

Abstract
An unprecedented NHC catalyzed annulation of enals to form 3,4-disubstituted cyclopentanones has been discovered. Aryl enals undergo dimerization in the presence of a single-electron oxidant to form C2 symmetric cyclopentanones. A cross-reaction has also been developed, allowing for the synthesis of differentially substituted cyclopentanones. Mechanistically, the reaction is thought to proceed through radical intermediates, further establishing the synthetic utility of this class of reactivity.

Over the past decade, N-heterocyclic carbene (NHC) catalyzed reactions of the homoenolate equivalent have been the focus of intense investigation.1 Glorius and Bode reported the first examples of NHC-generated homoenolate equivalents in 2004 independently and concurrently.2 These seminal reports showed the synthetic utility of the homoenolate in the context of synthesizing 2,3-disubstituted γ-lactones (eq 1). Soon after these reports Bode and He showed that γ-lactams are easily accessed via this pathway through the coupling of an enal and an imine (eq 2).3 In 2006 Nair and co-workers demonstrated the coupling of enals with chalcones to furnish 1,3,4-trisubstituted cyclopentene (eq 3).4 Further developments in this field have broadened the partners that participate in these annulations and investigated asymmetric version of the reaction.5 These annulation reactions are all presumed to proceed via 2-electron pathways.
 |
(1) |
 |
(2) |
 |
(3) |
In 2014, we described the NHC-catalyzed conversion of enals to aldols using nitroarenes as oxidants in methanol. The net transformation converts enals and nitroarenes to β-hydroxy esters. Evidence points to one electron oxidation of the homoenolate equivalent6 by an electron deficient nitroarene oxidant.7,8 During our investigations, we observed the formation of cyclopentanone products9 when the reaction is conducted in a non-nucleophilic solvent. For example, the reaction of 4-methoxycinnamaldehyde 1a with 4-nitropyridine N-oxide 2 in dichloromethane at room temperature in the presence of NHC 3a forms cyclopentanone 4a in 49 % yield as a single diastereomer. Intrigued by this previously unknown reactivity, we began to explore the reaction parameters.
An initial screen of chiral NHC catalysts at room temperature proved discouraging with only backbone-fluorinated NHC 3b providing any desired product in a meager 12 % ee.10 Switching the solvent to toluene afforded an increase to 60 % ee. Increasing in the reaction temperature to 70 °C improved the reactivity and allowed us to screen a variety of chiral catalysts. Ultimately, NHC 3c proved optimal, providing the desired product in 51 % yield, 84 % ee, as a single diastereomer. A high-throughput experimentation (HTE) screen revealed that trifluorotoluene was the best solvent and the beneficial effect of lithium chloride on yield.11
With optimized conditions identified, we explored the scope of the dimerization. Electron-rich and electron-deficient aryl enals are tolerated in the reaction. Electron-rich substrates proved to provide higher yields than their electron-poor counterparts. The heteroaromatic 2-furylcinnamaldehyde works in the reaction as well. Unfortunately, aliphatic enals do not participate. To our delight, 4-bromocinnamaldeyde provides product in reasonable yield, and contains a potential handle for cross coupling reactions. In all cases, the cyclopentanone products are formed as a single diastereomer. The remainder of the mass balance in these reactions is the α,β-unsaturated acid, the result of a 2-electron oxidation of the Breslow intermediate to form the acyl azolium, followed by attack by adventitious water.
Mechanistically, we believe that this transformation is related to the β–hydroxylation of enals that we previously reported.7 We postulate that the reaction proceeds by formation of Breslow intermediate I,12 which transfers a single electron to 4-nitropyridine N-oxide 2 to generate a radical cation II and radical anion III. Radical cation II is then attacked at the β-carbon by a second, native, Breslow intermediate I to form IV.13 IV then undergoes a second single electron transfer to radical anion III to form acyl azolium V. The acyl azolium is then attacked intramolecularly by the pendant enolate azolium to liberate one equivalent of NHC and form cyclopentanone VI. This cyclopentanone is attacked by water to liberate the second equivalent of NHC and form β-ketoacid VII. This β-ketoacid then undergoes decarboxylation to furnish cyclopentanone product 4.14 Once radical anion III abstracts the second electron from IV, intermediate VIII is formed, which decomposes to nitroso IX (observed by LCMS). This reactivity represents a rare example of carbon turnover of the acyl azolium.15 Typically, exogenous alcohol, a tethered alcohol, or a tethered amine attacks the acyl azolium. Even in the case of the cyclopentene forming reactions1 the acyl azolium is turned over by a tethered alcohol to form a β-lactone, which then extrudes CO2.
We also entertained the idea that the reaction proceeds by one Breslow intermediate undergoing a 2-electron oxidation to form an acyl azolium which is in turn attacked via a [1,4] addition by a native Breslow intermediate. To test this hypothesis, we subjected 3-phenylpropiolaldehyde 5, a precursor for the acyl azolium, and 4-methoxycinnamaldehyde 1a to reaction with carbene 3c but observed no cyclopentanone products.16 Further, employing the known 2-electron oxidant phenazine 7 in the reaction also does not lead to cyclopentanone products.17
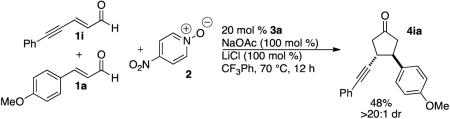
In an effort to broaden the synthetic utility of the transformation, we investigated the use of a hetero-coupling partner that would deliver unsymmetrical adducts. All attempts at using other electron-deficient alkenes (such as nitroalkenes, enoates, enones) met with failure; the reaction appears to require an enal on both sides. As a partial solution, we found that biasing the reaction with an excess of one of the enals delivers the cross product in good yields. Best results are achieved when the more electron rich (more reactive) enal is used in excess. In these reactions, the dimer of the more reactive enal is always formed, but the dimer of the limiting enal is observed in only trace amounts.
To demonstrate the utility of the cylopentanone products, a Baeyer-Villager oxidation and a Beckman rearrangement were carried out. In both cases, retention of stereochemistry was observed and lactone 8 and lactam 9 products were formed in good yields.
In conclusion, we have developed a methodology for the enantioselective synthesis of 3,4-disubstituted cyclopentanones. The protocol allows for the dimerization of aryl enals18 to form C-2 symmetric cyclopentanones. Further, a cross reaction has been developed to synthesize non-symmetric 3,4-disubstituted cyclopentanones. The proposed reaction mechanism invokes radical intermediates. Furthering the substrate scope and investigating new modes of reactivity enabled by these intermediates is the subject of ongoing investigations.
Supplementary Material
Figure 1.

