

Graphical Abstract
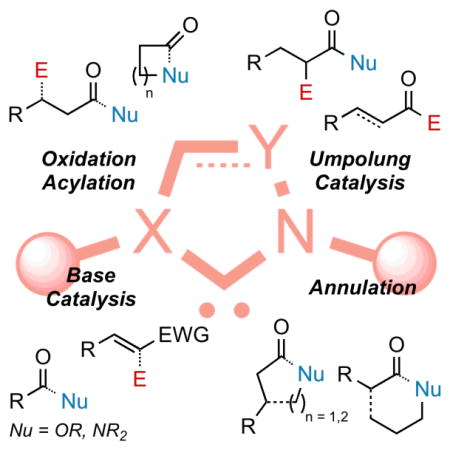
1. INTRODUCTION
The seminal independent reports of stable carbenes by Bertrand1 and Arduengo2 are rightly judged to have stimulated broad interest in their properties and their reactivity.3 The impact of neutral 2-electron donor ligands for transition metals that are distinct in their electronic properties and steric demand to the broadly used phosphines and imines have relegated carbenes to a special place in transition metal catalysis.4 These ligands are dominated by the imidazolylidene framework with Grubbs’ second generation metathesis catalyst a gold standard example.5 Today, carbenes as ligands on transition metals are ubiquitous spanning the entire periodic table and impacting a plethora of transformations.6
On the other hand, imidazolylidene carbenes are a minor player in organocatalysis using stable carbenes. The genesis of reactivity in this field can be ascribed to Ukai’s demonstration that thiazolium salts catalyze the benzoin reaction7 and Breslow’s subsequent determination of its mechanism.8 Indeed, the evolution of umpolung catalysis dates back to the original discovery of the cyanide-catalyzed benzoin reaction in 1832.9
Sheehan’s efforts at rendering the benzoin reaction enantioselective focused on a chiral thiazolium precatalyst, and date to 1966,10 the same year that Noyori described a chiral ligand on Cu for enantioselective cyclopropanation.11 Following the key insight advanced by Breslow that the azolium salts are acidic at the C-H bond and a weak base can deprotonate it, each of these early reports depicted the active catalyst as the ylide, but this is largely a semantic argument. These catalysts are stable carbenes, generated in situ from the azolium salt and base.
Today, organocatalysis using stable carbenes is dominated not by thiazolylidene or imidazolylidene carbenes, but triazolylidene carbenes. These were first described in 1995 in a seminal report by the groups of Enders at Aachen and Teles at BASF.12 The sporadic effort at chiral azolium salts for the benzoin reaction turned abruptly from the thiazolium to the triazolium scaffold following this seminal report.
Since then, several groups reported chiral variants of the triazolylidene carbene eventually leading to the development of chiral bicyclic triazolylidene scaffolds by Knight and Leeper,13 which drastically improved the achievable stereoselectivity in a variety of NHC-catalyzed reactions. These reports culminated with the introduction of the aminoindanol-derived scaffold in 200214 that has proven to be broadly efficacious and dominates the field. From its beginning in the catalysis of the benzoin reaction, these catalysts have been investigated in many different transformations, and major advancements in umpolung catalysis are summarized in Figure 1. The field has periodically been reviewed, with the most recent such comprehensive review appearing in this journal in 2007.15 This review will thus focus on developments since the last review with the caveat that seminal contributions are acknowledged.
Figure 1.

Evolution of Umpolung Catalysts
2. SYNTHESIS AND PROPERTIES OF N-HETEROCYCLIC CARBENES
2.1 Structures of N-Heterocyclic Carbenes
As a result of the proliferation of nucleophilic carbenes in transition metal catalysis and organocatalysis, many different types of carbenes have been synthesized. As mentioned earlier, the advent of NHC organocatalysis was dominated by thiazolium-based carbenes, and as the field matured, imidazolium and triazolium scaffolds have also become popular. We have compiled a list of chiral azolium pre-catalysts in Figure 2 to demonstrate the immense diversity of catalysts used (counter-ions are omitted for clarity). In addition to the vast number of chiral catalysts reported, many achiral azoliums have been prepared. These can be found in Figure 3.
Figure 2.


Literature Examples of Chiral Carbene Precursors
Figure 3.

Literature Examples of Achiral Carbene Precursors
2.2 Synthesis of Azolium-Based Carbene Precursors
With a wide variety of catalysts reported, several methods have been described for their synthesis. The synthesis of azoliums as pre-carbenes has been recently reviewed in this journal.16 This section will focus on the carbene precursors found most often in organocatalyzed processes (thiazol-, imidazol-, imidazolin-, and triazolium). For thiazoliums, condensation of an α-chloroketone with an N-substituted thioformamide has been demonstrated.17 Thiazoliums have also been prepared from their corresponding thiazolin-2-thione when treated with hydrogen peroxide under acidic conditions (Scheme 1).18
Scheme 1.

Synthesis of Thiazolium Salts via Condensation of Thioformamide
The synthesis of imidazoliums and imidazoliniums adheres to two broad strategies: annulation by introduction of the backbone onto an aminal, or cyclization of a diamine or diimine with a one-carbon component. The annulation of an aminal or derivative was the original method used by Arduengo to synthesize the first characterizable carbene.2 Since then, a variety of methods have been reported to increase the structural diversity of NHCs. These preparations usually use formamidines as the diamine component, summarized in Scheme 2.19 Imidazoliums may also be prepared by alkylation of the parent heterocycle.20
Scheme 2.

Synthesis of Imidazoliums by Cyclization of the Backbone
The most widely used approach for the synthesis of imidazolium and imidazolinium salts is the introduction of the pre-carbenic carbon in the cyclization step. Using this strategy, imidazoliums have been generated from a variety of 1,1-biselectrophiles, including diiodomethane,21 Weiss’ reagent,22 chloromethyl ethers,23 and chloromethyl pivalates24 (Scheme 3). In a similar vein, cyclization of formaldehyde onto a diamine, followed by oxidation has also been demonstrated.25
Scheme 3.

Synthesis of Imidazoliums by One Carbon Annulation
The synthesis of imidazoliniums can be achieved by cyclization with trialkylorthoformates26 or formaldehyde27 (coupled with oxidation) onto a diamine (Scheme 4). Another strategy is the intramolecular cyclization of formamides.28
Scheme 4.

Synthesis of Imidazoliniums by One Carbon Annulation
While there are a variety of methods to prepare thiazolium and imidazolium scaffolds, fewer methods are usually employed to generate triazolium-based precatalysts. First introduced by Enders and Teles in 1995,12 triazolium salts for catalysis can be prepared using a 5-step procedure starting from an aroyl chloride (Scheme 5).29 A similar strategy for the preparation of aliphatic substituted triazolium salts has also been reported.30
Scheme 5.

Preparation of Trisaryl-Triazolium Salts
Leeper first introduced chiral bicyclic triazolium salts in 1998, using a three-step sequence from the chiral morpholinone or γ-lactam.13 Enders showed utilized this procedure to synthesize triazolium salts with an oxygen in the backbone starting from oxazolidinones.31 Rovis synthesized a variety of chiral and achiral amino-acid and amino-indanol based triazolium scaffolds32 using a simpler three step sequence starting with chiral lactams or 2-pyrrolidinone (Scheme 6).33 As can be seen by the enormous number of catalysts from Figures 2 and 3, this method is reliable and competent for a wide variety of lactams.
Scheme 6.

Preparation of Bicyclic Triazolium Salts
Bode used a similar strategy to synthesize chiral triazolium A4. Problems with the stability of mesityl-hydrazine led these workers to instead condense the hydrochloride salt with free-based hydrazide 1 (Scheme 7). This strategy is more convenient when using less stable, electron-rich aryl hydrazines.34
Scheme 7.

Synthesis of Mesityl Substituted Triazolium Salts
2.3 pKa Measurements of N-Heterocyclic Carbenes
Since many NHC catalyzed reactions generate the active carbene in situ by deprotonation of the corresponding azolium, a discussion of the acidity of these pre-catalysts is important.35 For ease of reference, reported computational and experimental pKa values in DMSO and H2O are summarized in 1. Among the most common organocatalysts, imidazoliums36 are generally the least acidic (pKa ~ 20–24 in water), while thiazoliums37 and triazoliums are more acidic (pKa ~ 16–19 in water), mirroring the trends found in the parent azoles. The most studied motifs relevant to organocatalysts are imidazoliums and imidazoliniums, and several important trends are apparent. 1,3-Dimethylimidazolium has a pKa of 22.0 in DMSO, similar to the value found in water (pKa = 23.0 in H2O).38,39 Substituting a methyl group for longer aliphatic chains (e.g. Ethyl, nButyl, nOctyl) has little effect on the acidity; however, an increase in pKa (~ 0.5 pKa units) occurs when a tbutyl substituent is present. Intriguingly, this effect appears additive; di-t-butylimidazolium (G13) gives a pKa of 23.2 (1.2 pKa units higher than the dimethyl analogue). This trend is also present when the pKa’s are determined in water. The effect of the N-aryl substituent was also found to have a significant impact on the acidity of triazoliums.40 Achiral catalyst G18 with a phenyl substituent is one pKa unit less acidic than the analogous azolium G19 bearing a pentafluorophenyl substituent. The popular pyrrolidinone based scaffolds also appear to be slightly more acidic than the morpholinone based NHCs (e.g. B2 versus A11). While substituents on the azolium can have a drastic impact on the acidity, the counterion appears to have only a negligible effect.41 Choice of solvent likely determines the importance of counterion.38
2.4 Lewis Basicity and Nucleophilicity of N-Heterocyclic Carbenes
Although studies on the reactivity of NHCs in organocatalysis (vide infra) and fundamental studies on their impact as ligands on metals35 have vastly increased our understanding of NHCs, few reports have been directed toward the intrinsic reactivity of NHCs. Elegant work by Mayr experimentally evaluated the nucleophilicity of different carbenes with reference electrophiles (Scheme 8).42 Importantly, Mayr determined that the intrinsic nucleophilicity of imidazolinylidine is 3 orders of magnitude greater than that of triazolinylidine. Mayr also has calculated the Lewis basicity of these species which parallels that of its nucleophilicity (calculated Methyl Cation Affinities [MCAs]: 2: 712.2 kJ/mol; 3: 767.2 kJ/mol; 4: 768.9 kJ/mol). Importantly, the weakest of these is still more than 100 kJ/mol more Lewis basic than prototypical Lewis bases such as PPh3, DBU and DMAP.
Scheme 8.

Nucleophilicity of NHCs
3. UMPOLUNG ACYL-ANION CATALYSIS
3.1 Characterization of the Breslow Intermediate
Early on, one of the most studied reactions employing NHCs as organocatalysts was the benzoin reaction. The observed benzoin product requires the conversion of the aldehyde carbon from an electrophile to a nucleophile. The newly generated nucleophile or acyl-anion equivalent can then couple with another equivalent of aldehyde to give the observed product. This inversion of reactivity (in contrast to the more common polar reactivity) has since become known as umpolung, a concept first described by Wittig in 1951 and later popularized by Seebach.43
Many research groups have experimentally and computationally investigated the mechanism of the NHC-catalyzed benzoin reaction and it is widely believed to begin with initial addition of the NHC to an aldehyde followed by proton transfer to give neutral enaminol 5 (Scheme 9).44 This neutral species or acyl-anion equivalent 5 was first proposed by Breslow and has since become known as the Breslow intermediate.8 This key intermediate has been exploited in a number of other reactions (vide infra); as a result, the isolation and characterization of the Breslow intermediate has been a widely sought after goal.
Scheme 9.

Postulated Generation of Acyl-Anion Equivalents
Early work by Jordan investigated alkyl-thiazolium species closely related to the postulated Breslow intermediate.45 More recently, cyclic aza-Breslow intermediates were synthesized by Douthwaite and co-workers.46 While this work provided insight into these species, little structural information could be gleaned. The keto tautomer of the Breslow intermediate (6) was reported by Berkessel and Teles, and postulated to be a catalyst deactivation pathway; however, attempts to trap the Breslow intermediate with silylating agents met with failure (Scheme 10).
Scheme 10.
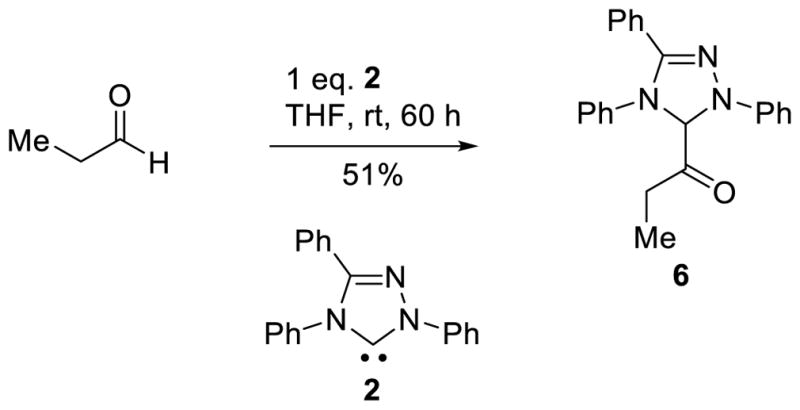
Generation of Keto-tautomer
Rovis reported stable, catalytically relevant nitrogen analogues of the triazolylidene derived Breslow intermediate.47 The reaction of azolium A9 with iminiums 7 or 8 in the presence of base gives the corresponding aza-Breslow intermediates 9 and 10 in 68 and 77% yield, respectively. Oxidation potentials and UV-Vis spectra were collected, in addition to NMR characterization and X-ray crystal structures, revealing 9 has a reduction potential of −0.17 V vs SCE, while 10 has a reduction potential of −0.49 V. Solid state analysis showed a preference for the E-enetriamine geometry, despite calculations showing a preferred Z-geometry for the putative Breslow intermediate itself.44c,48 In addition, 1H NMR showed a mixture of 4 compounds, likely arising from the two enetriamine geometries being present, along with C-N bond rotamers (Scheme 11).
Scheme 11.

Synthesis and Properties of aza-Breslow Analogues
Importantly, mixing catalytic amounts of 9 with a substrate for the intermolecular Stetter reaction in the presence of acetic acid delivers the expected Stetter product in 99% yield and 97% ee confirming the catalytic relevance of this type of intermediate in carbene organocatalysis (Scheme 12). The acid is proposed to protonate the enetriamine followed by release of carbene and generation of the iminium, the microscopic reverse of the pathway described in Scheme 11 above.
Scheme 12.

Aza-Breslow Catalyzed Intramolecular Stetter Reaction
Mayr isolated O-methylated Breslow intermediates with several azolylidenes and investigated the nucleophilicity of these species with benzhydrilium ions.49 The authors found a Z-enolate geometry is favored by 2:1 for the thiazole-derived intermediates, while the triazole based Breslow prefers the E-geometry (E:Z = 10:1). The nucleophilicity of these compounds was then compared using benzhydrilium ion 11 as the reference electrophile. The thiazolylidene enol ether 13 reacted at a similar rate to the imidazolylidene enol ether 14. Interestingly, the triazolylidene enol ether 15 and benzothiazolylidene enol ether 12 reacted much slower compared to the other two olefins studied (Scheme 13).
Scheme 13.

Nucleophilicity of O-methylated Breslow intermediates
Berkessel and Teles successfully characterized Breslow intermediates derived from imidazolinylidenes and aromatic aldehydes.50 As predicted, the authors determined the aldehydic proton is the source of the enol proton in the Breslow intermediate. Mixing one eqivalent of deutero-benzaldehyde with NHC 16 generates the corresponding tetrahedral intermediate, which then undergoes proton transfer to form the deuterated Breslow intermediate (Scheme 14).
Scheme 14.

Formation of Breslow intermediates
The equilibrium between the Breslow intermediate and the free carbene and the reversibility of its formation was demonstrated by crossover experiments with enaminol 17, in equilibrium with free carbene (4) and benzaldehyde. When mixed with more electrophilic aldehyde 18, rapid formation of enaminol 19 is observed. Interestingly, in spite of the greater stability of 19 relative to 17, the sole benzoin adducts are those accessed from enaminol 17 (cross-benzoin product 20 is the major product along with trace amounts of homo-benzoin product 21). The other possible products 22 and 23 are not observed, suggesting full reversibility between 17 and 19 as well as a lower barrier for the benzoin addition involving 18 (Scheme 15).
Scheme 15.

Reversibility of Breslow Intermediate Formation
Other studies carried out on the formation of the Breslow intermediate were aimed at investigating the influence of the N-aryl substituent on reactivity. At issue is the origin of selectivity for benzoin and Stetter type reactions (c.f. section 3) displayed by pentafluorophenyl substituted trazoliums (e.g. B10, B13) and the rate acceleration noted for the reactions of α-reducible aldehydes (annulations, oxidations, redox reactions; c.f. sections 4 and 5) when N-mesityl triazoliums are employed (e.g. A4, A12). Bode investigated the impact on catalysis of N-mesityl triazolylidene NHCs.51 Through control experiments and kinetic studies, he proposed that the formation of the Breslow intermediate is reversible when sterically small N-arylsubstituents are present on the NHC, while bulkier N-aryl groups on the NHC lead to irreversible formation of the Breslow intermediate. Thus, the rate-limiting step in these processes could change from Breslow intermediate formation (in the case of pentafluorophenyl substituents) to the reaction of the Breslow intermediate with an electrophile (in the case of mesityl substituents) leading to the observed rate increase with the mesityl-derived NHCs with certain reactions (Figure 4). Smith and O’Donoghue carried out similar studies using the intramolecular Stetter reaction as the model reaction.52 In this study, the authors found that the formation of the initial NHC-aldehyde adduct is reversible regardless of which aryl substituent is used followed by relatively slow formation of the Stetter products. However, a large rate increase is observed when 2,6-electron withdrawing groups are on the aryl ring.
Figure 4.

Impact of N-substituent on Reversibility of Breslow Intermediate Formation
3.2 The Benzoin Reaction
3.2.1 Enantioselective Benzoin Reactions
One of the most investigated reactions employing NHC catalysts is the benzoin reaction. Originally discovered by Wöhler and Liebig in 1832 using cyanide as the catalyst,9 the reaction was later shown by Ukai with thiazolium catalysts in the presence of base.7 After these initial reports, several research groups began investigating more efficient and selective catalysts for this biomimetic transformation53; the work up to 2007 is nicely summarized in previous reviews.15,54 Despite this, many challenges remained; here we will focus on the advances made since then. As a result of the success of this reaction, it has become a benchmark reaction for the application of new NHC manifolds, and several groups continue to evaluate new scaffolds in this way (Scheme 16).55
Scheme 16.

NHCs Tested in the Benzoin Reaction (2007–2014)
Connon and Zeitler have reported the most efficacious conditions for the enantioselective benzoin reaction to date.56 Using just 4 mol% of triazolium precatalyst B11, they effected the homo-coupling of benzaldehyde in 90% yield and >99% ee. The catalytic efficiency of B11 has been attributed to the incorporation of an H-bonding group, which helps control the selectivity. A variety of other aryl aldehydes also participate with high levels of stereocontrol, although with less consistent results (Scheme 17).
Scheme 17.

Highly Efficient Catalytic System for the enantioselective Benzoin Reaction
3.2.2 Cross Aldehyde-Aldehyde Benzoin Reactions
The cross-benzoin reaction has also been studied, allowing access to a wider variety of products. Cookson first demonstrated the intramolecular cross-coupling of two aldehydes in 1976.57 This work highlighted a lack of chemo-selectivity for the two aldehydes, which plagues many of the cross-benzoin reactions reported to date. Thus, when two products can result from an intramolecular cross-benzoin reaction, a 1:1 product ratio is observed. In the intermolecular reaction, four different products can result; however, it is possible to control the product ratio by increasing the equivalents of one aldehyde, as originally demonstrated by Stetter.58 Since these seminal reports, many groups have investigated the cross-coupling of aldehydes.59 Connon and Zeitler found that by using triazolium precatalyst G19 product selectivites for the aliphatic, aromatic cross-benzoin reaction could be biased to synthetically useful amounts (63 – 70% yield with branched aliphatic aldehydes and 4-halo benzaldehydes).57a Glorius demonstrated the selective coupling of aromatic aldehyde with 2-substituted benzaldehyde derivatives relying on steric hindrance to drive product selectivity.57b
Yang noted an interesting difference in reactivity between thiazolium G5 and triazolium G19; the thiazolylidene favors Breslow intermediate formation with the aromatic aldehyde and subsequent reaction with the aliphatic aldehyde (up to 98:2 product selectivity), whereas the triazolylidene preferentially forms the Breslow intermediate with the aliphatic aldehyde, followed by reaction with the aromatic aldehyde (up to 11:89 product selectivity).60
A conceptually different approach was recently advanced by Gravel who addressed the cross-benzoin problem by catalyst design. Gravel engineered a catalytic system that selectively forms the cross-benzoin products between aliphatic aldehydes and aryl aldehydes.61 Remarkably, the catalyst selectively forms the Breslow intermediate with the aliphatic aldehyde, and then adds to the aromatic aldehyde, obviating the need for a large excess of one aldehyde. Furthermore, using related chiral triazolium A20, Gravel effected the reaction in a promising 40% ee. Aside from enzymatic catalysts,62 this is one of two examples of an enantioselective intermolecular cross-benzoin reaction between two aldehydes (Scheme 19). The other example was reported by Yang but used a large excess of the aliphatic aldehyde (10 equiv, delivering cross-benzoin adduct in 41% yield and 60% ee).58a While very important precedents, they showcase the need for further catalyst development to address this problem.
Scheme 19.

Cross-Benzoin Reaction of Aliphatic and Aryl Aldehydes
The coupling of aldehydes with formaldehyde has proven more selective. Inoue first demonstrated this reaction in 1985 using paraformaldehyde and aryl aldehydes.63 This reaction could also be effected with enzymes.64 Glorius developed catalyst G24 to increase the utility of this reaction.65 These conditions afford the desired hydroxyketone in good yields across a broad substrate scope (Scheme 20).
Scheme 20.

Hydroxymethylation of Aldehydes
3.2.3 Cross Aldehyde-Ketone Benzoin Reactions
Suzuki reported the intramolecular hetero-coupling of aldehydes and ketones in 2003, synthesizing preanthraquinones in high yields.66 Ema and Sakai examined the synthesis of bicyclic tertiary alcohols, where both bridgehead carbons contain a stereogenic center. Initially, asymmetric induction was modest; however, tuning the catalyst and conditions increased the yield and enantioselectivity across a broad range of aldehydes (Scheme 21).67
Scheme 21.

Synthesis of Bicyclic Tertiary Alcohols
Vadde and Vasam employed a thiazolium salt for the synthesis of naphthalenones from o-phthalaldehydes via an intramolecular cross aldehyde-ketone benzoin reaction.68 This protocol is tolerant of a broad range of electron withdrawing or donating groups on the aryl ketone, as well as differing substitution patterns, delivering the naphthalenone in high yields (75 – 94%). In a similar method, You employed a camphor-derived triazolium catalyst (A22) to the synthesis of dihydroisoquinolones (Scheme 22).69
Scheme 22.

Enantioselective Synthesis of Dihydroisoquinolones
A Michael addition – intramolecular benzoin cascade reaction strategy was developed by Rovis to synthesize highly functionalized cyclopentanones with high enantioselectivity.70 Two approaches were pursued relying on iminium catalysis with enals or enamine catalysis with aliphatic aldehydes to generate the intramolecular benzoin substrate. In the case of the iminium cascade, the reaction proceeds via initial generation of α,β-unsaturated iminium 25 from the enal and secondary amine catalyst 24. After conjugate addition of the diketone to 25, protonation and hydrolysis affords aldehyde 26 with concomitant release of 24. The carbene then catalyzes the diastereoselective intramolecular benzoin reaction to give the observed products (Scheme 23). In the case of the enamine cascade reaction, the secondary amine catalyst reacts with the aliphatic aldehyde generating enamine 29, which then adds to the activated Michael acceptor 28. Hydrolysis of this intermediate gives aldehyde 30, which then undergoes NHC-catalyzed intramolecular benzoin reaction to form the observed cyclopentanol products (Scheme 24). Interestingly, chiral precatalyst A3 is required in order to generate the products with high diastereoselectivity. Using achiral precatalyst G19 in this reaction gives the product in 89% yield and 96% ee, but in only 5:1 dr, while the opposite antipode of precatalyst (ent-A3) affords the product in 59% yield, 93% ee, and 4:1 dr. Enders used iminium catalysis with α-oxo-sulfones to generate cyclopentanones bearing three contiguous stereocenters.71
Scheme 23.

Synthesis of Cyclopentanols via Dual Secondary Amine/NHC Catalysis
Scheme 24.

Synthesis of Cyclopentanols via Michael Addition/NHC Cascade Reaction
In addition to research on the intramolecular cross aldehyde-ketone benzoin reaction, the intermolecular variant has also been investigated. Although it does not involve carbenes as catalysts, Johnson demonstrated acyl silanes are competent acyl-anion precursors in the cross aldehyde-ketone benzoin reaction using lanthanum tricyanide as the catalyst.72 This allowed access to a wide variety of silyl protected benzoin adducts in generally good yields. Demir investigated the use of acyl-phosphonates as acyl-anion precursors in the cross aldehyde-ketone benzoin reaction.73 These reactions work well when using trifluoromethyl ketones as the acceptor. Cyclohexanone affords the product in 54% yield (Scheme 25).
Scheme 25.

Acyl-Silanes and Phosphonates as Acylanion Equivalents
Enders further investigated these acceptors using an achiral triazolium NHC precursor.74 This protocol couples aryl aldehydes with aryl-trifluoromethyl ketones in high yield (64 – 99%), and with high chemoselectivity. Although initial attempts at using a chiral triazolium were hindered by low chemoselectivity between homo-benzoin and the desired cross product, Enders and coworkers were able to overcome this issue with chiral triazolium B15 (Scheme 26).75
Scheme 26.

Enantioselective Cross-Benzoin Reaction with Trifluoromethyl Ketones
Connon and Zeitler examined α-ketoester acceptors in the direct intermolecular cross-benzoin reaction.76 With an achiral triazolium catalyst the reaction proved remarkably general across aliphatic and aryl aldehydes, as well as tolerating substitution on the α-ketoester. In addition, the authors provided one example of an enantioselective reaction, delivering product in 48% yield and 76% ee. Using chiral catalyst A20, Gravel was successful in expanding the scope of the enantioselective protocol, although aliphatic α-ketoesters do not participate under these reaction conditions.77 However, aliphatic aldehydes react smoothly to give the tertiary alcohol in good yields, with excellent stereocontrol (77 – 93% ee) (Scheme 27).
Scheme 27.

Enantioselective Coupling of Aliphatic Aldheydes with α-Arylketoesters
3.2.4 Cross Aldehyde-Imine Benzoin Reactions
Murry and Frantz first reported the coupling of the Breslow intermediate to imines.78 You, subsequently, demonstrated the cross-aza-benzoin reaction of aryl-aldehydes with aryl imines to give α-amino ketone products using thiazolium G3.79 The reaction works well with several different aryl groups (Scheme 28).
Scheme 28.

Cross-Aza-Benzoin Reaction
Enders reported the first cross-aza-benzoin reaction of furfural derivatives with trifluoromethyl ketimines in moderate to good yields (32 – 87%), using an achiral triazolium pre-catalyst.80 Ye demonstrated the coupling of enals with trifluoromethyl ketones with excellent enantioselectivity, using chiral triazolium B23.81 Interestingly, these conditions tolerated other electron withdrawing groups on the imine (Scheme 29).
Scheme 29.

Cross-Aza-Benzoin Reaction with Ketimines
Rovis and DiRocco effected the enantioselective reaction of aliphatic aldehydes with N-Boc imines.82 Straight-chain aliphatic aldehydes give the products in good yields with a high degree of stereocontrol. Branched aliphatic aldehydes participate with excellent enantioselectivity, but deliver the product in lower yields (Scheme 30).
Scheme 30.

Cross Aza-Benzoin Reaction with N-Boc Imines
Rovis reported an interesting cross-aza-benzoin reaction using photocatalysis to generate an iminium electrophile as the acceptor. Surprisingly, the reaction was tolerant of oxygen, and the scope of the reaction was carried out without any special precautions to avoid ambient oxygen (Scheme 31).
Scheme 31.

Dual Photocatalytic NHC Cross Aza-Benzoin Reaction
3.3 The Stetter Reaction
3.3.1 Intramolecular Stetter Reactions
Ciganek reported the first general intramolecular Stetter reaction in 1995,83,84 nearly 20 years after Stetter’s contributions to the intermolecular reaction. The enantioselective intramolecular Stetter reaction, first reported by Enders,12 has been the object of much research in recent years and progress in this field has been prevsiouly reviewed (Scheme 32).85
Scheme 32.

