

Abstract
The Campylobacter lari group is a phylogenetic clade within the epsilon subdivision of the Proteobacteria and is part of the thermotolerant Campylobacter spp., a division within the genus that includes the human pathogen Campylobacter jejuni. The C. lari group is currently composed of five species (C. lari, Campylobacter insulaenigrae, Campylobacter volucris, Campylobacter subantarcticus, and Campylobacter peloridis), as well as a group of strains termed the urease-positive thermophilic Campylobacter (UPTC) and other C. lari-like strains. Here we present the complete genome sequences of 11 C. lari group strains, including the five C. lari group species, four UPTC strains, and a lari-like strain isolated in this study. The genome of C. lari subsp. lari strain RM2100 was described previously. Analysis of the C. lari group genomes indicates that this group is highly related at the genome level. Furthermore, these genomes are strongly syntenic with minor rearrangements occurring only in 4 of the 12 genomes studied. The C. lari group can be bifurcated, based on the flagella and flagellar modification genes. Genomic analysis of the UPTC strains indicated that these organisms are variable but highly similar, closely related to but distinct from C. lari. Additionally, the C. lari group contains multiple genes encoding hemagglutination domain proteins, which are either contingency genes or linked to conserved contingency genes. Many of the features identified in strain RM2100, such as major deficiencies in amino acid biosynthesis and energy metabolism, are conserved across all 12 genomes, suggesting that these common features may play a role in the association of the C. lari group with coastal environments and watersheds.
Keywords: flagella, hemagglutination, methylome, UPTC
Introduction
Campylobacter lari (formerly Campylobacter laridis) strains were originally described as nalidixic acid-resistant thermophilic Campylobacter (NARTC; Skirrow and Benjamin 1980). Subsequently, the urease-positive thermophilic Campylobacter (UPTC), the nalidixic-acid susceptible (NASC) group, and the urease-producing NASC were identified as C. lari variants (Endtz et al. 1997; Megraud et al. 1988; Owen et al. 1988; Vandamme et al. 1991). Taxonomic placement of the UPTC strains remains undetermined. UPTC strains were originally proposed to be a biovar of C. lari (Owen et al. 1988). However, multilocus enzyme electrophoresis (MLEE) and amplified fragment length polymorphism (AFLP) typing demonstrated that the UPTC strains were related to but distinct from C. lari (Matsuda et al. 2003; Duim et al. 2004; Debruyne et al. 2009), forming two separate clusters following phylogenetic analysis (Duim et al. 2004; Debruyne et al. 2009). Some strains originally identified as C. lari were later defined as novel taxa, including: Campylobacter peloridis (Debruyne et al. 2009, also NARTC cluster IV in Duim et al. 2004) and Campylobacter volucris (Debruyne et al. 2010b). Campylobacter lari was also divided into two novel subspecies, C. lari subsp. lari (Debruyne et al. 2009) and C. lari subsp. concheus (Debruyne et al. 2009, also NASC cluster III in Duim et al. 2004). Additional lari-like species were described, such as Campylobacter insulaenigrae (Foster et al. 2004) and Campylobacter subantarcticus (Debruyne et al. 2010a). Together, these taxa comprise the C. lari group.
Although the C. lari group is a phylogenetically distinct clade within the genus Campylobacter (supplementary fig. S1, Supplementary Material online), taxa within this clade are highly related, and this similarity may reflect, in part, the similar hosts and environments from which these strains are isolated. Campylobacter lari was isolated originally from gulls (Skirrow and Benjamin 1980; Benjamin et al. 1983). Other members of the C. lari group are also isolated from gulls (UPTC, C. volucris [Kaneko et al. 1999; Debruyne et al. 2010b]) and other shorebirds, such as plovers, redshanks, dunlins, sandpipers, skuas, albatrosses, and penguins (C. lari, UPTC, C. subantarcticus [Waldenstrom et al. 2002, 2007; Leotta et al. 2006; Debruyne et al. 2010a; Ryu et al. 2014]). The C. lari group strains are also isolated from marine mammals (C. insulaenigrae, C. lari [Foster et al. 2004; Stoddard et al. 2007; Garcia-Pena et al. 2010; Gonzalez et al. 2011]), shellfish (C. lari, C. peloridis [Endtz et al. 1997; Van Doorn et al. 1998; Debruyne et al. 2009]), and seawater/fresh water (C. lari, UPTC [Obiri-Danso and Jones 1999; Obiri-Danso et al. 2001; Meinersmann et al. 2013; Khan et al. 2014]). Even though members of the C. lari group have been isolated from livestock (Tresierra-Ayala et al. 1994; Aarestrup et al. 1997; Harvey et al. 1999; Scanlon et al. 2013), this clade is primarily associated with coastal regions and watersheds.
Within Campylobacter, C. jejuni is the primary agent in nearly all Campylobacter-related human illnesses. However, C. lari has been associated occasionally with human illness, causing gastroenteritis with abdominal pain, fever, and diarrhea (Broczyk et al. 1987; Lin et al. 1998; Prasad et al. 2001; Otasevic et al. 2004) or bacteremia in immunocompromised (Nachamkin et al. 1984; Martinot et al. 2001) or otherwise debilitated patients (Morris et al. 1998). Isolation of other members of the C. lari group is even more infrequent, although in some cases this may be due to the relative novelty of some taxa. Nevertheless, C. insulaenigrae (Chua et al. 2007), C. peloridis and C. lari subsp. concheus (Debruyne et al. 2009), and UPTC strains (Megraud et al. 1988) have also been isolated from human clinical and fecal samples. Further studies, however, are required to determine the pathogenicity of members of this clade.
The genome of C. lari subsp. lari strain RM2100 (ATCC-BAA 1060; CDC strain D67, “case 6” [Tauxe et al. 1985]), isolated from an 8-month-old girl with watery diarrhea, has been sequenced to completion (Miller et al. 2008). Although 90% of the genes predicted in the strain RM2100 genome are similar to genes present in the genomes of other thermotolerant Campylobacter spp., a substantial number of genes, especially those associated with amino acid biosynthesis and energy metabolism, and identified previously in other Campylobacter genomes, are absent from the C. lari subsp. lari strain RM2100 genome. Citrate synthase, a key component of the TCA cycle, is not encoded by the C. lari subsp. lari strain RM2100 genome. Polymerase chain reaction (PCR) analysis indicated that citrate synthase is not encoded by C. insulaenigrae (Stoddard et al. 2007) or other members of the C. lari group (data not shown), suggesting that the genomic features identified in C. lari subsp. lari may apply to the clade as a whole. To determine whether the genomic features identified in C. lari subsp. lari strain RM2100 are common to the C. lari group and can help to explain the host and environmental association of this clade, the genomes of the remaining validly named taxa within the C. lari group (table 1) were sequenced to completion. Also, to provide further evidence for the taxonomic placement of the UPTC strains, the genomes of four UPTC strains were also sequenced. Here, we present the comparative genomic analysis of 12 C. lari group strains.
Table 1.
Strains Sequenced in This Study
| Coverage |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Strain | Type Strain | Source | Location | Optical Map | 454 | Illumina | PacBio | Total | Reference | Accession Number(s) | |
| C. insulaenigrae NCTC 12927 | Y | Marine mammal | Scotland | Y | 107× | H; 1,347× | N/A | 1,454× | Foster et al. 2004 | CP007770 | |
| C. lari concheus LMG 11760 | N | Human | Canada (ONT) | Y | 71× | H; 1,287× | N/A | 1,358× | Debruyne et al. 2009 | CP007771 | |
| UPTC CCUG 22395 | N | Human | France | N | 61× | H; 1,478× | N/A | 1,539× | N/A | CP007776 | |
| UPTC NCTC 11845 | N | River water | United Kingdom | Y | 123× | H; 1,048× | 182× | 1,352× | N/A | CP007775 | |
| UPTC RM16701 | N | River water | United States (CA) | N | 82× | M; 708× | N/A | 790× | N/A | CP007777 | |
| UPTC RM16712 | N | River water | United States (CA) | N | 110× | M; 551× | N/A | 661× | N/A | CP007778 | |
| C. peloridis LMG 23910 | Y | Shellfish | The Netherlands | Y | 63× | H; 1,175× | 222× | 1,459× | Debruyne et al. 2009 | CP007766CP007767 (pPEL1)CP007768 (pPEL2) | |
| C. subantarcticus LMG 24374 | N | Gentoo penguin | S. Georgia, Antarctica | Y | 35× | H; 1,146× | 141× | 1,323× | Debruyne et al. 2010a | CP007772 | |
| C. subantarcticus LMG 24377 | Y | Gray-headed albatross | S. Georgia, Antarctica | Y | 136× | H; 1,086× | 138× | 1,360× | Debruyne et al. 2010a | CP007773 | |
| C. volucris LMG 24379 | N | Black-headed gull | Sweden | Y | 57× | H; 1,381× | N/A | 1,438× | Debruyne et al. 2010b | CP007774 | |
| Campylobacter spp. RM16704 | Na | River water | United States (CA) | N | 84× | M; 475× | 164× | 723× | N/A | CP007769 | |
Note.—H, HiSeq sequencing; M, MiSeq sequencing; N/A, not applicable.
aIf Campylobacter spp. strain RM16704 represents a novel species, then it would likely be designated as the type strain.
