

Abstract
α,β-Unsaturated oxime pivalates are proposed to undergo reversible C(sp2)-H insertion with cationic Rh(III) complexes to furnish five-membered metallacycles. In the presence of 1,1-disubstituted olefins, these species participate in irreversible migratory insertion to give, after reductive elimination, 2,3-dihydropyridine products in good yields. Catalytic hydrogenation was then used to convert these molecules into piperidines, which are important structural components of numerous pharmaceuticals.
Graphical Abstract
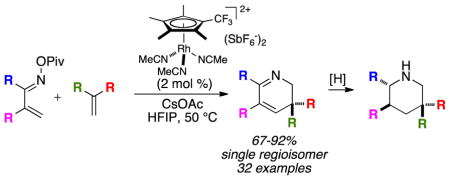
Nitrogen heterocycles are among the most abundant structural components of pharmaceuticals.1 Due to their omnipresence in biologically active molecules, the synthesis of nitrogen heterocycles has attracted considerable attention in the synthetic community. Pyridines represent one of the most prevalent scaffolds encountered in medicinal chemistry.1b,c Removal of one unsaturation from a pyridine ring affords dihydropyridines.2 These molecules come as three double-bond isomers: 1,4-dihydropyridine 1, 2,3-dihydropyridine 2, and 1,2-dihydropyridine 3 (Figure 1). The first synthesis of a 1,4-dihydropyridine is attributed to Arthur Hantzsch for work done over a century ago.3 The importance of this motif is highlighted by its presence in nicotinamide adenine dinucleotide (NADH), a universal biological reducing agent. Interest in 1,4-dihydropyridines has been further stimulated after their incorporation into Ca-channel blockers.4
Figure 1.

Structures of Dihydropyridines
In stark contrast to 1,4-dihydropyridines, which are stable structures, the 2,3- and 1,2-congeners are kinetically labile and thus not found in bioactive molecules as such. Nevertheless, they have been implicated as versatile intermediates in biosynthetic pathways leading to alkaloid natural products. Recent examples include the proposed biosynthesis of the marine natural product Symbioimine,5 as well as isoquinuclidine alkaloids Keramaphidin B and Manzamine A.6
While N-alkyl 2,3-dihydropyridinium salts are well-documented and even isolable,7 there is scarce information on the synthesis of N-unprotected 2,3-dihydropyridines.8 This constitutes a handicap to the synthetic community, given the potential number of transformations that such intermediates can engage in.
In recent years, Rh(III)-catalyzed C-H activation has become a method of choice for constructing nitrogen heterocycles.9,10 In particular, coupling of α,β-unsaturated imines and oximes with alkynes represents a useful approach to pyridines.11 We have recently demonstrated that high regioselectivities are obtainable when electronically-biased alkenes are used in place of alkynes as coupling partners (Scheme 1, eq 1).11a Furthermore, substituted acrylic acids were shown to give the complementary regioisomer of the pyridine products, where the carboxylate acts as a traceless directing group (eq 2).11b In a conceptually distinct approach, Bergman and Ellman have shown that a Rh(I) catalyst can couple unsaturated imines with internal alkynes followed by imminium formation and reduction to deliver complementary tetrahydropyridines depending on nature of the acid (eq 3).11c This latter reaction was proposed to proceed through dihydropyridine intermediates.
Scheme 1.