Seminal Intramolecular Stetter Reactions
There has also been a large amount of work since 2007, many expanding the scope of the reaction or using the process for synthesis of biologically relevant compounds. The Rovis group was successful varying the heteroatom linker of the popular chromanone scaffold, as well as the Michael acceptors86, including vinylphosphonates, vinylphosphine oxides,87 and alkynyl phosphonates88 (Scheme 33). McErlean has further demonstrated that the reaction provides rapid access to fused pyrans.89
Scheme 33.

Vinyl Phosphonates as Coupling Partners
The NHC catalyzed coupling of aldehydes to alkynes was reported, giving chromene products.90 This was also used in an intramolecular/intermolecular Stetter cascade reaction giving 1,4-dicarbonyl species, which are easily converted into their corresponding pyrroles through a Paal-Knorr synthesis.91 Significantly, McErlean expanded the reaction to include 1,6-acceptors in a formal vinylogous Stetter reaction.92 Using achiral precatalyst G19, he effected the reaction with several different tethered, electron-poor dienes (Scheme 34).
Scheme 34.

McErlean’s Extended Stetter Reaction
As a result of the success of the Stetter reaction involving salicylaldehyde-derived substrates, it has become a benchmark reaction for many new carbene catalysts. Zeitler immobilized thiazolium, imidazolium, and triazolium precursors on polyethylene glycol beads, and tested their catalytic efficiency in the intramolecular Stetter and redox esterification reactions.93 Similarly, Hara synthesized thiazolium catalysts with perfluorinated side chains to aid in purification and recovery of the carbene catalyst.94 Both of these catalysts showed similar reactivity to their previously reported counterparts in the intramolecular Stetter reaction. The use of alkyl-thiazolium ionic liquids has also been demonstrated in catalyzing the intramolecular Stetter reaction in the presence of 15 mol% Et3N.95 These ionic liquids provide Stetter products with short reaction times and high yields when microwave irradiation is used. Chiral catalysts have also been reported and tested in the enantioselective intramolecular Stetter reaction. You reported camphor-derived triazolium scaffolds (e.g. A22) that catalyze the reaction in high yields and with excellent stereocontrol.96 These authors also reported imidazolium and triazolium catalysts based on (1R,2R)-(+)-diphenyl ethylenediamine (e.g. A19),97 which perform well in this reaction, albeit with less consistent results. The synthesis of chiral triazolium scaffolds incorporating a pyridine as the N-aryl substituent has also been demonstrated (e.g. B20).98 A β-pinene-derived triazolium catalyst A23, reported by Rafinski, has also proven efficient for the intramolecular Stetter reaction (Scheme 35).99
Scheme 35.

Recent Chiral Catalysts for the Intramolecular Stetter Reaction.
3.3.2 Intermolecular Stetter Reactions
The intermolecular reaction has been useful in the synthesis of many organic precursors. While the reaction was extensively studied with achiral catalysts by Stetter in the 1970’s, new coupling partners have been reported since. You demonstrated the coupling of aryl aldehydes with arylsulfonyl-indoles.100 The reaction proceeds via initial expulsion of tosylate by the enamine, generating the corresponding α,β-unsaturated iminium ion, which acts as the Michael acceptor for the intermolecular Stetter reaction. A broad range of aryl aldehydes perform well, but aliphatic aldehydes deliver the products in lower yield. Substitution at various positions of the indole were also tolerated; however, N-methyl substituted indole gives no reaction (Scheme 36). Interestingly, the enantioselective reaction was attempted, giving the product with low conversion (14 – 36%), but with high levels of enantioselectivity (90 – 97% ee). Biju expanded the scope of Michael acceptors further to include vinyl-sulfones and phosphonates (Scheme 37).101
Scheme 36.

Coupling of Aldehydes with Arylsulfonyl Indoles
Scheme 37.

Vinyl-Sulfones and Vinyl-Phosphonates in the Intermolecular Stetter Reaction
Another interesting application is the reaction between heteroaromatic aldehydes and 2-nitroglucal.102 Two products are observed, which could each be isolated in good yield by judicious selection of the reaction conditions (Scheme 38). The use of amine bases leads to the Stetter reaction, while an excess of stronger carbonate bases results in elimination of nitrous acid, forming the corresponding enone.
Scheme 38.

Stetter Reaction with 2-Nitroglucal
Along with new acceptors, new donor partners have also been investigated. Massi reported the use of alkyl α-diketones as acetaldehyde donors in the Stetter reaction with chalcones (Scheme 39).103 Miyashita originally demonstrated this strategy using catalytic Bu4NCN to effect the double acylation of Michael acceptors. Acrylonitrile, as well as vinyl ketones and vinyl esters are competent in the reaction affording 1,4-diketones in modest to good yields (37–93%).104
Scheme 39.

Diketones as Acyl-anion Precursors
In a similar vein, the double acylation of aryl-vinyl ketones has also been accomplished using thiazolium catalyst G24.105 This reaction is proposed to proceed through addition of the NHC to the diketone, giving tetrahedral intermediate 33. The acyl group then transfers to the alkoxide forming the O-acylated Breslow intermediate (34), which then adds to the enone forming enolate 35. The acyl-group then transfers to the α-carbon of the arylketone, to generate the observed product, and regenerate the active catalyst (Scheme 40).
Scheme 40.

Benzils as Nucleophilic Coupling Partners
Chi reported the catalytic activation of carbohydrates with thiazoliums, generating formaldehyde acyl-anion equivalents, which then could couple with chalcones.106 This reaction proceeds through initial retro-formoin of the carbohydrate, followed by an intermolecular Stetter reaction (Scheme 41).
Scheme 41.

Hydroformylation of Chalcone Derivatives
The synthesis of 1,4-dicarbonyls via intermolecular Stetter reaction coupled with a Paal-Knorr synthesis has been the object of several reports. The ease of generating furans, pyrroles, and thiophenes has been demonstrated from several research groups, and much of the literature in this area has been previously reviewed.15 This strategy has recently been employed to generate 2,3-diaryl furans (Scheme 42).107 A similar transformation could be effected with malononitrile and two different aromatic aldehydes giving the same type of products.108 This presumably follows the same reaction pathway after an in situ Knoevenagel reaction has occurred between the more electrophilic aldehyde and malononitrile.
Scheme 42.

Synthesis of Diaryl-Aminofurans
The intermolecular Stetter reaction has also been coupled with Michael and Aldol reactions to generate complex products from simple starting materials. This strategy has been applied to a two-step protocol for the synthesis of highly substituted cyclopentenes and cyclopentanols (Scheme 43).109 Different substitution patterns control the outcome of this reaction. Thus, when aryl aldehydes are used in the Stetter reaction, the subsequent Michael-aldol sequence preferentially forms the cyclopentene product. However, when heteroaromatic aldehydes are used, the cyclopentanol product forms.
Scheme 43.

Stetter-Michael Cascade Reaction
One-pot intermolecular Stetter reactions coupled with intramolecular Michael or aldol reactions have also been developed. These reactions trap the enolate, formed after conjugate addition of the Breslow intermediate to the Michael acceptor, with either an aldehyde or exogenous Michael acceptor (Scheme 44).
Scheme 44.

General Stetter-Aldol-Michael Reactions
This reactivity was first demonstrated by Gravel, exploiting an intermolecular Stetter reaction, followed by a diastereoselective, intramolecular Michael reaction, furnishing indane products.110 The reaction works well when electron-deficient aryl-aldehydes are used, but still delivers product when aliphatic or electron-rich aryl-aldehydes are used (Scheme 45). Gravel also expanded this to spiroindanes (Scheme 46).111
Scheme 45.

Diastereoselective Indane Synthesis by Stetter-Michael Cascade
Scheme 46.

Synthesis of Spiroindanes
Ye further exploited a Stetter-aldol sequence to generate 4-hydroxytetralones diastereoselectively (Scheme 47).112 Aliphatic or aromatic vinyl ketones performed well; however, disubstituted Michael acceptors were found to be less reactive. Ye expanded the utility of this approach to generate highly substituted indanones (Scheme 48).113
Scheme 47.

Synthesis of Hydroxytetralones
Scheme 48.

Synthesis of Indanones
3.3.3 Enantioselective Intermolecular Stetter Reactions
Despite the impressive works devoted to the achiral intermolecular reaction and the enantioselective intramolecular version, the enantioselective intermolecular Stetter reaction remained elusive for many years. Enders first attempted this in 1989, coupling n-butanal with chalcone to deliver the corresponding 1,4-diketone in a modest 30% ee and 29% yield with a chiral thiazolium precatalyst. The ee could be improved slightly to 39% ee using thiazolium precatalyst F4, although at the expense of yield (4%).114 However, it was not until 2008 that Enders and Rovis reported the first major advances in this arena. Enders described the coupling of aryl aldehydes with chalcones,115 and later arylidene malonates,116 in good yields with promising enantioselectivity (up to 78% ee). The coupling of acetaldehyde to chalcones was also demonstrated, albeit with somewhat diminished enantioselectivity.117 Concurrently with Enders’ work, Rovis demonstrated the Stetter reaction between glyoxamides and alkylidene malonates in good yield with excellent levels of stereocontrol, which was immediately followed with a diastereoselective example using alkylidene ketoamides (Scheme 49), with the second stereocenter insulated against epimerization by allylic 1,3-strain.118
Scheme 49.

Catalytic Enantioselective Intermolecular Stetter Reaction
Shortly after, Rovis demonstrated the efficient coupling of hetero-aromatic aldehydes to nitroalkenes.119 Interestingly, fluorinated catalyst B13 gives an increase in yield and selectivity, compared to the des-fluoro analogue (B12) or the trans-fluoro analogue (22% yield, 88% ee) (Scheme 50). The origin of fluorine’s effect on the catalyst was later studied computationally. These studies indicate the cis-fluorine increases attractive interactions between the developing positive charge of the Breslow intermediate and the developing negative charge on the nitroalkene.120
Scheme 50.

Stetter Reaction with Nitroalkenes
Fluorinated catalyst B13 was used again to expand the reaction to enals and nitroalkenes by Rovis.121 The use of catechol as an additive greatly improved the reactivity, allowing for lower catalyst loadings (as low as 0.1 mol%). The increase in reactivity is postulated to arise from catechol assisting in the proton transfer forming the Breslow intermediate, previously demonstrated to be the rate determining step in the intramolecular reaction.122 Kinetic isotope measurements with deutero-aldehyde or deutero-catechol support this argument (Scheme 51). Gravel further demonstrated that fluorinated catalyst B13 was effective in the intermolecular Stetter using alkylidine α-ketoester substrates as acceptors.123
Scheme 51.

Rate Increase with Catechol Additive
Continuing this work, aliphatic aldehydes were demonstrated as competent coupling partners with nitro-olefins, although trans-fluorinated catalyst B19 proved more efficient than the cis-analogue (15% yield, 74% ee) or the des-fluoro catalyst (16% yield, 74% ee).124 The surprising fluorine effect, in this case, was again found computationally to result from electrostatic interaction between the catalyst and nitroalkene (Scheme 52).
Scheme 52.

Aliphatic Aldehyde Coupling Partners
Chi demonstrated the use of enal coupling partners with modified chalcones in good yield and enantioselectivity.125 Glorius effected an enantioselective intermolecular Stetter reaction with aryl-aldehydes and methyl-2-acetamidoacrylate, also providing a single example of an aliphatic aldehyde participating in the reaction (Scheme 53).126 Unfortunately, attempts to use other 2-acetamidoacrylates were unsuccessful, instead, completely inhibiting reactivity.127 Rather than an enantioselective addition of the acyl-anion to the Michael acceptor, this approach relies on asymmetric protonation of the corresponding enolate, formed after conjugate addition of the Breslow intermediate to the Michael acceptor.
Scheme 53.

Stetter Reaction of Methyl-2-acetamidoacrylate
Elegant work by Glorius expanded the scope of Michael acceptors, using 2-substituted acrylates containing no other activating groups.127 There was a single example of a β-substituted acrylate (Z-methyloct-2-enoate) used in this reaction, with diminished, yet promising, selectivity and reactivity (59% yield, 80% ee). This represents the only reported example of an NHC catalyst effecting an enantioselective intermolecular Stetter reaction with β-substituted, unactivated Michael acceptors (Scheme 54).
Scheme 54.

Stetter Reaction with Simple Acrylates
3.4 Hydroacylation of Double and Triple Bonds
While the Stetter reaction couples aldehydes to Michael acceptors, acyl-anion reactivity has been extended to electron neutral olefins. She and Pan first demonstrated this reactivity using tethered alkyl tosylates.128 Interestingly, the authors found the selectivity changes when their native substrate 36 is substituted with a phenyl group (37) (Scheme 55).
Scheme 55.
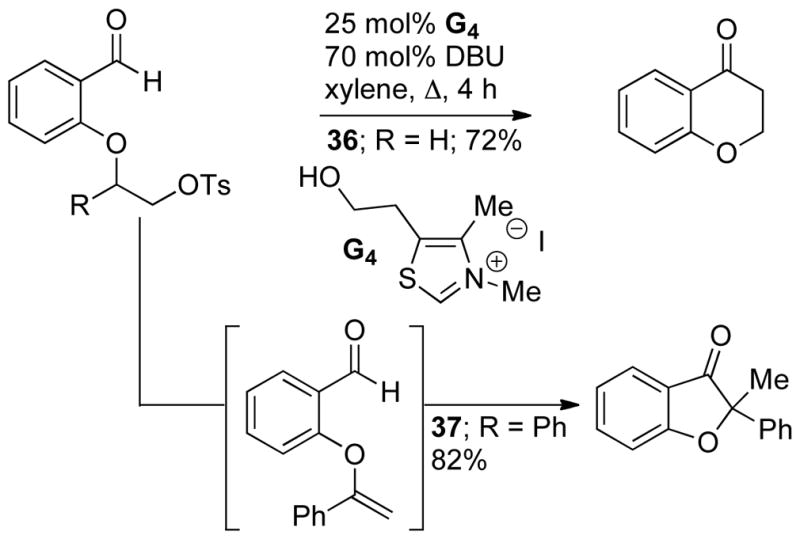
Hydroacylation Precedent
These authors later found the reaction works with enol ethers, formed by elimination of the tosyl group.129 Based on deuterium labeling studies, the authors proposed the following mechanism: after addition of the NHC to the aldehyde and subsequent proton transfer, the Breslow intermediate (38) adds to the enol ether, while the enol ether deprotonates the hydroxyl group of 38, generating tetrahedral intermediate 39. Collapse of 39 releases the observed product (40) and regenerates active catalyst (Scheme 56).
Scheme 56.

Mechanism of Hydroacylation of Enol Ethers
After these seminal reports, Glorius reported that the reaction is capable of coupling aldehydes with alkynes in good yields using thiazolium catalyst G24. When terminal alkynes are used, the resulting chromanone proved a suitable substrate for an intermolecular Stetter reaction with aromatic aldehydes (Scheme 57).130
Scheme 57.

Hydroacylation of Alkynes
In the presence of a different base and under slightly higher temperatures, Glorius found that the reaction delivers benzofuranone products, rather than the expected chromanone.131 He proposes this product arises from a retro-Michael reaction, giving the corresponding enone, which then isomerizes to diketone 41. This can then undergo Michael addition with the phenol moiety to generate the observed benzofuranone product (Scheme 58). Zeitler also reported a similar strategy using almost identical conditions to those shown in Scheme 57 to give a mixture of the chromanone and benzofuranone products. Treatment of the crude mixture with DBU and further heating converts the chromanone product to the desried benzofuranone.132
Scheme 58.

Proposed Mechanism for the Formation of Benzofuranones
In an elegant piece of work, Glorius described the enantioselective hydroacylation of tethered styrenes, using chiral triazolium A12, generating the chromanone products in good yield and with excellent selectivity (Scheme 59).133 DFT studies of this reaction were conducted and suggest the C-C bond formation, O-H deprotonation is concerted.
Scheme 59.

Enantioselective Hydroacylation of Styrenes
The mechanism of this and related transformations (vide infra) was the subject of some debate with the central issue being whether the C-C bond forming step is concerted or stepwise. DFT studies133 indicate the C-C bond forming reaction occurs in a single step, and it has been suggested the reaction is reminiscent of a Conia-ene reaction. It has also been suggested the reaction could take place by a reverse-Cope elimination type pathway, from a resonance form of the Breslow intermediate (Figure 5),134 a mechanism first proposed to account for the high diastereoselectivity in the intramolecular enantioselective Stetter reaction on trisubstituted Michael acceptors.135 Computational studies by Domingo support a two stage, single step mechanism for C-C bond formation in the intermolecular reaction.136 He proposes initial hydrogen atom transfer from the hydroxyl group of the Breslow intermediate to the terminal alkene carbon, followed by barrierless recombination of the pseudodiradical species to form the C-C bond.
Figure 5.

Proposed Single Step Mechanisms for the Hydroacylation of Alkenes
The intermolecular reaction has also been reported. Glorius showed the efficient coupling of aryl aldehydes to arynes was possible using achiral thiazolium catalyst G24.137 Shortly after, the coupling of cyclopropenes was also demonstrated, using achiral triazolium G20 with aryl aldehyde coupling partners (Scheme 60).34b
Scheme 60.

Intermolecular Hydroacylation of Cyclopropenes
120 mol% catalyst was used
The enantioselective intermolecular hydroacylation of cyclopropenes was demonstrated.138 Although the achiral mesityl catalyst G20 was efficient at mediating the racemic reaction, using chiral mesityl catalysts delivered the expected cyclopropanes in low yields, but with promising enantioselectivity. In order to increase the efficiency of this reaction, Glorius designed chiral catalyst A17, bearing a dimethoxy phenyl ring, which vastly increased the yield of the desired product, and proved beneficial to selectivity as well (Scheme 61).
Scheme 61.

Enantioselective Hydroacylation of Cyclopropenes
Recently, Glorius demonstrated the reaction with less activated styrenes, using achiral dimethoxyphenyl triazolium G27.139 The linear product is favored over the possible branched product, but to varying degrees. Some substrates, especially electron-rich ones, selectively form the branched product (Scheme 62).
Scheme 62.

Hydroacylation of Styrenes
4. CATALYSIS INVOLVING EXTENDED BRESLOW INTERMEDIATES
As seen above, α,β-unsaturated aldehydes are often competent coupling partners in prototypical acyl anion reactivity such as the Stetter reaction, but they also have other unique reactivity compared to aryl or aliphatic aldehydes. The typically electrophilic β-carbon of the enal can become nucleophilic, as a result of the conjugated alkene, and react in an a3 to d3 umpolung, generating β-functionalized carbonyl compounds. The mechanism for this type of transformation is shown in Scheme 63. The free carbene first adds in a 1,2-fashion to an enal to form tetrahedral intermediate 42, which undergoes a proton transfer to generate the extended Breslow intermediate 43, typically referred to as the homoenolate equivalent. This homoenolate equivalent is in resonance with carbanion 44, where negative charge is localized three atoms away from the azolium, demonstrating the origin of the nucleophilicity of that carbon. The carbanion can then add to an electrophile to give enol azolium 45, which can then tautomerize to acylazolium 46. Nucleophilic substitution of 46 then generates the product 47 and regenerates the NHC catalyst.
Scheme 63.

Prototypical NHC-Catalyzed Homoenolate Reaction Pathway
Walia, Rao, and Singh first reported this type of umpolung reactivity in the context of a cyanide-catalyzed transformation in 1964.140 It was found that α,β-unsaturated aldimines are transformed to the corresponding saturated amide in the presence of a catalytic amount of cyanide in water. A subsequent report showed that enals afford the corresponding saturated methyl esters when treated with a catalytic amount of cyanide in methanol (Scheme 64).141
Scheme 64.
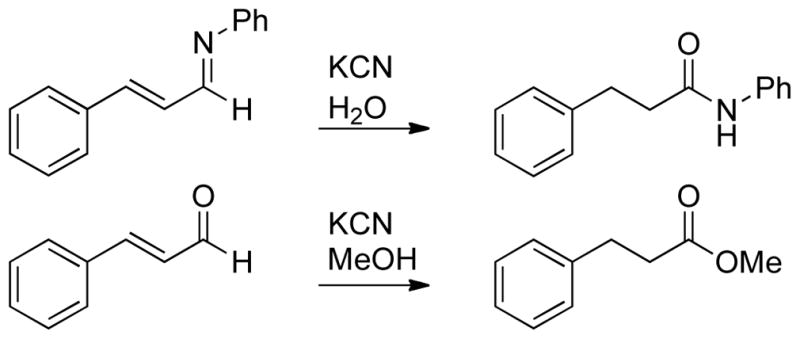
Cyanide Catalyzed a3 to d3 Umpolung
The cyanide-catalyzed formation of saturated esters is believed to follow a similar path as the NHC catalyzed a3 to d3 umpolung shown in Scheme 63. Cyanide initially adds to the aldehyde forming tetrahedral intermediate 48, which then undergoes proton transfer to form carbanion 49. This is in resonance with carbanion 50 that can be protonated to generate enol 51, which then tautomerizes to acyl cyanide 52. Methanol addition to 52 releases methyl β-phenylpropionate 53 and the active cyanide catalyst (Scheme 65).
Scheme 65.
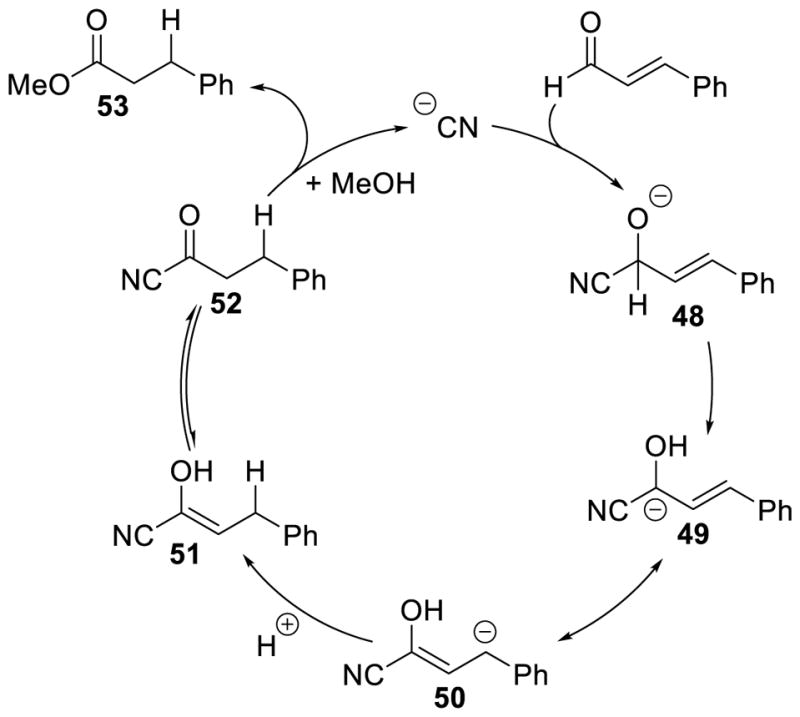
Mechanism of the Cyanide Catalyzed β-Protonation of Enals
This type of reactivity lay dormant until 40 years later when Bode142 and Glorius143 independently reported the NHC-catalyzed variant of the homoenolate reaction in 2004. Since these seminal reports, the field of NHC-catalyzed homoenolate reactivity has blossomed, leading to a plethora of methods to synthesize useful molecules from simple enal starting materials.
4.1 Annulation Reactions
4.1.1 Oxygen Heterocycle Synthesis
Glorius143 and Bode142 reported the first examples of NHC catalyzed homoenolate reactivity in the context of an annulation reaction between enals and aryl aldehydes affording γ-lactone products. Diarylimidazolium precatalysts proved most efficient while thiazolium salts give undesired benzoin side products. This reaction is generally accepted to proceed via initial generation of extended Breslow intermediate 54 from the carbene and enal, which can then add [1,2] across the aryl aldehyde giving enol azolium 55. Tautomerization of 55 gives acylazolium 56, which then cyclizes to deliver the γ-lactone product and liberate the catalyst (Scheme 66).
Scheme 66.

Proposed Mechanism for γ-Lactone Formation
Using this methodology, Bode showed a variety of aryl and propargyl enals are competent coupling parners with aryl aldehydes, forming the desired γ-lactone products 57 in moderate to good yields (41 to 87%) and moderate diastereoselectivity (3:1 to 5:1 dr) (Scheme 67).
Scheme 67.

Bode’s Annulation of Enals and Aryl Aldehydes
Glorius’ protocol allows for the coupling of aryl enals with benzaldehyde derivatives to form γ-lactones in similar yields (32 to 70%) and diastereoselectivity (3:1 to 4:1 dr). This protocol also incorporated aryl-trifluoromethyl ketone substrates, providing yields from 74 to 92% and diastereoselectivity from 2:1 to 3:1 (Scheme 68). Glorius further demonstrated the use of chiral triazolium C2 delivers products 58a and 58b in 12 and 25% ee, respectively (Scheme 69).
Scheme 68.

Glorius’ Annulation of Enals and Aryl Aldehydes
Scheme 69.

Enantioselective Lactonization with Trifluoromethyl Ketones
Scheidt and Cohen used a different strategy to render this reaction asymmetric, relying on a chiral Lewis acid in the presence of an achiral NHC.144 Cinnamaldehyde undergoes dimerization to γ-lactone products in 60% yield, 20:1 dr, and 60% ee in the presence of Taddol-based titanium complex 59, imidazolium precalyst G7, and DBU (Scheme 70).
Scheme 70.

Ti-Taddol/NHC Cooperative Catalysis
Scheidt subsequently reported an enantioselective homoenolate addition to acyl phosphonates to generate γ-lactone 60.145 In a collaboration with the Cheong group, computer modeling led to the identification of chiral NHC D7 as the optimal catalyst for the reaction. When catalyst D7 and MTBD (7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene) were used experimentally, the products form in good yields (54 to 93%) and enantioselectivity (78 – 91% ee), but with modest diastereoselectivity (up to 3:1 dr) (Scheme 71).
Scheme 71.

Enantioselective Acyl Phosphonate Annulation
In 2006, Nair and coworkers reported the homoenolate addition of enals to 1,2-dicarbonyl compounds to synthesize γ-lactones.146 Cyclohexanedione 61 and isatins 62 are competent electrophiles for this process, as well as electron-rich or electron-deficient enals. Yields for the spiro-cyclohexanone products range from 60–74% while spiro-oxindole γ-lactones are formed in 85–98% yield. Unfortunately, the diastereoselectivity remained low (1:1) (Scheme 72). The scope of this methodology was later expanded to include diaryl diones.147 Since this report several other methods have been reported rendering this reaction asymmetric.148 Most notably, Scheidt coupled cinnamaldehyde derivatives with isatins in 70 – 93% yield, 1.6:1 – 20:1 dr, and 86 – 99% ee.149 The authors further provided one example of an aliphatic enal (trans-butenal) participating in the reaction to give the product in 76% yield, >20:1 dr, and 78% ee using chiral triazolium A14.
Scheme 72.
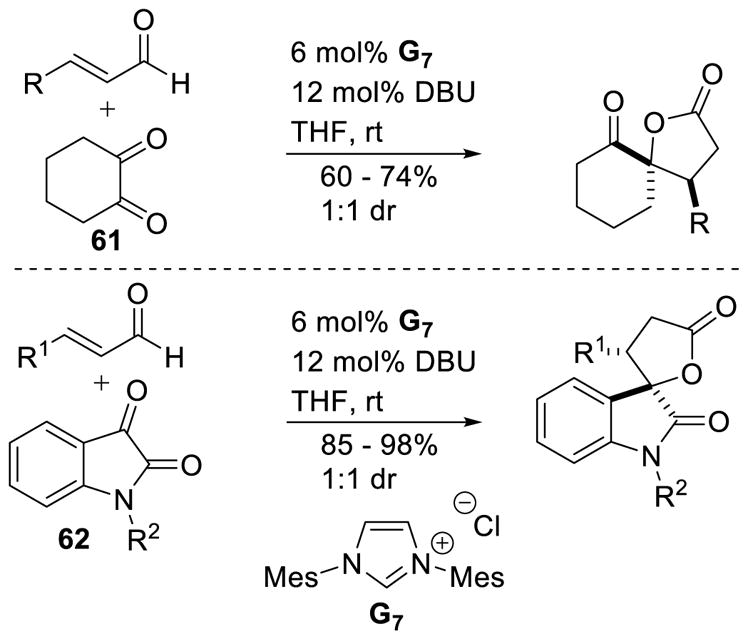
Synthesis of Spirocyclic-γ-Lactones
In 2008, You showed that glyoxylate derivatives 63 are also capable of undergoing the annulation reaction.150 A variety of chiral NHC catalysts were investigated and NHC ent-A15 was shown to provide the best enantioselectivity, giving up to 78% ee for the trans isomer, although the diastereoselectivity remained low (1.5:1 dr). This could be improved with chiral catalyst ent-A4 (up to 5:1 dr), but leads to an erosion of enantioselectivity (Scheme 73).
Scheme 73.