Materials and Methods
Growth Conditions and Chemicals
Campylobacter strains were cultured at 37 °C on Brain Heart Infusion agar (Becton Dickinson, Sparks, MD) amended with 5% (v/v) laked horse blood (Hema Resource & Supply, Aurora, OR). The incubation atmosphere was 5% H2, 10% CO2, and 85% N2. PCR enzymes and reagents were purchased from New England Biolabs (Beverly, MA) or Epicentre (Madison, WI). All chemicals were purchased from Sigma-Aldrich Chemicals (St. Louis, MO) or Fisher Scientific (Pittsburgh, PA). DNA sequencing reagents and capillaries were purchased from Applied Biosystems (Foster City, CA), Roche Life Science (Indianapolis, IN), Illumina Inc. (San Diego, CA), or Pacific Biosciences (Menlo Park, CA).
Isolation of Campylobacter Strains from River Water Samples
In a six-well plate (Corning, Corning, NY), 7.2 ml of a river water sample was added to 0.8 ml 10× ABB (Anaerobe Basal Broth, Oxoid [Remel], Lenexa, KS) + 10 × Preston supplement (amphotericin B [10 μg/ml], rifampicin [10 μg/ml], trimethoprim lactate [10 μg/ml], and polymyxin B [5 UI/ml]; Oxoid) to achieve a final 1 × ABB + Preston concentration. These six-well plates were placed inside a plastic zip-loc bag containing 1–2% O2 + Bioblend gas (10% CO2, 10% H2, 80% N2; Praxair, Danbury, CT) and incubated for 24 h at 37 °C and 40 rpm. For each enriched sample, a 10-μl loop was struck onto an ABA (Anaerobe Basal Agar) plate (Oxoid) amended with 5% laked horse blood (Hema) and CAT supplement (cefoperazone [8 μg/ml], amphotericin B [10 μg/ml], and teicoplanin [4 μg/ml]; Oxoid). Plates were incubated in a microaerobic gas jar (AnaeroJar 3.5 L System; Oxoid) at 37 °C under 1–2% O2 + Bioblend gas for 24–48 h. All positive cultures were examined under a 1,000 × microscope. Cultures positive for Campylobacter were then filtered through a 0.6 -μ mixed cellulose filter onto an ABA plate. After growth for 24 h, single colonies were picked onto a new ABA plate, then incubated 24–48 h. Pure cultures were stored at −80 °C; species identification of the isolates was achieved by 16 S rRNA gene sequencing. All strains were tested for urease and nitrate reductase activity.
Urease and Nitrate Tests
Urease and nitrate reductase activities were detected using the API-Campy system (bioMerieux, France). All tests were repeated at least twice. Campylobacter jejuni subsp. doylei (Cjd) strain SSI 5384 (ure−, nap−) was used as a negative control.
Polymerase Chain Reactions
Genomic DNA was prepared as described previously (Miller et al. 2005). Standard amplifications were performed on a Tetrad thermocycler (Bio-Rad, Hercules, CA) with the following settings: 94 °C for 30 s, 53 °C for 30 s, and 72 °C for 2 min (30 cycles). Each amplification mixture contained 50 ng genomic DNA, 1× PCR buffer (Epicentre), 1× PCR enhancer (Epicentre), 2.5 mM MgCl2, 250 μM each dNTP, 50 pmol each primer, and 1 U polymerase (New England Biolabs). Amplicons were purified using ExoSAP-IT (Affymetrix, Santa Clara, CA). Sequencing and PCR oligonucleotides were designed using Primer Premier (v 5.0; Premier Biosoft, Palo Alto, CA) and purchased from Eurofins (Huntsville, AL).
Sanger Sequencing
Sanger cycle sequencing reactions were performed on a 96-well Tetrad thermocycler (Bio-Rad) using the ABI PRISM BigDye terminator cycle sequencing kit (version 3.1) and standard protocols. Cycle sequencing extension products were purified using BigDye XTerminator (Applied Biosystems). DNA sequencing was performed on an ABI PRISM 3730 DNA Analyzer (Applied Biosystems), using POP-7 polymer and ABI PRISM Genetic Analyzer Data Collection and ABI PRISM Genetic Analyzer Sequencing Analysis software. Sequences were trimmed, assembled, and analyzed in SeqMan (v 8.0.2; DNASTAR, Madison, WI).
Roche and Illumina Next-Generation Sequencing
Shotgun and paired-end (8–12 kb) 454 reads were obtained on a Roche 454 GS-FLX+ Genome Sequencer with Titanium chemistry using standard protocols. Illumina libraries were prepared using the KAPA Low-Throughput Library Preparation Kit with Standard PCR Amplification Module (Kapa Biosystems, Wilmington, MA), following manufacturer’s instructions except for the following changes: 750 ng DNA was sheared at 30 psi for 40 s and size selected to 700–770 bp following Illumina protocols. Standard desalting TruSeq LT and PCR Primers were ordered from Integrated DNA Technologies (Coralville, IA) and used at 0.375 and 0.5 µM final concentrations, respectively. PCR was reduced to 3–5 cycles. Libraries were quantified using the KAPA Library Quantification Kit (Kapa), except with 10 µl volume and 90-s annealing/extension PCR, then pooled and normalized to 4 nM. Pooled libraries were requantified by ddPCR on a QX200 system (Bio-Rad), using the Illumina TruSeq ddPCR Library Quantification Kit and following manufacturer’s protocols, except with an extended 2-min annealing/extension time. The libraries were sequenced 2 × 250 bp paired end v2 on a MiSeq instrument (Illumina) at 13.5 pM, following manufacturer’s protocols. Illumina HiSeq reads were obtained from SeqWright (Houston, TX).
Single Molecule, Real-Time Sequencing
Single Molecule, Real-Time (SMRT) sequencing was performed on the Pacific Biosciences (PacBio) RS sequencing platform using 10- or 20-kb SMRTbell libraries, and C2/C2 (most strains) or P5/C3 (strain RM16704) sequencing chemistry. The two chemistries used the 90- or 120-min data collection protocols, respectively. The SMRTbell libraries were prepared from 5 to 10 μg of bacterial genomic DNA, using the standard protocol from Pacific Biosciences as described in the 10-kb Template Preparation and Sequencing and Procedure (http://www.smrtcommunity.com/SamplePrep), or the 20-kb procedure for strain RM16704, and processed for sequencing as recommended by the supplier. A FASTQ file was generated from the PacBio reads using SMRTanalysis (ver. 2.1), and the reads were error-corrected using pacBioToCA with self-correction (Koren et al. 2013). The longest 20× of the corrected reads was assembled with Celera Assembler 7.0 (Koren et al. 2012). The resulting contigs were polished using Quiver (Chin et al. 2013).
Genome Sequencing and Assembly
The 11 genomes characterized in this study (table 1) were all sequenced initially using Roche 454 technology. Shotgun and paired-end 454 reads were assembled using the Roche Newbler assembler (v2.6) into one or two chromosomal scaffolds, providing draft genome sequences with a coverage of 35–136× (table 1). Intrascaffold gaps were filled using the 454 repeat contigs and the Perl script contig_extender3 (Merga et al. 2013). Contig gaps were closed/validated using PCR amplification and Sanger sequencing, generating draft pseudomolecules for each genome. Illumina HiSeq or MiSeq reads were assembled within Newbler as reference assemblies using the draft pseudomolecules as templates: These Illumina contigs were assembled into the 454 assembly within Seqman (v. 8.0.2) to validate all 454 base calls. The Illumina reads also provided an additional 475–1,478× coverage (table 1). The presence/absence of single nucleotide polymorphisms (SNPs) within the repeat contigs was assessed using Geneious (v. 7.1.2; Biomatters, Auckland, New Zealand) and the Illumina reads or by using the 454 paired-end reads to link SNPs to adjacent unique contigs. For five strains (table 1), the genomes could not be closed using standard 454/Sanger/Illumina technology, due to a complex topography consisting of multiple repeat contigs, and two of these genomes could not be assembled into a single scaffold. Therefore, these genomes were sequenced using a PacBio RS sequencer (Pacific Biosciences), which produced sequencing reads long enough to uniquely span each of the repeat regions. Using only the PacBio data, all five strains were closed into single, circularly closed chromosomes. Illumina reads were used to validate the PacBio base calls as described above. Seven assemblies were verified (table 1) using a bacterial optical restriction map (OpGen, Gaithersburg, MD).