Rh(III)-Catalyzed Synthesis of 2,3-Dihydropyridines
In our initial work (eq 1), we never observed the presumed intermediate dihydropyridine adducts even in the absence of external oxidants. We speculated that the instability of the 2,3-dihydropyridine was responsible, and rapid in situ oxidation to the pyridine was faster than the initial annulation.12 We speculated that the use of 1,1-disubstituted alkenes would prevent oxidation and deliver 2,3-dihydropyridines as isolable outcomes of the reaction (eq 4). Herein we disclose our results.
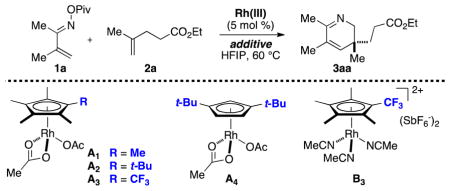
We commenced our investigation by subjecting α,β-unsaturated oxime pivalate 1a and alkene 2a to catalytic amounts of various CpRh(III) diacetates13 in HFIP at 60 °C (Table 1). As can be seen from the Table, replacing the Cp* ligand (A1) with its more sterically congested tert-butyl-substituted congener (A2) increases the yield of the 2,3-dihydropyridine product (3aa) from 81% to 91%. Product yield is further enhanced when electron-deficient Cp-ligand A3 bearing a trifluoromethyl10b substituent is used. Provided that CsOAc was employed as an additive, product 3aa was isolated in 99% yield when 5 mol % of A3 was used.
Table 1.
Optimization of the Reaction Conditionsa
| |||
|---|---|---|---|
| entry | Rh(III) | additive | yield (%)d |
| 1 | [Cp*RhCl2]2 | AgOAcb | <12c |
| 2 | A1 | none | 55 |
| 3 | A1 | p-TsOHb | 72 |
| 4 | A1 | CsOAc | 81 |
| 5 | A2 | CsOAc | 91 |
| 6 | A3 | CsOAc | 99 |
| 7 | A4 | CsOAc | 92 |
| 8 | B3 | CsOAc | 99 |
| 9e | B3 | CsOAc | 99 |
Reaction Conditions: 1a (1.0 equiv), 2a (1.2 equiv), additive (2.0 equiv), Rh(III) catalyst (5 mol %) in HFIP (0.3 M), at 60 °C for 16 h.
10 mol % of additive was used.
Determined by 1H NMR.
Yield of isolated product.
Catalyst loading was decreased to 2 mol %, reaction temperature was decreased to 50 °C.
The use of Rh(III) diacetates is problematic for some Cp-ligands due to their hygroscopicity. Fortunately, use of the more easily handled cationic tris(acetonitrile) Rh(III) precatalyst B3, bearing a trifluoromethyl-substituted Cp*-ligand, did not deteriorate the yield of the 2,3-dihydropyridine. Interestingly, the dinuclear [Cp*RhCl2]2 complex did not promote the title transformation with either CsOAc or AgOAc as additives.
With the optimal conditions in hand, we examined the scope of the reaction. α,β-Unsaturated oxime pivalates 1 bearing various linear or branched alkyl substituents at positions 2 and 3 are tolerated (Scheme 2, 3aa-3da). Substrates bearing an aromatic ring at position 3 are also competent (3ha-3na), as well as functional groups like silylated alcohols (3fa) and halides (3ga, 3la).
Scheme 2.

α,β-Unsaturated Oxime Pivalate Scope
Turning our attention to the scope of the alkenes, we found that various 1,1-disubstituted olefins are competent substrates for 2,3-dihydropyridine synthesis (Scheme 3). Functional groups including esters (3ab), ketones (3ac), ethers (3ad), and protected amines (3am) are tolerated. With methylene cyclopentanes and cyclohexanes as substrates, spiro-bicyclic 2,3-dihydropyridines are obtained in good yields (3dj-3ao). Finally, exo-methylene cyclohexanes bearing a prostereogenic carbon at the 4-position furnished the corresponding 2,3-dihydropyridines (3an and 3ao) with high levels of diastereocontrol (up to 10:1 dr).
Scheme 3.

Alkene Scope
To demonstrate the synthetic utility of the 2,3-dihydropyridines, we turned our attention to derivation of the products. Stimulated by the wide abundance of piperidines in biologically active copounds,1a attention was placed on the reduction of C-C and C-N double bonds. We successfully developed a single-pot protocol wherein the Rh(III)-catalyzed coupling of imines and alkenes was sequenced with Pd-catalyzed hydrogenation (Scheme 4) to deliver high overall yield of piperidine products. Surprisingly, the major diastereomer formed proved to have the trans relationship between the substituents at the 2 and 3 positions, although the level of selectivity seems to be modestly substrate dependent (compare 6mi vs 6hi). The relative configuration of products 6am and 6ao was unambiguously assigned by X-ray crystallography.13
Scheme 4. One-Pot Synthesis of Piperidines.