Background
Scheme 2.

Control Experiments
Scheme 3.

Derivatization
Table 1.
Reaction Optimization

| Entry | NHC | Additive | Temp | Solvent | Yield (%)b | ee (%)c |
|---|---|---|---|---|---|---|
| 1 | 3a | - | 23 °C | CH2Cl2 | 49 | - |
| 2 | 3b | - | 23 °C | CH2Cl2 | 25 | 12 |
| 3 | 3b | - | 23 °C | PhMe | 28 | 60 |
| 4 | 3c | - | 23 °C | CH2Cl2 | <5 | - |
| 5 | 3d | - | 23 °C | CH2Cl2 | <5 | - |
| 6 | 3e | - | 23 °C | CH2Cl2 | <5 | - |
| 7 | 3b | - | 70 °C | PhMe | 46 | 50 |
| 8 | 3c | - | 70 °C | PhMe | 51 | 84 |
| 9 | 3d | - | 70 °C | PhMe | 30 | 50 |
| 10 | 3e | - | 70 °C | PhMe | 44 | 76 |
| 11 | 3f | - | 70 °C | PhMe | 26 | −78 |
| 12 | 3c | - | 70 °C | PhCF3 | 64 | 84 |
|
| ||||||
| 13 | 3c | LiCl | 70 °C | PhCF3 | 79 | 84 |

Reactions were conducted with 1.0 equiv of 1 and 0.66 equiv of 2.
Isolated yields after chromatography.
Enantiomeric excess was determined by HPLC analysis on a chiral stationary phase.
Table 2.
Dimerization Scope