Genome Annotation and Analysis
Putative coding sequences (CDSs) were determined using GeneMark (Besemer and Borodovsky 2005). Initial annotation was accomplished by comparing the predicted proteins with the proteome of the C. lari subsp. lari strain RM2100 (Miller et al. 2008) and with the NCBI (National Center for Biotechnology Information) nonredundant (nr) database using BLASTP; positive matches had an identity of ≥50%, and an alignment length of ≥75% across both the query and match sequences. Annotation was also assisted through the detection of characteristic Pfam motifs (Punta et al. 2012). The list of putative CDSs was then used to create a preliminary GenBank-formatted (.gbk) file that was entered into Artemis (release 16.0; Rutherford et al. 2000). Annotation/curation within Artemis included the fusion of split CDSs into pseudogenes and the identification of genes overlooked in the initial GeneMark analysis. The start codon of each putative CDS was curated manually, either through visual inspection within Artemis or through BLAST comparison of each CDS to its orthologs within the C. lari group, where present. transfer RNAs (tRNAs) were annotated using tRNAscan-SE (v 1.23; Lowe and Eddy 1997). rRNA loci were identified through RNAmmer (Lagesen et al. 2007). Comparative genomic analysis was performed through a pairwise BLASTP analysis of the C. lari group proteome against itself. For each protein, a custom Perl script was used to identify the top match within the other proteomes, where present, using the match parameters described above. This Perl script also determined the core proteome for the C. lari group and calculated the pairwise average amino acid identities (AAI) between the core proteomes of any two strains. Further comparative analyses were performed using 1) the BLAST Ring Image Generator (BRIG v0.95; Alikhan et al. 2011) and BLASTN, with a default minimum threshold of 50% and an E value of 0.0001; or 2) JSpecies (v. 1.2.1; Richter and Rossello-Mora 2009), using default parameters, to determine average nucleotide identity (ANI) values.
Determination of Variability at the Homopolymeric G:C Tracts
A set of Illumina HiSeq or MiSeq reads were available for each of the 12 genomes characterized in this study, with a coverage ranging from 475× to 1,478×. A custom Perl script was designed to 1) identify the position of all G:C tracts (≥8 bp) within a genome, 2) determine the genome sequences that flank each identified G:C tract 15 nt upstream and downstream, 3) bin each Illumina read as positive if the flanking sequences within the read match 15/15 nt to the genomic flanking sequences and if the G:C tract within the read is homopolymeric, and 4) tabulate the tract lengths within the positive reads. This script was run using the entire Illumina read set for each genome. If the results accurately reflected true variation of the genomic G:C tracts and were not due to, for example, poor Illumina read quality, then hypervariability would not be observed in the more stable homopolymeric A:T tracts. Thus, the script was modified to identify A:T tracts ≥9 bp (9 bp was chosen over 8 bp due to the extremely large number of 8 bp A:T tracts per genome); this modified script was tested on the C. insulaenigrae and C. subantarcticus genomes. As expected, the A:T tracts were quite stable and demonstrated an average variability of approximately 0.4% (data not shown), far lower than that observed in the G:C tracts (for C. insulaenigrae, average variability = 7%, range = 0–41%). Therefore, an arbitrary cutoff value of 2% (>2 SD) was selected as a base value: Tract lengths whose proportion was less than 2% were not included in the determination of hypervariability.
Phylogenetic Analysis
Sequence alignments were performed using CLUSTALX (ver. 2.1). Dendrograms were constructed using the neighbor-joining method and Poisson correction. Bootstraps were conducted with 500 replicates. Phylogenetic analyses were performed using MEGA version 6.05 (Tamura et al. 2013).
Methylome Analysis Using SMRT Sequencing
As described in previous publications, the kinetic information contained in the same SMRT sequencing data used for the de novo assembly can be utilized to characterize the methylome of bacteria (Flusberg et al. 2010; Clark et al. 2012; Murray et al. 2012). The methylomes of the four strains sequenced and closed on the PacBio RS were determined using the RS_Modification_and Motif_Analysis.1 protocol included in SMRT Portal. The de novo PacBio-only assemblies of the corresponding strains from this study were used as the reference genomes in the base modification analysis. The methylome results are deposited in New England Biolab’s REBASE (http://rebase.neb.com/cgi-bin/pacbiolist).
Accession Numbers
The complete nucleotide sequences and annotations of the strains characterized in this study were deposited in GenBank (table 1). The complete nucleotide sequence and annotation of C. lari subsp. lari strain RM2100 (Miller et al. 2008) was deposited previously in GenBank under the accession numbers CP000932 (chromosome) and CP000933 (megaplasmid pCL2100).
Results and Discussion
General Features
The genome of C. lari subsp. lari strain RM2100 was determined to contain 1,525,460 bp (Miller et al. 2008). The genomes of 11 other strains, representing at least five additional taxa within the C. lari group, were sequenced in this study. A summary of the features of these 11 genomes, with inclusion of the previously sequenced C. lari subsp. lari strain RM2100, is presented in table 2. The genome sizes of these 11 strains ranged from 1.465 (C. insulaenigrae) to 1.853 Mb (C. subantarcticus strain LMG 24377). The size differential between the genomes can be explained in part by the presence of genomic islands and prophage (see below). Consistent with the %G+C content of C. lari subsp. lari strain RM2100 (29.7), the %G+C content of the 11 genomes ranges from 28.19 to 29.94.
Table 2.
General Features of the Campylobacter lari Group Genomes
| C. lari subsp. lari | C. lari subsp. concheus | UPTC NCTC 11845 | UPTC CCUG 22395 | UPTC RM16701 | UPTC RM16712 | C. subantarcticus LMG 24374 | C. subantarcticus LMG 24377 | Campylobacter spp. RM16704 | C. peloridis | C. volucris | C. insulaenigrae | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| General features | Chromosome | ||||||||||||
| Size (kb) | 1,525.46 | 1,502.10 | 1,791.51 | 1,523.03 | 1,516.46 | 1,565.01 | 1,782.54 | 1,853.00 | 1,557.54 | 1,711.37 | 1,517.95 | 1,465.08 | |
| % G+C content | 29.70 | 29.73 | 29.36 | 29.86 | 29.90 | 29.74 | 29.94 | 29.75 | 28.47 | 28.51 | 28.57 | 28.19 | |
| CDS numbersa | 1,495 | 1,451 | 1,702 | 1,481 | 1,475 | 1,507 | 1,675 | 1,770 | 1,490 | 1,591 | 1,478 | 1,440 | |
| Assigned function (% CDS) | 836 (56) | 821 (57) | 837 (49) | 835 (56) | 822 (56) | 834 (55) | 840 (50) | 832 (47) | 817 (55) | 827 (52) | 830 (56) | 812 (56) | |
| Pseudogenes | 18 | 20 | 21 | 15 | 9 | 20 | 51 | 56 | 30 | 19 | 19 | 22 | |
| General function (% CDS) | 373 (25) | 378 (26) | 426 (25) | 390 (26) | 387 (26) | 382 (25) | 458 (27) | 485 (27) | 388 (26) | 404 (25) | 383 (26) | 360 (25) | |
| Hypothetical (% CDS) | 286 (19) | 252 (17) | 439 (26) | 256 (17) | 266 (18) | 291 (19) | 377 (23) | 453 (26) | 285 (19) | 360 (23) | 265 (18) | 268 (19) | |
| Prophage/genetic islands | 1 | 1 | 2 | 2 | 1 | 2 | 2 | 3 | 0 | 2 | 1 | 1 | |
| Ribosomal RNA operons | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | |
| CRISPR | N | N | Y | Yb | N | N | Y | N | Y | Y | N | N | |
| G:C tracts ≥8 nt (# HV) | 15 (15) | 15 (14) | 23 (22) | 10 (10) | 14 (14) | 18 (16) | 50 (46) | 44 (43) | 41 (39) | 26 (26) | 17 (17) | 27 (23) | |
| Plasmids (size kb) | 46.2 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | 47.8; 3.6 | N/A | N/A | |
| Gene classes | |||||||||||||
| Gene classes | Signal transduction | ||||||||||||
| Che/Mot proteins | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | |
| MCP | 12 | 11 | 14 | 14 | 14 | 11 | 10 | 10 | 9 | 13 | 9 | 8 | |
| 2CS response regulator | 6 | 6 | 6 | 6 | 5 | 6 | 6 | 6 | 6 | 6 | 6 | 4 | |
| 2CS histidine kinase | 6 | 6 | 6 | 6 | 5 | 6 | 6 | 6 | 6 | 6 | 6 | 4 | |
| Other | 7 | 7 | 7 | 7 | 8 | 6 | 7 | 6 | 6 | 6 | 7 | 6 | |
| R/M systems | |||||||||||||
| Type I (hsd) | 0 | 0 | 0 | 1 | 0 | 1 | 2 | 0 | 1 | 1 | 0 | 0 | |
| Type II/IIS | 2 | 1 | 1 | 1 | 2 | 2 | 2 | 3 | 2 | 3 | 4 | 3 | |
| Type III | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | |
| Other (mcrBC) | 1 | 1 | 1 | 0 | 2 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | |
| DNA methylases | 2 | 3 | 3 | 4 | 4 | 3 | 4 | 4 | 2 | 3 | 1 | 3 | |
| Transcription | |||||||||||||
| Regulatory proteins | 18 | 17 | 16 | 16 | 17 | 17 | 13 | 18 | 15 | 15 | 12 | 14 | |
| σ factors | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | |
| Hag proteins | 1 | 3 | 7 | 2 | 3 | 2 | 3 | 3 | 10 | 15 | 0 | 0 | |
| Motility | |||||||||||||
| FlaAB (class; orient.) | 1;→→ | 1;→→ | 2;→→ | 2;←→ | 2;←→ | 2;←→ | 1;→→ | 1;→→ | 2;←→ | 2;←→ | 1;→→ | 1;→→ | |
| MAF (class) | 1 | 1 | 2 | 2 | 2 | 2 | 1 | 1 | 2 | 2 | 1 | 1 | |
| Pse/Leg | Y/Y | Y/(Y) | Y/N | Y/N | Y/N | Y/N | Y/Y | Y/Y | Y/N | Y/N | Y/Y | Y/Y | |
Note.—HV, hypervariable; Che/Mot, chemotaxis/motility; MCP, methyl-accepting chemotaxis protein; 2CS, two component system; MAF, motility accessory factor; Pse/Leg, pseudaminic acid/legionaminic acid.