Enal Addition to Glyoxylates
Glorius reported the use of sterically hindered β,β-disubstituted enals in a dual catalytic NHC/Brønsted acid manifold to generate spirocyclic lactones 64.151 The reaction tolerates aliphatic, aryl and dienyl enals, as well as a variety of substitution patterns on the isatin component. Interestingly, the authors found employing a Brønsted acid in the reaction increases both the yield and diastereoselectivity, a strategy that had been previously demonstrated beneficial to imine annulations by Rovis152 (vida infra). Glorius proposes the acid prearranges the transition state via a hydrogen bonding network while simultaneously activating the isatin (Scheme 74).
Scheme 74.

Dual Catalytic NHC/Brønsted Acid Protocol for Spiroxindole Synthesis
Larger ring sizes can also be generated using NHC-catalyzed homoenolate processes. Ye reported an enantioselective formal [3+4] annulation between enals and o-quinone methides in 2013.153 While both aryl and aliphatic enals participate in the reaction, the scope was limited to dioxolane-fused o-quinone methides bearing an aryl substituent off the exo-olefin. This protocol addresses the potential [3+2] side reaction by taking advantage of the driving force of rearomatization from the [3+4] reaction. Yields range from 79 – 97%, diastereoselectivity is modest to excellent (3:1 to >20:1 dr), and enantioselectivity is high (81–98% ee) (Scheme 75).
Scheme 75.

NHC-catalyzed [3+4] Annulation
Nair also discovered an NHC-catalyzed annulation of enals and tropone.154 Termed an [8+3] annulation, the reaction likely proceeds by conjugate addition to the more electrophilic position followed by cyclization of the resultant alkoxide. The reaction affords the fused δ-lactone 65 in 39–62% yield (Scheme 76).
Scheme 76.

[8+3] Annulation of Tropone
4.1.2 Nitrogen Heterocycle Synthesis
The homoenolate intermediate has also been exploited in a number of cases to generate nitrogen containing heterocycles. Bode and He first demonstrated this reactivity in 2005 with the homoenolate addition of enals to imines.155 The reaction tolerates a broad scope of aromatic enals and aryl imines 66 to generate γ-lactams in good yields (5 – 73%) and modest to good diastereoselectivity (1.7:1 to 10:1 dr) (Scheme 77). However, the scope of the imine N-substituent was limited to a 4-methoxyphenylsulfonamide. When replaced with a more common tosyl substituent, the carbene irreversibly adds to the imine deactivating the catalyst.
Scheme 77.

Homoenolate Addition to Sulfonylimines
Later work by Bode overcame some of the issues associated with the acyclic sulfonylketimines by instead using a cyclic variant.156 Aryl and aliphatic enals are tolerated in the reaction, as well as a wide variety of saccharin-derived ketimines bearing both aryl and aliphatic groups; yields of 67 range from 55–95% with diastereoselectivity varying from 1:1 to >20:1 dr. Remarkably aryl-enals require just 0.5 mol% of catalyst for quantitative yield at room temperature (Scheme 78).
Scheme 78.

Cyclic Sulfonylketimine Scope
The large enhancement in reactivity was attributed to an alternative reaction mechanism where hydrogen bonding between the sulfonyl oxygen and the hydroxyl group of the initial carbene-aldehyde adduct form a prearranged transition state 68. From this transition state, an ene-like reaction can occur, presumably facilitating proton transfer from the formal acyl-proton to the imine nitrogen with concomitant C-C bond formation generating enol azolium 69. Tautomerization to the acylazolium is followed by intramolecular trapping with nitrogen to furnish γ-lactam 67 (Scheme 79). However, a more traditional homoenolate pathway proceeding via an open transition state cannot be discounted. The authors’ also demonstrated the reaction with chiral NHC A15, giving product in an encouraging 91% yield, 73% ee, and 6:1 dr (Scheme 80).
Scheme 79.

Ene-like Transition State of Cyclic Sulfonylketimine
Scheme 80.

Enantioselective Cyclic Sulfonylketimine Annulation
Bode also demonstrated the homoenolate is capable of effecting a 1,4-addition to α,β-unsaturated N-sulfonyl ketimines to generate cyclopentane fused β-lactams 70.157 This reaction is notable because it favors β-lactam formation, despite competing enal dimerization and hetero Diels-Alder pathways. The scope is broad including both aryl and aliphatic enals, but requires the use of diaryl N-sulfonyl ketimines. Yields are moderate to excellent (45–94%), diastereoselectivity is good to excellent (5:1 to >20:1), and enantioselectivity is uniformly high (88–99% ee) (Scheme 81).
Scheme 81.

Scope of β-Lactam Formation
Bode proposed this reaction proceeds via a cross aza-benzoin/oxy-Cope rearrangement cascade where the Breslow intermediate adds to the ketimine, followed by an oxy-Cope rearrangement to furnish enolate 71. This enolate then adds to the imine via a Mannich reaction and the nitrogen anion cyclizes on the acyl azolium, liberating catalyst and producing the β-lactam product (Scheme 84). However, it is possible that the homoenolate 72 adds 1,4 to the ketimine directly furnishing intermediate 71a under a more traditional NHC-homoenolate pathway (Scheme 82).
Scheme 84.

Scheidt’s N-Acyl Hydrazone Annulation
Scheme 82.

Aza-Benzoin-Oxy-Cope Rearrangement Mechanism Proposed by Bode
Scheidt and Chan showed that diazenes are competent electrophiles for the NHC-generated homoenolate to afford pyrazolidinones.158 Electron rich or deficient aryl as well as aliphatic enals are tolerated, yet the diazene component is limited to electron rich aromatic substituents. The reaction was rendered asymmetric with the use of chiral NHC A15 providing pyrazolidinone 73 in 61% yield and 90% ee (Scheme 83).
Scheme 83.

Synthesis of Pyrazolidinones
Scheidt further demonstrated N-acyl hydrazones are competent coupling partners for the homoenolate in a cooperative Lewis acid/NHC manifold.159 It is proposed that the Mg(OtBu)2 coordinates to the acyl oxygen and hydrazone nitrogen, activating the acyl hydrazone toward nucleophilic addition. The addition of Mg(OtBu)2 also allowed for lower catalyst loading. Using 5 mol % triazolium ent-A7 in the presence of 15 mol % 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), the authors’ demonstrated aliphatic and aryl enals deliver γ-lactam products in 61–85% yield, 85–97% ee, and 5:1 up to >20:1 dr (Scheme 84).
Rovis and co-workers developed a cooperative Brønsted acid/NHC catalytic system to generate γ-lactams from enals and aza-dienes.152 It is proposed that a small amount of weak base 74 deprotonates triazolium salt B22 to form the free carbene, which then adds to the enal generating the extended Breslow intermediate. The conjugate acid of 74 then protonates aza-diene 75, making it more electrophilic (Scheme 85). Evidence for this mode of activation was provided by the observation of asymmetric induction when a chiral carboxylate base was used in the presence of an achiral NHC catalyst. Interestingly, this methodology delivers the trans γ-lactam in contrast to the products formed by the methods of Bode and Scheidt.
Scheme 85.

Rovis’ γ-Lactam Synthesis
Scheidt and Chan reported azomethine ylides are capable coupling partners for the NHC-generated homoenolate in a formal [3+3] to provide pyridazinones 76.160 A variety of electron-rich, aliphatic, and dienyl enals participate in the reaction. The imine tolerates electron rich and electron poor substituents, but enolizable and 2-substituted aryl imines do not participate. The reaction proceeds with good yields and excellent diastereoselectivity in all cases. The high diastereoselectivity is attributed to a hydrogen bond between the Breslow intermediate 77 and azomethine ylide 78 which preorganizes the transition state for a syn-addition (Scheme 86).
Scheme 86.

Azomethine Ylide Annulation
In a related [3+3] annulation, Scheidt showed nitrones readily react with the homoenolate to generate heterocyclic lactones.161 The NHC-generated homoenolate intercepts the nitrone and then tautomerizes to the acylazolium. Intramolecular addition of the nitrone to the acylazolium then generates lactone 79, which undergoes alcoholysis to generate linear ester 80. This reaction efficiently delivers product in 69–80% yield, 81–93% ee, and 20:1 dr. Aliphatic and aryl enals are tolerated, but the nitrone moiety is limited to aryl substitution at carbon and nitrogen (Scheme 87).
Scheme 87.

Homoenolate Addition to Nitrones
Nitroso compounds have also been shown to be viable acceptors in homoenolate processes. Ying first demonstrated this reactivity in 2008 with an isoxazolidinone synthesis via the coupling of enals and nitrosobenzene.162 The reaction is believed to proceed via an NHC-generated homoenolate equivalent adding to the nitrogen of nitrosobenzene to give intermediate 81, which then tautomerizes to acylazolium 82. Interception of 82 by the alkoxide affords the isoxazolidinone product 83 (Scheme 88).
Scheme 88.

Mechanism of Isoxazolidinone Formation
The oxazolidinone product could be further elaborated to the β-amino ester upon treatment with methanol and acid. The β-amino ester is isolated in yields ranging from 30–85%. Aromatic enals are efficient coupling partners, while aliphatic aldehydes participate in slightly diminished yields (Scheme 89). Recently, Takemoto used this same catalyst to synthesize 3,3-disubstituted indolin-2-thiones from a tethered enal and isothiocyanate in 43 – 78% yield.163
Scheme 89.

Scope of the Nitroso Coupling Reaction
In a fascinating extension of this reactivity, nitroso compounds were shown to couple with the homoenolate equivalent via a formal [4+3] annulation.164 Mechanistically, this reaction is thought to proceed in the same manner as the [3+2] isoxazolidinone formation above, but then undergoes 1,2-Bamberger-type rearrangement to furnish the 7-membered lactone 84 (Scheme 90). Electron-rich and electron-deficient aryl and heteroaryl enals are tolerated in the reaction (45 – 81% yield), but the nitroso coupling partner is limited to 1-methyl-4-nitrosobenzene (Scheme 91).
Scheme 90.

Proposed Mechanism of Nitroso [4+3] Annulation
Scheme 91.

Scope of the NHC-catalyzed Nitroso [4+3] Annulation
Siddiqui reported an interesting cascade reaction in 2013, synthesizing 1,3-diazapane derivatives 85 via a three-component coupling of enals, aryl aldehydes, and ureas.165 The reaction is proposed to proceed by initial condensation of the aryl aldehyde with the urea to form intermediate 86, which then couples to the NHC-generated homoenolate to furnish 87. After proton transfer and tautomerization, the distal urea nitrogen can cyclize onto the acylazolium, generating the 1,3-diazapane product 88 (Scheme 92). The scope of this reaction proved tolerant of a variety of aryl enals, aryl aldehydes, and ureas (including thioureas). Yields range from 65–91% and diastereoeselectivity is generally excellent (>20:1) (Scheme 93).
Scheme 92.

Mechanism of 1,3-Diazapane Synthesis
Scheme 93.

Scope of 1,3-Diazapane Synthesis
4.1.3 Carbocycle Synthesis
Im addition to heterocycle synthesis, the NHC-generated homoenolate has also been utilized in the generation of five-membered carbocycles. Nair first introduced this reactivity in 2006, coupling enals with chalcones to furnish 1,3,4-trisubstituted cyclopentenes.166 The accepted mechanism for this transformation begins with formation of the extended Breslow intermediate followed by a 1,4 addition of the homoenolate to the chalcone furnishing intermediate 89. Tautomerization of 89 leads to ketone 90, which then undergoes an aldol reaction with the enol-azolium to provide alkoxide 91. Cyclization of the alkoxide onto the acylazolium liberates the active catalyst and furnishes β-lactone 92, which decarboxylates to provide the observed cylopentene product (93) (Scheme 94).
Scheme 94.

Mechanism of Cyclopentene Formation
A variety of aryl enals and chalcones are competent partners in this reaction with generally good yields (55 to 88%) and excellent diastereoselectivity; only the trans diastereomer is observed in all cases. Importantly, aliphatic substitution is tolerated on each partner (Scheme 95).
Scheme 95.

Nair’s Cyclopentene Synthesis
Interestingly, Nair also demonstrated the acylazolium can be intercepted by an external alcohol to give cyclic ester 94 or straight chain ester 95.167 The yields in this reaction range from 57–69%, with products obtained as single diastereomers, in product ratios from 2:1 to 5:1 (94:95). However, the scope of this reaction is limted to aryl enals and bis-aryl enones (Scheme 96).
Scheme 96.
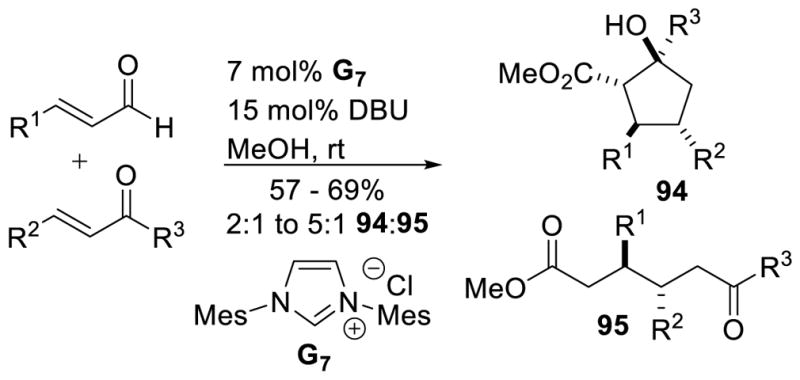
Interception of Acylazolium with Methanol
The reaction of dialkylidine ketones with enals has also been demonstrated, generating cyclopentanone and cyclopentene products.168 The origin of product selectivity is believed to arise from C-acylation of the acylazolium leading to the cyclopentanone product (Scheme 97), while the cyclopentene product results from an intramolecular aldol / decarboxylation pathway (vide supra). The product selectivity appears to be substrate controlled, but generally gives a distribution of up to 2:1, favoring the cyclopentene product (Scheme 98).
Scheme 97.

Mechanism of Cyclopentanone Formation
Scheme 98.

Cyclopentanone vs Cyclopentene Formation
Shortly after Nair’s seminal report,166 Bode reported an enantioselective cyclopentene forming reaction, coupling enals to 4-oxoenoates.169 Bode’s reaction is notable because it provides the cis-diastereomer, in contrast to the methodology reported by Nair which delivers the trans product. The authors propose the reaction proceeds via an intermolecular cross aldehyde-ketone benzoin reaction followed by an NHC-promoted oxy-Cope rearrangement (Scheme 99).
Scheme 99.

Bode’s Proposed Mechanism for Cyclopentene Formation
Similar to Bode’s work, Scheidt and co-workers demonstrated that selective formation of the cis-cyclopentene isomer, from the same starting materials as in Nair’s reaction, can be achieved using titanium isopropoxide as a Lewis acid co-catalyst with a chiral NHC, but the reaction delivers enantiomeric product from the same antipode of aminoindanol catalyst.170 The authors argue that pre-organization of the s-cis transition state 99 via coordination of the enal and chalcone oxygens to the Lewis acid is operative (Scheme 101).
Scheme 101.
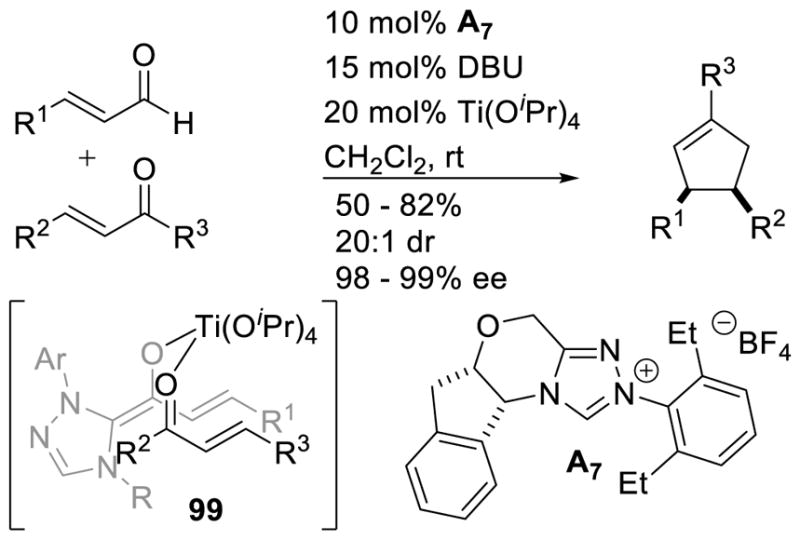
Dual Lewis Acid/NHC Mediated Cyclopentene Formation
In 2011, Scheidt illustrated that cinnamaldehyde derivatives and α-ketoesters participate in an NHC/Lewis acid annulation to form cyclopentanols 100.171 These products are obtained in good yields (52–85%), moderate to excellent diastereoselectivity (5:1 to 20:1 dr), and excellent enantioselectivity (91–99% ee) (Scheme 102).
Scheme 102.

Cyclopentanol Formation
The mechanistic nuances in this reaction have been the subject of some debate. It is possible this reaction proceeds via initial 1,4-addition of the homoenolate to the chalcone providing direct access to intermediate 98 (Scheme 100), as argued by Scheidt.170 However, when chalcones are subjected to the same conditions as the 4-oxoenoates, the trans-cyclopentene forms selectively with diminished enantioselectivity (55% ee – not shown), while the cis-cyclopentene forms in 99% ee.169 Bode argues that the differential stereochemical outcomes between 4-oxoenoates and chalcones, coupled with the diminished levels of enantioselectivity in the trans-product suggest the benzoin-oxy-Cope pathway is operative for the 4-oxoenoates. Furthermore, it is postulated that oxoenoates prefer to react through an s-cis conformation 101 whereas chalcones prefer to react through an s-trans conformation 102, providing stereochemical divergence (Figure 6).
Scheme 100.

Enantioselective Synthesis of Cyclopentenes
Figure 6.
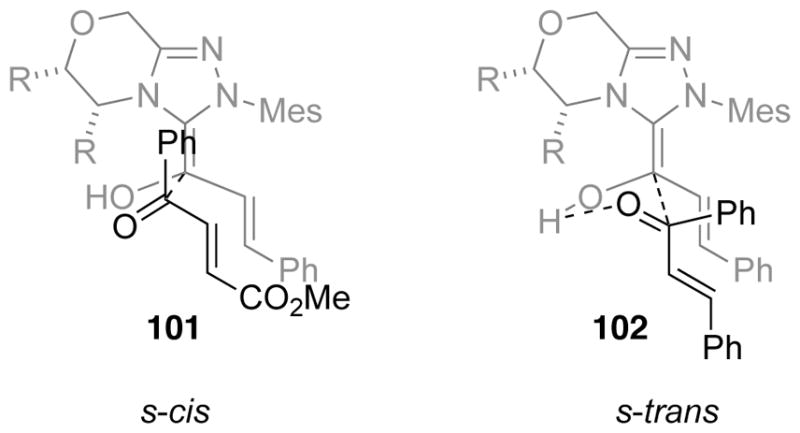
s-cis vs s-trans Transition States
Recently, Nair and co-workers employed tethered enal/chalcone substrates in the cyclopentene reaction to generate cyclopentene-fused macrocycles 103.172 This methodology allows for the formation of 10, 11, 12, and 13 membered macrocycles. Yields are moderate to good with excellent diastereoselectivity across all substrates (Scheme 103).
Scheme 103.

Macrocyclization via Cyclopentene Synthesis
Glorius demonstrated an NHC-catalyzed carbocyclic annulation reaction furnishing spirocycles 104 from aurones and aza-aurones in a formal [3+2] annulation.173 This is believed to follow initial conjugate addition of the homoenolate to the Michael acceptor. The resultant enol azolium tautomerizes to the acylazolium, and after cyclization of the pendant enolate liberates free carbene and furnishes the desired product (Scheme 104).
Scheme 104.

Spirocycle Formation from Aza-aurones
This protocol is tolerant of aryl and aliphatic enals, as well as aza-aurones bearing a variety of substitution patterns on the aryl ring and the β-carbon of the Michael acceptor. The yields in this transformation range from 42 to 83%, diastereoselectivity is generally good (3:1 to >20:1 dr), and enantioelectivity ranges from 88 to 94 % ee (Scheme 105).
Scheme 105.

Scope of Spirocyclic [3+2]
4.2 Non-Annulative Processes
4.2.1 β–Functionalization of Enals
NHC-catalyzed homoenolate reactions are not limited to annulative processes. In 2005, Scheidt and Bode independently reported the first β-functionalization of enals that did not result in an annulation, demonstrating enals can undergo β-protonation to generate an acylazolium.174,175 In 2009, Nair and co-workers demonstrated the NHC-generated homoenolate addition to nitroalkenes to furnish δ-nitroesters.176 Mechanistically, the reaction is believed to proceed via 1,4-addition of the extended Breslow intermediate to the nitroalkene. The resultant nitronate 105 is protonated and the enol azolium tautomerizes to form acylazolium 106. The acylazolium is then intercepted by methanol to furnish product and liberate catalyst (Scheme 106). A variety of aryl enals and nitrostyrene derivatives readily participate in the reaction with yields ranging from 40–70% and diastereoselectivity ranging from 3:1 to 15:1 in favor of the anti isomer (Scheme 107).
Scheme 106.

Catalytic Cycle for δ-Nitroester Formation
Scheme 107.

Nair’s δ-Nitroester Methodology
Liu and Rovis reported enantioselective variations of this reaction in 2012 and 2013, respectively.177 Liu and co-workers found chiral NHC ent-A7 provides product in 48–86 % yield, 81–99 % ee, and 4:1 to 12:1 dr favoring the anti isomer.177a A variety of nitrostyrene derivatives, including nitro dienes and nitroenynes, were coupled with aryl and aliphatic enals (Scheme 108).
Scheme 108.

Liu’s δ-Nitroester Methodology
Rovis’ protocol, which tolerates aliphatic and aryl nitroalkenes as well as aryl enals, is unique in that it provides access to the syn isomer, directly contrasting the work of Nair and Liu where the anti isomer forms selectively.177b Rovis also reported a one-pot protocol for the synthesis of δ-lactams via a reductive workup of the δ-nitroester (Scheme 109).
Scheme 109.

Rovis’ δ-Nitroester and δ-Lactam Methodology
The dichotomy in diastereoselectivity between Liu and Rovis is proposed to arise from an inversion in Breslow intermediate geometry. Thus, while Liu and Rovis use pseudo-enantiomeric catalysts, C-C bond formation occurs from the same enantiotopic face of the enal. An inversion in Breslow intermediate geometry would allow a simple explanation of this stereochemical anomaly. Further, it is proposed that Liu’s reaction proceeds via an open-transition state 107 to afford the anti diastereomer, while Rovis’ method proceeds through a closed transition state 108 giving the syn product (Scheme 110).
Scheme 110.

Proposed Transition States for Nitroester Formation
4.3 Alternate Access to Homoenolate Reactivity
In 2009, Bode reported that α-hydroxy enones are efficient bench-stable surrogates of enals for NHC-catalyzed homoenolate additions to various electrophiles.178 A limitation is that the increased steric demand of these substrates inhibits their use with bulky chiral catalysts. In 2013, Chi and co-workers demonstrated that saturated esters are potential homoenolate precursors.179 This reactivity is notable because it functionalizes a traditionally non-reactive β-carbon of a saturated ester. The reaction is proposed to proceed by initial addition of the carbene to the electron deficient aryl ester generating acylazolium 109, which then tautomerizes to enolate azolium 110. This intermediate can then undergo a proton transfer from the β-carbon to the enolate oxygen furnishing extended Breslow intermediate 111 (Scheme 111).
Scheme 111.

Mechanism for β-Functionalization of Saturated Esters
Using this methodology, cyclopentene products form in 8–76% yield, 5:1 to 17:1 dr, and 82–96% ee. Aliphatic and aryl esters are tolerated as the homoenolate precursor and bis-aryl enones are used for the Michael acceptor. γ-Lactones may be synthesized using this methodology by coupling CF3/aryl ketones with hydrocinnamates to furnish products in 29–80% yield, 68–92% ee, and 1.3:1 to 4.5:1 dr. This method was also used to synthesize nitrogen heterocycles. γ-Lactams are formed in 55–76% yield, 90–96% ee, and 4:1 to 7:1 dr (Scheme 112).
Scheme 112.

Scope of β-Activation of Aliphatic Esters
4.4 Single-Electron Pathways
In 2008, Studer reported the first example of the Breslow intermediate undergoing a single electron oxidation, demonstrating the NHC-catalyzed TEMPO oxidation of aldehydes to esters (cf. section 5.1.3).180 Enals, on the other hand, offer the potential to oxidize the β-position leading to β-oxidized carbonyls. Rovis discovered a single electron oxidation of the Breslow intermediate, reporting the β-hydroxylation of enals using electron-deficient nitroarene oxidants.181 Aryl and aliphatic enals were found capable of engaging in an oxygen atom transfer from an aryl-nitro group to the β-position of an enal in the presence triazolylidenes. Mechanistically, it is believed the Breslow intermediate transfers a single electron to the nitroarene to generate Breslow derived radical cation 112 and nitroarene centered radical anion 113. A resonance structure exists which places the radical on the oxygen of the nitro group. This oxygen centered radical can then combine at the β-position of the enal to generate 114, which can then collapse to expel nitroso compound 115. The subsequent acylazolium is then intercepted with methanol to provide the β-hydroxy ester 116 and regenerate the carbene catalyst. The major side-product of the reaction is the two-electron oxidation of the Breslow intermediate to the unsaturated acylazolium. It is hypothesized this product forms via deprotonation of the hydroxyl group on intermediate 112, followed by a second single electron oxidation (Scheme 113).
Scheme 113.

Proposed Mechanism of β-Hydroxylation of Enals
Aliphatic enals are competent in this reaction providing the desired product in generally good yields, while aryl enals participate with lower yields. Overall, the β-hydroxylation products are isolated in 20–74% yield, with enantioselectivity ranging from 63–92% ee. This protocol is tolerant of protected amines as well as ether functional groups. Tri-substituted enals are also competent, albeit with diminished yield and selectivity (40%, 63% ee) (Scheme 114).
Scheme 114.

Scope of β-Hydroxylation
Isolation of nitroso derivatives from the reaction medium implicate the nitro-group as the source of oxygen in this reaction. As a result of the puzzling reactivity, Rovis and White performed several mechanistic studies. A stereochemical convergence was noted when either the cis or trans enal was subjected to the reaction conditions at different temperatures. At room temperature, the cis enal gives the opposite enantiomer of product as when the trans enal is employed. However, at 65 °C, both the cis and trans enals give the same enantiomer of product. The use of stoichiometric catalyst with the cis enal at 65 °C gives nearly the same result as the catalytic reaction, suggesting thermal isomerization of the cis enal does not outcompete the productive carbene catalyzed process (Scheme 115).
Scheme 115.

Stereochemical Probe
Concurrently, Chi reported a related strategy for the dimerization of nitroalkenes,182 wherein it is proposed the nitroalkene acts a single electron oxidant to generate radical anion 117 and Breslow centered radical cation 118. At this time, the Breslow radical undergoes a deprotonation to generate neutral radical 119. The nitroalkene derived radical anion couples with another equivalent of nitroalkene to generate radical anion 120. Radical anion 120 abstracts a second single electron from neutral Breslow radical and accepts two protons to furnish product and to generate acylazolium 121. The acyl azolium is then intercepted by methanol to regenerate catalyst and liberate ester product (Scheme 116). This mechanistic proposal is supported by EPR analysis of the nitroalkene centered radical anion.
Scheme 116.

Proposed Mechanism of Nitroalkene Dimerization
Electron-rich and electron-poor aryl and aliphatic nitroalkenes undergo the dimerization with yields ranging from 33–92% and dr ranging from 2:1 to 9:1. β,β-disubstituted nitroalkenes also participate in this reaction. Aryl aldehydes are required as the electron donor in this reaction (Scheme 117).
Scheme 117.

Scope of Nitroalkene Dimerization
5. CATALYSIS INVOLVING ACYLAZOLIUM INTERMEDIATES
5.1 Reactions of α,β-Unsaturated Acylazoliums
In addition to umpolung chemistry, NHCs have also been shown as competent catalysts in a variety of non-umpolung processes. Notably, NHC-bound acylazolium and azolium enolate intermediates have attracted considerable attention over the last decade.183 As in the case of NHC catalyzed umpolung reactions, the origins of α,β-unsaturated acylazoliums is biomimetic. In a set of cleverly designed experiments, Townsend and co-workers showed a thiamine diphosphate (ThDP, vitamin B1)-derived α,β-unsaturated acylazolium ion 123 is an intermediate in the biosynthesis of clavulanic acid (122), a potent β-lactamase inhibitor (Scheme 118).184
Scheme 118.

