 5 PaLoc postacquisition by large-scale deletions. A comparison between the PaLoc regions of strains L033 (top) and M120 (bottom). The insertion of Tn6218 (indicated in black box) has resulted in a deletion relative to M120 of 51,379 bp comprising most of the PaLoc and a large region upstream. Only tcdC and the 3′-terminal 2,456 bp of tcdA remain of the PaLoc in L033.
Fig. 8.—

Genetic organization of the toxin-encoding loci of C. difficile and C. sordellii. The typical C. difficile PaLoc genetic organization (A) compared with the organization in C. sordellii strains VPI 9048 (B) and the toxin variant (tcsH−tcsL+) ATCC 9714 (C).
Clostridium difficile clade 5 exhibits significantly more genetic diversity than previously acknowledged. The level of diversity among Australian clade 5 isolates is intriguing. Certain clades are associated with particular geographical areas (e.g., clade 4 with South East Asia, clade 2 with North America). It is possible that clade 5 may have originated in Australia; however, further investigation is required.
The two main lineages within clade 5 appear to have acquired the PaLoc independently, but possibly from a similar source. The loss of the PaLoc via interclade homologous recombination has occurred on several occasions, resulting in nearly all the nontoxigenic strains in this study. A number of evolutionary events resulting in significant changes to the clade 5 PaLoc were identified in this study, showing that the PaLoc is continually evolving. In order to better understand C. difficile and its evolution as a pathogen, we must continue to study its phylogeny and the evolutionary history of the PaLoc.
Supplementary Material
Supplementary figures S1–S3 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
This work was supported in part by the Oxford Partnership Comprehensive Biomedical Research Centre with funding from the Department of Health's NIHR Biomedical Research Centres funding scheme. The views expressed in this publication are those of the authors and not necessarily those of the Department of Health. The work was also supported in part by the UKCRC Modernising Medical Microbiology Consortium, funded under the UKCRC Translational Infection Research Initiative supported by Medical Research Council, Biotechnology and Biological Sciences Research Council, and the National Institute for Health Research on behalf of the Department of Health (grant G0800778) and the Wellcome Trust (grant 087646/Z/08/Z). D.W.C. is the recipient of a NIHR Senior Investigator award. X.D. would like to acknowledge the NIHR for Health Protection Research Unit funding. This publication made use of the C. difficile Multilocus Sequence Typing website, http://pubmlst.org/cdifficile/, sited at the Department of Zoology, University of Oxford.
Literature Cited
- Amimoto K, Noro T, Oishi E, Shimizu M. A novel toxin homologous to large clostridial cytotoxins found in culture supernatant of Clostridium perfringens type C. Microbiology. 2007;153:1198–1206. doi: 10.1099/mic.0.2006/002287-0. [DOI] [PubMed] [Google Scholar]
- Bakker D, Smits WK, Kuijper EJ, Corver J. TcdC does not significantly repress toxin expression in Clostridium difficile 630ΔErm. PLoS One. 2012;7:e43247. doi: 10.1371/journal.pone.0043247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer MP, et al. Clostridium difficile infection in Europe: a hospital-based survey. Lancet. 2011;377:63–73. doi: 10.1016/S0140-6736(10)61266-4. [DOI] [PubMed] [Google Scholar]
- Bentley DR, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman RA, Riley TV. Isolation of Clostridium difficile from stored specimens and comparative susceptibility of various tissue cell lines to cytotoxin. FEMS Microbiol Lett. 1986;34:31–35. [Google Scholar]
- Braun V, et al. A chimeric ribozyme in Clostridium difficile combines features of group I introns and insertion elements. Mol Microbiol. 2000;36:1447–1459. doi: 10.1046/j.1365-2958.2000.01965.x. [DOI] [PubMed] [Google Scholar]