aCDS numbers do not include pseudogenes.
bcas1 gene nonfunctional.
The C. lari strain RM2100 genome is predicted to contain 1,495 CDSs with an additional 18 fragmented CDSs, containing frameshift mutations or other point mutations, designated as putative pseudogenes (table 2). Excluding those CDSs contained in prophage or genomic islands, similar numbers of CDSs (1,384–1,499) were identified within the C. lari group. The number of pseudogenes per genome was also generally consistent (9–22; table 2); however, a higher number of pseudogenes was identified in Campylobacter spp. strain RM16704 (30) and in both C. subantarcticus strains (51 and 56). The percentages of CDSs annotated with an assigned function, general function or as hypothetical are similar between all 12 genomes (table 2).
With the exception of Campylobacter spp. strain RM16704, all of the genomes characterized here contain at least one genomic island (C. lari subsp. concheus, C. subantarcticus, C. peloridis, UPTC) or putative prophage (C. lari subsp. lari, C. insulaenigrae, C. subantarcticus, C. volucris) (supplementary table S1, Supplementary Material online). All of the genomic islands/prophage are inserted immediately adjacent to a tRNA, with two-thirds located next to a leucyl-tRNA. Additionally, each of the C. lari group genomes (with the exception of Campylobacter spp. strain RM16704) contains a genomic insertion, ranging in size from 2 to 192 kb, at the same location, adjacent to flgL. At flgL, Campylobacter spp. strain RM16704 also contains several predicted pseudogenes and a CRISPR (clustered regularly interspaced short palindromic repeats)-Cas locus; therefore, it is possible that this strain contains a degenerate genomic insertion in this region. Although the insertion points of the integrated elements within the C. lari group are strongly conserved, each of the integrated elements is unique with regard to gene content and size. Five of the integrated elements encode putative type VI secretion systems (T6SS). Two such elements are contained within the two C. subantarcticus strains. Each of these T6SS-encoding elements in C. subantarcticus also contains an integrated mu phage; however, the mu phage gene content and insertion point are different in each strain.
Similar to other Campylobacter spp., the 12 genomes characterized in this study contain homopolymeric G:C tracts (table 2 and supplementary table S2, Supplementary Material online). High-depth Illumina HiSeq and MiSeq sequencing was used to determine the hypervariability of these G:C tracts; 98% of these G:C tracts were variable, under the parameters used in this study. Most strains contain between 10 and 26 variable G:C tracts; however, three strains, the two C. subantarcticus strains and Campylobacter spp. RM16704, contain 38–47 variable G:C tracts. The higher number of variable G:C tracts in these strains may also correlate with their high number of predicted pseudogenes. Nearly all of the G:C tracts identified in the C. lari group have lengths between 8 and 12 bp, consistent with the G:C tracts identified in C. jejuni. Also, as in other Campylobacter spp., many of the G:C tracts are contained within known surface structure-related genes (i.e., lipooligosaccharide [LOS], capsule, and flagellar modification); however, many are also located within genes encoding hemagglutination domain (Hag) proteins or hemagglutination-associated genes (see below).
Conservation of Gene Content and Gene Order within the C. lari Group
Comparative analysis of the 12 genomes characterized here indicates that the C. lari group is a highly related clade within Campylobacter (fig. 1). Of the 1,495 protein-encoding genes identified in C. lari subsp. lari, orthologs of 1,145 (77%) genes were identified in each of the other 11 genomes; these 1,145 genes are termed here the C. lari group core genome. In total, 390 of these have no defined function or only a general function (e.g., metallophosphatase). The majority of the noncore genes can be placed in categories typically associated with variability in Campylobacter, that is prophage, genomic islands, and integrated elements; signal transduction (two-component systems and methyl-accepting chemotaxis proteins); surface structure (LOS, capsule, flagella, and flagellar modification regions); restriction/modification; respiratory enzymes; and transporters/efflux proteins.
Fig. 1.—

BRIG plot of the Campylobacter lari group. The BRIG image was created using BLASTN and a minimum default threshold of 50%. The reference strain was C. lari subsp. lari strain RM2100. White areas correspond to sequences with similarity values below the minimum threshold.
In addition to gene content, gene order is also remarkably conserved within the C. lari group. Pairwise BLASTP comparisons between the core proteins of C. lari subsp. lari and their orthologs in the other 11 strains were performed. The chromosomal location of each core protein, relative to the replication origin, in C. lari subsp. lari strain RM2100 was plotted against the chromosomal location of its ortholog (fig. 2). An unbroken diagonal line would represent perfect synteny between any two genomes. As shown in figure 2, the C. lari group genomes are strongly syntenic; for many plots, the only substantial discontinuity occurs around cla_0825, which is the approximate site of the flgL-adjacent genomic island in C. lari subsp. lari strain RM2100. However, four genomes demonstrate minor chromosomal rearrangements relative to C. lari subsp. lari. The C. insulaenigrae, C. peloridis, and C. volucris genomes contain the same rearrangement, where two chromosomal segments (cla_0466-cla_0516 and cla_0522-cla_0606) have exchanged positions relative to the second ribosomal RNA locus (cla_0517-cla_0521). The major rearrangement in Campylobacter spp. strain RM16704 is an inversion of the chromosomal segment bounded by hemN1 and flgL. This strain also contains a minor translocation and inversion of the segment bounded by ychF and cjaB; this translocation is likely due to recombination between Hag genes. Together, these results are consistent with the phylogenetic relationships observed within the C. lari group, where C. peloridis, C. volucris, C. insulaenigrae, and Campylobacter spp. strain RM16704 are the most distantly related to C. lari subsp. lari.
Fig. 2.—

Colinearity of the Campylobacter lari group genomes. Each core protein in the C. lari subsp. lari strain RM2100 genome (x axis) was compared with the core proteins of other C. lari group members and with those of C. jejuni strain RM11168 (y axis) by BLASTP analysis. Each protein represents a match above 50% similarity. The x and y axis values represent gene numbers.
Genes Absent within the C. lari Group
Multiple genes encoding enzymes involved in amino acid/cofactor biosynthesis and energy metabolism/respiration were predicted to be absent in C. lari subsp. lari strain RM2100 (Miller et al. 2008). These included genes involved in the biosynthesis of acetyl-coenzyme A (acs); arginine (argBCDFO), glutamate (gltBD); leucine (leuABCD); methionine (metABEF); pantothenate (panBCD); proline (proAB); and tryptophan (trpACDEF) (Miller et al. 2008). Additionally, the respiratory enzyme-encoding genes gltA (citrate synthase), acn (aconitase), icd (isocitrate dehydrogenase), sucCD (succinyl-CoA synthetase), sdhABC (succinate dehydrogenase), and cioAB (terminal oxidase) were not identified in strain RM2100 (Miller et al. 2008). Other genes, such as the quorum sensing-associated gene luxS and those encoding the CeuBCDE enterochelin and ChuABCD hemin ABC transporters, were also absent. With a few exceptions, these genes are absent also within the other 11 genomes analyzed in this study, making their absence a general characteristic of the C. lari group, and indicating that multiple auxotrophy is a general feature of this clade. The pantothenate biosynthetic genes were identified in both C. subantarcticus strains and argF was identified in Campylobacter spp. strain RM16704, C. lari subsp. concheus and UPTC strain NCTC 11845. As carA and carB are core to the C. lari group, those strains that encode ArgF would be able to synthesize arginine in the presence of ornithine; the remainder of the C. lari group could presumably synthesize arginine in the presence of citrulline, given the presence of argG and argH in all C. lari group strains.