Biomimetic Origins of α,β-Unsaturated Acylazolium Reactivity
The most commonly used method to access NHC-derived α,β-unsaturated acylazolium intermediates (e.g. 124) relies on internal redox activation of α-oxidizable aldehydes (e.g. ynals 125 and α-bromoenals 126) with N-heterocyclic carbenes (Scheme 119, method A).185,186 Alternative methods include the addition of nucleophilic carbenes to α,β-unsaturated acyl fluorides 127,187a,b esters 128,187a,c in situ generated mixed anhydrides (method B),187d and α,β-unsaturated aldehydes 129 in conjunction with an external oxidant (method C).188, 180
Scheme 119.

Access Routes to α,β-Unsaturated Acylazolium Ions
5.1.1 Generation From Ynals and α,β-Unsaturated Acyl Fluorides
In 2006, Zeitler reported the NHC catalyzed generation of α,β-unsaturated acylazoliums, converting ynals 125 to α,β-unsaturated esters 130 (Scheme 120).185a The reaction tolerates aryl and aliphatic ynals as well as a range of primary alcohols, though secondary alcohols give lower yields.
Scheme 120.

Synthesis of α,β-Unsaturated Esters from Ynals
The proposed mechanism for this transformation is shown in Scheme 121. N-heterocyclic carbene 3 first adds to the carbonyl of ynal 125 forming the corresponding tetrahedral intermediate, which can then tautomerize to the unsaturated Breslow intermediate 131. Protonation of 131 affords the allenol, which tautomerizes rapidly into the α,β-unsaturated acylazolium ion 132. Interception of 132 by an alcohol gives the desired α,β-unsaturated ester.
Scheme 121.
Postulated Mechanism for the NHC-Catalyzed Synthesis of α,β-Unsaturated Esters
Scheidt and co-workers reported a similar methodology for the NHC-promoted synthesis of α,β-unsaturated esters 133 from ynals 125 (Scheme 122).185b,c As noted previously by Zeitler, the choice of catalyst had a pronounced effect on the yield and stereoselectivity of the reaction. A combination of imidazolium salt G7 with a bulky proton source (BHT = 2,6-di-tert-butyl-4-methylphenol) is optimal.
Scheme 122.

Effect of Carbene Structure on the Synthesis of α,β-Unsaturated Esters
In addition to their susceptibility towards interception by alcohols in a direct 1,2-fashion, α,β-unsaturated acylazoliums can also undergo 1,4-addition of enolic carbon-based nucleophiles. Lupton187a,b and Bode185d independently reported the first examples of these “Claisen-type” reactions. Lupton and co-workers propose a carbene-induced fragmentation of α,β-unsaturated enol esters to the corresponding acylazolium/enolate ion pair with subsequent recombination forming 2,3-dihydropyranones (Scheme 123). Aromatic, heteroaromatic, and aliphatic α,β-unsaturated esters all react smoothly to give the corresponding dihydropyranones. β-Disubstituted acylazoliums are good substrates as well, allowing for an efficient construction of quaternary carbon centers. Lupton and co-workers were also successful in accessing similar reactivity from a combination of α,β-unsaturated acyl fluorides and TMS enol ethers (Scheme 124).
Scheme 123.

NHC-Catalyzed Rearrangement of α,β-Unsaturated Enol Esters
Scheme 124.

Scope of the Intermolecular NHC-Catalyzed Michael Addition
The authors proposed a mechanism similar to the one previously proposed for enol esters (Scheme 125). Carbene 16 initially intercepts the acyl fluoride liberating a fluoride ion and generating α,β-unsaturated acylazolium species 134. Desilylation of the TMS enol ether by the fluoride ion forms enolate 135, which then undergoes 1,4-addition to 134 affording azolium enolate 136. Subsequent tautomerization of 136 gives acylazolium 137, which undergoes lactonization to furnish the observed dihydropyranone product and regenerate the the carbene catalyst. The same authors later expanded their methodology to silylated dienol ethers (Scheme 126).189 The formal all-carbon [4+2] cycloaddition with α,β-unsaturated acyl fluorides produced 1,3-cyclohexadienes in high yield with excellent diastereocontrol (dr >20:1) using NHC 138 as the catalyst.
Scheme 125.

Postulated Mechanism for Dihydropyranone Synthesis
Scheme 126.

[4+2] Cycloaddition of α,β-Unsaturated Acyl Fluorides and Silyldienol Ethers
In 2013, Lupton and co-workers reported an interesting NHC-catalyzed Ireland-Coates-Claisen rearrangement of α,β-unsaturated acyl fluorides with silylated push-pull cyclopropanes. These reactions provide rapid access to highly functionalized β-lactones with exquisite stereoselectivity (>20:1 dr in all cases) (Scheme 127).190a Electron-rich and electron-poor α,β-unsaturated acyl fluorides are tolerated, giving the desired lactones in good yields (41–93%).
Scheme 127.

Annulation of α,β-Unsaturated Acyl Fluorides and Push-Pull Cyclopropanes
Careful mechanistic studies performed by Lupton et al., including observation of a secondary kinetic isotopic effect at the β-position, culminated in a reasonable picture of the reaction mechanism (Scheme 128). Addition of the free carbene 3 to α,β-unsaturated acyl fluoride forms the corresponding α,β-unsaturated acylazolium. Desilylation and retro-aldol of the silylated cyclopropane produces bifunctional enolate 139, which then intercepts the acylazolium forming hemiacetal 140. After turnover-limiting Ireland-Coates-Claisen rearrangement, the resultant alkoxide (141) then undergoes aldol cyclization and lactonization to afford the desired product and regenerate 3.
Scheme 128.

Postulated Mechanism for the Ireland-Coates-Claisen Rearrangement
Shortly after the achiral reaction appeared, an enantioselective variant was reported using chiral triazolium A21.190b The authors found N-aryl triazolylidenes deliver the desired product in lower yield and diminished ee compared to the analogous N-alkyl triazolylidenes. A variety of aliphatic and aryl-substituted cinnamyl fluorides are competent coupling partners. However, the use of electron deficient aryl-substituted cinammyl fluorides deliver the product with lower ee. Thus, the 2,6-dimethoxyphenol lactones (142 and 143) invariably form in higher ee. The electronic nature of the aryl ester also displayed an impact on enantioselectivity (Scheme 129).
Scheme 129.

Enantioselective Ireland-Coates-Claisen Rearrangement
In 2010, Bode and co-workers described a similar transformation, proposing a Claisen rearrangement as the key step.185d Using chiral precatalyst A4, the authors efficiently coupled ynals to enolic C-nucleophiles such as Kojic acids with good enantioselectivity (Scheme 130). Aliphatic and aryl ynals are both competent in the reaction, and aliphatic groups on the Kojic acid component are tolerated. The authors probed the effect of the counterion of the triazolium precatalyst, and found that more basic counterions like Cl− and AcO− were sufficient to generate the free carbene to some extent without any added base.
Scheme 130.

NHC-Catalyzed Enantioselective Claisen Rearrangement
A possible catalytic cycle describing this transformation is depicted in Scheme 131. Based on kinetic studies, the authors propose initial formation of the Breslow intermediate followed by protonation to give α,β-unsaturated acylazolium 144. Enol 145 then undergoes initial reversible 1,2-addition to 144, forming a kinetically important hemiacetal intermediate 146. The latter is poised for a [3,3]-sigmatropic Coates-Claisen rearrangement resulting, after tautomerization and lactonization, in the desired dihydropyranone (methanolysis occurs on workup). However, with this data, the authors could not exclude a 1,4-addition pathway.
Scheme 131.

Postulated Reaction Mechanism of Coates-Claisen Rearrangement
The origin of reactivity and high stereoinduction in these annulations of α,β-unsaturated acylazoliums with enolic nucleophiles was studied computationally and experimentally by Mayr and Studer.191 Kinetic studies were employed to elucidate the inherent electrophilicity of α,β-unsaturated acylazoliums. Combining this knowledge with high-level DFT calculations, the authors suggest that enol and enamine nucleophiles 147 react with imidazolylidene-derived α,β-unsaturated acylazoliums (148) by means of a 1,4-Michael-type addition reaction via a contact ion pair (Scheme 132, path A). On the other hand, DFT computations from Schoenebeck and Bode with a triazolylidene-derived acylazolium (149) suggest a two-step mechanism beginning with 1,2-addition followed by [3,3]-sigmatropic rearrangement as the key C-C bond formation step (Scheme 132, path B).185e,192 In this mechanism, the nucleophile reversibly pre-associates with 149 to form a kinetically competent hemiacetal intermediate (as in Scheme 131). However, a loose ion pair between the nucleophile and the electrophile, as suggested by Mayr and Studer, could not be excluded. Lupton and co-workers observed 1,2-addition byproducts in their reaction and concluded the Coates-Claisen mechanism may be operative with certain substrate classes. This was later demonstrated by a cross-over experiment.187a
Scheme 132.

Mechanistic Dichotomy for the Reaction of Acylazoliums with Enols/Enamines
Xiao and co-workers developed a synthetically useful version of the “Claisen-type” reaction between ynals and 1,3-dicarbonyl compounds, allowing for the mild construction of 3,4-dihydropyranones in moderate-to-good yields (Scheme 133).185g,h Notably, the use of non-symmetric 1,3-diketones, such as 1-phenylbutane-1,3-dione, selectively provide a single regioisomer of product.
Scheme 133.

NHC-Catalyzed Synthesis of 3,4-Dihydropyranones
Moreover, when employing the enantiopure aminoindanol-derived triazolium salt precatalyst A4, the desired 3,4-dihydropyranones were obtained in good yields (34–87%) and up to 98% ee for a variety of substrates (aryl and aliphatic, Scheme 134).185h The authors note the importance of adding 4 Å molecular sieves to the reaction mixture. Presumably, the intermediate α,β-unsaturated acylazolium species is highly sensitive to residual moisture. Curiously, the use of a base was not necessary to generate the active carbene species, as previously demonstrated by Bode et al.185d
Scheme 134.

Enantioselective Annulation of Ynals and 1,3-Dicarbonyl Compounds
In 2012, Du, Lu and co-workers published the synthesis of chiral spirooxindole 4H-pyran-2-one derivatives by the reaction of oxindoles with ynals.193 The authors successfully explored different substitution patterns on both the ynal and oxindole components (Scheme 135). The reaction gives moderate-to-high yields (40–93%) and good diastereoselectivity (up to >95:5 dr). Interestingly, N-unprotected oxindoles afford similar results.
Scheme 135.

NHC-Catalyzed Spirooxindole Synthesis
The isomeric indolin-3-ones were employed by the same research group as nucleophilic reaction partners in an annulation with α,β-unsaturated acylazolium ions (Scheme 136).194 The reaction tolerates a variety of aryl-substituted ynals and gives the desired tricyclic indole products in moderate-to-good yields (19–91%), although Knoevenagel side products were also observed in most cases.
Scheme 136.

Scope of the NHC-Catalyzed Reaction between Indolin-3-ones and Ynals
Alexakis and co-workers prepared highly functionalized 5,6-fused bicyclic acetals by an NHC-catalyzed reaction between ynals and α-cyano-1,4-diketones (Scheme 137).195 Under these conditions, a wide range of substrates gives the product in good yields (61–90%) and modest-to-good diastereoselectivity (up to 20:1 dr).
Scheme 137.

NHC-Catalyzed Synthesis of 5,6-Fused Bicyclic Acetals
Combining all of the available experimental data, the authors proposed the following reaction mechanism for the annulation between α-cyano-1,4-diketones and ynals. Initially, an NHC-promoted internal redox converts ynal into the α,β-unsaturated acylazolium. In parallel, the α-cyano-1,4-diketone undergoes a double keto-enol tautomerization to give the nucleophilic bis-enol 150, which then adds 1,4 to the acylazolium through its most nucleophilic (less conjugated) carbon atom, giving intermediate 151 after tautomerization. A reversible 1,2-addition of the enol oxygen on to the second carbonyl group leads to cyclic hemiacetal 152, which can now readily cyclize to the observed 5,6-fused bicyclic acetal (Scheme 138).
Scheme 138.

Postulated Mechanism for the Reaction of α-Cyano-1,4-Diketones and Ynals
5.1.2 Generation from α-Bromoenals and α,β-Unsaturated Esters
Ye and co-workers expanded the arsenal of α,β-unsaturated acylazolium precursors with the introduction of α-bromoenals.186a These bench-stable reagents were successfully coupled with 1,3-dicarbonyl compounds to afford 3,4-dihydropyranones in excellent isolated yields, using achiral imidazolium salt precatalyst G7 (Scheme 139).
Scheme 139.

3,4-Dihydropyranone Synthesis using α-Bromoenals
The transformation was rendered enantioselective when enantiopure triazolium salt precatalysts B7 and B23 were employed (Scheme 140). Furthermore, both enantiomers of the product could be obtained by choosing N-heterocyclic carbenes with the same absolute configuration but different stereodirecting substituents, under the optimized reaction conditions. It is interesting to note that a triazolylidene-derived carbene devoid of an N-aryl substituent was used to promote a highly stereoselective “Claisen-type” reaction.186a
Scheme 140.

Enantiodivergent Cycloaddition of α-Bromoenals and 1,3-Dicarbonyl Compounds
In 2014, Enders reported an enantioselective version of the reaction developed previously by Du and Lu193 between indolin-3-ones and α,β-unsaturated acylazolium intermediates (Scheme 141).196 The authors subjected α-bromoenals to indolinones in the presence of the chiral aminoindanol-derived triazolium A4 to afford dihydropyranoindol-2-ones in moderate-to-good yields (60–72%) and excellent ee values (up to 98% ee). Electron-donating or -withdrawing groups on the aromatic ring of the α-bromoenal component are well tolerated, as well as heteroaromatic rings. Disappointingly, alkyl-substituted α-bromoenals are not suitable precursors of α,β-unsaturated acylazoliums. In a related reaction, Hui coupled α-bromoenals with chalcone derivatives to give tetrahydroquinolines in 93 – 98% yield, >25:1 dr, and 90 – 99% ee. Aliphatic substituents are tolerated on either coupling partner maintaining the excellent stereoselectivity, but in lower yields.197
Scheme 141.

NHC-Catalyzed Enantioselective Annulation of Indolin-3-ones with α-Bromoenals
The reaction between an α,β-unsaturated aldehyde with another enolizable aldehyde under NHC catalysis can result in eight possible products (four benzoin products, two Stetter products, and two γ-butyrolactones resulting from homoenolate reactivity). Very recently, Biju and co-workers achieved a chemoselective NHC-catalyzed reaction between two different aldehydes (Scheme 142).186c The highly enolic phenylacetaldehyde derivatives were reacted with α,β-unsaturated acylazoliums, generated in situ from α-bromoenal and chiral triazolium carbene precursor A4, resulting in formation of 2-unsubstituted 3,4-dihydropyranones in good yields (41–96%) and very high enantioselectivity (87–99% ee).
Scheme 142.

Enantioselective Annulation of α-Bromoenals with Enolizable Aldehydes
Phenylacetaldehyde derivatives with electron-releasing and -withdrawing groups on the aromatic ring underwent smooth annulation reaction with α-bromoenal-derived α,β-unsaturated acylazoliums. Moreover, heteroaromatic acetaldehyde derivatives turned out to be competent substrates as well. Additionally, this reaction is not limited to aromatic acetaldehyde derivatives and tolerates alkenyl-substituted acetaldehydes. Looking at the α-bromoenal component, it was noted that aromatic, alkenyl as well as alkyl groups are all tolerated.
As discussed in Section 4.3, carbenes add to activated esters. Very recently, Chi and co-workers reported an organocatalytic activation of α,β-unsaturated esters for enantioselective reaction with imines.187c The key step involves the addition of the NHC catalyst to the ester substrate to form an electrophilic α,β-unsaturated acylazolium intermediate (Scheme 143).
Scheme 143.

Enantioselective [3+3] Annulation of α,β-Unsaturated Esters and Enamides
Both electron-donating and -withdrawing substituents at the para-position of the phenyl group on the α,β-unsaturated ester are well tolerated. Among the arylimines examined, replacement of the methyl group with another alkyl substituent (ethyl, n-butyl, or homoallyl) invariably led to effective formation of product. In all cases, the desired dihydropyridinones were obtained in good-to-excellent yields (51–83%) and with very high ee values (91–99% ee). β,β-Disubstituted unsaturated esters are also tolerated in this reaction when less sterically hindered triazolium precatalyst A12 is employed. Products are formed in moderate-to-good yields (62–79%) and high enantioselectivity (up to 94% ee) (Scheme 144).
Scheme 144.

Generation of Dihydropyridinones from β,β-Disubstituted Unsaturated Esters
The authors propose that a 1,2-addition of free carbene 153 to the α,β-unsaturated ester leads to formation of the corresponding acylazolium intermediate. Concurrently, the imine isomerizes to nucleophilic enamide 154. As proposed by Bode and co-workers,185d formation of the key C-C bond likely occurs through initial 1,2-addition of 154 to the acylazolium, followed by a concerted Claisen rearrangement to give intermediate 155. The enamine nitrogen can then cyclize onto the acylazolium affording the desired product and regenerating 153 (Scheme 145).
Scheme 145.

Postulated Mechanism of Dihydropyridinone Formation
In 2014, Wang, Ye and co-workers disclosed a highly enantioselective synthesis of γ-butyrolactams employing α-aminoketones as 1,2-bis(nucleophiles) and α,β-unsaturated carboxylic acids as acylazolium precursors (Scheme 146).187d When employing the chiral aminoindanol-derived NHC precursor A4 bearing an N-mesityl group, the desired γ-butyrolactams are obtained in moderate-to-good yields (52–73%), moderate diastereoselectivity (3:1 to 12:1 dr), and very high enantioselectivity (88–97% ee). A wide variety of substituents on the aryl rings of both reaction components are tolerated in this reaction.
Scheme 146.

Enantioselective [3+2] Annulation of α,β-Unsaturated Acids with α-Aminoketones
The NHC-catalyzed [3+2] cyclocondensation of α,β-unsaturated carboxylic acids and α-aminoketones was then extended by the same authors to the [3+3] annulation with sulfamate-derived cyclic imines, affording chiral dihydropyridinones (Scheme 147).187d The optimized conditions for the [3+2] annulation also work well for the [3+3] reaction with cyclic imines, leading to the desired tricyclic dihydropyridinones in high yields (57–86%) and exquisite enantioselectivity (95–98% ee).
Scheme 147.

Enantioselective [3+3] Annulation of Sulfamate-Derived Imines
Finally, sultam-derived cyclic imines were also investigated as potential substrates for the [3+3] cyclocondensation with α,β-unsaturated acid-derived acylazoliums (Scheme 148). As expected, a range of α,β-unsaturated carboxylic acids react well to give the desired tricyclic cycloadducts in good yields (52–90%) and with high enantioselectivity (77–98% ee). In addition, the reaction of β,β-disubstituted as well as α,β-disubstituted α,β-unsaturated acids afford products with multiple stereogenic centers and with high diastereo- and enantioselectivity.
Scheme 148.

Enantioselective [3+3] Annulation of Sultam-Derived Imines
The postulated mechanism of the reaction is depicted in Scheme 149. The addition of the free carbene to the mixed anhydride 156 (formed in situ by the reaction between α,β-unsaturated acid and pivaloyl chloride) drives the formation of the α,β-unsaturated acylazolium intermediate. Michael addition of α-amino ketone to the acylazolium affords adduct 157 which, upon tautomerization, undergoes intramolecular lactamization to give the observed product and regenerate the NHC catalyst.
Scheme 149.

Plausible Mechanism of Cyclocondensation
5.1.3 Oxidative Methods for α,β-Unsaturated Acylazolium Generation
As shown by the recent work of Lupton and co-workers, efficient generation of α,β-unsaturated acylazoliums from precursors at the ester oxidation state (e.g. 159) require alkyl substituted N-heterocyclic carbenes (Scheme 150). It is important to note many of the reactions involving α,β-unsaturated acylazolium intermediates generated from precursors at the aldehyde oxidation state 158 employ aryl substituted N-heterocyclic carbenes (e.g. 16). This divergence in chemoselectivity can be correlated to the electronic density of the free carbenes. The latter property can be traced back to the Tolman Electronic Parameter (TEP) and the 13C chemical shift of the carbenic carbon atom. The combination of NHC catalysis with organic or inorganic oxidants has been recently reviewed by Studer.198
Scheme 150.

Electronic Requirements for Acylazolium Generation
Oxidative NHC-catalyzed transformations of aldehydic substrates are also inherently biomimetic in origin.199 Thus, the thiamine pyrophosphate (TPP)-dependent enzyme pyruvate ferredoxin oxidoreductase (PFOR) catalyzes the oxidative decarboxylation of pyruvate 161 to form acetyl-CoA 162 and CO2 (Scheme 151). This anaerobic decarboxylation reaction is a reversible process and the electrons produced are transferred to ferredoxin via Fe4S4 clusters. The key step of this biological transformation involves single-electron transfer from the Breslow intermediate to Fe4S4 cluster, leading to formation of a postulated radical cation 163.
Scheme 151.

TPP-Mediated Enzymatic Transformation of Pyruvate to Acetyl-CoA
From a historical perspective, similar to the discovery of the benzoin reaction, the initial findings for redox NHC activation of α,β-unsaturated aldehydes was made with cyanide-catalyzed transformations.188i,j,k,203,204 In 1873, Wallach and co-workers reported the redox neutral, cyanide-promoted conversion of chloral 164 to dichloroacetic acid 165 in water (Scheme 152).200 Kötz and co-workers first proposed a mechanistic rationale for this reaction in 1913.201 Upon initial formation of cyanohydrin 166, two distinct reaction intermediates were postulated. The nucleophilic enol intermediate 167 might arise from elimination of HCl. Although, the oxirane intermediate 168 could arise from cyanohydrin 166 by intramolecular nucleophilic displacement of the chloride ion. Intermediates 167 and 168 are then converted to the acyl cyanide 169, which is subsequently hydrolyzed to 165.
Scheme 152.

The Wallach Reaction and the Kötz Mechanistic Proposal
Corey and co-workers have shown that the cyanide ion catalyzes the oxidation of allylic or benzylic alcohols to esters, when carried out in presence of stoichiometric MnO2 as oxidant (Scheme 153).188g The reaction was proposed to take place via cyanohydrin 170 that is oxidized to the acyl cyanide 171. This last species then acts as an activated carboxylate towards nucleophilic interception by MeOH solvent. In 1977, Castells reported the oxidation of aryl aldehydes to the corresponding methyl esters with thiazolium salts and nitrobenzene oxidants.202
Scheme 153.
Cyanide-Catalyzed Oxidation of Allylic Alcohols
Scheidt and co-workers disclosed a similar NHC-catalyzed protocol for the oxidation of allylic and benzylic alcohols188e employing MnO2 as a mild oxidant (Scheme 154). A variety of activated alcohols were smoothly oxidized to the respective α,β-unsaturated esters in good-to-excellent yields (65–93%). Electron-rich alcohols, such as 4-methoxybenzyl alcohol, show no reactivity in this system. Finally, the authors attempted a desymmetrization of meso-diol employing a chiral triazolium catalyst. An encouraging 80% ee was obtained under the optimized conditions.188e
Scheme 154.

NHC-Catalyzed Oxidation of Allylic Alcohols
One year later, Studer and co-workers reported an oxidative NHC-catalyzed process where TEMPO was employed as the stoichiometric oxidant.188f Both electron-rich and electron-poor benzaldehyde derivatives are oxidized to the corresponding esters in excellent yields (91–96%). Enals and aliphatic aldehydes are also tolerated by this reaction, albeit in slightly lower yields, likely owing to a fundamental difference in the mechanism relative to that when using MnO2 (Scheme 155).
Scheme 155.

Biomimetic NHC-Catalyzed Oxidation of Aldehydes
In 2010, Studer and co-workers were the first to propose that an in situ oxidation of the tetrahedral intermediate 172 to the corresponding α,β-unsaturated acylazolium species (173) could be accomplished with mild organic oxidant 174.188a According to the authors, under optimal conditions, this reaction is faster than the proton transfer that would lead to the extended Breslow intermediate 175 (Scheme 156). This methodology is complementary to the use of α,β-unsaturated acyl fluorides or ynals in the NHC-catalyzed 3,4-dihydropyranone synthesis (Scheme 121, vide supra). In their study, Studer and co-workers employed 1,3-dicarbonyl compounds as the enolic, nucleophilic coupling partner (Scheme 157).
Scheme 156.

Postulated Mechanism for Oxidative NHC-Catalyzed Annulation of Enals
Scheme 157.

NHC-Catalyzed Synthesis of 3,4-Dihydropyranones by Redox Activation
You and co-workers reported the first enantioselective oxidative NHC-catalyzed annulation between enals and 1,3-dicarbonyl compounds (Scheme 158).188b A broad substrate scope was accesible in moderate-to-high yields (17–90%) and high enantioselectivity (up to 96% ee) with a camphor-derived triazolium salt A22 together with a catalytic amount of NaBF4.
Scheme 158.

Enantioselective Redox-Type Michael Addition of 1,3-Dicarbonyl Compounds to Enals
Du, Lu and co-workers studied the NHC-catalyzed annulation of indolin-3-ones with oxidatively-generated α,β-unsaturated acylazolium intermediates (Scheme 159).193 The reaction conditions differed markedly from those employed by the same research group in their redox-neutral reaction between indolin-3-ones and ynals (vide supra).194 Thus, a triazolium-derived carbene G20 was employed in the former reaction, while an imidazolium-derived carbene G7 was employed in the latter. Both reactions display comparable tolerance to substrate scope, albeit the oxidative protocol affords consistently higher yields.
Scheme 159.

Scope of the Oxidative Annulation of Indolin-3-ones and Enals
Recently, Studer and co-workers described an enantioselective cyclopropanation reaction of α,β-unsaturated aldehydes using oxidative NHC catalysis (Scheme 160).203
Scheme 160.

Enantioselective Cyclopropanation of Enals by Oxidative NHC Catalysis
In these reactions, NHC-catalyzed redox activation of α,β-unsaturated aldehydes is used for generation of the α,β-unsaturated acylazolium, which can then be intercepted by the sulfur ylide to form enolate 176. This intermediate can then cyclize to afford acylazolium 177, which reacts with alcohol to give the corresponding cyclopropanecarboxylic acid ester (Scheme 161). When using the chiral triazolium A4 as the carbene precursor, moderate-to-good yields (40–74%), good-to-excellent diastereo- (up to 20:1 dr) and enantioselectivity (up to 96% ee) are obtained. The authors observed that aryl groups bearing electron-withdrawing substituents on the enal component afford products with higher enantioselectivity than those bearing electron-releasing groups.
Scheme 161.

Postulated Catalytic Cycle for NHC-Catalyzed Cyclopropanation of Enals
In 2011, Studer and co-workers reported an organocascade reaction involving an oxidative NHC-catalyzed conjugate addition as a key step (Scheme 162).204 In this work, redox activation of enals was applied for the construction of the core structure of highly substituted indanes. Under optimized conditions, using chiral triazolium ent-A4 as the carbene precursor, keto-enals reacted with β-diketones in moderate-to-good yields (38–74%), high diastereo- (from 5.6:1 to > 98:2 dr) and exquisite enantioselectivity (> 99% ee in most cases) for a wide range of electronically distinct substrates.
Scheme 162.

Oxidative NHC-Catalyzed Synthesis of Trisubstituted Indanes
The authors proposed a catalytic cycle for their redox NHC cascade (Scheme 163). The increase of initial selectivity by a subsequent second stereoselective reaction was proposed as the origin of the observed high enantioselectivity.205 Initial reaction of the keto-enal with the free carbene in the presence of oxidant 174 generates the reactive α,β-unsaturated acylazolium ion 178. This redox-activated Michael acceptor then undergoes conjugate addition by the β-diketone to form azolium enolate 179, which can then undergo intramolecular 1,4-addition yielding enolate 180. Subsequent O-acylation affords the desired indane and liberates the carbene catalyst.
Scheme 163.

Suggested Catalytic Cycle for the Oxidative NHC Cascade Reaction
The in situ generated α,β-unsaturated acylazoliums were successfully trapped with numerous other pronucleophiles including vinylogous amides.188c Bode described an aza variant of the “Claisen-type” reaction with α,β-unsaturated acylazolium ions (Scheme 164). A large number of unprotected, conjugated enamines containing cyano, ester and nitro groups cleanly afford 3,4-dihydropyridinones in good yields (58–99%) and high enantioselectivity (79–96% ee). A broad range of enals (aliphatic, alkenyl and aromatic), in conjunction with the chiral triazolium salt A4 and the mild organic oxidant 174, served as suitable α,β-unsaturated acylazolium precursors.
Scheme 164.