- Braun V, Hundsberger T, Leukel P, Sauerborn M, von Eichel-Streiber C. Definition of the single integration site of the pathogenicity locus in Clostridium difficile. Gene. 1996;181:29–38. doi: 10.1016/s0378-1119(96)00398-8. [DOI] [PubMed] [Google Scholar]
- Braun V, von Eichel-Streiber C. Virulence-associated mobile elements in bacilli and clostridia. In: Kaper JB, Hacker J, editors. Pathogenicity islands and other mobile virulence factors. Washington, D.C.: ASM Press; 1999. pp. 233–264. [Google Scholar]
- Brouwer MS, et al. Horizontal gene transfer converts non-toxigenic Clostridium difficile strains into toxin producers. Nat Commun. 2013;4:2601. doi: 10.1038/ncomms3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter GP, et al. The anti-sigma factor TcdC modulates hypervirulence in an epidemic BI/NAP1/027 clinical isolate of Clostridium difficile. PLoS Pathog. 2011;7:e1002317. doi: 10.1371/journal.ppat.1002317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartman ST, Kelly ML, Heeg D, Heap JT, Minton NP. Precise manipulation of the Clostridium difficile chromosome reveals a lack of association between the tcdC genotype and toxin production. Appl Environ Microbiol. 2012;78:4683–4690. doi: 10.1128/AEM.00249-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver T, Thomson N, Bleasby A, Berriman M, Parkhill J. DNAPlotter: circular and linear interactive genome visualization. Bioinformatics. 2009;25:119–120. doi: 10.1093/bioinformatics/btn578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver TJ, et al. ACT: the Artemis Comparison Tool. Bioinformatics. 2005;21:3422–3423. doi: 10.1093/bioinformatics/bti553. [DOI] [PubMed] [Google Scholar]
- Collins MD, et al. The phylogeny of the genus Clostridium: proposal of five new genera and eleven new species combinations. Int J Syst Bacteriol. 1994;44:812–826. doi: 10.1099/00207713-44-4-812. [DOI] [PubMed] [Google Scholar]
- Davies AH, Roberts AK, Shone CC, Acharya KR. Super toxins from a super bug: structure and function of Clostridium difficile toxins. Biochem J. 2011;436:517–526. doi: 10.1042/BJ20110106. [DOI] [PubMed] [Google Scholar]
- Didelot X, et al. Microevolutionary analysis of Clostridium difficile genomes to investigate transmission. Genome Biol. 2012;13:R118. doi: 10.1186/gb-2012-13-12-r118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didelot X, Falush D. Inference of bacterial microevolution using multilocus sequence data. Genetics. 2007;175:1251–1266. doi: 10.1534/genetics.106.063305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingle KE, et al. Clinical Clostridium difficile: clonality and pathogenicity locus diversity. PLoS One. 2011;6:e19993. doi: 10.1371/journal.pone.0019993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingle KE, et al. Recombinational switching of the Clostridium difficile S-layer and a novel glycosylation gene cluster revealed by large-scale whole-genome sequencing. J Infect Dis. 2013;207:675–686. doi: 10.1093/infdis/jis734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingle KE, et al. Evolutionary history of the Clostridium difficile pathogencity locus. Genome Biol Evol. 2014;6:36–52. doi: 10.1093/gbe/evt204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert C, et al. Clinical and microbiological features of Clostridium difficile infections in France: the ICD-RAISIN 2009 national survey. Med Mal Infect. 2013;43:67–74. doi: 10.1016/j.medmal.2013.01.004. [DOI] [PubMed] [Google Scholar]
- Eklund MW, Poysky FT, Peterson ME, Meyers JA. Relationship of bacteriophages to alpha toxin production in Clostridium novyi types A and B. Infect Immun. 1976;14:793–803. doi: 10.1128/iai.14.3.793-803.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott B, Reed R, Chang BJ, Riley TV. Bacteremia with a large clostridial toxin-negative, binary toxin-positive strain of Clostridium difficile. Anaerobe. 2009;15:249–251. doi: 10.1016/j.anaerobe.2009.08.006. [DOI] [PubMed] [Google Scholar]
- Elliott B, et al. New types of toxin A-negative, toxin B-positive strains among clinical isolates of Clostridium difficile in Australia. J Med Microbiol. 2011;60:1108–1111. doi: 10.1099/jmm.0.031062-0. [DOI] [PubMed] [Google Scholar]