Genomic Variation within the C. lari Group
Although, as described above, many genes are absent generally within the lari clade, the presence/absence of other genes is restricted to one or a few strains. cysE and cysK were not identified in C. insulaenigrae, and the thiH, thiG, thiS and thiN genes were not identified in Campylobacter spp. strain RM16704, suggesting cysteine and thiamine auxotrophy, respectively. The NrdDG anaerobic nucleoside-triphosphate reductase activating system is not encoded by C. insulaenigrae, nor is the Mdh NAD-dependent malate dehydrogenase. The PhosSR-PstSCAB phosphate two-component system/ABC transporter was not encoded by C. peloridis or UPTC strain RM16701 (fig. 1). The napDLBGHA nitrate reductase gene cluster was not identified in strain Campylobacter spp. RM16704; additionally, the sorAB molybdenum-containing sulfite:cytochrome c oxidoreductase (Myers and Kelly 2005) was only in the C. insulaenigrae, C. lari subsp. lari, C. peloridis, C. volucris and UPTC NCTC 11845 genomes, and the lactate dehydrogenase/lactate permease gene cluster (Thomas et al. 2011) was identified only within the C. lari subsp. lari, C. insulaenigrae, C. volucris, and C. subantarcticus genomes (fig. 1). Finally, C. insulaenigrae appears to contain a gene cluster similar to the C. jejuni l-fucose utilization genomic island (cj0480c–cj0490) (Stahl et al. 2011).
Phylogenetic Placement of the UPTC Strains and Campylobacter spp. Strain RM16704
The genomes of four UPTC strains were examined to see whether the data could more accurately place the UPTC strains within the taxonomic structure of the C. lari group. The strains that we sequenced were two from culture collections and two isolated in 2013 from a watershed survey of the Salinas Valley of California. The genomes of these four strains, while not identical, were quite similar and several features observed here, for example, absence of nalidixic acid resistance, arsenate resistance (Nakajima et al. 2013), truncated flagellar subunits (Sekizuka et al. 2004, 2007), and the organization of the urease operon (Kakinuma et al. 2007), were consistent with those characterized previously in other UPTC strains. Average AAI analysis of the C. lari group (table 3) places the UPTC strains in the same cluster as the two C. lari subspecies; however, finer taxonomic distinctions could not be achieved. ANI analysis similarly indicated that the UPTC strains were most closely related to C. lari subsp. lari and C. lari subsp. concheus (supplementary table S3, Supplementary Material online). Here, however, ANI analysis also clearly distinguished the UPTC strains from both C. lari subsp. lari and C. lari subsp. concheus. Although UPTC strains have been shown to form two distinct clusters following AFLP analysis (Duim et al. 2004; Debruyne et al. 2009), in this study the UPTC genomes examined formed a single cluster in both AAI and ANI analyses. It is unknown whether the genomes of four strains within the same cluster were sequenced here or whether the two AFLP clusters are indistinguishable following genomic analysis. To provide a more definitive taxonomic placement of the UPTC strains, the genomes of additional strains will need to be sequenced. Genome sequencing of 24 additional UPTC strains, representing both AFLP clusters, is currently in progress.
Table 3.
Average Amino Acid Identities of the Campylobacter lari Group Core Proteome
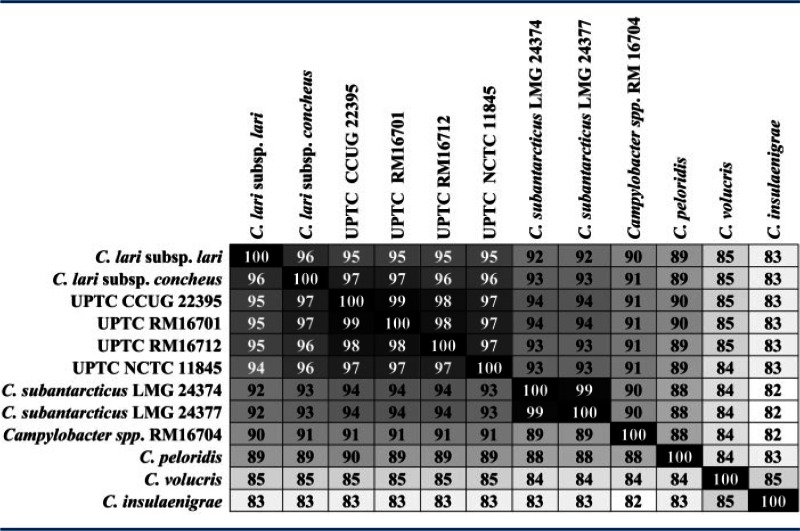 |
Note.—To more easily visualize amino acid similarities of the proteomes, we utilized a gradient heat map with black = 100%.
Campylobacter spp. strain RM16704 can be more readily placed within the taxonomy of the C. lari group. Campylobacter spp. strain RM16704 was isolated during the 2013 watershed survey described above. AAI and ANI analyses (table 3 and supplementary table S3, Supplementary Material online) clearly place this strain as a species distinct from the other validly named taxa within this clade. These results were verified through AtpA and 16S rRNA phylogenetic analyses (data not shown). Campylobacter spp. strain RM16704 does not encode the Nap nitrate reductase, and the absence of nitrate reductase activity in this strain was confirmed through biochemical tests (data not shown). The nap− phenotype is a useful defining characteristic, as the inability to reduce nitrate is unique to the C. lari group and nearly unique among Campylobacter. Cjd is the only other campylobacter unable to reduce nitrate (Steele and Owen 1988) and Campylobacter spp. strain RM16704 could be easily distinguished from Cjd through the use of a hippuricase test. Although it is likely that Campylobacter spp. strain RM16704 represents a novel taxon within Campylobacter, characterization of a single strain does not meet the current minimum standards for the definition of a new species; therefore, additional strains will need to be isolated and characterized before this taxon can be officially described.
The Flagellar and Flagellar Modification Loci
The flagella of the C. lari group clearly divide this clade into two distinct subgroups. Sekizuka et al. (2007) first identified flagellin genes in some UPTC isolates that were truncated relative to those of C. lari subsp. lari strain RM2100; furthermore, in these isolates flaA and flaB were divergently transcribed. With the exception of UPTC strain NCTC 11845, whose truncated flagellar genes are transcribed in the same direction, the UPTC isolates characterized in this study all contain truncated, divergently transcribed flagellar genes (table 4), as do C. peloridis and Campylobacter spp. strain RM16704. The flagellar genes of C. subantarcticus, C. volucris, C. insulaenigrae, and C. lari subsp. concheus are similar in size and orientation to those of C. lari subsp. lari strain RM2100 (table 4).
Table 4.
Characteristics of the Campylobacter lari Group Flagellar and Flagellar Modification Genes
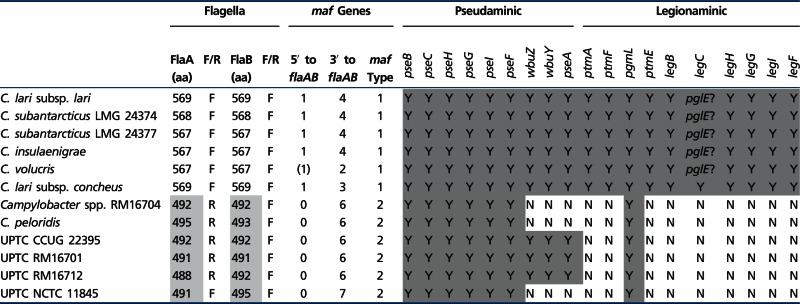 |
Note.—aa, amino acids; (1) indicates a partial maf gene; truncated flagellar subunits are shaded light grey; Y (shaded grey), putative flagellar modification genes encoded by the C. lari group genomes. In five genomes, the missing LegC function may be substituted by PglE, thus these are also shaded grey.