Enantioselective Synthesis of 3,4-Dihydropyridinones
The authors employed α′-hydroxyenones as enal surrogates to expand the scope of this carbene-catalyzed aza-Claisen rearrangement (Scheme 165).178,206 A wide variety of aromatic and heteroaromatic substituents are tolerated as reaction partners for annulation with vinylogous amides. The product 3,4-dihydropyridinones are isolated in good-to-excellent yields (59–99%). The increased steric hindrance of these substrates precludes the use of chiral NHCs, but smaller and achiral variants like G20 proved to be efficient catalysts. Mechanistically, the title reaction was proposed to occur via a “Claisen-like” mechanism, similar to the reaction between ynals and Kojic acids reported by the same research group.185d
Scheme 165.

NHC-Catalyzed Annulation between α′-Hydroxyenones and Enamines
One year later, the same research group employed cyclic N-sulfonylimines as the nucleophilic coupling partners for oxidatively-generated α,β-unsaturated acylazolium Michael acceptors.188d Cyclic N-sulfonylimines are known to be potent nucleophiles through enamine generation. Furthermore, contrary to the previously described N-unprotected enamines, these nucleophiles are unable to give 1,2-addition products (Scheme 166).
Scheme 166.

Redox NHC-Catalyzed Annulation between Cyclic N-Sulfonylimines and Enals
The chiral N-mesityl substituted triazolium salt A4, used together with Hünig’s base, proved to be the optimal catalyst for these annulation reactions. Aromatic as well as aliphatic enals are competent precursors of α,β-unsaturated acylazolium ions. Interestingly, this catalyst system was the first that achieved highly enantio- and diastereoselective “Claisen-type” annulations of α- and β,β′-disubstituted enals. The high diastereoselectivity observed in these reactions is proposed to arise from substrate-directed protonation, rather than catalyst-directed protonation.
In 2013, Chi and co-workers discovered that direct functionalization of β C(sp3)-H bonds in saturated aldehydes was possible using oxidative NHC catalysis (Scheme 167).207 For comparison, in the canonical oxidative NHC pathway, an enal first reacts with the free carbene to form an extended Breslow intermediate 181. The latter is then oxidized to the α,β-unsaturated acylazolium ion 182 with aid of a mild organic oxidant. Chi’s approach differs from the canonical pathway in that it relies on direct functionalization of the β C(sp3)-H bond of a saturated aldehyde substrate. In this scenario, following an initial oxidation of the saturated Breslow intermediate 183, the resultant NHC-bound saturated acylazolium ion 184 undergoes deprotonation of α-CH protons, leading to formation of an azolium enolate 185. An additional oxidative process then transforms the ester enolate intermediate to 182. Chi and co-workers reacted α,β-unsaturated acylazolium intermediates, generated oxidatively from saturated aldehydes, with enolic 1,3-dicarbonyl compounds to afford dihydropyranones (Scheme 168).
Scheme 167.

Oxidative Pathways Towards α,β-Unsaturated Acylazoliums
Scheme 168.

Direct β-Functionalization of Aliphatic Aldehydes
The scope of the reaction is limited to β-aryl-substituted propionaldehydes, but both electron-withdrawing and -releasing substituents are tolerated, and the products are obtained in moderate-to-good yields (53–98%). On the other hand, 1,3-dicarbonyl compounds with both aryl and alkyl substituents are tolerated. High enantiomeric excesses are observed in these reactions (up to 94% ee) when the chiral triazolium precatalyst A4 is used.
Two distinct mechanistic pathways were proposed to explain the oxidation of the NHC-bound azolium enolate 185 to the α,β-unsaturated acylazolium intermediate 182 (Scheme 169). The first pathway involves two consecutive single-electron transfer processes and passes through a radical cation intermediate 186. To probe this pathway, the authors carried out their reaction in presence of TEMPO. The yield and the enantioselectivity of product formation were not affected by the presence of TEMPO. However, in light of more recent work by Rovis and co-workers, showing that nitroarenes oxidize the Breslow intermediate by single-electron pathways and that radical recombination is faster than TEMPO trapping, it seems reasonable to conclude that a similar situation may be operative here.181 A second mechanistic scenario involving electron-pair transfer processes seems somewhat less likely.
Scheme 169.

Two Possible Pathways for the Oxidation of the Saturated Azolium Enolate
5.2 Reactions Involving NHC-Bound Allenoate Intermediates
As shown previously, an NHC-bound allenoate intermediate was invoked as an intermediate in the synthesis of unsaturated esters (Scheme 123, vide supra); however, during the two years following the discovery of α,β-unsaturated acylazolium reactivity trapping this allenoate remained elusive (Scheme 170).
Scheme 170.

Reactivity Pathways Involving an NHC and an Ynal
Sun reported a breakthrough in this area in 2012 with the isomerization of ynals to allenoates by an NHC-catalyzed internal redox reaction (Scheme 171).208 Alkynyl aldehydes bearing a γ-leaving group 272 are used as the substrates, and a mesityl-substituted thiazolium salt G27 is required for optimal reactivity.
Scheme 171.

NHC-Catalyzed Isomerization of Ynals to Allenoates
The reaction works well with aryl and alkyl groups at the R1, R2 positions and good-to-excellent yields of the product allenoates are obtained (73–89%). The use of the chiral N-heterocyclic carbene precursor ent-A3 provides low enantioselectivity (up to 30% ee, Scheme 172).
Scheme 172.
Attempt at an Enantioselective Variant of Allenoate Synthesis
The authors propose that the unsaturated Breslow intermediate 187 (formed by nucleophilic addition of carbene on to the ynal) expels the leaving group and leads to a cumulenol intermediate 188 (Scheme 173). This species then tautomerizes to allenoyl azolium 189, which is finally intercepted by a solvent molecule.
Scheme 173.

Postulated Mechanism for NHC-Catalyzed Isomerization of Ynals
More recently, She and co-workers carried out a formal [3+2] annulation between ynals and β,γ-unsaturated α-ketoesters using a cooperative NHC/Lewis acid strategy (Scheme 174).209 The reaction affords γ-butenolides as the sole products in moderate-to-good yields (27–80%).
Scheme 174.

Formal [3+2] Annulation by A Cooperative NHC/Lewis Acid Strategy
In a rare occurence for an organocatalytic reaction, a saturated, imidazolinium-derived carbene precursor G9 proved to be the optimal catalyst structure. Additionally, the use of stoichiometric amounts of LiCl was crucial to obtain good conversions. Substrates with either electron-withdrawing or -donating groups on the aromatic ring are well tolerated. When the aryl group of the ester was changed to an alkenyl substituent, the yield of the [3+2] annulation decreased significantly. Unfortunately, replacing the aryl group with an alkyl chain gives no conversion. The authors also probed an enantioselective version of their formal [3+2] annulation strategy. Up to 53% ee was obtained with a commercially-available chiral NHC precursor A13.
The authors proposed a cooperative catalytic cycle to describe their transformation, where the NHC generates the nucleophilic allenoate intermediate 190, while the lithium cation acts as a Lewis acid and lowers the LUMO energy of the α-ketoester (Scheme 175). The lithium cation is proposed to pre-organize the transition state of reaction by bridging both reactants together.
Scheme 175.

Proposed Mechanism of the Formal [3+2] Annulation between Ynals and α-Ketoesters
Independently, Snyder and co-workers shed light on the potential application of the NHC-bound allenoate reactivity in a diastereoselective cycloisomerization to the Securinega alkaloids.210 Their synthetic strategy was based on an intramolecular [3+2] annulation between an NHC-bound allenoate and a ketone, again using a cooperative NHC/Lewis acid strategy (Table 1). The N-protected tetrahydropyranone rac-191 was chosen as the model substrate. It was then established that the bicyclic triazolium chloride G15, 2 eq. of titanium(IV) isopropoxide and DBU were the best catalyst combination, affording the tricyclic γ-butenolide in 38% yield.
Table 1.
Initial Optimization of the NHC/Lewis Acid-Catalyzed Annulation
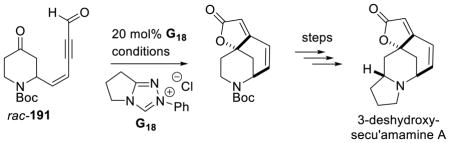
| ||||
|---|---|---|---|---|
| Entry | Base (15 mol%) | Lewis acid (eq.) | Solvent | Yield (%) |
| 1 | none | Ti(OiPr)4 (2 eq.) | CH2Cl2 | 37 |
| 2 | none | Ti(OiPr)4 (1 eq.) | CH2Cl2 | 30 |
| 3 | DBU | Ti(OiPr)4 (2 eq.) | CH2Cl2 | 38 |
| 4 | DBU | Ti(OiPr)4 (1 eq.) | CH2Cl2 | 37 |
| 5 | DBU | none | CH2Cl2 | 12 |
A further improvement in yield (up to 91%) was made when switching the solvent from methylene chloride to toluene, and introducing the tetrahydropyranone via slow addition over 4 h. Snyder and co-workers then embarked on the synthesis of a natural product analog 3-deshydroxy-secu’amamine A using their cooperative NHC/Lewis acid methodology.
A highly enantioselective annulation of NHC-bound allenoates and α-ketoesters was developed in 2014 by Scheidt and co-workers.211 The authors built on the results obtained earlier by She and likewise employed a cooperative NHC/Lewis acid catalytic system. Notably, it became rapidly apparent that chiral triazolium-based NHC catalysts were ineffective and the authors turned their attention towards C1-symmetric biaryl saturated-imidazolium-derived NHCs, originally developed by the Hoveyda group.212 However, because the chiral carbene was not sufficient to induce high enantioselectivity by itself, a chiral counterion strategy for the lithium cation was chosen as a means to enhance the enantioselectivity (Scheme 176).
Scheme 176.

A Matched Chiral NHC/Chiral Lithium Phosphate Combination as Means of High Stereoinduction
Following this paradigm, the [3+2] annulation of ynals and α-ketoesters was rendered highly enantioselective when combining the chiral C2-symmetric Brønsted acid 192 and the chiral C1-symmetric imidazolidinium precatalyst D8 (Scheme 177).
Scheme 177.

Enantioselective Cooperative NHC/Chiral Phosphate-Catalyzed Allenoate Annulation
The reaction is tolerant of both electron-deficient and electron-rich aromatic ynals and α-ketoesters. The desired γ-butenolides are isolated in good-to-excellent yields (62–92%) and with very high enantioselectivity (up to 92% ee). Of note, ortho-substituted aromatic ynals are not suitable substrates. Additionally, alkyl-substituted ynals and α-ketoesters were not described. Concurrently, Ma and co-workers, building on previous work,213 reported a highly enantioselective [3+2] annulation of β-bromoenals and N-protected isatins to furnish spirooxindole-butenolides, catalyzed by a N-heterocyclic carbene catalyst bearing a hydroxyl moiety B28 (Scheme 178).214
Scheme 178.

Enantioselective Spiroannulation of β-Bromoenals and Isatins
The reaction proceeds smoothly for a wide scope of N-substituted isatins bearing electron-releasing or electron-withdrawing groups at 4-, 5-, 6-, and 7-positions. The spirooxindole-butenolides are obtained in excellent yields (85–99%) and high enantioselectivity (88–92% ee). Electron-rich isatins undergo the reaction slowly and require extended reaction times. Additionally, the β-bromoenal component tolerates either electron-withdrawing or electron-releasing groups at the para- or meta-positions of the benzene ring. Alkyl-substituted β-bromoenals were not reported as substrates. The authors propose that the unsaturated Breslow intermediate 193 undergoes nucleophilic addition to the ketone moiety of the isatin (Scheme 179). A postulated transition structure for this step might involve a bifurcated hydrogen bond between the hydroxyl group of the carbene catalyst and the two carbonyl groups of the isatin substrate, guiding the approach of the homoenolate through its Si face. However, the involvement of the Cs+ ion as a Lewis acid could not be excluded.
Scheme 179.

Proposed Reaction Pathway for the Spiroannulation Reaction
5.3 Reactions Involving Saturated Acylazoliums and Azolium Enolates
5.3.1 Transesterification and Amidation Reactions from Esters
Saturated NHC-bound acylazoliums are important reactive intermediates for carbene-promoted transesterification reactions, as well as precursors of azolium enolates that have a rich chemistry of their own (vide infra).215 Saturated acylazoliums were first proposed as intermediates in NHC-catalyzed transesterifications. Hedrick initially investigated N-heterocyclic carbenes as catalysts for the polymerization of L-lactide.216 Soon afterwards, independently and concurrently, the research groups of Nolan as well as Hedrick and Waymouth reported the NHC-catalyzed transesterification reaction of esters with simple alcohols.217 Nolan and co-workers used low catalyst loadings (0.5–5 mol%) of N-aryl or N-alkyl imidazolium salts (G7 and G14, respectively) as precatalysts in the presence of molecular sieves to achieve uniformly high yields of transesterification products (93–100%). Hedrick and Waymouth used the free carbenes 3 and 195 to effect the transesterification equilibrium of benzoate esters, and showed that secondary alcohols are tolerated when the N-methyl-substituted imidazolylidene catalyst 195 is used (Scheme 180).
Scheme 180.

NHC-Catalyzed Transesterification Reactions
The initially proposed mechanism for these NHC-catalyzed transesterification reactions involved the formation of acylazolium intermediate 196.217 However, a follow-up study by Movassaghi implicated the NHC as a general base catalyst 197 (Scheme 181).217d
Scheme 181.
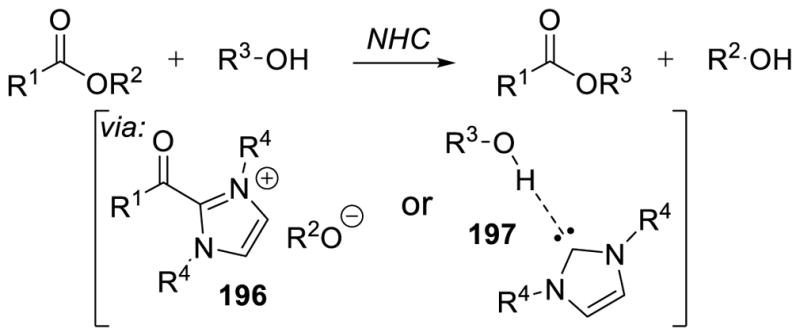
Mechanistic Dichotomy for NHC-Catalyzed Transesterification Reactions
Hu and co-workers provided computational evidence in support of a Brønsted base mediated pathway.217e Two reaction pathways were inspected: one involving a neutral tetrahedral intermediate 198 and the other involving a zwitterionic tetrahedral intermediate 199 (Scheme 182). It was shown that the NHC facilitates proton transfer from the incoming alcohol to the leaving group, without ionization of the tetrahedral intermediate. Furthermore, acyl substitution of the NHC into the ester carbonyl, leading to an acylazolium ion 200, was shown to be energetically unfavorable.
Scheme 182.

Computational Study of the Brønsted Base Mediated Pathway
Movassaghi converted a range of aliphatic, aromatic esters and lactones to amides via NHC-catalyzed ester amidations.217d The amine component of the reaction was limited to 1,2- or 1,3-aminoalcohols (Scheme 183), presumably because of an internal O to N acyl shift after the initial transesterification. The chemical yields were moderate-to-high (16–100%) depending on the structure of the ester starting material.
Scheme 183.

Amidation of Esters with Aminoalcohols
In addition to its synthetic utility, these studies had a profound influence on the understanding of reaction mechanism of NHC-catalyzed transesterification reactions (Scheme 184). Based on characterization of the carbene-alcohol adduct 201 by NMR, the authors concluded that their reaction took place via Brønsted base-mediated pathway. More specifically, it was shown that the carbene catalyzes the reaction by partially deprotonating the alcohol and thus rendering it more nucleophilic. Nucleophilic addition on the ester generates the tetrahedral intermediate 202. Finally, elimination of the alcoholate leaving group then provides ester 203, which undergoes O → N acyl-transfer to provide the desired amide.
Scheme 184.

Activation of Aminoalcohols by NHCs for Brønsted Base Catalysis
Suzuki218 and Maruoka219 independently developed NHC-catalyzed kinetic resolution of secondary alcohols. Suzuki and co-workers employed chiral, C2-symmetric imidazolium salts E1-4, 6 and vinyl acetate. Under the optimized conditions, moderate enantiomeric excess (up to 58% ee) was obtained in the first reported NHC-catalyzed kinetic resolution of chiral secondary alcohols (Scheme 185).
Scheme 185.

Suzuki’s NHC-Catalyzed Kinetic Resolution of Secondary Alcohols
Maruoka reported an improved, highly enantioselective protocol for carbene-catalyzed kinetic resolution of secondary alcohols (Scheme 186). Thus, by employing a more sterically demanding acylation agent, such as vinyl diphenyl acetate, the authors achieved far higher selectivity (s values up to 80).
Scheme 186.

Maruoka’s NHC-Catalyzed Kinetic Resolution of Secondary Alcohols
α-Reducible aldehydes have also been shown to lead to acylazolium intermediates. To put this work in historical context, Wallach described the cyanide-catalyzed conversion of chloral to dichloracetic acid (Scheme 187, top). In 2004, the Bode and Rovis groups independently and concurrently reported a Wallach-type transformation with N-heterocyclic carbenes. Bode and co-workers described the thiazolium-catalyzed conversion of epoxy- and aziridinyl-aldehydes to β-hydroxy/amino esters (Scheme 187, middle).220 Rovis and co-workers reported the triazolylidene-catalyzed conversion of α-haloaldehydes to aliphatic esters (Scheme 187, bottom).221 In both cases, the reducible functionality at the alpha-position is eliminated and results in a net oxidation of the carbonyl group to the acid oxidation state, with a concomitant in situ formation of the acylazolium intermediate. Exogenous alcohol nucleophile results in catalyst turnover.
Scheme 187.

The Wallach Reaction and Related NHC-Promoted Redox Reactions
Since this seminal work, several other families of electrophiles or acyl partners have been developed. The nucleophilic partner is largely dominated by alcohols with amines being much more problematic (vide infra). The embedded reactivity has led to several reactions already described in other parts of this review (cf. for example, Sections 4.1, 4.2, 5.1 and 5.2). In these cases an alpha-reducible aldehyde functionality is converted to an alpha-reduced ester group under redox neutral conditions.222 In 2005, Scheidt and co-workers reported a related reaction where the saturated acylazolium ion, generated from α,β-unsaturated aldehydes and benzimidazolium salt G1, is trapped by a nucleophilic alcohol.174 A broad scope of the corresponding saturated esters is obtained by employing phenol as the proton source (Scheme 188). Later, Enders described a similar strategy for the synthesis of oxime esters and hydroxamic esters using triazolium catalyst G19.223
Scheme 188.

Trapping of Saturated Acylazoliums with Alcohols
A postulated reaction mechanism is depicted in Scheme 189 below. The free carbene first adds to the carbonyl compound to generate the conjugated Breslow intermediate 205. This formal homoenolate equivalent is then protonated at the β-position, leading to formation of the azolium enol 206. After tautomerization of 206 to the saturated acylazolium 207, nucleophilic addition of the alcohol furnishes the desired product and regenerates the carbene catalyst.
Scheme 189.
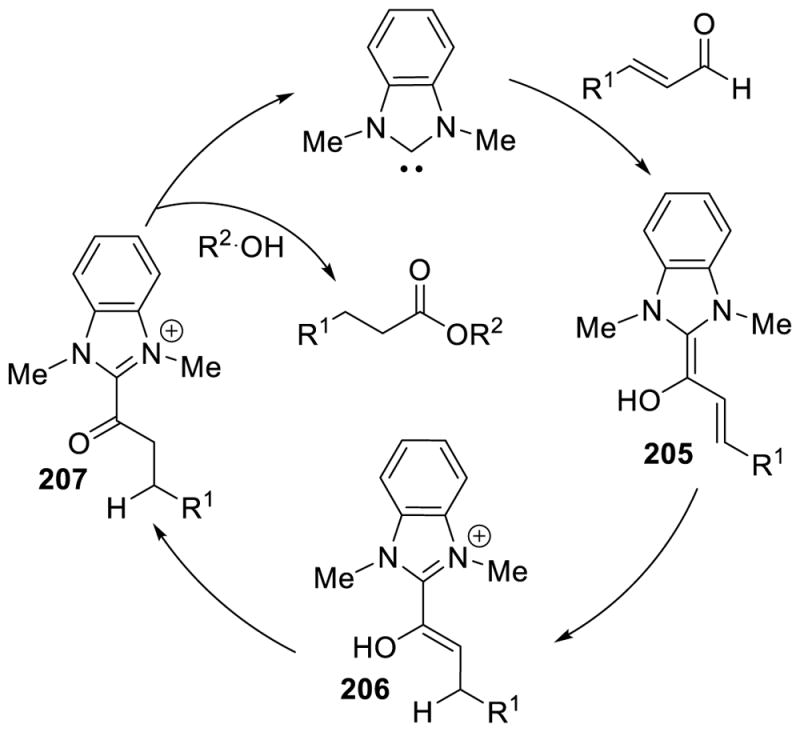
Postulated Mechanism for the NHC-Promoted Esterification of Enals
Importantly, Bode showed that the chemical structures of the carbene precursor and the catalytic base had a profound effect on the outcome of NHC-promoted reaction with α,β-unsaturated aldehydes.175 Thus, for imidazolium-type NHC precursors G7, higher loadings of a strong base, such as tBuOK, promote carbon-carbon bond formation via homoenolate reactivity and lead to enal dimerization (cf. Section 4.1). On the other hand, lower loadings of a weaker base such as diisopropylethylamine (DIPEA) favor β-protonation of the extended Breslow intermediate and the formation of a saturated acylazolium (Scheme 190). In contrast, when employing the bicyclic triazolium precatalyst G20 in combination with DIPEA as base, saturated acylazolium reactivity was observed exclusively.
Scheme 190.

Chemodivergent Reactivity of Enals
In 2005, Rovis reported a highly enantioselective esterification reaction involving NHC-bound azolium enolate inermediates (Scheme 191).224 The redox esterification of α,α-dichloroaldehydes in the presence of the chiral triazolium salt A3 provides the corresponding α-haloesters in moderate-to-good yields (65–86%) and high enantiomeric excesses (76–93% ee).
Scheme 191.

Enantioselective Synthesis of α-Chloroesters
The key step in the catalytic cycle is the diastereoselective protonation of the NHC-bound azolium chloroenolate. Furthermore, two important additives were used in this reaction; 18-crown-6 and 2,6-dibromo-4-methyl-phenol 208. Their role was to assure a homogeneous reaction mixture and suppress racemization of the product α-chloroaldehyde.
Rovis subsequently extended this chemistry to the enantioselective hydration of haloaldehydes.225 More specifically, α,α-dichloroaldehydes and α-fluoro enals are employed as precursors of saturated acylazolium intermediates that are subsequently intercepted by water (Scheme 192). Furthermore, the developed reaction allows for installation of an alpha-deuterium using D2O as the deuteron source.
Scheme 192.

Enantioselective Hydration of α-Haloaldehydes
Most of the above examples involve alcohols as nucleophiles. Amine nucleophiles were reported in two isolated cases: Rovis and co-workers showed that aniline participates with haloaldehyde,221 while Scheidt and co-workers illustrated an electron-deficient vinylogous carbamate as a partner for enals.174 When more typical amines are used as partners, amides are formed in trace amounts with imine formation and redox hydration both competitive. This problem is entirely circumvented if a peptide-coupling co-catalyst is used in the NHC catalyzed transformation, as shown by Rovis and co-workers in 2007 (Scheme 193).226 HOAt, HOBt, C6F5OH among others may be used in co-catalytic amounts to enable the coupling of alpha-reducible aldehydes such as haloaldehydes, epoxyaldehydes, aziridinyl aldehydes and enals with a variety of aliphatic primary and secondary amines as well as anilines. Bode reported a conceptually related approach using imidazole as a partner and adding the amine moiety at the end of reaction.227
Scheme 193.

Dual Catalytic Cycle for Amidation of α-Reducible Aldehydes
As a mechanistic probe, Rovis and co-workers showed that a chiral NHC catalyst induces asymmetry in the protonation event adjacent to the acyl moiety but is incapable of resolving racemic amines, implicating the achiral co-catalyst in the amidation event. An ingenious approach to resolving chiral, racemic amines was reported in 2011 by Bode and co-workers.228 The optimized catalytic system was composed of an achiral, bicyclic triazolium precatalyst G20 and a chiral, aminoindanol-derived hydroxamic acid 209. These conditions were used to resolve a range of chiral, racemic cyclic amines bearing a stereogenic carbon α to the nitrogen atom (Scheme 194). The mesityl-substituted α′-hydroxyenone led to highest selectivity in the acylation reactions.
Scheme 194.

Kinetic Resolution of Cyclic Amines using NHC/Hydroxamic Acid Cooperative Catalysis
These optimized conditions were tested in the resolution of a range of α-substituted piperidines. All the reactions gave moderate-to-good selectivity factors (s-factor of ca. 8–74). Additionally, substituted piperazines and morpholines were resolved with s-factors ranging from 11 to 23. Unexpectedly, tetrahydroisoquinolines were resolved with very high levels of selectivity (up to s = 74).
In this catalytic resolution of amines, two independent catalytic cycles work in concert, each with its own unique chemoselectivity. Thus, the free NHC adds to the α′-hydroxyenone, followed by a retro-benzoin reaction leading to the formation of an extended Breslow intermediate 210. β-Protonation of the latter, followed by tautomerization affords the saturated acylazolium intermediate 211. A chiral N-acylating reagent 212 is then generated by the reaction between 211 and 209. The secondary amine is then acylated by 212 to generate the enantioenriched amide (Scheme 195).
Scheme 195.

Synergistic Catalytic Cycles for Kinetic Resolution of Racemic Amines
5.3.2 [4+2] Cycloadditions of NHC-Bound Azolium Enolates
In 2006, Bode reported the first highly stereoselective [4+2] hetero-Diels-Alder reaction proceeding via a NHC-bound azolium enolate intermediate.242a A nucleophilic azolium enolate derived from 4-oxo-2-butenoate undergoes cyclization with N-protected α,β-unsaturated imines to form dihydropyridinones in moderate-to-high yields (52–90%) and excellent enantioselectivity (97–99% ee). The catalytic reaction tolerates aromatic, heteroaromatic, and alkyl groups on the imine component (Scheme 196).
Scheme 196.

Enantioselective Hetero-Diels-Alder Reaction
An attempt to rationalize the observed reactivity was made by the authors when they proposed a mechanism for their transformation (Scheme 197). The free carbene first adds to the carbonyl function of the 4-oxo-2-butenoate leading to formation of extended Breslow intermediate 213, which then undergoes intra- (or inter-)molecular proton transfer to afford the chiral azolium enolate 214. A concerted, albeit highly asynchronous transition structure was proposed for the [4+2] cycloaddition step. The resultant azolium-attached hemiaminal intermediate 215 then expels the free carbene with concomittant formation of the desired dihydropyridinone cycloaddition product. Rovis developed an important alternate access to these azolium enolates involving an oxidative protocol and utilizing aliphatic aldehydes as substrates.229 Very recently, Enders has generalized [4+2] cycloadditions to incorporate indolyl substrates.230
Scheme 197.

Postulated Catalytic Cycle for NHC-Catalyzed Hetero-Diels-Alder Reaction
The addition of NHCs to ketenes, or related compounds, provides an approach to azolium enolates that is alternative to the methods involving rearrangements of acyl anion equivalents. Although the availability of the more reactive ketenes restricts their potential impact, clear advantages have emerged from this process.
Azolium enolates, generated in situ by nucleophilic addition of a N-heterocyclic carbene to an aryl(alkyl)-disubstituted ketene, were employed as dienophiles in [4+2] annulations with electron-deficient dienes. In 2008, Ye and co-workers demonstrated that ketenes react with enones to afford 2,3-dihydropyranones in moderate-to-good yields (57–93%), high diastereo-(15:1 to 99:1 dr) and enantioselectivity (84–92% ee) (Scheme 198).231
Scheme 198.

[4+2] Annulation of Ketenes with Enones
One year later, this type of reactivity was extended by the same research group to benzoyldiazenes in place of enones as the electrophiles (Scheme 199).232 Of note, the latter [4+2] cycloaddition reaction displays reactivity different to an analogous reaction catalyzed by DMAP, which afforded [2+2] annulation products under otherwise similar reaction conditions.233
Scheme 199.