|
See footnotes a–c in Table 1.
Table 3.
Cross-Reaction

|
Reactions were carried out with 1.0 equivalent of 1, 4.0 equivalents of 1', and 4.0 equivalent of 2.
See footnotes b–c in Table 1.
ACKNOWLEDGMENT
We thank NIGMS for generous support (GM72586) and Daniel DiRocco (Merck Research Laboratories) for a generous gift of aminoindanol. We thank Daniel Reuss (CSU) for help with assaying CO2 levels.
Funding Sources NIGMS (GM72586).
Footnotes
Supporting Information. Experimental procedures and compound characterization. This material is available free of charge via the Internet at http://pubs.acs.org.”
The authors declare no competing financial interest.
REFERENCES
- 1.For a review see: Flanigan DM, Michailidis-Romanov F, White NA, Rovis T. Chem. Rev. 2015;115 doi: 10.1021/acs.chemrev.5b00060. doi: 10.1021/acs.cev.5bhemr0060.
- 2.(a) Burstein C, Glorius F. Angew. Chem. Int. Ed. 2004;43:6205. doi: 10.1002/anie.200461572. [DOI] [PubMed] [Google Scholar]; (b) Sohn SS, Rosen EL, Bode JW. J. Am. Chem. Soc. 2004;126:14370. doi: 10.1021/ja044714b. [DOI] [PubMed] [Google Scholar]
- 3.He M, Bode JW. Org. Lett. 2005;7:3131. doi: 10.1021/ol051234w. [DOI] [PubMed] [Google Scholar]
- 4.Nair V, Vellath S, Poonoth M, Suresh J. J. Am. Chem. Soc. 2006;128:8736. doi: 10.1021/ja0625677. [DOI] [PubMed] [Google Scholar]
- 5.(a) Li Y, Zhao Z-A, He H, You S-L. Adv. Synth. Catal. 2008;350:1885. [Google Scholar]; (b) Cohen DT, Scheidt KA. Chem. Sci. 2012;3:53. doi: 10.1039/C1SC00621E. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jang KP, Hutson GE, Johnston RC, McCusker EO, Cheong PH–Y, Scheidt KA. J. Am. Chem. Soc. 2014;136:76. doi: 10.1021/ja410932t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Rommel M, Fukuzumi T, Bode JW. J. Am. Chem. Soc. 2008;130:17266. doi: 10.1021/ja807937m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Chan A, Scheidt KA. J. Am. Chem. Soc. 2007;129:5334. doi: 10.1021/ja0709167. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Phillips EM, Reynolds TE, Scheidt KA. J. Am. Chem. Soc. 2008;130:2416. doi: 10.1021/ja710521m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Zhao X, DiRocco DA, Rovis T. J. Am. Chem. Soc. 2011;133:12466. doi: 10.1021/ja205714g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Chiang P-C, Kaeobamrung J, Bode JW. J. Am. Chem. Soc. 2007;129:3520. doi: 10.1021/ja0705543. [DOI] [PubMed] [Google Scholar]; (i) Cardinal-David B, Raup DEA, Scheidt KA. J. Am. Chem. Soc. 2010;132:5345. doi: 10.1021/ja910666n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.In the context of our characterization of the aza-Breslow intermediate, we conducted cyclic voltammetry studies that revealed oxidation potentials for this intermediate (DiRocco DA, Oberg KM, Rovis T. J. Am. Chem. Soc. 2012;134:6143. doi: 10.1021/ja302031v.). Two-electron oxidation of the Breslow intermediate has been extensively described including the use of two equivalents of one-electron oxidants such as TEMPO (Guin J, De Sarkar S, Grimme S, Studer A. Angew. Chem. Int. Ed. 2008;47:8727. doi: 10.1002/anie.200802735.).
- 7.White NA, Rovis T. J. Am. Chem. Soc. 2014;136:14674. doi: 10.1021/ja5080739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Y, Du Y, Huang Z, Xu J, Wu X, Wang Y, Wang M, Yang S, Webster RD, Chi YR. J. Am. Chem. Soc. 2015;137:2416. doi: 10.1021/ja511371a. [DOI] [PubMed] [Google Scholar]
- 9.(a) Singh M, Mandal AK. Heterocycles. 1982;18:291. 3,4-Diphenyl cyclopentanone has been synthesized in low yield by the cyanide catalyzed dimerization of cinnamaldehyde imine followed by acidic hydrolysis of the generated azepine product; see: [Google Scholar]; (b) Kise E, Itaka S, Iwasaki K, Ueda N. J. Org. Chem. 2002;67:8305. doi: 10.1021/jo026183k. Electro-chemical reduction of cinnamates has been found to generate 3,4-diarylcyclopentanone adducts after acid induced decarboxylation; see: [DOI] [PubMed] [Google Scholar]
- 10.DiRocco DA, Oberg KM, Dalton DM, Rovis T. J. Am. Chem. Soc. 2009;131:10872. doi: 10.1021/ja904375q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.See supporting information.
- 12.Breslow RS. J. Am. Chem. Soc. 1958;80:3719. [Google Scholar]
- 13.An alternate hypothesis raised by a reviewer involves dimerization of radical cation II to form a protonated form of V (Scheme 1). The use of a sub-stoichiometric amount of oxidant (0.66 equiv relative to enal) suggests we only need to oxidize one of the two enal-derived partners prior to dimerization. However, given that we have seen overreduction of the nitroarene in our hydroxylation chemistry (ref. 7), we cannot rigorously exclude this possibility.
- 14.Analysis of the headspace of a reaction conducted under N2 at-mosphere reveals 120,000 ppm CO2.
- 15.(a) Nair V, Babu BP, Vellalath S, Suresh E. Chem. Commun. 2008:747–749. doi: 10.1039/b715733a. [DOI] [PubMed] [Google Scholar]; (b) Verma P, Patni PA, Sunoj RB. J. Org. Chem. 2011;76:5606. doi: 10.1021/jo200560t. [DOI] [PubMed] [Google Scholar]; (c) Menon RS, Biju AT, Nair V. Chem. Soc. Rev. 2015;44:5040. doi: 10.1039/c5cs00162e. For a review encompassing this topic, see: [DOI] [PubMed] [Google Scholar]
- 16.Zeitler K. Org. Lett. 2006;8:637. doi: 10.1021/ol052826h. [DOI] [PubMed] [Google Scholar]
- 17.Zhao X, Ruhl KE, Rovis T. Angew. Chem. Int. Ed. 2012;51:12330. doi: 10.1002/anie.201206490. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
18.Under these conditions, aliphatic enals and dienals do not afford dimer product. An yne-enal participates in the cross-reaction with achiral catalyst, but not with chiral catalyst.

Scheme 1.

Proposed Catalytic Cycle
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.