- Geric Stare B, Rupnik M. Clostridium difficile toxinotype XI (A−B−) exhibits unique arrangement of PaLoc and its upstream region. Anaerobe. 2010;16:393–395. doi: 10.1016/j.anaerobe.2010.04.001. [DOI] [PubMed] [Google Scholar]
- Goorhuis A, et al. Emergence of Clostridium difficile infection due to a new hypervirulent strain, polymerase chain reaction ribotype 078. Clin Infect Dis. 2008;47:1162–1170. doi: 10.1086/592257. [DOI] [PubMed] [Google Scholar]
- Griffiths D, et al. Multilocus sequence typing of Clostridium difficile. J Clin Microbiol. 2010;48:770–778. doi: 10.1128/JCM.01796-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: an RNA family database. Nucleic Acids Res. 2003;31:439–441. doi: 10.1093/nar/gkg006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber AR, Lorenz R, Bernhart SH, Neubock R, Hofacker IL. The Vienna RNA websuite. Nucleic Acids Res. 2008;36:W70–74. doi: 10.1093/nar/gkn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker J, et al. Pathogenomics of mobile genetic elements of toxigenic bacteria. Int J Med Microbiol. 2004;293:453–461. doi: 10.1078/1438-4221-00290. [DOI] [PubMed] [Google Scholar]
- He M, et al. Evolutionary dynamics of Clostridium difficile over short and long time scales. Proc Natl Acad Sci U S A. 2010;107:7527–7532. doi: 10.1073/pnas.0914322107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolley KA, Maiden MC. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics. 2010;11:595. doi: 10.1186/1471-2105-11-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, et al. Identification of toxin A-negative, toxin B-positive Clostridium difficile by PCR. J Clin Microbiol. 1998;36:2178–2182. doi: 10.1128/jcm.36.8.2178-2182.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato N, et al. Identification of toxigenic Clostridium difficile by the polymerase chain reaction. J Clin Microbiol. 1991;29:33–37. doi: 10.1128/jcm.29.1.33-37.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight DR, Riley TV. Prevalence of Clostridium difficile gastrointestinal carriage in Australian sheep and lambs. Appl Environ Microbiol. 2013 doi: 10.1128/AEM.01888-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight DR, Thean S, Putsathit P, Fenwick S, Riley TV. Cross-sectional study reveals high prevalence of Clostridium difficile non-PCR ribotype 078 strains in Australian veal calves at slaughter. Appl Environ Microbiol. 2013;79:2630–2635. doi: 10.1128/AEM.03951-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanis JM, Heinlen LD, James JA, Ballard JD. Clostridium difficile 027/BI/NAP1 encodes a hypertoxic and antigenically variable form of TcdB. PLoS Pathog. 2013;9:e1003523. doi: 10.1371/journal.ppat.1003523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig W, Schleifer K-H, Whitman WB. Revised road map to the phylum Firmicutes. In: De Vos P, et al., editors. Bergey's Manual of Systematic Bacteriology. New York: Springer; 2009. pp. 1–14. [Google Scholar]
- Magill SS, et al. Multistate point-prevalence survey of health care-associated infections. N Engl J Med. 2014;370:1198–1208. doi: 10.1056/NEJMoa1306801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monot M, et al. Reannotation of the genome sequence of Clostridium difficile strain 630. J Med Microbiol. 2011;60:1193–1199. doi: 10.1099/jmm.0.030452-0. [DOI] [PubMed] [Google Scholar]
- O'Neill GL, Ogunsola FT, Brazier JS, Duerden BI. Modification of a PCR ribotyping method for application as a routine typing scheme for Clostridium difficile. Anaerobe. 1996;2:205–209. [Google Scholar]
- Pelaez T, et al. Characterization of swine isolates of Clostridium difficile in Spain: a potential source of epidemic multidrug resistant strains? Anaerobe. 2013 doi: 10.1016/j.anaerobe.2013.05.009. [DOI] [PubMed] [Google Scholar]
- Pirs T, Ocepek M, Rupnik M. Isolation of Clostridium difficile from food animals in Slovenia. J Med Microbiol. 2008;57:790–792. doi: 10.1099/jmm.0.47669-0. [DOI] [PubMed] [Google Scholar]