[4+2] Annulation of Ketenes with Benzoyldiazenes
In both of these transformations, a nucleophilic, NHC-bound azolium enolate is first formed by addition of the free carbene to the ketene. This step is then followed by annulation with the appropriate electrophile (enone or benzoyldiazene). Finally, elimination of the NHC catalyst affords the cycloannulated products. Interestingly, the sense of absolute induction was inverted when the benzoyldiazene reaction was carried out with NHC precatalyst B14, which had the same absolute configuration but bore an unprotected alcohol function and a N-mesityl substituent. Recently, Enders has expanded this reactivity to include the annulation of (E)-2-strylbenzothiazoles and α-chloroaldehydes generating the products in 44 – 97% yield, 4:1 to 9:1 dr, and 92 – 98% ee.234 Interestingly, the chemistry was also competent with N-(benzothiazolyl)imines as the coupling partner. generating benzothiazolo-pyrimidinones in 34–78% yield, 4:1 – >20:1 dr, and 87–99% ee.235
5.3.3 [2+2] Cycloadditions of NHC-Bound Azolium Enolates
In 2008, Ye236 and Smith237 independently and concurrently described an interesting triazolylidene-catalyzed Staudinger-type [2+2] cycloaddition between ketenes and N-protected imines. Ye and co-workers employed the bulky L-pyroglutamate-derived NHC precursor B6, in combination with aryl(alkyl)-disubstituted ketenes and N-Boc imines (Scheme 200). Smith and coworkers utilized diphenylketene with N-tosyl imines and aminoindanol-derived precatalyst ent-A1. Smith also found the rarely used imidazolinium precatalyst D6 was also efficacious. A wide range of electron-releasing or -withdrawing substituents on the aromatic rings of both the ketene and the imine reaction components is tolerated, providing the desired β-lactams in moderate-to-good yields (53–78%) and exquisite enantioselectivity under Ye’s conditions (91–99% ee).
Scheme 200.

NHC-Catalyzed Staudinger Reaction
The reaction mechanism was proposed to involve initial addition of the free carbene to the ketene, leading to formation of an NHC-bound azolium enolate intermediate 216 (Scheme 201). This enolate then undergoes stepwise [2+2] annulation with the imine, giving intermediate 217 which, after release of the NHC catalyst, finally affords the desired β-lactam product.
Scheme 201.
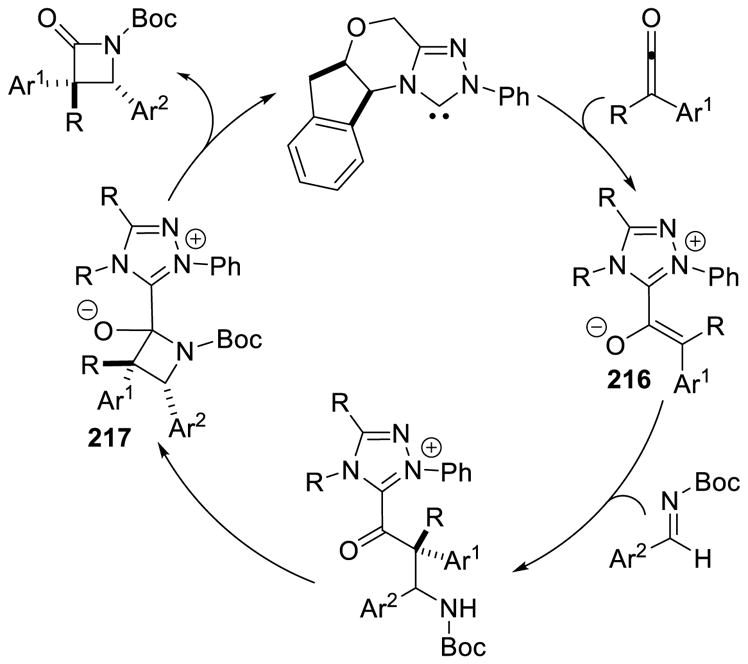
Mechanism of the [2+2] Staudinger-Type Cycloaddition of Ketenes and Imines
Since then, Ye and co-workers demonstrated that other electrophiles could react with in situ generated NHC-bound azolium enolates in related [2+2] cycloadditions.238 Thus, activated α-ketoaldehydes reacted with aryl(alkyl)-disubstituted ketenes to provide β-lactones in good yields (78–99%) and high diastereo-(4:1 to > 20:1 dr) and enantioselectivity (4–99% ee) (Scheme 202).
Scheme 202.

[2+2] Annulation of Ketenes and α-Ketoaldehydes
In addition, N-sulfinylanilines also proved competent substrates for [2+2] annulation with ketenes (Scheme 203). The reaction provides enantioenriched thiazetidinone oxides in high yields (73–96%) and enantioselectivity (16–99% ee).
Scheme 203.

[2+2] Annulation of Ketenes and N-Sulfinylanilines
5.3.4 [3+2] Cycloadditions of NHC-Bound Azolium Enolates
In 2010, an enantioselective formal [3+2] cycloaddition of NHC-bound azolium enolates and oxaziridines was described by Ye and co-workers. Aryl(alkyl)-disubstituted ketenes were used as precursors of azolium enolates (Scheme 204). A bifunctional NHC precatalyst B27 bearing a free hydroxyl group was employed. Ketenes with a para-substituted aryl group (with both electron-withdrawing and -releasing substituents) and a meta-substituted aryl group provide the corresponding oxazolin-4-ones in good moderate-to-yields (38–78%) and high diastereo- and enantioselectivity (up to 15:1 dr and 95% ee).
Scheme 204.

Formal [3+2] Cycloaddition of Disubstituted Ketenes and Oxaziridines
Interestingly, cycloaddition reactions catalyzed by NHC precursor B17 bearing a TMS-protected hydroxyl group furnished the desired opposite enantiomer of product in 79–90% ee, albeit in somewhat lower chemical yields (36–65%). Mechanistically, the reaction is proposed to be initiated by nucleophilic addition of the free carbene to the ketene, which results in formation of the NHC-bound azolium enolate intermediate 218 (Scheme 205). Subsequent oxidation of 218 by the oxaziridine affords the zwitterionic intermediate 219 together with a tosylimine. Epoxide opening followed by nucleophilic addition of the imine by the resultant alkoxide leads to the second zwitterionic intermediate 220. Finally, intramolecular lactamization of 220 affords the desired oxazolin-4-one and liberates the NHC catalyst.
Scheme 205.

Proposed Catalytic Cycle for NHC-Catalyzed [3+2] Cycloaddition of Ketenes and Oxaziridines
Very recently, Enders and co-workers described an interesting formal [3+2] cycloaddition of NHC-bound azolium enolates with 2-nitrovinylindoles.239 Chiral azolium enolates were generated in situ from α-reducible chloroaldehydes and chiral triazolium salt A4 (Scheme 206). The electron-withdrawing nitrovinyl group reverses the polarity at C3, leading to an umpolung of this three-carbon unit. The reaction proceeds smoothly for a variety of 2-nitrovinylindole substrates bearing either electron-withdrawing or -releasing substituents. The corresponding tricyclic products are obtained in moderate-to-good yields (30–78%) and excellent diastereo- (>20:1 dr) and enantioselectivity (96–99% ee). Remarkably, a pyrrole derivative is also a suitable substrate and affords 1H-pyrrolizin-3(2H)-one in 35% yield and 91% ee.
Scheme 206.

Formal [3+2] Annulation of α-Chloroaldehydes with Nitrovinylindoles
Regarding the mechanism of reaction, the free carbene performs a nucleophilic addition on α-chloroaldehyde leading to formation of the chlorinated Breslow intermediate 221 (Scheme 207). Subsequent elimination of chloride leads to NHC-bound azolium enolate 222, which undergoes Michael addition to 2-nitrovinylindole. Finally, the catalytic cycle is completed by intramolecular lactamization of intermediate 223, affording the desired 1H-pyrroloindol-3(2H)-one and liberating the carbene catalyst.
Scheme 207.

Proposed Mechanism of Reaction between Azolium Enolates and 2-Nitrovinylindoles
5.3.5 [2+2+2] Cycloadditions of NHC-Bound Azolium Enolates
Following the seminal reports by Louie and co-workers, who described the first NHC-catalyzed [2+2+2] annulation of isocyanates,240 Ye and co-workers reported that ketene reacts with either isothiocyanate or carbon disulfide under NHC catalysis to afford heterocycles (Scheme 208). This reaction represents the first enantioselective trimerization between dissimilar ketene equivalents.241 The cycloaddition reaction was tested between N-benzoyl isothiocyanate and various aryl(alkyl)-disubstituted ketenes. Both electron-releasing and -withdrawing substituents are tolerated on the aromatic ring. Furthermore, ketenes with methyl, n-propyl, and n-butyl groups work well and afford the desired [2+2+2] cycloadducts in good-to-excellent yields (50–99%) and high levels of enantioinduction (61–97% ee).
Scheme 208.

Non-Symmetrical [2+2+2] Annulation
The mechanism was proposed to involve a chemoselective addition of the NHC to the ketene giving azolium enolate 224 (Scheme 209). Subsequently, reaction of 224 with isothiocyanate provides intermediate 225, which is poised for nucleophilic addition on a second equivalent of ketene. Cyclization followed by elimination of the carbene catalyst finally furnishes cycloadduct.
Scheme 209.

Postulated Mechanism of the [2+2+2] Cycloaddition
Interestingly, by simply changing the catalyst structure to B23 ketenes underwent a Staudinger-type [2+2] cycloaddition with para-nitrophenyl-protected isothiocyanates (Scheme 210). A variety of aryl(alkyl)ketenes were tested in the title reaction. Both, electron-withdrawing and - releasing substituents on the aromatic ring work well to give the corresponding 4-thioxo-2-azetidinones in good yields (62–85%) and very high enantioselectivity (92–97% ee). Moreover, ketenes with various alkyl groups (methyl, n-propyl, and n-butyl) also worked well and afford the desired products with high yields and enantioselectivity. However, a ketene with a branched isopropyl substituent gave no cycloadduct. Mechanistically, formation of the observed β-lactam product can be explained by intramolecular lactamization of the acylazolium intermediate 225 prior to addition of the second equivalent of the ketene (cf. Scheme 109)
Scheme 210.

Staudinger-Type [2+2] Annulation
5.3.6 Electrophilic Trapping of Azolium Enolates Followed by Esterification
In addition to NHC-promoted annulation reactions of ketenes with various electrophiles, the in situ generated azolium enolate intermediates can tautomerize to the corresponding saturated azolium enolates, which undergo esterification reactions.
Under appropriate reaction conditions, saturated NHC-bound acylazoliums undergo tautomerization to azolium enolates (Scheme 211). The reactivity of these two intermediates diverges markedly. Thus, while saturated acylazoliums undergo transesterification reactions (vide supra), in situ generated azolium enolates participate in hetero-Diels-Alder reactions.242 Importantly, there is mounting evidence that all three NHC-bound reactive intermediates (homoenolate equivalents, acylazoliums, and azolium enolates) are interconnected and can be generated from common starting meterials such as ketenes, α-functionalized aldehydes, enals, and activated esters (cf. Sections 4.3, 5.3).183
Scheme 211.

The Common Extended Breslow Intermediate for Acylazolium/Azolium Enolate Reactivity
In 2009, Ye243 and Smith244 independently disclosed chiral NHC-catalyzed enantioselective esterifications of ketenes (Schemes 212 and 214, respectively). In Ye’s reaction, aryl(alkyl)- disubstituted ketenes were reacted with benzhydrol in the presence of precatalyst B6 to afford chiral esters in low-to-moderate yields (24–75%) and moderate-to-high enantioselectivity (11–95% ee). Unfortunately, decent levels of enantioselectivity were obtainable when benzhydrol was used as a nucleophile.
Mechanistically speaking, nucleophilic addition of the NHC to the ketene leads to formation of azolium enolate 226 (Scheme 213). The latter species then undergoes enantioselective protonation with benzhydrol to give the corresponding acylazolium/alcoholate ion pair 227. Esterification by the resultant alkoxide then provides a zwitterionic tetrahedral intermediate 228 that collapses to the ester product.
Scheme 213.
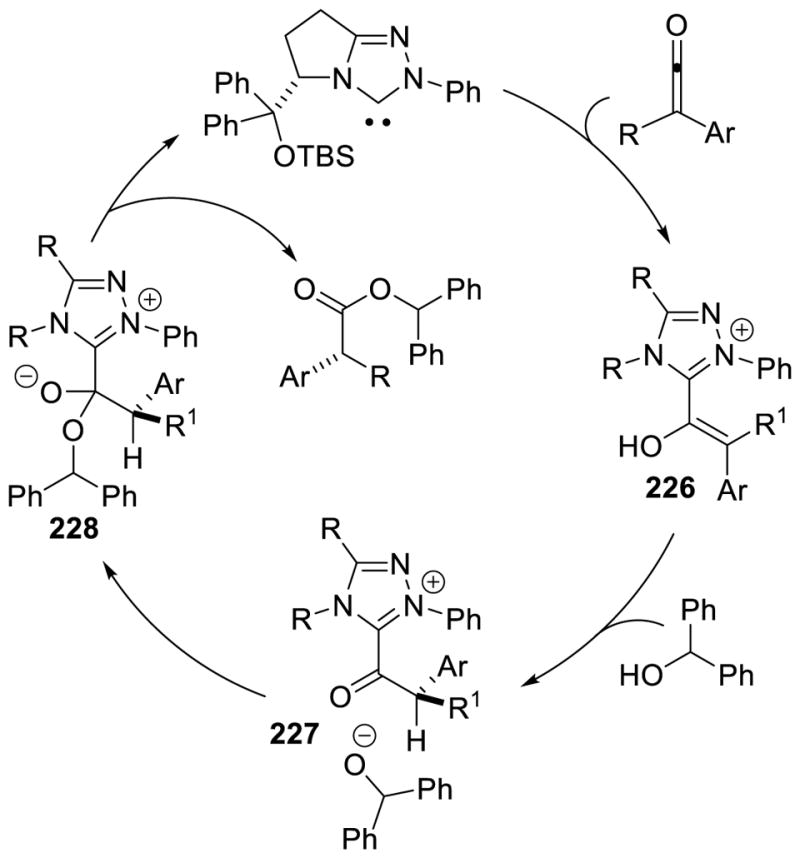
Possible Mechanism of NHC-Catalyzed Esterification of Ketenes
The NHC-promoted ketene esterification reactions by Smith and co-workers used a different alcohol (2-phenyl phenol) in combination with a slightly different triazolylidene precursor C3. These reactions occur with good yields (65–91%) and enantioselectivities (33–84% ee).
In 2010, Smith and co-workers demonstrated that the azolium enolate, formed in situ by the addition of a nucleophilic carbene to a ketene, could be trapped by other electrophiles instead of a proton. The authors were particularly interested in performing chlorinative esterifications of ketenes (Scheme 215). To this end, aryl(alkyl)-disubstituted ketenes were converted to enantioenriched α-chloro esters with chlorinating reagent 229 and employing the chiral triazolylidene-type catalyst B6.245 The desired α-chloro esters were isolated in good-to-excellent yields (67–97%) and with low-to-moderate enantioselectivity (26–61% ee).
Scheme 215.

Enantioselective Chlorinative Esterification of Ketenes
The authors proposed enantioselective chlorination of the NHC-bound azolium enolate 230 leads to the acylazolium/pentachlorophenolate ion pair 231 (Scheme 216). Subsequent esterification then provides product and liberates the NHC catalyst.
Scheme 216.

Plausible Mechanism of Enantioselective Chlorination of Ketenes
In 2011, Wang and co-workers described an interesting NHC-initiated domino reaction of formylcyclopropanes and indolecarbaldehydes (Scheme 217).246,247 The benzimidazolium salt G1 gave the best result in terms of chemical yield. Unfortunately, the addition of imidazole, which is a commonly used nucleophilic co-catalyst in acylation reactions, failed to improve the reaction yield. Substrates with either electron-donating or -withdrawing substituents on the indole ring reacted with formylcyclopropyl 1,1-diesters to provide the products in low-to-moderate yields (14–62%). Interestingly, monoactivated formylcyclopropyl esters reacted successfully as well, albeit in lower yields.
Scheme 217.

NHC-Catalyzed Domino Reaction of Formylcyclopropane and Indolecarbaldehyde
Initial interception of the formylcyclopropane by the free carbene leads to formation of the Breslow intermediate 232 (Scheme 218). Subsequent NHC-promoted ring-opening of the cyclopropane ring affords azolium enolate 233 that reacts with indolecarbaldehyde to give the zwitterionic intermediate 234. Intramolecular proton transfer followed by lactamization through the indole nitrogen atom yields the hydroxy pyrrolo-indole and liberates the carbene catalyst. Subsequent carbene-independent dehydration then affords the desired product.
Scheme 218.

Postulated Mechanism of the Domino Reaction
In 2014, Sun and co-workers reported an enantioselective, oxidative α-fluorination of aliphatic aldehydes under NHC catalysis.248 Accordingly, saturated aldehydes are converted to α-fluoroesters using NFSI as both the oxidant and the fluorine source (Scheme 219). Various aliphatic aldehydes react smoothly and afford the corresponding α-fluoroesters in moderate yields (52–75%) and with excellent enantioselectivity (87–95% ee). Various oxidizable functional groups such as an alkyne, pyridine and thiophene were also compatible with the strongly oxidizing reaction conditions.
Scheme 219.

α-Fluorination of Saturated Aldehydes using NHC in Conjunction with NFSI
In addition to saturated aldehydes, the authors also examined α-chloroaldehydes as azolium enolate precursors for their fluorination reaction (Scheme 220). In this case, only one equivalent of NFSI was sufficient because an external oxidant was no longer required. Under otherwise identical reaction conditions, comparable results in terms of yields and enantioselectivity of products were observed.
Scheme 220.

α-Fluorination of α-Chloroaldehydes
Furthermore, the catalytic enantioselective fluorination was extended to syntheses of α-fluoro amides and thioesters (Scheme 221). Due to the intrinsic reluctance of acylazoliums to acylate amines and thiols (vide supra), a relay shuttle additive had to be introduced with pyrazole (236) proving optimal, allowing access to highly enantioenriched α-fluoro amides and thioesters in moderate-to-good yields (52–72% ee).
Scheme 221.

One-Pot Synthesis of α-Fluoro Amides and Thioesters
5.3.7 Generation of Azolium Enolates from Saturated Esters
Very recently, Chi and co-workers reported a “backward” pathway for azolium enolate generation.249 In stark contrast to the “forward” process that starts from an enal (or other α-functionalized aldehydes) and proceeds to the azolium enolate, the reverse process starts from a stable carboxylic ester bearing a good leaving group OR2 (Scheme 222). The increased acidity of α C-H bonds of the saturated acylazolium intermediate would then lead to facile deprotonation and formation of the NHC-bound azolium enolate.
Scheme 222.

NHC-Mediated Activation of Saturated Esters
The authors exploited the nucleophilicity of saturated ester-derived azolium enolates in hetero-Diels-Alder cycloadditions with N-protected α,β-unsaturated imines (Scheme 223). The reaction proceeds well when aryl-substituted α,β-unsaturated imines and activated arylacetic esters are employed as reaction components. Electron-releasing or -withdrawing substituents are well tolerated on the aryl ring. In all cases, the product dihydropyridinones are generated in moderate-to-excellent yields (51–94%). High enantioselectivity is observed (60–99% ee) when enantiomerically pure triazolium B2 is used as precatalyst.
Scheme 223.

Enantioselective Hetero-Diels-Alder Reaction
5.3.8 Reactions Involving Azolium Dienolates
In cases where a γ-proton is available on the α,β-unsaturated acylazolium intermediate (e.g. 236), its removal would lead to formation of a conjugated NHC-bound azolium enolate (or dienolate 237). The reactivity of this species mirrors its nucleophilic nature and contrasts sharply with reactivity patterns of “normal” NHC-bound azolium enolates 238. Thus, azolium dienolates can undergo electrophilic addition on either the α- or γ-positions (Scheme 224). In addition, catalytically-generated dienolates participate in formal [4+2] cycloaddition reactions.
Scheme 224.

Approaches to Azolium Enolates and Dienolates
In 2011, Ye reported the first access to catalytically-generated azolium dienolates involved the addition of a N-heterocyclic carbene to an α,β-unsaturated ketene.250 More specifically, the intermediate α,β-unsaturated ketenes are generated in situ from the corresponding α,β-unsaturated acyl chlorides by dehydrohalogenation. These were reacted with trifluoromethyl ketones under NHC catalysis to afford chiral 6-trifluoromethyl-5,6-dihydropyran-2-ones (Scheme 225). The best results were obtained with the bicyclic triazolium precatalyst B27, cocatalytic amounts of Cs2CO3 and an excess of Et3N. Under these optimized conditions, acyl chlorides with aromatic rings bearing electron-withdrawing substituents afford the corresponding cycloadducts in good-to-excellent yields (76–94%) and with high enantioselectivity (72–93% ee). On the contrary, acyl chlorides with electron-donor-substituted phenyl rings or electron-rich heteroaromatic rings gave the products with slightly decreased ee values (67–86% ee). It is also noteworthy that the reaction of an alkyl-substituted acyl chloride delivers a single regioisomer of the corresponding cycloadduct in high yield (50%) but with moderate enantioselectivity (57% ee). Finally, the trifluoromethyl ketones used were limited to aromatic and heteroaromatic substrates exclusively.
Scheme 225.

Enantioselective [4+2] Annulation of α,β-Unsaturated Ketenes with Trifluoromethyl Ketones
The same authors later expanded their carbene-catalyzed methodology to isatins as the activated ketones (Scheme 226). It was found that the reaction conditions optimized for trifluoromethyl ketones were also applicable to isatins.251 Thus, a set of isatins with electron-withdrawing or - releasing substituents react well with α,β-unsaturated acyl chlorides to give spirocyclic oxindole-dihydropyranones in moderate-to-good yields (55–84%) and with good-to-high enantioselectivity 226 (71–95% ee). An N-benzyl protecting group proved equally effective, without loss of enantioselectivity.
Scheme 226.

Enantioselective [4+2] Annulation of α,β-Unsaturated Ketenes with Isatins
The authors proposed a mechanistic scenario for their transformation (Scheme 227). The catalytic cycle is presumably initiated by 1,2-addition of the free carbene to the α,β-unsaturated ketene 239, generated from the α,β-unsaturated chloride by the action of base. The resultant azolium dienolate intermediate 240 then undergoes nucleophilic addition on the activated ketone, regioselectively through its γ terminus. Lactonization of the transient alkoxide 241 finally furnishes the product spirocyclic dihydropyranone, while regenerating the carbene catalyst.
Scheme 227.

Proposed Catalytic Cycle for [4+2] Annulation of α,β-Unsaturated Ketenes and Activated Ketones
In 2012, Chi reported an oxidative NHC-catalyzed [4+2] cycloaddition of β,β′-disubstituted enals with trifluoromethyl ketones (Scheme 228).252 The reaction was proposed to take place via an oxidatively-generated, NHC-bound dienolate intermediate. The typical NHC-mediated enal reactions involving homoenolate intermediates were suppressed by introducing an extra substituent at the enal β-carbon. Bis-quinone 174, previously explored in oxidations of Breslow intermediates, was found to be a good oxidant in this reaction as well. Phenyl rings with electon-withdrawing or -releasing substituents, heteroaromatic as well as alkenyl groups were all tolerated on the enal reaction partner. The reaction of alkyl-substituted enals was not reported. On the other hand, trifluoromethyl ketones bearing an aromatic or heteroaromatic rings proved competent substrates as well. Encouragingly, replacing these aryl groups by an alkyl substituent also leads to effective [4+2] annulations. In all cases, the desired dihydroquinolones are isolated in moderate-to-good yields (52–81%). In addition, very high enantioselectivity (up to 94% ee) is attained with chiral triazolium precatalyst A4 in conjunction with Sc/Mg-based Lewis acid cocatalyst. The ee values decrease when alkyl-substituted trifluoromethyl ketones are employed as substrates (60–80% ee).
Scheme 228.

Enantioselctive Oxidative γ-Addition of Enals to Trifluoromethyl Ketones
The proposed mechanism involves the free carbene reacting with α,β-unsaturated aldehyde to afford an extended Breslow intermediate 242 (Scheme 229). Oxidation of the latter with bis-quinone 174 gives an α,β-unsaturated acylazolium ion 243. The key dienolate intermediate 244 likely comes from γ-deprotonation of the oxidatively-generated α,β-unsaturated acylazolium 243. Dienolate 244 then performs nucleophilic addition to the trifluoromethyl ketone, affording the product dihydropyranone and regenerating the NHC catalyst. The Lewis acidic Sc(III) cocatalyst is likely involved in multipoint coordination, bringing the reactants together and organizing the transition structure.
Scheme 229.

Proposed Mechanism for Reaction of NHC-Bound Dienolate with a Carbonyl Compound
Similar NHC-bound azolium dienolate intermediates were effectively generated in the absence of external oxidants by using α-bromoenals as precursors. Along these lines, Yao and co-workers reported an efficient synthesis of spirocyclic oxindole-dihydropyranones by reaction of tetrasubstituted α-bromoenals with isatins (Scheme 230).253 The best precatalyst proved to be the achiral N-mesityl-substituted imidazolium salt G7. When employed in THF as solvent and with addition of Cs2CO3 as base, the reaction tolerates both electron-withdrawing and -releasing substituents on the isatin component.
Scheme 230.

Annulation of α-Bromoenals with Isatins
Electron-rich isatins require longer reaction times, but provide better isolated yields of products relative to their electron-poor counterparts. Moreover, different substituents on the N-atom of isatins influenced the yield markedly; when the methyl group is replaced with an allyl or benzyl group, the reaction yield decreases slightly. Dissappointingly, low enantiomeric excess (28% ee) and reduced chemical yield (42%) are obtained when a chiral NHC precatalyst is employed, under otherwise identical reaction conditions.
Accordingly, the bromo-Breslow intermediate 245, generated by addition of nucleophilic carbene to α-bromoenal, is proposed to undergo transformation to α,β-unsaturated acylazolium ion 246 through a3 to d3 umpolung followed by debromination (Scheme 231). Deprotonation at the γ-position then affords an NHC-bound azolium dienolate 247. Next, intermediate 247 adds to the isatin followed by intramolecular cyclization to deliver the target product, with concomitant release of carbene.
Scheme 231.

A Proposed Mechanism for Reaction of α-Bromoenals with Isatins
Lin, Sun and co-workers first reported an α-fluorination reaction of transiently-generated NHC-bound dienolates.254 The same authors have previously described an NHC-catalyzed redox reaction of ynals bearing a leaving group at the γ-position (vide supra).208 Their new strategy consisted in employing enals that bear a leaving group at the γ-position as precursors of dienolate intermediates (Scheme 232).
Scheme 232.

α-Fluorination of NHC-Bound Dienolate Intermediate
Enantioselective α-fluorinations were achieved by employing NFSI as the electrophilic fluorine source, in conjunction with the chiral triazolium precatalyst A4 (Scheme 233). Competing side reactions such as difluorination and protio-redox were successfully suppressed by using 1.05 equivalents of NFSI together with 2.0 equivalents of NaOAc. The authors also noted that the steric nature of the carbonate leaving group (OMe, OEt, or OtBu) had little effect on yield and enantioselectivity.
Scheme 233.

Scope of the α-Fluorination Reaction of Dienolates
The NHC-catalyzed enantioselective α-fluorination proceeds in good yields (61–82%) for a range of aryl-substituted enals bearing a carbonate leaving group, with either electron-releasing or -withdrawing substituents on the phenyl ring. The enantioselectivity is consistently high (85–92% ee). The reaction of alkyl-substituted enals takes place with comparatively high yields and enantioselectivity, albeit mixtures of E/Z isomers are obtained in these cases. Moreover, the presence of a trisubstituted carbon atom at the γ-position of the enal does not affect the reaction efficiency (67% yield, 77% ee). In sharp contrast, substitution at the α-position of the enal reduces the reaction efficiency significantly (5% yield after 48 h), even though the desired product containing a quaternary carbon center is isolated with a moderately high ee value (55% ee).
The authors proposed the following reaction mechanism (Scheme 234): the addition of the free carbene to an enal generates the extended Breslow intermediate 248. The latter then undergoes elimination of the carbonate leaving group followed by deprotonation to afford the NHC-bound dienolate intermediate 249. Subsequent C-F bond formation takes place selectively from the Re-face. Acyl substitution by methanol proceeds via a tetrahedral intermediate forms the desired fluorinated product and releases the NHC catalyst. On the basis of a series of NMR experiments, the authors propose that the free NHC reacts faster with the enal substrate than with NFSI, which explains catalyst turnover.
Scheme 234.