Division of the C. lari group into two subgroups is also maintained following analysis of the flagellar glycosylation loci. Campylobacter flagella are often modified through the addition of the nine carbon sugars pseudaminic acid (PseAm) and/or legionaminic acid (LegAm) and their derivatives (Thibault et al. 2001; Logan et al. 2008; Schoenhofen et al. 2009; Morrison and Imperiali 2014). The C. lari group in general is predicted to contain the complete PseAm biosynthetic pathway (pseBCHGIF; table 4). However, three strains (Campylobacter spp. strain RM16704, C. peloridis, and UPTC strain NCTC 11845) do not contain pseA (table 4), indicating that these strains cannot synthesize the acetamidino derivative of PseAm (Thibault et al. 2001). Moreover, an ortholog of cj1295, necessary for the modification of PseAm with di-O-methyl glyceric acid (Hitchen et al. 2010), is found throughout the C. lari group, though in some strains this ortholog is a contingency gene, in others not, and in C. volucris the gene is present as a contingency gene in two copies. Therefore, it is likely that the C. lari group flagella are glycosylated by a wide variety of PseAm derivatives. Truncation of the UPTC flagellar subunits was predicted to remove most of the flagellar glycosylation sites (Sekizuka et al. 2007). Nevertheless, the UPTC subgroup is predicted to synthesize PseAm; thus, either glycosylation occurs at different sites in the C. lari group or the UPTC subgroup flagella maintain a lower glycosylation density. Although PseAm and its derivatives are synthesized by the C. lari group, only the C. lari subsp. lari flagellar subgroup is predicted to encode the LegAm biosynthetic pathway (table 4). In C. jejuni and Campylobacter coli, genes encoding both biosynthetic pathways are located immediately adjacent to flaAB. However, in the C. lari group, the PseAm genes are unlinked to flaAB, and the LegAm genes in the C. lari subsp. lari subgroup are located either within the LOS (C. lari subsp. lari, C. subantarcticus, C. insulaenigrae, C. volucris) or capsular (CPS) (C. lari subsp. concheus) loci. The placement of the LegAm genes within these loci is interesting, suggesting perhaps that the flagellar subunits in these strains are not modified with LegAm, but rather the LOS or CPS structures. The C. lari subsp. concheus LegAm biosynthetic genes have much lower similarity to other Campylobacter LegAm genes; therefore, it is unknown whether LegAm or another nine-carbon sugar is synthesized in this strain. Also, no legC ortholog was identified in C. lari subsp. lari, C. subantarcticus, C. insulaenigrae, and C. volucris; however, it is possible that the LegC function may be substituted by PglE. Finally, maf (motility accessory factor) genes in Campylobacter have been associated with glycosylation of the flagellar subunits (Guerry et al. 2006; van Alphen et al. 2008). Consistent with this role, the maf genes also form two distinct clades within the C. lari group (table 4). Additionally, in the UPTC subgroup, the maf genes are present only downstream of flaAB in 6–7 copies, whereas in the C. lari subsp. lari subgroup, one maf gene is located upstream of flaAB and 2–4 copies are located downstream.
LOS and CPS Biosynthesis Loci
The LOS and CPS genomic loci encode enzymes involved in the biosynthesis of surface polysaccharide structures that function in the interactions of the bacteria with the environment. The LOS and CPS biosynthesis loci of the C. lari group strains are organized as previously observed in C. jejuni strains (Karlyshev et al. 2005; Parker et al. 2008). At either end of the LOS loci are the conserved heptosyltransferase genes, waaC and waaF, with additional LOS biosynthesis genes in between. The CPS loci possess the conserved CPS transporter genes (kpsMTEDF and kpsCS) flanking polysaccharide biosynthesis genes. Although the structure and content of these molecules is beyond the scope of this manuscript, these genomic loci of the C. lari group suggest diverse structures, based on variable oligo/polysaccharide biosynthesis genes. Indeed, all four of the UPTC strains and both C. subantarcticus strains possess their own unique set of LOS and CPS biosynthesis genes, suggesting interstrain variability. Interestingly, C. insulaenigrae and C. subantarcticus strain LMG 24374 possess genes (cstIIneuBCA) responsible for the production of sialylated LOS that are ganglioside mimics, and thus, certain C. lari group strains could lead to the development of Guillain–Barré and Fisher syndromes following infection.
Hag Proteins in the C. lari Group
A unique feature of the lari clade is the presence of multiple genes encoding Hag proteins that range in size from 96 to 166 kDa. These genes have been identified in other Campylobacter spp. (Fouts et al. 2005; Asakura et al. 2012); however, in many strains the Hag genes are highly fragmented and likely nonfunctional. All members of the C. lari group, with the exception of C. insulaenigrae and C. volucris, contain at least one presumably functional Hag gene. Notably, Campylobacter spp. strain RM16704 and C. peloridis strain LMG 23910 contain 10 and 15 Hag genes, respectively; analysis of an additional C. peloridis strain identified a similar number of Hag genes (data not shown), suggesting that the high number of Hag genes might be typical of this species. The Hag genes of the lari clade fall into two groups: “Stand-alone” Hag genes and those linked to genes encoding a hypothetical protein and a putative hemolysin activation/secretion protein, the latter group termed here a “Hag triad.” It is also noteworthy that most of the stand-alone Hag genes and Hag triads are associated with hypervariable G:C tracts: 15 of the 18 stand-alone Hag genes identified here contain G:C tracts and in all but one of the 27 Hag triads, the hypothetical protein-encoding gene contains a G:C tract at its 5′-end. Phylogenetic analysis identified a large amount of diversity within the C. lari group Hag proteins (supplementary fig. S2, Supplementary Material online). In comparison, the hypothetical protein and hemolysin activation/secretion protein components of the Hag triads were nearly identical within any given strain (supplementary fig. S3, Supplementary Material online, and data not shown), suggesting that intrastrain Hag diversity is not due to acquisition by lateral transfer with subsequent homologous recombination at the flanking loci. The role of the Hag proteins, or for that matter the role of either of the Hag-associated proteins within the Hag triad, in C. lari group biology is unknown. Nevertheless, the Hag genetic diversity and the association of these genes, alone or within the context of the Hag triad, with hypervariable G:C tracts indicate that further study of these genes is warranted.
Methylome Diversity in the C. lari Group
The methylomes of the five strains sequenced on the PacBio RS sequencer (C. peloridis, UPTC strain NCTC 11845, Campylobacter spp. strain RM16704, and both C. subantarcticus strains) reveal a diverse range of N6-methyladenine (m6A) methyltransferase (MTase) activities within the C. lari group. As shown in supplementary table S4, Supplementary Material online, of the 24 different detected sequence motifs targeted by m6A MTases, only one (5′-RAm6ATTY-3′, underlined base indicates methylation on the opposite DNA strand) is shared across all five strains and another (5′-Gm6ATC-3′) is shared across four strains but not detected in the Campylobacter spp. strain RM16704 genome. Campylobacter lari strain RM2100 is predicted to have several restriction-modification (R-M) systems but only one has a putative Type II N4-methylcytosine or m6A MTase with a predicted recognition sequence of 5′-GAATTC-3′ (http://tools.neb.com/∼vincze/genomes/). The absence of detection of N4-methylcytosine, which is typically easily detected with SMRT sequencing, in this study suggests that this putative MTase more likely targets adenine and may have the slightly less restrictive recognition motif of 5′-RAm6ATTY-3′. Methylation of the 5′-RAm6ATTY-3′ motif is likely related to the presence of cj0208 orthologs within the five strains characterized here. The Cj0208 DNA methyltransferase is a core protein within the C. lari group and is encoded by several other Campylobacter species (Fouts et al. 2005). Noteworthy in the C. insulaenigrae and C. volucris genomes is the presence of the cognate EcoRI family endonuclease gene adjacent to the cj0208 orthologs. Methylation of the 5′-Gm6ATC-3′ motif is consistent with the presence of a DpnII family R-M system in the C. peloridis genome and in both C. subantarcticus genomes; the UPTC strain NCTC 11845 genome contains the DpnII family methyltransferase but not the cognate endonuclease. Although a thorough investigation into all of the R-M systems observed and their potential biological roles is beyond the scope of this study, this work provides the first comprehensive list of different sequence motifs targeted by MTases in members of the C. lari group.
Conclusions
The C. lari group is a highly related phylogenetic clade within the genus Campylobacter, whose members often share similar hosts (e.g., shore birds, marine mammals, and shellfish) and environments (i.e., coastal regions and watersheds). Consistent with this association, genomic analyses of C. lari group members indicate that these strains are very similar in terms of gene content and organization. Seventy-seven percent of the genes identified in the previously sequenced C. lari subsp. lari strain RM2100 genome are conserved throughout the C. lari group. Furthermore, the genomic topography of this group is also quite conserved, with minor rearrangements identified within the C. insulaenigrae, C. peloridis, C. volucris, and Campylobacter spp. strain RM16704 genomes. Additionally, many features, such as profound defects in the amino acid biosynthetic and respiratory machinery, first identified in strain C. lari subsp. lari RM2100, are also conserved within the C. lari group as a whole. Many of the taxa characterized in this study are represented by a single strain, and it should be noted that variation identified in a single strain does not imply that the same variation exists in the species as a whole. Nevertheless, genes conserved or absent across the entire C. lari group should be taken seriously as candidates for future research into the host- or environmental-association of C. lari group strains.
The goal of this study was to identify genes related to adaptation to the coastal or watershed environment, and to provide additional data to help determine whether UPTC strains represent a possible third subspecies within C. lari. Although putative halotolerance genes were identified in C. lari subsp. lari strain RM2100 in previous work, leading to the hypothesis that their presence might be critical to the colonization of shorebirds and shellfish, these genes were not consistently found in isolates characterized in this study. In fact, no obvious genes or pathways were identified within the C. lari group that could be implicated in the host- or environmental-association of members of the clade. The present data suggest the possibility that it is the absence of amino acid biosynthetic pathways in some strains that may be more important in determining host/environment range, rather than the presence of pathways for increased halotolerance. For example, defects in biosynthetic pathways might restrict colonization of these organisms to hosts that can supply the missing metabolites. With respect to the UPTC strains, the present data are consistent with the hypothesis that they represent a third subspecies within C. lari, but are not sufficient to prove the hypothesis. The 11 high-quality closed and completed genomes reported here form a critical basis for expanded studies to further illuminate the complex basis of Campylobacter speciation and ecology, especially for the C. lari group.