- Roberts DE, Ascherman D, Kleckner N. IS10 promotes adjacent deletions at low frequency. Genetics. 1991;128:37–43. doi: 10.1093/genetics/128.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Palacios A, Stämpfli HR, Stalker M, Duffield T, Weese JS. Natural and experimental infection of neonatal calves with Clostridium difficile. Vet Microbiol. 2007;124:166–172. doi: 10.1016/j.vetmic.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupnik M. Heterogeneity of large clostridial toxins: importance of Clostridium difficile toxinotypes. FEMS Microbiol Rev. 2008;32:541–555. doi: 10.1111/j.1574-6976.2008.00110.x. [DOI] [PubMed] [Google Scholar]
- Rupnik M, Avesani V, Janc M, von Eichel-Streiber C, Delmée M. A novel toxinotyping scheme and correlation of toxinotypes with serogroups of Clostridium difficile isolates. J Clin Microbiol. 1998;36:2240–2247. doi: 10.1128/jcm.36.8.2240-2247.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupnik M, Widmer A, Zimmermann O, Eckert C, Barbut F. Clostridium difficile toxinotype V, ribotype 078, in animals and humans. J Clin Microbiol. 2008;46:2146. doi: 10.1128/JCM.00598-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford K, et al. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- Schneeberg A, Neubauer H, Schmoock G, Grossmann E, Seyboldt C. Presence of Clostridium difficile PCR ribotype clusters related to 033, 078 and 045 in diarrhoeic calves in Germany. J Med Microbiol. 2013 doi: 10.1099/jmm.0.056473-0. [DOI] [PubMed] [Google Scholar]
- Schneeberg A, Rupnik M, Neubauer H, Seyboldt C. Prevalence and distribution of Clostridium difficile PCR ribotypes in cats and dogs from animal shelters in Thuringia, Germany. Anaerobe. 2012;18:484–488. doi: 10.1016/j.anaerobe.2012.08.002. [DOI] [PubMed] [Google Scholar]
- Sebaihia M, et al. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat Genet. 2006;38:779–786. doi: 10.1038/ng1830. [DOI] [PubMed] [Google Scholar]
- Sirigi Reddy AR, Girinathan BP, Zapotocny R, Govind R. Identification and characterization of Clostridium sordellii toxin gene regulator. J Bacteriol. 2013;195:4246–4254. doi: 10.1128/JB.00711-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song KP, Ow SE, Chang SY, Bai XL. Sequence analysis of a new open reading frame located in the pathogenicity locus of Clostridium difficile strain 8864. FEMS Microbiol Lett. 1999;180:241–248. doi: 10.1111/j.1574-6968.1999.tb08802.x. [DOI] [PubMed] [Google Scholar]
- Squire MM, et al. Novel molecular type of Clostridium difficile in neonatal pigs, Western Australia. Emerg Infect Dis. 2013;19:790–792. doi: 10.3201/eid1905.121062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stabler RA, et al. Macro and micro diversity of Clostridium difficile isolates from diverse sources and geographical locations. PLoS One. 2012;7:e31559. doi: 10.1371/journal.pone.0031559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbs S, et al. Production of actin-specific ADP-ribosyltransferase (binary toxin) by strains of Clostridium difficile. FEMS Microbiol Lett. 2000;186:307–312. doi: 10.1111/j.1574-6968.2000.tb09122.x. [DOI] [PubMed] [Google Scholar]
- Tamura K, et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritt A, Eisen JA, Facciotti MT, Darling AE. An Integrated Pipeline for de Novo Assembly of Microbial Genomes. PLoS One. 2012;7:e42304. doi: 10.1371/journal.pone.0042304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Eichel-Streiber C, Zec-Pirnat I, Grabnar M, Rupnik M. A nonsense mutation abrogates production of a functional enterotoxin A in Clostridium difficile toxinotype VIII strains of serogroups F and X. FEMS Microbiol Lett. 1999;178:163–168. doi: 10.1111/j.1574-6968.1999.tb13773.x. [DOI] [PubMed] [Google Scholar]
- Walker AS, et al. Relationship between bacterial strain type, host biomarkers, and mortality in Clostridium difficile infection. Clin Infect Dis. 2013;56:1589–1600. doi: 10.1093/cid/cit127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaiß NH, et al. Typing Clostridium difficile strains based on tandem repeat sequences. BMC Microbiol. 2009;9:6. doi: 10.1186/1471-2180-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.