Proposed Mechanism for α-Fluorination of NHC-Bound Dienolates with NFSI
Concurrently, Ye employed α,β-unsaturated aldehydes bearing a γ-leaving group as precursors of azolium dienolates for reaction with azodicarboxylates as electrophiles (Scheme 235).255 The yields of the resultant dihydropyridazinones are moderate-to-high (49–86%) for an entire range of enals bearing aromatic or alkenyl substituents. It is noteworthy that aliphatic enals also work well to give the desired annulation products in moderate yields. The reaction of various azodicarboxylates is also successful. Furthermore, the formal [4+2] hetero-Diels-Alder cycloaddition proceeds with excellent enantioselectivity (94–99% ee) when the chiral NHC precursor A4 is employed.
Scheme 235.

NHC-Catalyzed [4+2] Annulation of Oxidized Enals and Azodicarboxylates
A possible catalytic cycle is depicted in Scheme 236 below. The addition of the in situ generated NHC to the enal gives the extended Breslow intermediate 250. The latter then collapses to azolium dienolate 251 by removal of the γ leaving group. Conjugate addition of the dienolate to azadicarboxylate affords α,β-unsaturated acylazolium 252, which finally cyclizes to give the dihydropyridazinone and regenerate the carbene catalyst.
Scheme 236.

Postulated Mechanism for the [4+2] Annulation of Azolium Dienolates and Azodicarboxylates
6. LEWIS BASE CATALYSIS
6.1 Nucleophilic Catalysis
6.1.1 Morita-Baylis-Hillman and Rauhut-Currier Reactions
The Morita-Baylis-Hillman reaction catalyzed by NHCs was first reported by Ye in 2007, coupling cyclic enones to aryl N-tosyl imines (Scheme 237).256 The enantioselective variant was also reported, coupling cyclopent-2-enone with N-tosylphenylmethanimine in up to 44% ee.257
Scheme 237.

Enantioselective Morita-Baylis-Hillman reaction
Cross-over experiments with imine 253 and azolium adduct 254 gives a 1:1.3 mixture of 255:256, implying the carbene adds to the tosyl-imine reversibly; however, formation of the MBH adduct is not reversible under the reaction conditions (Scheme 238).
Scheme 238.

Cross-over experiments for the MBH reaction
Ye also demonstrated the MBH reaction of nitrostyrenes with azodicarboxylates using thiazolium precatalyst G19 in good yields (Scheme 239).258 A broad range of aryl-nitrostyrenes are competent in the reaction; however, aliphatic nitroalkenes do not participate because of competitive polymerization.
Scheme 239.

MBH Reaction of Nitroalkenes and Diazodicarboxylates
In a similar reaction, Ye reported a [4+2] annulation of nitroalkenes and α,β–unsaturated ketones.259 The reaction is believed to proceed via a Rauhut-Currier type mechanism, where the nitroalkene-carbene adduct adds 1,4 to the enone, followed by displacement of the alkyl azolium by the enolate oxygen. Interestingly, the desired [4+2] cycloadduct is formed in only trace amounts when DABCO or PPh3 are used instead of thiazolium G30. Another interesting observation made during this work was the reversal of diastereoselectivity by varying the N-substituent of the thiazolium pre-catalyst. 2,4,6-trimethylphenyl precatalyst G30 gives 2,3-trans product (257) selectively, while the formation of the 2,3-cis product (cis-257) is favored by electron poor 3.5-bis(trifluoromethyl)phenyl thiazolium G29 (Scheme 240).
Scheme 240.

[4+2] Annulation of Nitroalkenes and Oxodienes
Scheidt reported the NHC catalyzed Rauhut-Currier reaction, coupling 1,1-bis(phenylsulfonyl)ethylene to α,β–unsaturated aldehydes.260
Matsuoka further expanded this chemistry to include the dimerization of methacroleins in the presence of an alcohol (Scheme 242).261 Based on mass spectrometry experiments, the authors propose the reaction proceeds via initial conjugate addition of the carbene to the acrolein, followed by conjugate addition to another equivalent of acrolein. The catalyst is regenerated by methanol displacement of the azolium.
Scheme 242.

Dimerization of Methacroleins
6.1.2 Umpolung of Michael Acceptors
The conjugate addition of the carbene to Michael acceptors has also been demonstrated to result in β-functionalization. This likely proceeds through a formal deoxy-Breslow intermediate, which transforms the normally electrophilic β-carbon to a nucleophile. This reactivity was first reported by Fu in 2006, who found it while exploring an intramolecular Heck reaction with alkyl tosylates (Scheme 243).262
Scheme 243.

β-Alkylation of Deoxy-Breslow Intermediates
Scheidt reported a similar reaction using vinyl-sulfones, which rearrange after treatment with the NHC, and then undergo a cycloaddition with nitrones to give isoxazolines (Scheme 244).263 Interestingly, only one diastereomer is generated (measured by 1H NMR) in the reaction.
Scheme 244.

Synthesis of Isoxazolines
Matsuoka and Glorius independently reported the tail-to-tail dimerization of acrylates with carbene catalysts (Scheme 245).264 Since these two important contributions, much work has been devoted to the β-coupling of acrylates to form dimers, trimers, and tetramers. This reactivity has also been exploited in polymerization catalysis.265
Scheme 245.

Dimerization of Acrylates
Matsuoka later expanded this chemistry to include the dimerization of methacrylonitrile in the presence of an alcohol.266 Matsuoka also noted an interesting reaction between previously generated deoxy-Breslow intermediates and isocyanates (Scheme 246).267 Attempts at a one-step three component coupling were hindered by carbene addition to the isocyanate, leading to the formation of a urea side product. Later, Glorius demonstrated the selective tail-to-tail homo-coupling of electron-deficient styrenes (Scheme 247).268 Aside from dimerization reactions proceeding through deoxy Breslow intermediates, Nair demonstrated the reaction of unsaturated cinnamils to give vinyl fulvene derivatives rather than the expected lactone.269
Scheme 246.

Coupling of Deoxy-Breslow Intermediates and Isocyanates
Scheme 247.

Dimerization of styrenes
136% yield can be obtained using 20 mol% G23.
In most of these reactions, to date, the most efficient catalytic systems are triaryl triazolium based. This is surprising, given that the triaryl triazolylidenes are not often used in other NHC-catalyzed reaction (bicyclic triazolium precatalysts are dominant - cf. figure 2) and that triazolylidenes are generally less nucleophilic than imidazolylidenes or thiazolylidenes. Mayr investigated the reactivity difference of several deoxy-Breslow intermediates through kinetic measurements with reference electrophiles.270 The nucleophilicity of a triazolylidene derived deoxy-Breslow interemediate was found to be intermediate compared to the more nucleophilic imidazolylidene deoxy-Breslow and the less nucleophilic imidazolinylidene variant. This data suggests the nucleophilicity of the deoxy-Breslow can not explain the preference for triazolium pre-catalysts in these reactions.
6.1.3 Activation of Boryl Groups
In 2009, Hoveyda and co-workers demonstrated that imidazole based carbenes are capable of catalyzing carbon-boron bond formation between enones and bis(pinacolato)diboron to synthesize β-boron substituted ketones via a conjugate addition.271 It is believed that the free carbene coordinates to one boron atom of bis(pinacolato)diboron to generate activated species 258. This activated species then adds to the enone via a 1,4-addition to generate 259, which then undergoes a tautomerization to liberate carbene and boron-enolate 260. The boron-enolate generates ketone product 261 upon aqueous workup. Marder and co-workers have characterized a crystal structure of intermediate 258 (Scheme 248).272
Scheme 248.

Mechanism of NHC Catalyzed β-Boron Addition
A crystal structure of intermediate 258 shown in Figure 7 clearly demonstrates the boron-NHC interaction as the B-B bond distance increases by 0.039 Å as the NHC-bound boron atom undergoes pyraminalization from sp2 to sp3. Further, the distorted tetrahedral geometry of the NHC-bound boron atom is clearly visible in the crystal structure.
Figure 7.
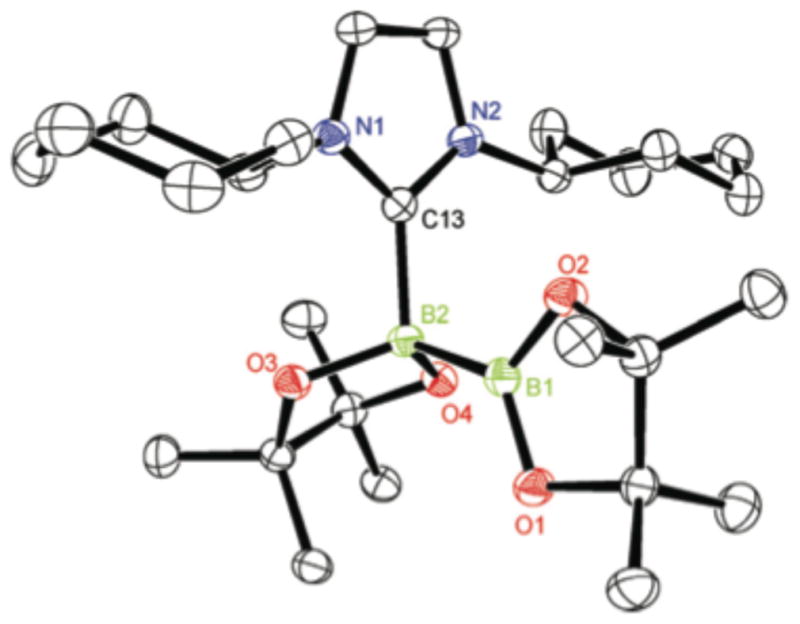
X-ray Crystal Structure of 258
Hoveyda’s initial report on the NHC-catalyzed boron conjugate addition reaction showed that a variety of cyclic and acyclic ketones and esters are acceptable Michael acceptors. It was also shown that β,β-disubstituted Michael acceptors participate in the reaction to form quaternary substituted carbons. Yields are high (44 – >98 %) and diastereoselectivity ranges from 1.6:1 to 7.2:1 (Scheme 249).
Scheme 249.

Hoveyda’s Initial NHC-Catalyzed Conjugate Boron Addition
In 2012, Hoveyda expanded the utility of NHC-catalyzed conjugate borylation by developing the enantioselective transformation and expanding the types of Michael acceptors that participate in the reaction.212g Initial attempts at an enantioselective reaction were met with limited conversion. It was hypothesized that this may be due to difficulties associated with the sterically demanding NHC coordinating to the bulky bis(pinacolato)diboron 262. The use of methanol as an additive provided a solution to this problem and the reaction was shown to proceed efficiently. It is proposed that methanol exchanges with the pinacol unit of the diboron to generate a more sterically accessible diboron unit 263, which may coordinate the carbene (Scheme 250).
Scheme 250.
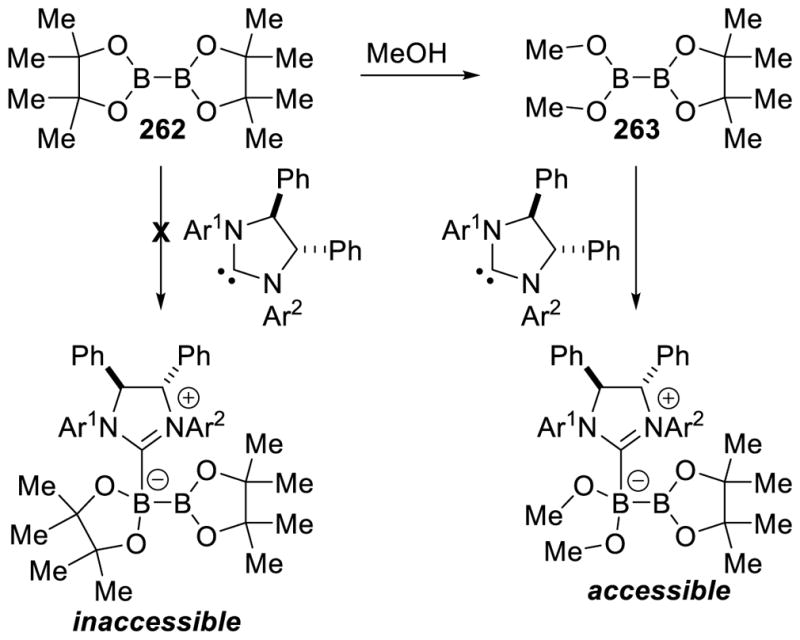
Use of Methanol to Generate Less Hindered Diboron Reagent
A wide variety of Michael acceptors including enones, enals, enoates, and α,β-unsaturated amides are tolerated in the reaction. Of particular note is that enals, substrates that have been demonstrated extensively to undergo dimerization reactions in the presence of NHC-catalysts (cf. sections 3.2, 3.3 and 4.2), are well tolerated in the reaction and undergo minimal undesired NHC catalyzed pathways. Yields in this reaction range from 43 to 94% and enantioselectivity ranges from 42 to 96% ee (Scheme 251).
Scheme 251.

Enantioselective Conjugate Boron Addition
6.1.4 Activation of Silyl and Stannyl Groups
Similar to the NHC-catalyzed boron conjugate addition, Hoveyda has developed an enantioselective NHC-catalyzed silicon conjugate addition.273 The reaction proceeds via activation of dimethylphenylsilylpinacolatoboron [Me2PhSi-B(pin)] 264 by an NHC catalyst to selectively transfer the silicon to the β-position of a Michael acceptor (Figure 8). The selectivity of the transfer arises from the high Lewis acidity of boron, which selectively coordinates the NHC, thus activating the silicon atom for transfer to the β-position.
Figure 8.

NHC Activation of Silicon-Boron Complex
In this reaction, cyclic enones, lactones, acyclic α,β-substituted ketones, esters, and aldehydes are tolerated as substrates to provide β-silylated products in up to 98% yield and 70 – 96% ee (Scheme 252). Of note, enals perform well in this reaction, suggesting the NHC catalyst selectively interacts with the silicon-boron complex rather than the aldehyde, which would provide products arising from Breslow intermediate reactivity (cf. Section 3). A similar reaction was reported by Commeiras and Parrain activating tributyl(trimethylsilyl)stannane for the 1,2 addition or 1,4 addition of tributyl tin to aldehydes or enals.274 Aliphatic, heteroaromatic, and electron rich aryl aldehydes proceed in good yields (85–100%); however electron deficient aryl aldehydes are much less consistent (often resulting in no reaction). Acrolein and its derivatives also participate, generating the 1,4-tin addition adduct as the only observable addition product, while β-substituted enals favored 1,2 addition.
Scheme 252.

NHC Catalyzed Conjugate Silicon Addition
6.2 Base Catalysis
N-Heterocyclic carbenes have also found a role as a general base for the activation of alcohols, amines, and 1,3-diketones. Scheidt and co-workers showed in 2010 that NHC catalysts are capable of activating alcohols for a 1,4-addition to enones to generate β-alkoxy ketones (Scheme 253).275 Aryl and alkyl ketones, as well as enoates participate in the reaction. A variety of primary and secondary alcohols are competent nucleophiles. The reaction is thought to proceed via activation of the alcohol by the NHC catalyst to form complex 265. This active complex then transfers the alcohol to the β-position of the enone and then subsequently transfers a proton to the α-position for a net hydroalkoxylation reaction (Scheme 254), similar to the role of NHCs in transesterification (cf. Section 5.3.1).
Scheme 253.

Scope of NHC-Catalyzed Conjugate Addition of Alcohols to Enones
Scheme 254.
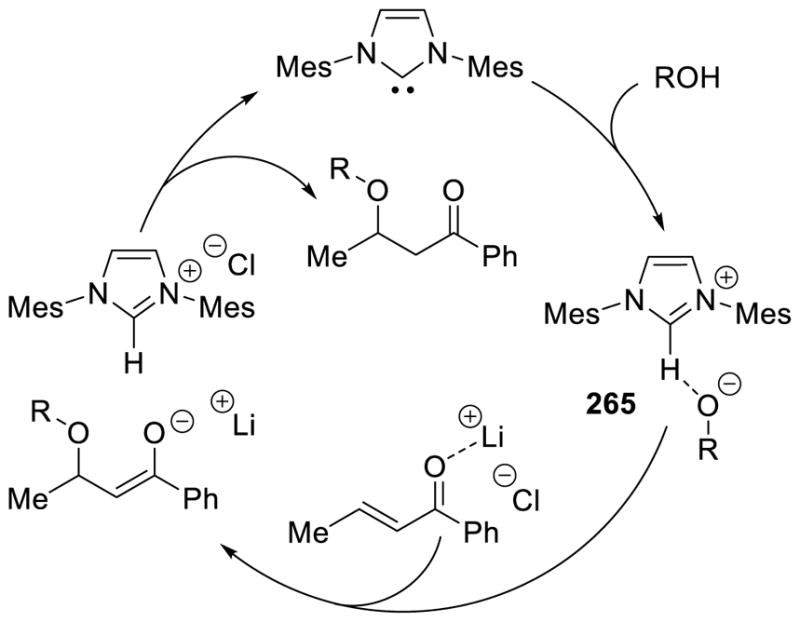
NHC-Catalyzed Conjugate Addition of Alcohol to Enone
Zhang and Kang reported the aza-variant of the NHC-catalyzed conjugate addition in 2011.276 Aryl and aliphatic amines readily yield product in the reaction and aryl as well as aliphatic substitution on the enone is acceptable (Scheme 255). Recently, NHC CO2 Adducts adducts were also shown as competent precatalysts for the Michael additions of thiols, diphenylphosphine oxide, diethylphosphonic acid, and 1,3-dicarbonyl compounds.277
Scheme 255.

Scope of the NHC-Catalyzed Aza-Conjugate Addition
In addition to conjugate additions of alcohols and amines, this type of reactivity has been extended to 1,3-dicarbonyls as the nucleophilic coupling partner as well as efficient catalysts for D-H exchange reactions on these substrates.278 In 2009, Rodriguez and Coquerel reported the synthesis of α-spirolactones and α-spirolactams through an olefin cross-metathesis/NHC Michael addition cascade reaction.279 The first step involves installing an electron withdrawing group on an olefin tethered to the 1,3-dicarbonyl, followed by an NHC-catalyzed Michael-addition of 1,3-dicarbonyl compounds to the tethered Michael acceptor. Interestingly, a one pot procedure was developed where the crude reaction mixture from the cross-metathesis reaction could be directly treated with P(nBu)3 to decomplex the free carbene 16, which then catalyzes the subsequent Michael addition. The same group also demonstrated the intermolecular coupling of 1,3-dicarbonyls with vinyl electron-withdrawing groups.280 Recently, Chen and Huang demonstrated the stereoselective 1,4-addition of 1,3-dicarbonyl compounds to nitrostyrene derivatives.281 Mechanistically, the reaction is believed to proceed via complexation between the free carbene and the cyclic enol form of the 1,3-dicarbonyl 266, activating it. Then, the nitrostyrene approaches the complex and reacts via an ene-type mechanism. The high stereoselectivity of this reaction is attributed to the highly organized transition state 267 that is accessed via the carbene coordination to the 1,3-dicarbonyl (Scheme 256). Further, a negative non-linear effect was observed, implying that two molecules of catalyst are involved in the transition state.
Scheme 256.
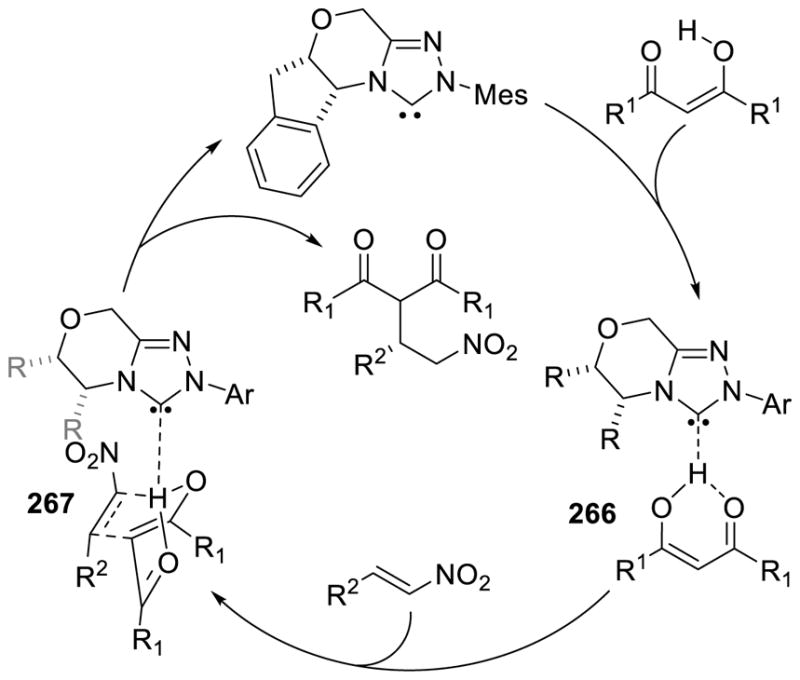
Mechanism of NHC-Promoted Conjugate Addition of 1,3-Dicarbonyls
The authors note careful selection of reaction parameters allows for the proper pKa balance between carbene catalyst and substituted 1,3-dicarbonyl product. The reaction requires a fine balance of pKa to ensure that all of the reactive moieties are in their proper reactive form to proceed. It was found that a mixture of LHMDS and hexafluoroisopropanol (HFIP) provides optimal reactivity. A variety of electron-rich and electron-deficient nitrostyrenes are tolerated as Michael acceptors, with a single aliphatic nitroalkene included in the substrate scope. Symmetrical aliphatic and aryl 1,3-diketones work well in the reaction, and a variety of unsymmetrical 1,3-dicarbonyl species, including 1,3-diketones and β- α-ketoesters, work well, but because the products epimerize easily in basic conditions, diastereoselectivity is minimal (Scheme 257).
Scheme 257.

Scope of Conjugate 1,3-Dicarbonyl Addition
7. CONCLUSION
With the broad diversity of chemistry demonstrated in this review, it is clear N-heterocyclic carbenes have had a broad impact on the field of organic chemistry, often allowing for the mild construction of complex molecules from simple starting materials. As the number of reactivity manifolds has increased, so too has the popularity of these unique catalysts in the community, as evidenced by Figure 9 showing the number of publications appearing between 1970 and 2014 relating to heterocyclic carbenes.
Figure 9.

Histogram of Scifinder Search of “Heterocyclic Carbene” (January 2015)
Despite the appearance of several landmark papers hinting at the enormous potential of N-heterocylic carbene catalysts, it was not until the year 2000 that a drastic increase in the field began. It is remarkable to think many of the advances presented came largely during the last 15 years. A closer inspection of Figure 9 reveals that 8627 publications appeared between 2000 and 2014 relating to heterocylic carbenes, in sharp contrast to 149 appearing between 1970 and 1999.
Along with an increase in the number of reactivity manifolds presented, there has also been an increase in their use in an industrial setting and toward natural product syntheses.282 This review highlights the accomplishments made in this time-frame, but also points out the challenges still in need of a solution. The development of new transformations will no doubt continue to provide new challenges; nevertheless, designing more efficient N-heterocyclic carbenes that are more reactive and selective in the established reactivity remains a major focus.
Scheme 18.

Yang’s Cross-Benzoin Reaction
Scheme 212.

Enantioselective Esterification of Ketenes
Scheme 214.

Enantioselective Esterification of Ketenes (Smith)
Scheme 241.

Rauhut-Currier Reactions of Enals
Chart 1.

Acidity of Azolium Based NHC Precursors*
*pKa values determined in H2O (DMSO) [computed]
Acknowledgments
We are grateful to the NIGMS (GM72586) for generous support of our work in this area. TR acknowledges the many coworkers on this project, past and present, whose experimental and intellectual contributions have made our advances in this project possible.
Biographies

Darrin Flanigan was born in Springfield, Oregon in 1989. He received his B. S. in chemistry from Oregon State University in 2011 where he performed undergraduate research with Professor Paul R. Blakemore. In 2011, he joined the Rovis research group where he is currently a fourth year graduate student. His research in the Rovis group has been focused on the development of new N-heterocyclic carbene-catalyzed reactions.

Fedor Romanov-Michailidis was born in 1987 in Moscow, in the former USSR. He moved to Ferney-Voltaire, France in 1998 where he received his high school education. He then moved to Geneva, Switzerland where he obtained his Bachelor degree in Biochemistry in 2008. He obtained his Masters degree in Chemistry in 2010 after working at Nestle, Lausanne. He then returned to Geneva where he received his PhD degree in 2014 under the guidance of Prof. Alexandre Alexakis. He is currently performing his post-doctoral studies as a SNSF fellow in Fort Collins, Colorado in the laboratories of Prof. Tomislav Rovis. His research interests gravitate around discovering new reactions for organic synthesis.

Nicholas White was born in Iowa in 1987. He received his bachelor’s degree in chemistry from Loyola University Chicago in 2010 where he performed undergraduate research under the guidance of Professor James Babler. In 2010 Nick joined Tomislav Rovis’ research group at Colorado State University where he is currently a fifth year graduate student. His research in the Rovis group has focused on the development of N-heterocyclic carbene catalyzed reactions of enals. Upon the completion of his thesis work, Nick will be moving to MIT to pursue postdoctoral research in the labs of Professor Stephen Buchwald.