Supplementary Material
Supplementary figures S1–S3 and tables S1–S4 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
This work was funded by the United States Department of Agriculture, Agricultural Research Service, CRIS project 5325-42000-230-047. The authors thank Birgitta Duim for critical reading of this manuscript.
Literature Cited
- Aarestrup FM, Nielsen EM, Madsen M, Engberg J. Antimicrobial susceptibility patterns of thermophilic Campylobacter spp. from humans, pigs, cattle, and broilers in Denmark. Antimicrob Agents Chemother. 1997;41:2244–2250. doi: 10.1128/aac.41.10.2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. 2011;12:402. doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asakura H, et al. Molecular evidence for the thriving of Campylobacter jejuni ST-4526 in Japan. PLoS One. 2012;7:e48394. doi: 10.1371/journal.pone.0048394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin J, Leaper S, Owen RJ, Skirrow MB. Description of Campylobacter laridis, a new species comprising the Nalidixic Acid Resistant Thermophilic Campylobacter (NARTC) group. Curr Microbiol. 1983;8:221–238. [Google Scholar]
- Besemer J, Borodovsky M. GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 2005;33:W451–W454. doi: 10.1093/nar/gki487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broczyk A, Thompson S, Smith D, Lior H. Water-borne outbreak of Campylobacter laridis-associated gastroenteritis. Lancet. 1987;1:164–165. doi: 10.1016/s0140-6736(87)92003-4. [DOI] [PubMed] [Google Scholar]
- Chin CS, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods. 2013;10:563–569. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- Chua K, et al. Campylobacter insulaenigrae causing septicaemia and enteritis. J Med Microbiol. 2007;56:1565–1567. doi: 10.1099/jmm.0.47366-0. [DOI] [PubMed] [Google Scholar]
- Clark TA, et al. Characterization of DNA methyltransferase specificities using single-molecule, real-time DNA sequencing. Nucleic Acids Res. 2012;40:e29. doi: 10.1093/nar/gkr1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debruyne L, et al. Campylobacter subantarcticus sp. nov., isolated from birds in the sub-Antarctic region. Int J Syst Evol Microbiol. 2010a;60:815–819. doi: 10.1099/ijs.0.011056-0. [DOI] [PubMed] [Google Scholar]
- Debruyne L, et al. Campylobacter volucris sp. nov., isolated from black-headed gulls (Larus ridibundus) Int J Syst Evol Microbiol. 2010b;60:1870–1875. doi: 10.1099/ijs.0.013748-0. [DOI] [PubMed] [Google Scholar]
- Debruyne L, On SL, De Brandt E, Vandamme P. Novel Campylobacter lari-like bacteria from humans and molluscs: description of Campylobacter peloridis sp. nov., Campylobacter lari subsp. concheus subsp. nov. and Campylobacter lari subsp. lari subsp. nov. Int J Syst Evol Microbiol. 2009;59:1126–1132. doi: 10.1099/ijs.0.000851-0. [DOI] [PubMed] [Google Scholar]
- Duim B, et al. Identification of distinct Campylobacter lari genogroups by amplified fragment length polymorphism and protein electrophoretic profiles. Appl Environ Microbiol. 2004;70:18–24. doi: 10.1128/AEM.70.1.18-24.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endtz HP, et al. Genotypic diversity of Campylobacter lari isolated from mussels and oysters in The Netherlands. Int J Food Microbiol. 1997;34:79–88. doi: 10.1016/s0168-1605(96)01174-9. [DOI] [PubMed] [Google Scholar]
- Flusberg BA, et al. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods. 2010;7:461–465. doi: 10.1038/nmeth.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster G, et al. Campylobacter insulaenigrae sp. nov., isolated from marine mammals. Int J Syst Evol Microbiol. 2004;54:2369–2373. doi: 10.1099/ijs.0.63147-0. [DOI] [PubMed] [Google Scholar]
- Fouts DE, et al. Major structural differences and novel potential virulence mechanisms from the genomes of multiple campylobacter species. PLoS Biol. 2005;3:e15. doi: 10.1371/journal.pbio.0030015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Pena FJ, et al. Isolation and characterization of Campylobacter spp. from Antarctic fur seals (Arctocephalus gazella) at Deception Island, Antarctica. Appl Environ Microbiol. 2010;76:6013–6016. doi: 10.1128/AEM.00316-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez M, Paz Villanueva M, Debruyne L, Vandamme P, Fernandez H. Campylobacter insulaenigrae: first isolation report from South American sea lion (Otaria flavescens, (Shaw, 1800)) Braz J Microbiol. 2011;42:261–265. doi: 10.1590/S1517-83822011000100033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerry P, et al. Changes in flagellin glycosylation affect Campylobacter autoagglutination and virulence. Mol Microbiol. 2006;60:299–311. doi: 10.1111/j.1365-2958.2006.05100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey RB, et al. Prevalence of Campylobacter spp isolated from the intestinal tract of pigs raised in an integrated swine production system. J Am Vet Med Assoc. 1999;215:1601–1604. [PubMed] [Google Scholar]
- Hitchen P, et al. Modification of the Campylobacter jejuni flagellin glycan by the product of the Cj1295 homopolymeric-tract-containing gene. Microbiology. 2010;156:1953–1962. doi: 10.1099/mic.0.038091-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakinuma Y, et al. Cloning, sequencing and characterization of a urease gene operon from urease-positive thermophilic Campylobacter (UPTC) J Appl Microbiol. 2007;103:252–260. doi: 10.1111/j.1365-2672.2006.03212.x. [DOI] [PubMed] [Google Scholar]
- Kaneko A, Matsuda M, Miyajima M, Moore JE, Murphy PG. Urease-positive thermophilic strains of Campylobacter isolated from seagulls (Larus spp.) Lett Appl Microbiol. 1999;29:7–9. doi: 10.1046/j.1365-2672.1999.00565.x. [DOI] [PubMed] [Google Scholar]
- Karlyshev AV, et al. Analysis of Campylobacter jejuni capsular loci reveals multiple mechanisms for the generation of structural diversity and the ability to form complex heptoses. Mol Microbiol. 2005;55:90–103. doi: 10.1111/j.1365-2958.2004.04374.x. [DOI] [PubMed] [Google Scholar]
- Khan IU, et al. A national investigation of the prevalence and diversity of thermophilic Campylobacter species in agricultural watersheds in Canada. Water Res. 2014;61:243–252. doi: 10.1016/j.watres.2014.05.027. [DOI] [PubMed] [Google Scholar]
- Koren S, et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat Biotechnol. 2012;30:693–700. doi: 10.1038/nbt.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, et al. Reducing assembly complexity of microbial genomes with single-molecule sequencing. Genome Biol. 2013;14:R101. doi: 10.1186/gb-2013-14-9-r101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagesen K, et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leotta G, Vigo G, Giacoboni G. Isolation of Campylobacter lari from seabirds in Hope Bay, Antarctica. Pol Polar Res. 2006;27:303–308. [Google Scholar]
- Lin CW, Yin PL, Cheng KS. Incidence and clinical manifestations of Campylobacter enteritis in central Taiwan. Zhonghua Yi Xue Za Zhi (Taipei). 1998;61:339–345. [PubMed] [Google Scholar]
- Logan SM, Schoenhofen IC, Guerry P. O-linked flagellar glycosylation in Campylobacter. In: Nachamkin I, Szymanski CM, Blaser MJ, editors. Campylobacter. Washington (DC): ASM Press; 2008. pp. 471–481. [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinot M, et al. Campylobacter lari bacteremia. Clin Microbiol Infect. 2001;7:96–97. doi: 10.1046/j.1469-0691.2001.00212.x. [DOI] [PubMed] [Google Scholar]
- Matsuda M, et al. Characterization of urease-positive thermophilic Campylobacter subspecies by multilocus enzyme electrophoresis typing. Appl Environ Microbiol. 2003;69:3308–3310. doi: 10.1128/AEM.69.6.3308-3310.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megraud F, Chevrier D, Desplaces N, Sedallian A, Guesdon JL. Urease-positive thermophilic Campylobacter (Campylobacter laridis variant) isolated from an appendix and from human feces. J Clin Microbiol. 1988;26:1050–1051. doi: 10.1128/jcm.26.5.1050-1051.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinersmann RJ, Berrang ME, Little E. Campylobacter spp. recovered from the Upper Oconee River Watershed, Georgia in a 4-year study. Microb Ecol. 2013;65:22–27. doi: 10.1007/s00248-012-0117-8. [DOI] [PubMed] [Google Scholar]