*numbers in parantheses denoted yields and dr’s after recrystallization.
Labeling studies provided valuable insight into the mechanism of the reaction. A small primary kinetic isotopic effect (KIE = 1.41) was observed when deuterated oxime pivalate d5-1j was used (Scheme 5). This observation may be explained by evoking a reversible C-H activation event and incorporation of deuterium from solvent rather than a direct indicator of rate difference.14 A rather large inverse KIE (0.74) was observed with deuterated alkene d2-2f, which is consistent with rate-determining olefin insertion.
Scheme 5.

Observation of KIE Effects
A Hammett study provided further support for olefin insertion being the slow step.13 A large and negative value of the reaction constant (ρ = −1.2) indicates that positive charge builds up at the reaction site during the rate-limiting step. Preliminary kinetic analysis15 of the reaction revealed a first order dependence of rate on both catalyst and oxime. Furthermore, first order dependence on alkene was also seen at low concentrations; higher concentrations demonstrated some evidence of an inhibitory effect.14
In order to account for the observed mechanistic data, the following catalytic cycle is proposed (Scheme 6). Following the formation of the active Rh(III) diacetate complex, C-H activation takes place reversibly through a concerted metallation deprotonation mechanism. The resultant five-membered metallacycle II then undergoes rate-limiting migratory insertion to give the seven-membered metallacycle IV. Reductive elimination/N-O bond cleavage furnishes the 2,3-dihydropyridine and regenerates the catalyst.
Scheme 6.