- Merga JY, Winstanley C, Williams NJ, Yee E, Miller WG. Complete genome sequence of the Arcobacter butzleri cattle isolate 7h1h. Genome Announc. 2013;1:e00655–13. doi: 10.1128/genomeA.00655-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller WG, et al. Diversity within the Campylobacter jejuni type I restriction-modification loci. Microbiology. 2005;151:337–351. doi: 10.1099/mic.0.27327-0. [DOI] [PubMed] [Google Scholar]
- Miller WG, Wang G, Binnewies TT, Parker CT. The complete genome sequence and analysis of the human pathogen Campylobacter lari. Foodborne Pathog Dis. 2008;5:371–386. doi: 10.1089/fpd.2008.0101. [DOI] [PubMed] [Google Scholar]
- Morris CN, Scully B, Garvey GJ. Campylobacter lari associated with permanent pacemaker infection and bacteremia. Clin Infect Dis. 1998;27:220–221. doi: 10.1086/517683. [DOI] [PubMed] [Google Scholar]
- Morrison MJ, Imperiali B. The renaissance of bacillosamine and its derivatives: pathway characterization and implications in pathogenicity. Biochemistry. 2014;53:624–638. doi: 10.1021/bi401546r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray IA, et al. The methylomes of six bacteria. Nucleic Acids Res. 2012;40:11450–11462. doi: 10.1093/nar/gks891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers JD, Kelly DJ. A sulphite respiration system in the chemoheterotrophic human pathogen Campylobacter jejuni. Microbiology. 2005;151:233–242. doi: 10.1099/mic.0.27573-0. [DOI] [PubMed] [Google Scholar]
- Nachamkin I, et al. Campylobacter laridis causing bacteremia in an immunosuppressed patient. Ann Intern Med. 1984;101:55–57. doi: 10.7326/0003-4819-101-1-55. [DOI] [PubMed] [Google Scholar]
- Nakajima T, et al. Molecular identification of an arsenic four-gene operon in Campylobacter lari. Folia Microbiol (Praha). 2013;58:253–260. doi: 10.1007/s12223-012-0207-5. [DOI] [PubMed] [Google Scholar]
- Obiri-Danso K, Jones K. Distribution and seasonality of microbial indicators and thermophilic campylobacters in two freshwater bathing sites on the River Lune in northwest England. J Appl Microbiol. 1999;87:822–832. doi: 10.1046/j.1365-2672.1999.00924.x. [DOI] [PubMed] [Google Scholar]
- Obiri-Danso K, Paul N, Jones K. The effects of UVB and temperature on the survival of natural populations and pure cultures of Campylobacter jejuni, Camp. coli, Camp. lari and urease-positive thermophilic campylobacters (UPTC) in surface waters. J Appl Microbiol. 2001;90:256–267. doi: 10.1046/j.1365-2672.2001.01239.x. [DOI] [PubMed] [Google Scholar]
- Otasevic M, Lazarevic-Jovanovic B, Tasic-Dimov D, Dordevic N, Miljkovic-Selimovic B. The role of certain Campylobacter types in the etiology of enterocolitis. Vojnosanit Pregl. 2004;61:21–27. doi: 10.2298/vsp0401021o. [DOI] [PubMed] [Google Scholar]
- Owen RJ, Costas M, Sloss L, Bolton FJ. Numerical analysis of electrophoretic protein patterns of Campylobacter laridis and allied thermophilic campylobacters from the natural environment. J Appl Bacteriol. 1988;65:69–78. doi: 10.1111/j.1365-2672.1988.tb04319.x. [DOI] [PubMed] [Google Scholar]
- Parker CT, Gilbert M, Yuki N, Endtz HP, Mandrell RE. Characterization of lipooligosaccharide-biosynthetic loci of Campylobacter jejuni reveals new lipooligosaccharide classes: evidence of mosaic organizations. J Bacteriol. 2008;190:5681–5689. doi: 10.1128/JB.00254-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad KN, Dixit AK, Ayyagari A. Campylobacter species associated with diarrhoea in patients from a tertiary care centre of north India. Indian J Med Res. 2001;114:12–17. [PubMed] [Google Scholar]
- Punta M, et al. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter M, Rossello-Mora R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A. 2009;106:19126–19131. doi: 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford K, et al. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- Ryu H, et al. Intestinal microbiota and species diversity of Campylobacter and Helicobacter spp. in migrating shorebirds in Delaware Bay. Appl Environ Microbiol. 2014;80:1838–1847. doi: 10.1128/AEM.03793-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanlon KA, et al. Occurrence and characteristics of fastidious Campylobacteraceae species in porcine samples. Int J Food Microbiol. 2013;163:6–13. doi: 10.1016/j.ijfoodmicro.2013.02.004. [DOI] [PubMed] [Google Scholar]
- Schoenhofen IC, Vinogradov E, Whitfield DM, Brisson JR, Logan SM. The CMP-legionaminic acid pathway in Campylobacter: biosynthesis involving novel GDP-linked precursors. Glycobiology. 2009;19:715–725. doi: 10.1093/glycob/cwp039. [DOI] [PubMed] [Google Scholar]
- Sekizuka T, et al. Molecular cloning, nucleotide sequencing and characterization of the flagellin gene from isolates of urease-positive thermophilic Campylobacter. Res Microbiol. 2004;155:185–191. doi: 10.1016/j.resmic.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Sekizuka T, Murayama O, Moore JE, Millar BC, Matsuda M. Flagellin gene structure of flaA and flaB and adjacent gene loci in urease-positive thermophilic Campylobacter (UPTC) J Basic Microbiol. 2007;47:63–73. doi: 10.1002/jobm.200610194. [DOI] [PubMed] [Google Scholar]
- Skirrow MB, Benjamin J. “1001” Campylobacters: cultural characteristics of intestinal campylobacters from man and animals. J Hyg (Lond). 1980;85:427–442. doi: 10.1017/s0022172400063506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl M, et al. L-fucose utilization provides Campylobacter jejuni with a competitive advantage. Proc Natl Acad Sci U S A. 2011;108:7194–7199. doi: 10.1073/pnas.1014125108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele TW, Owen RJ. Campylobacter jejuni subsp. doylei subsp. nov., a subspecies of nitrate-negative campylobacters isolated from human clinical specimens. Int J Syst Bacteriol. 1988;38:316–318. [Google Scholar]
- Stoddard RA, et al. Campylobacter insulaenigrae isolates from northern elephant seals (Mirounga angustirostris) in California. Appl Environ Microbiol. 2007;73:1729–1735. doi: 10.1128/AEM.01816-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauxe RV, et al. Illness associated with Campylobacter laridis, a newly recognized Campylobacter species. J Clin Microbiol. 1985;21:222–225. doi: 10.1128/jcm.21.2.222-225.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault P, et al. Identification of the carbohydrate moieties and glycosylation motifs in Campylobacter jejuni flagellin. J Biol Chem. 2001;276:34862–34870. doi: 10.1074/jbc.M104529200. [DOI] [PubMed] [Google Scholar]
- Thomas MT, et al. Two respiratory enzyme systems in Campylobacter jejuni NCTC 11168 contribute to growth on L-lactate. Environ Microbiol. 2011;13:48–61. doi: 10.1111/j.1462-2920.2010.02307.x. [DOI] [PubMed] [Google Scholar]
- Tresierra-Ayala A, Bendayan ME, Bernuy A, Pereyra G, Fernandez H. Chicken as potential contamination source of Campylobacter lari in Iquitos, Peru. Rev Inst Med Trop Sao Paulo. 1994;36:497–499. doi: 10.1590/s0036-46651994000600004. [DOI] [PubMed] [Google Scholar]
- van Alphen LB, et al. A functional Campylobacter jejuni maf4 gene results in novel glycoforms on flagellin and altered autoagglutination behaviour. Microbiology. 2008;154:3385–3397. doi: 10.1099/mic.0.2008/019919-0. [DOI] [PubMed] [Google Scholar]
- Van Doorn LJ, et al. Rapid identification of diverse Campylobacter lari strains isolated from mussels and oysters using a reverse hybridization line probe assay. J Appl Microbiol. 1998;84:545–550. doi: 10.1046/j.1365-2672.1998.00378.x. [DOI] [PubMed] [Google Scholar]
- Vandamme P, Pot B, Kersters K. Differentiation of Campylobacter and Campylobacter-like organisms by numerical analysis of one-dimensional electrophoretic protein patterns. Syst Appl Microbiol. 1991;14:57–66. [Google Scholar]
- Waldenstrom J, et al. Prevalence of Campylobacter jejuni, Campylobacter lari, and Campylobacter coli in different ecological guilds and taxa of migrating birds. Appl Environ Microbiol. 2002;68:5911–5917. doi: 10.1128/AEM.68.12.5911-5917.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldenstrom J, On SL, Ottvall R, Hasselquist D, Olsen B. Species diversity of campylobacteria in a wild bird community in Sweden. J Appl Microbiol. 2007;102:424–432. doi: 10.1111/j.1365-2672.2006.03090.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}