Postulated Catalytic Cycle
In conclusion, we have described an efficient Rh(III)-catalyzed synthesis of semi-saturated nitrogen heterocycles. A significant ligand effect on reactivity was observed, the electron-deficient trifluoromethyl-substituted Cp*CF3 being optimal. The scope of the transformation is large16 and many of the obtained 2,3-dihydropyridines were successfully reduced to piperidines with good levels of diastereocontrol. Mechanistically, it was established that the reaction follows overall second-order kinetics, migratory insertion being the turnover-limiting step.
Supplementary Material
Acknowledgments
We thank NIGMS (GM80442) for support. FRM thanks the Swiss National Science Foundation for fellowship support. KFS thanks NIGMS for a Supplement. We thank Johnson Matthey for a generous loan of Rh salts, Christopher D. Rithner and Karolien Denef (CSU) for assistance with NMR, Brian Newell (CSU) for solving X-ray crystal structures, and Maria Romanova-Michaelides (University of Geneva) for assistance with mathematical analysis.
Footnotes
Notes
The authors declare no competing financial interest.
Supporting Information. Experimental procedures, spectroscopic characterization of compounds, X-ray crystal structures, and detailed mechanistic studies. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Vitaku E, Smith DT, Njardarson JT. J Med Chem. 2014;57:10257. doi: 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]; (b) Roughley SD, Jordan AM. J Med Chem. 2011;54:3451. doi: 10.1021/jm200187y. [DOI] [PubMed] [Google Scholar]; (c) Carey JS, Laffan D, Thomson C, Williams MT. Org Biomol Chem. 2006;4:2337. doi: 10.1039/b602413k. [DOI] [PubMed] [Google Scholar]
- 2.For reviews on dihydropyridine synthesis, see: Stout DM, Meyers AI. Chem Rev. 1982;82:223.Lavilla R. J Chem Soc, Perkin Trans. 2002;1:1141.Bull JA, Mousseau JJ, Pelletier G, Charette AB. Chem Rev. 2012;112:2642. doi: 10.1021/cr200251d.
- 3.Hantzsch A. Justus Liebigs Ann Chem. 1882;215:1. [Google Scholar]
- 4.(a) Loev B, Goodman M, Snader K, Tedeschi R, Macko E. J Med Chem. 1974;17:956. doi: 10.1021/jm00255a010. [DOI] [PubMed] [Google Scholar]; (b) Vater W, Kroneberg G, Hoffmeister F, Kaller H, Meng K, Oberdorf A, Puls W, Schlossmann K, Stoepel K. Arzneim-Forsch. 1972;22:1. [PubMed] [Google Scholar]
- 5.(a) Kita M, Kondo M, Koyama T, Yamada K, Matsumoto T, Lee K, Woo J, Uemura D. J Am Chem Soc. 2004;126:4794. doi: 10.1021/ja049277f. [DOI] [PubMed] [Google Scholar]; (b) Zou Y, Che Q, Snider BB. Org Lett. 2006;24:5605. doi: 10.1021/ol062333s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kim J, Thomson RJ. Angew Chem, Int Ed. 2007;46:3104. doi: 10.1002/anie.200605160. [DOI] [PubMed] [Google Scholar]
- 6.(a) Baldwin JE, Whitehead RC. Tetrahedron Lett. 1992;33:2059. [Google Scholar]; (b) Yu LB, Chen D, Li J, Ramirez J, Wang PG, Bott SG. J Org Chem. 1997;62:208. doi: 10.1021/jo961175n. [DOI] [PubMed] [Google Scholar]; (c) Baldwin JE, Spring DR, Whitehead RC. Tetrahedron Lett. 1998;39:5417. [Google Scholar]; (d) Ruggeri RB, Hansen MM, Heathcock CH. J Am Chem Soc. 1988;110:8734. [Google Scholar]
- 7.Jakubowicz K, Abdeljelil KB, Herdemann M, Martin MT, Gateau-Olesker A, Al-Mourabit A, Marazano C, Das BC. J Org Chem. 1999;64:7381. [Google Scholar]
- 8.(a) Born S, Kobayashi Y. Synlett. 2008;16:2479. [Google Scholar]; (b) Fuks R, Merenyi R, Viehe H. Bull Soc Chim Belg. 1976;85:147. [Google Scholar]; (c) Hasan I, Fowler FW. J Am Chem Soc. 1978;100:6696. [Google Scholar]; (d) Lasne M, Ripoll J, Guillemin J, Denis J. Tetrahedron Lett. 1984;35:3847. [Google Scholar]
- 9.For recent reviews, see: Colby DA, Bergman RG, Ellman JA. Chem Rev. 2010;110:624. doi: 10.1021/cr900005n.Satoh T, Miura M. Chem-Eur J. 2010;16:11212. doi: 10.1002/chem.201001363.Patureau FW, Wencel-Delord J, Glorius F. Aldrichim Acta. 2012;45:31.Song G, Wang F, Li X. Chem Soc Rev. 2012;41:3651. doi: 10.1039/c2cs15281a.
- 10.(a) Hyster TK, Rovis T. J Am Chem Soc. 2010;132:10565. doi: 10.1021/ja103776u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hyster TK, Ruhl KE, Rovis T. J Am Chem Soc. 2013;135:5364. doi: 10.1021/ja402274g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hyster TK, TRovis T. Synlett. 2013:1842. doi: 10.1055/s-0033-1339510. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hyster TK, Dalton DM, Rovis T. Chem Sci. 2015;6:254. doi: 10.1039/c4sc02590c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Neely JM, Rovis T. J Am Chem Soc. 2013;135:66. doi: 10.1021/ja3104389. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Neely JM, Rovis T. J Am Chem Soc. 2014;136:2735. doi: 10.1021/ja412444d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Duttwyler S, Chen S, Takase MK, Wiberg KB, Bergman RG, Ellman JA. Science. 2013;339:678. doi: 10.1126/science.1230704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neely JM. PhD Dissertation. Colorado State University; Fort Collins, CO: 2014. Subjection of 1a and methyl acrylate to our optimized reaction conditions (see Scheme 2) delivered only pyridine adduct in 38% yield. No trace of 2,3-dihydropyridine was observed. [Google Scholar]
- 13.Boyer PM, Roy CP, Bielski JM, Merola JS. Inorg Chim Acta. 1996;245:7. [Google Scholar]
- 14.See the Supporting Information section for details regarding X-ray crystal structures and mechanistic studies.
- 15.Blackmond DG. Angew Chem Int Ed. 2005;44:4302. doi: 10.1002/anie.200462544. [DOI] [PubMed] [Google Scholar]
- 16.Terminal alkenes afford pyridine adducts in low yield. Internal alkenes are not suitable partners.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.