Tomislav Rovis was born in Zagreb in the former Yugoslavia but was largely raised in Southern Ontario, Canada. Following his undergraduate studies at the University of Toronto, he earned his Ph.D. degree at the same institution in 1998 under the direction of Professor Mark Lautens. From 1998–2000, he was an NSERC postdoctoral fellow at Harvard University with Professor David A. Evans. In 2000, he began his independent career at Colorado State University and was promoted in 2005 to Associate Professor and in 2008 to Professor. His group’s accomplishments have been recognized by a number of awards including an NSF CAREER and a Roche Excellence in Chemistry award. He has been named a GlaxoSmithKline Scholar, Amgen Young Investigator, Eli Lilly Grantee, Alfred P. Sloan Fellow, Monfort Professor at Colorado State University, Fellow of the American Association for the Advancement of Science, Katritzky Young Investigator in Heterocyclic Chemistry, and an Arthur C. Cope Scholar. He currently holds the John K. Stille Chair in Chemistry.
References
- 1.Igau A, Grutzmacher H, Baceiredo A, Bertrand G. Alpha., Alpha.′-bis-Carbenoid, Triply Bonded. Analogous. [Google Scholar]
- 2.Arduengo AJ, III, Harlow RL, Kline MA. Stable Crystalline Carbene. J Am Chem Soc. 1991;113:361–363. [Google Scholar]
- 3.Hopkinson MN, Richter C, Schedler M, Glorius F. An Overview of N-Heterocyclic Carbenes. Nature. 2014;510:485–496. doi: 10.1038/nature13384. [DOI] [PubMed] [Google Scholar]
- 4.(a) Hermann WA. N-Heterocyclic Carbenes: A New Concept in Organometallic Catalysis. Angew Chem Int Ed. 2002;41:1290–1309. doi: 10.1002/1521-3773(20020415)41:8<1290::aid-anie1290>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]; (b) Crabtree RH. NHC Ligands Versus Cyclopentadienyls and Phosphines as Spectator Ligands in Organometallic Catalysis. J Organomet Chem. 2005;690:5451–5457. [Google Scholar]
- 5.Samojłowicz C, Bieniek M, Grela K. Ruthenium-Based Olefin Metathesis Catalysts Bearing N-Heterocyclic Carbene Ligands. Chem Rev. 2009;109:3708–3742. doi: 10.1021/cr800524f. [DOI] [PubMed] [Google Scholar]
- 6.For recent reviews, see: Bellemin-Laponnaz S, Dagorne S. Group 1 and 2 and Early Transition Metal Complexes Bearing N-Heterocyclic Carbene Ligands: Coordination Chemistry, Reactivity, and Applications. Chem Rev. 2014;114:8747–8774. doi: 10.1021/cr500227y.Fliedel C, Braunstein P. Recent Advances in S-Functionalized N-Heterocyclic Carbene Ligands: From the Synthesis of Azolium Salts and Metal Complexes to Applications. J Organomet Chem. 2014;751:286–300.Izquierdo F, Manzini S, Nolan SP. The Use of the Sterically Demanding IPr* and Related Ligands in Catalysis. Chem Commun. 2014:14926–14937. doi: 10.1039/c4cc05289g.Nelson DJ, Nolan SP. Quantifying and Understanding the Electronic Properties of N-Heterocyclic Carbenes. Chem Soc Rev. 2013;42:6723–6753. doi: 10.1039/c3cs60146c.
- 7.Ukai T, Tanaka R, Dokawa T. A New Catalyst for the Acyloin Condensation. I. J Pharm Soc Jpn. 1943;63:296–304. (Chem Abstr 1951; 45:5148) [Google Scholar]
- 8.Breslow R. On the Mechanism of Thiamine Action. IV.1 Evidence from Studies on Model Systems. J Am Chem Soc. 1958;80:3719–3726. [Google Scholar]
- 9.Wöhler F, Liebig J. Untersuchungen über das Radikal der Benzoesäure. Ann Der Pharm. 1832;3:249–287. [Google Scholar]
- 10.Sheehan JC, Hunneman DH. Homogeneous Asymmetric Catalysis. J Am Chem Soc. 1966;88:3666–3667. [Google Scholar]
- 11.Nozaki H, Moriuti S, Takaya H, Noyori R. Asymmetric Induction in Carbenoid Reaction By Means of A Dissymmetric Copper Chelate. Tetrahedron Lett. 1966;7:5239–5244. [Google Scholar]
- 12.Enders D, Breuer K, Raabe G, Runsink J, Teles JH, Melder J-P, Ebel K, Brode S. Preparation, Structure, and Reactivity of 1,3,4-Triphenyl-4,5-dihydro-1H-1,2,4-triazol-5-ylidene, a New Stable Carbene. Angew Chem Int Ed. 1995;34:1021–1023. [Google Scholar]
- 13.Knight RL, Leeper FJ. Comparison of Chiral Thiazolium and Triazolium Salts as Asymmetric Catalysts for the Benzoin Condensation. J Chem Soc Perkin Trans 1. 1998:1891–1894. [Google Scholar]
- 14.Kerr MS, Read de Alaniz J, Rovis T. A Highly Enantioselective Catalytic Intramolecular Stetter Reaction. J Am Chem Soc. 2002;124:10298–10299. doi: 10.1021/ja027411v. [DOI] [PubMed] [Google Scholar]
- 15.Enders D, Niemeier O, Henseler A. Organocatalysis by N-Heterocyclic Carbenes. Chem Rev. 2007;107:5606–5655. doi: 10.1021/cr068372z. [DOI] [PubMed] [Google Scholar]
- 16.Benhamou L, Chardon E, Lavigne G, Bellemin-Laponnaz S, César V. Synthetic Routes to N-Heterocyclic Carbene Precursors. Chem Rev. 2011;111:2705–2733. doi: 10.1021/cr100328e. [DOI] [PubMed] [Google Scholar]
- 17.(a) Sheehan JC, Hara T. Asymmetric Thiazolium Salt Catalysis of the Benzoin Condensation. J Org Chem. 1974;39:1196–1199. [Google Scholar]; (b) Vougioukalakis GC, Grubbs RH. Synthesis and Activity of Ruthenium Olefin Metathesis Catalysts Coordinated with Thiazol-2-ylidene Ligands. J Am Chem Soc. 2008;130:2234–2245. doi: 10.1021/ja075849v. [DOI] [PubMed] [Google Scholar]
- 18.(a) Lebeuf R, Hirano K, Glorius F. Palladium-Catalyzed C-Allylation of Benzoins and an NHC-Catalyzed Three Component Coupling Derived Thereof: Compatibility of NHC- and Pd-Catalysts. Org Lett. 2008;10:4243–4246. doi: 10.1021/ol801644f. [DOI] [PubMed] [Google Scholar]; (b) Pesch J, Harms K, Bach T. Preparation of Axially Chiral N,N′-Diarylimidazolium and N-Arylthiazolium Salts and Evaluation of Their Catalytic Potential in the Benzoin and in the Intramolecular Stetter Reactions. Eur J Org Chem. 2004:2025–2035. [Google Scholar]
- 19.(a) Benhamou L, César V, Gornitzka H, Lugan N, Lavigne G. Imidazol-2-ylidene-4-olate: An Anionic N-Heterocyclic Carbene Pre-programmed for Further Derivatization. Chem Commun. 2009:4720–4722. doi: 10.1039/b907908d. [DOI] [PubMed] [Google Scholar]; (b) Kuhn KM, Grubbs RH. A Facile Preparation of Imidazolinium Chlorides. Org Lett. 2008;10:2075–2077. doi: 10.1021/ol800628a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) César V, Lugan N, Lavigne G. A Stable Anionic N-Heterocyclic Carbene and Its Zwitterionic Complexes. J Am Chem Soc. 2008;130:11286–11287. doi: 10.1021/ja804296t. [DOI] [PubMed] [Google Scholar]; (d) Jazzar R, Liang H, Donnadieu B, Bertrand G. A New Synthetic Method for the Preparation of Protonated-NHCs and Related Compounds. J Organomet Chem. 2006;691:3201–3205. doi: 10.1016/j.jorganchem.2006.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Sanderson MD, Kamplain JW, Beilawski CW. Quinone-Annulated N-Heterocyclic Carbene–Transition-Metal Complexes: Observation of π-Backbonding Using FT-IR Spectroscopy and Cyclic Voltammetry. J Am Chem Soc. 2006;128:16514–16515. doi: 10.1021/ja067475w. [DOI] [PubMed] [Google Scholar]; (f) Bertogg A, Camponovo F, Togni A. N-Ferrocenyl-Substituted Planar-Chiral N-Heterocyclic Carbenes and Their PdII Complexes. Eur J Inorg Chem. 2005:347–356. [Google Scholar]
- 20.Kamijo S, Yamamoto Y. Recent Progress in the Catalytic Synthesis of Imidazoles. Chem-Asian J. 2007;2:568–578. doi: 10.1002/asia.200600418. [DOI] [PubMed] [Google Scholar]
- 21.(a) Calder IC, Spotswood TM, Sasse WHP. The Dipyrido[1,2-e:2,1-e]imidazolium Cation, a New Aromatic Ring System. Tetrahedron Lett. 1963;4:95–100. [Google Scholar]; (b) Calder IC, Sasse WHF. Aromatic Nitrogen Bridgehead Compounds. I. The Dipyrido[1,2-c:2′,1′-e]-imidazolium and Related Cations. Aust J Chem. 1965;18:1819–1833. [Google Scholar]
- 22.Weiss R, Reichel S, Handke M, Hampel F. Generation and Trapping Reactions of a Formal 1:1 Complex between Singlet Carbon and 2,2-Bipyridine. Angew Chem Int Ed. 1998;37:344–347. doi: 10.1002/(SICI)1521-3773(19980216)37:3<344::AID-ANIE344>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 23.Arduengo AJ, III, Krafczyk R, Schmutzler R. Imidazolylidenes, Imidazolinylidenes and Imidazolidines. Tetrahedron. 1999;55:14523–14534. [Google Scholar]
- 24.(a) Glorius F, Altenhoff G, Goddard R, Lehmann C. Oxazolines as Chiral Building Blocks for Imidazolium Salts and N-Heterocyclic Carbene Ligands. Chem Commun. 2002:2704–2705. doi: 10.1039/b208045a. [DOI] [PubMed] [Google Scholar]; (b) Würtz S, Glorius F. Surveying Sterically Demanding N-Heterocyclic Carbene Ligands with Restricted Flexibility for Palladium-catalyzed Cross-Coupling Reactions. Acc Chem Res. 2008;41:1523–1533. doi: 10.1021/ar8000876. [DOI] [PubMed] [Google Scholar]
- 25.Bildstein B, Malaun M, Kopacka H, Wurst K, Mitterböck M, Ongania K-H, Opromolla G, Zanello P. N,N′-Diferrocenyl-N-Heterocyclic Carbenes and Their Derivatives. Organometallics. 1999;18:4325–4336. [Google Scholar]
- 26.Saba S, Brescia A, Kaloustian MK. One-pot Synthesis of Cyclic Amidinium Tetrafluoroborates and Hexafluorophosphates; The Simplest Models of N5,N100methenyltetrahydrofolate Coenzyme. Tetrahedron Lett. 1991;32:5031–5034. [Google Scholar]
- 27.(a) Donia RA, Shotton JA, Bentz LO, Smith GEP. Reactions of Mono- and Di-Amines with Carbon Disulfide. II. Methylenediamine and Imidazolidine-Carbon Disulfide Reactions. J Org Chem. 1949;14:952–961. [Google Scholar]; (b) Bildstein B, Malaun M, Kopacka H, Ongania KH, Wurst K. N-Heterocyclic Carbenes with N-Ferrocenyl-N-Methyl-Substitution: Synthesis, Reactivity, Structure and Electrochemistry. J Organomet Chem. 1999;572:177–187. [Google Scholar]
- 28.Alcarazo M, Roseblade SJ, Cowley AR, Fernández R, Brown JM, Lassaletta JM. Imidazo[1,5-a]pyridine: A Versatile Architecture for Stable N-Heterocyclic Carbenes. J Am Chem Soc. 2005;127:3290–3291. doi: 10.1021/ja0423769. [DOI] [PubMed] [Google Scholar]
- 29.Enders D, Breuer K, Kallfass U, Balensiefer T. Preparation and Application of 1,3,4-Triphenyl-4,5-dihydro-1H-1,2,4-triazol-5-ylidene, A Stable Carbene. Synthesis. 2003:1292–1295. [Google Scholar]
- 30.Enders D, Gielen H. Synthesis of Chiral Triazolinylidene and Imidazolinylidene Transition Metal Complexes and First Application in Asymmetric Catalysis. J Organomet Chem. 2001;617:70–80. [Google Scholar]
- 31.Enders D, Kallfass U. An Efficient Nucleophilic Carbene Catalyst for the Asymmetric Benzoin Condensation. Angew Chem Int Ed. 2002;41:1743–1745. doi: 10.1002/1521-3773(20020517)41:10<1743::aid-anie1743>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 32.Kerr MS, Read de Alaniz J, Rovis T. An Efficient Synthesis of Achiral and Chiral 1,2,4-Triazolium Salts: Bench Stable Precursors for N-Heterocyclic Carbenes. J Org Chem. 2005;70:5725–5728. doi: 10.1021/jo050645n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vora HU, Lathrop SP, Reynolds NT, Kerr MS, Read de Alaniz J, Rovis T. Preparation of Chiral and Achiral Triazolium Salts: Carbene Precursors with Demonstrated Synthetic Utility. Org Synth. 2010;87:350–361. [Google Scholar]
- 34.(a) Bode JW, Struble JR, Lian Y. Synthesis of an N-Mesityl Substituted Aminoindanol-Derived Triazolium Salt. Org Synth. 2010;87:362–376. [Google Scholar]; (b) Bugaut X, Liu F, Glorius F. N-Heterocyclic Carbene (NHC)-Catalyzed Intermolecular Hydroacylation of Cyclopropenes. J Am Chem Soc. 2011;133:8130–8133. doi: 10.1021/ja202594g. [DOI] [PubMed] [Google Scholar]
- 35.Dröge T, Glorius F. The Measure of All Rings—N-Heterocyclic Carbenes. Angew Chem Int Ed. 2010;49:6940–6952. doi: 10.1002/anie.201001865. [DOI] [PubMed] [Google Scholar]
- 36.Kim Y-J, Streitwieser A. Basicity of a Stable Carbene, 1,3-Di-tert-butylimidazol-2-ylidene, in THF. J Am Chem Soc. 2002;124:5757–5761. doi: 10.1021/ja025628j. [DOI] [PubMed] [Google Scholar]
- 37.Washabaugh MW, Jencks WP. Thiazolium C(2)-Proton Exchange: General-Base Catalysis, Direct Proton Transfer, and Acid Inhibition. J Am Chem Soc. 1989;111:674–683. [Google Scholar]
- 38.Chu Y, Deng H, Cheng J-P. An Acidity Scale of 1,3-Dialkylimidazolium Salts in Dimethyl Sulfoxide Solution. J Org Chem. 2007;72:7790–7793. doi: 10.1021/jo070973i. [DOI] [PubMed] [Google Scholar]
- 39.Amyes TL, Diver ST, Richard JP, Rivas FM, Toth K. Formation and Stability of N-Heterocyclic Carbenes in Water:3 The Carbon Acid pKa of Imidazolium Cations in Aqueous Solution. J Am Chem Soc. 2004;126:4366–4374. doi: 10.1021/ja039890j. [DOI] [PubMed] [Google Scholar]
- 40.Massey RS, Collett CJ, Lindsay AG, Smith AD, O’Donoghue AC. Proton Transfer Reactions of Triazol-3-ylidenes: Kinetic Acidities and Carbon Acid pKa Values for Twenty Triazolium Salts in Aqueous Solution. J Am Chem Soc. 2012;134:20421–20432. doi: 10.1021/ja308420c. [DOI] [PubMed] [Google Scholar]
- 41.Higgins EM, Sherwood JA, Lindsay AG, Armstrong J, Massey RS, Alder RW, O’Donoghue AC. pKas of the Conjugate Acids of N-Heterocyclic Carbenes in Water. Chem Commun. 2011:1559–1561. doi: 10.1039/c0cc03367g. [DOI] [PubMed] [Google Scholar]
- 42.Maji B, Breugst M, Mayr H. N-Heterocyclic Carbenes: Organocatalysts with Moderate Nucleophilicity but Extraordinarily High Lewis Basicity. Angew Chem Int Ed. 2011;50:6915–6919. doi: 10.1002/anie.201102435. [DOI] [PubMed] [Google Scholar]
- 43.(a) Bugaut X, Glorius F. Organocatalytic Umpolung N-Heterocyclic Carbenes and Beyond. Chem Soc Rev. 2012;41:3511–3522. doi: 10.1039/c2cs15333e. [DOI] [PubMed] [Google Scholar]; (b) Seebach D. Methods of Reactivity Umpolung. Angew Chem Int Ed. 1979;18:239–258. [Google Scholar]
- 44.(a) Hollóczki O, Kelemen Z, Nyulászi L. On the Organocatalytic Activity of N-Heterocyclic Carbenes: Role of Sulfur in Thiamine. J Org Chem. 2012;77:6014–6022. doi: 10.1021/jo300745e. [DOI] [PubMed] [Google Scholar]; (b) He Y, Xue Y. Theoretical Investigations on the Mechanism of Benzoin Condensation Catalyzed by Pyrido[1,2-a]-2-ethyl[1,2,4]triazol-3-ylidene. J Phys Chem A. 2011;115:1408–1417. doi: 10.1021/jp110352e. [DOI] [PubMed] [Google Scholar]; (c) Dudding T, Houk KN. Computational Predictions of Stereochemistry in Asymmetric Thiazolium- and Triazolium-Catalyzed Benzoin Condensations. Proc Nat Acad Sci. 2004;101:5770–5775. doi: 10.1073/pnas.0307256101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) White MJ, Leeper FJ. Kinetics of the Thiazolium Ion-Catalyzed Benzoin Condensation. J Org Chem. 2001;66:5124–5131. doi: 10.1021/jo010244h. [DOI] [PubMed] [Google Scholar]
- 45.Jordan F, Kudzin ZH, Rios CB. Generation and Physical Properties of Enamines Related to the Key Intermediate in Thiamin Diphosphate-Dependent Enzymic Pathways. J Am Chem Soc. 1987;109:4415–4416. [Google Scholar]
- 46.Simonovic S, Frison J-C, Koyuncu H, Whitwood AC, Douthwaite RE. Addition of N-Heterocyclic Carbenes to Imines: Phenoxide Assisted Deprotonation of an Imidazolium Moiety and Generation of Breslow Intermediates Derived from Imines. Org Lett. 2009;11:245–247. doi: 10.1021/ol802572n. [DOI] [PubMed] [Google Scholar]
- 47.DiRocco DA, Oberg KM, Rovis T. Isolable Analogues of the Breslow Intermediate Derived from Chiral Triazolylidene Carbenes. J Am Chem Soc. 2012;134:6143–6145. doi: 10.1021/ja302031v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hawkes KJ, Yates BF. The Mechanism of the Stetter Reaction – A DFT Study. Eur J Org Chem. 2008:5563–5570. [Google Scholar]
- 49.Maji B, Mayr H. Structures and Reactivities of O-Methylated Breslow Intermediates. Angew Chem Int Ed. 2012;51:10408–10412. doi: 10.1002/anie.201204524. [DOI] [PubMed] [Google Scholar]
- 50.Berkessel A, Elfert S, Yatham VR, Neudörfl J-M, Schlörer NE, Teles JH. Umpolung by N-Heterocyclic Carbenes: Generation and Reactivity of the Elusive 2,2-Diamino Enols (Breslow Intermediates) Angew Chem Int Ed. 2012;51:12370–12374. doi: 10.1002/anie.201205878. [DOI] [PubMed] [Google Scholar]
- 51.Mahatthananchai J, Bode JW. The Effect of the N-Mesityl Group in NHC-Catalyzed Reactions. Chem Sci. 2012;3:192–197. doi: 10.1039/C1SC00397F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Collett CJ, Massey RS, Maguire OR, Batsanov AS, O’Donoghue AC, Smith AD. Mechanistic Insights into the Triazolylidene-Catalyzed Stetter and Benzoin Reactions: Role of the N-Aryl Substituent. Chem Sci. 2013;4:1514–1522. [Google Scholar]
- 53.For reviews on other biomimetic reactions see: Enders D. Enzymemimetic C-C and C-N Bond Formations. In: Ottow E, Schöllkopf K, Schulz B-G, editors. Stereoselective Synthesis. Springer-Verlag; Berlin: 1993. pp. 63–90.
- 54.Bugaut X. Benzoin and Aza-Benzoin. In: Molander GA, Knochel P, editors. Comprehensive Organic Synthesis. 2. Vol. 1. Elsevier; Oxford: 2014. pp. 424–470. [Google Scholar]
- 55.(a) Enders D, Han J. Synthesis of Enantiopure Triazolium Salts from Pyroglutamic Acid and Their Evaluation in the Benzoin Condensation. Tetrahedron: Asymmetry. 2008;19:1367–1371. [Google Scholar]; (b) Ma Y, Wei S, Wu J, Yang F, Liu B, Lan J, Yang S, You J. From Mono-Triazolium Salt to Bis-Triazolium Salt: Improvement of the Asymmetric Intermolecular Benzoin Condensation. Adv Synth Catal. 2008;350:2645–2651. [Google Scholar]; (c) O’Toole SE, Connon SJ. The Enantioselective Benzoin Condensation Promoted by Chiral Triazolium Precatalysts: Stereochemical Control via Hydrogen Bonding. Org Biomol Chem. 2009;7:3584–3593. doi: 10.1039/b908517c. [DOI] [PubMed] [Google Scholar]; (d) Brand JP, Siles JIO, Waser J. Synthesis of Chiral Bifunctional (Thio)Urea N-Heterocyclic Carbenes. Synlett. 2010:881–884. [Google Scholar]; (e) Soeta T, Tabatake Y, Inomata K, Ukaji Y. Asymmetric Benzoin Condensation Promoted by Chiral Triazolium Precatalyst Bearing a Pyridine Moiety. Tetrahedron. 2012;68:894–899. [Google Scholar]; (f) Rafiński Z, Kozakiewicz A, Rafińska K. Highly Efficient Synthesis of Spirocyclic (1R)-Camphor-Derived Triazolium Salts: Application in the Catalytic Asymmetric Benzoin Condensation. Tetrahedron. 2014;70:5739–5745. [Google Scholar]
- 56.Baragwanath L, Rose CA, Zeitler K, Connon SJ. Highly Enantioselective Benzoin Condensation Reactions Involving a Bifunctional Protic Pentafluorophenyl-Substituted Triazolium Precatalyst. J Org Chem. 2009;74:9214–9217. doi: 10.1021/jo902018j. [DOI] [PubMed] [Google Scholar]
- 57.Cookson RC, Lane RM. Conversion of Dialdehydes into Cyclic α-Ketols by Thiazolium Salts: Synthesis of Cyclic 2-Hydroxy-2-Enones. J Chem Soc, Chem Commun. 1976:804–805. [Google Scholar]
- 58.Stetter H, Dämbkes G. Über die Präparative Nutzung der Thiazoliumsalz-Katalysierten Acyloin- und Benzoin-Bildung; II1. Herstellung Unsymmetrischer Acyloine und α-Diketone. Synthesis. 1977:403–404. [Google Scholar]
- 59.(a) Rose CA, Gundala S, Connon SJ, Zeitler K. Chemoselective Crossed Acyloin Condensations: Catalyst and Substrate Control. Synthesis. 2011:190–198. doi: 10.1021/jo101791w. [DOI] [PubMed] [Google Scholar]; (b) Piel I, Pawelczyk MD, Hirano K, Fröhlich R, Glorius F. A Family of Thiazolium Salt Derived N-Heterocyclic Carbenes (NHCs) for Organocatalysis: Synthesis, Investigation and Application in Cross-Benzoin Condensation. Eur J Org Chem. 2011:5475–5484. [Google Scholar]
- 60.(a) Jin MY, Kim SM, Han H, Ryu DH, Yang JW. Switching Regioselectivity in Crossed Acyloin Condensations between Aromatic Aldehydes and Acetaldehyde by Altering N-Heterocyclic Carbene Catalysts. Org Lett. 2011;13:880–883. doi: 10.1021/ol102937w. [DOI] [PubMed] [Google Scholar]; (b) Jin MY, Kim SM, Mao H, Ryu DH, Song CE, Yang JW. Chemoselective and Repetitive Intermolecular Cross-Acyloin Condensation Reactions Between a Variety of Aromatic and Aliphatic Aldehydes Using a Robust N-Heterocyclic Carbene Catalyst. Org Biomol Chem. 2014;12:1547–1550. doi: 10.1039/c3ob42486c. [DOI] [PubMed] [Google Scholar]
- 61.Langdon SM, Wilde MMD, Thai K, Gravel M. Chemoselective N-Heterocyclic Carbene-Catalyzed Cross-Benzoin Reactions: Importance of the Fused Ring in Triazolium Salts. J Am Chem Soc. 2014;136:7539–7542. doi: 10.1021/ja501772m. [DOI] [PubMed] [Google Scholar]
- 62.(a) Demir AS, Şeşenoglu O, Eren E, Hosrik B, Pohl M, Janzen E, Kolter D, Feldmann R, Dünkelmann P, Müller M. Enantioselective Synthesis of α-Hydroxy Ketones via Benzaldehyde Lyase-Catalyzed C–C Bond Formation Reaction. Adv Synth Catal. 2002;344:96–103. [Google Scholar]; (b) Iding H, Dünnwald T, Greiner L, Liese A, Müller M, Siegert P, Grötzinger J, Demir AS, Pohl M. Benzoylformate Decarboxylase from Pseudomonas putida as Stable Catalyst for the Synthesis of Chiral 2-Hydroxy Ketones. Chem Eur J. 2000;6:1483–1495. doi: 10.1002/(sici)1521-3765(20000417)6:8<1483::aid-chem1483>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]; (c) Dünkelmann P, Kolter-Jung D, Nitsche A, Demir AS, Siegert P, Lingen B, Baumann M, Pohl M, Müller M. Development of a Donor–Acceptor Concept for Enzymatic Cross-Coupling Reactions of Aldehydes: The First Asymmetric Cross-Benzoin Condensation. J Am Chem Soc. 2002;124:12084–12085. doi: 10.1021/ja0271476. [DOI] [PubMed] [Google Scholar]
- 63.Matsumoto T, Ohishi M, Inoue S. Selective Cross-Acyloin Condensation Catalyzed by Thiazolium Salt. Formation of 1-Hydroxy 2-One from Formaldehyde and Other Aldehydes. J Org Chem. 1985;50:603–606. [Google Scholar]
- 64.(a) Demir AS, Ayhan P, Igdir AC, Duygu AN. Enzyme Catalyzed Hydroxymethylation of Aromatic Aldehydes with Formaldehyde. Synthesis of Hydroxyacetophenones and (S)-Benzoins. Tetrahedron. 2004;60:6509–6512. [Google Scholar]; (b) Cosp A, Dresen C, Pohl M, Walter L, Röhr C, Müller M. α,β-Unsaturated Aldehydes as Substrates for Asymmetric C[BOND]C Bond Forming Reactions with Thiamin Diphosphate (ThDP)-Dependent Enzymes. Adv Synth Catal. 2008;350:759–771. [Google Scholar]
- 65.Kuhl N, Glorius F. Direct and Efficient N-Heterocyclic Carbene-Catalyzed Hydroxymethylation of Aldehydes. Chem Commun. 2011:573–575. doi: 10.1039/c0cc02416c. [DOI] [PubMed] [Google Scholar]
- 66.Hachisu Y, Bode JW, Suzuki K. Catalytic Intramolecular Crossed Aldehyde–Ketone Benzoin Reactions: A Novel Synthesis of Functionalized Preanthraquinones. J Am Chem Soc. 2003;125:8432–8433. doi: 10.1021/ja035308f. [DOI] [PubMed] [Google Scholar]
- 67.(a) Ema T, Oue Y, Akihara K, Miyazaki Y, Sakai T. Stereoselective Synthesis of Bicyclic Tertiary Alcohols with Quaternary Stereocenters via Intramolecular Crossed Benzoin Reactions Catalyzed by N-Heterocyclic Carbenes. Org Lett. 2009;11:4866–4869. doi: 10.1021/ol9019293. [DOI] [PubMed] [Google Scholar]; (b) Ema T, Akihara K, Obayashi R, Sakai T. Construction of Contiguous Tetrasubstituted Carbon Stereocenters by Intramolecular Crossed Benzoin Reactions Catalyzed by N-Heterocyclic Carbene (NHC) Organocatalyst. Adv Synth Catal. 2012;354:3283–3290. [Google Scholar]
- 68.Kankala S, Edulla R, Modem S, Vadde R, Vasam CS. N-Heterocyclic Carbene Catalyzed Intramolecular Crossed Aldehyde–Ketone Benzoin Condensation in the Chalcone of o-Phthalaldehyde: A Facile Synthesis of Naphthalenones. Tetrahedron Lett. 2011;52:3828–3831. [Google Scholar]
- 69.Jia M-Q, You S-L. N-Heterocyclic Carbene-Catalyzed Enantioselective Intramolecular N-Tethered Aldehyde–Ketone Benzoin Reactions. ACS Catal. 2013;3:622–624. [Google Scholar]
- 70.(a) Lathrop SP, Rovis T. Asymmetric Synthesis of Functionalized Cyclopentanones via a Multicatalytic Secondary Amine/N-Heterocyclic Carbene Catalyzed Cascade Sequence. J Am Chem Soc. 2009;131:13628–13630. doi: 10.1021/ja905342e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ozboya KE, Rovis T. Enamine/Carbene Cascade Catalysis in the Diastereo- and Enantioselective Synthesis of Functionalized Cyclopentanones. Chem Sci. 2011;2:1835–1838. doi: 10.1039/C1SC00175B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Enders D, Grossmann A, Huang H, Raabe G. Dual Secondary Amine/N-Heterocyclic Carbene Catalysis in the Asymmetric Michael/Cross-Benzoin Cascade Reaction of β-Oxo Sulfones with Enals. Eur J Org Chem. 2011:4298–4301. [Google Scholar]
- 72.Tarr JC, Johnson JS. Lanthanum Tricyanide-Catalyzed Acyl Silane–Ketone Benzoin Additions. Org Lett. 2009;11:3870–3873. doi: 10.1021/ol901314w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Demir AS, Esiringü I, Göllü M, Reis Ö. Catalytic Intermolecular Aldehyde–Ketone Coupling via Acyl Phosphonates. J Org Chem. 2009;74:2197–2199. doi: 10.1021/jo8026627. [DOI] [PubMed] [Google Scholar]
- 74.Enders D, Henseler A. A Direct Intermolecular Cross-Benzoin Type Reaction: N-Heterocyclic Carbene-Catalyzed Coupling of Aromatic Aldehydes with Trifluoromethyl Ketones. Adv Synth Catal. 2009;351:1749–1752. [Google Scholar]
- 75.Enders D, Grossmann A, Fronert G, Raabe G. N-Heterocyclic Carbene Catalysed Asymmetric Cross-Benzoin Reactions of Heteroaromatic Aldehydes with Trifluoromethyl Ketones. Chem Commun. 2010:6282–6284. doi: 10.1039/c0cc02013c. [DOI] [PubMed] [Google Scholar]
- 76.Rose AC, Gundala S, Fagan C-L, Franz JF, Connon SJ, Zeitler K. NHC-Catalysed, Chemoselective Crossed-Acyloin Reactions. Chem Sci. 2012;3:735–740. [Google Scholar]
- 77.Thia K, Langdon SM, Bilodeau F, Gravel M. Highly Chemo- and Enantioselective Cross-Benzoin Reaction of Aliphatic Aldehydes and α-Ketoesters. Org Lett. 2013;15:2214–2217. doi: 10.1021/ol400769t. [DOI] [PubMed] [Google Scholar]
- 78.Murry JA, Frantz DE, Soheili A, Tillyer R, Grabowski EJJ, Reider PJ. Synthesis of α-Amido Ketones via Organic Catalysis: Thiazolium-Catalyzed Cross-Coupling of Aldehydes with Acylimines. J Am Chem Soc. 2001;123:9696–9697. doi: 10.1021/ja0165943. [DOI] [PubMed] [Google Scholar]
- 79.Li G-Q, Dai L-X, You S-L. Thiazolium-Derived N-Heterocyclic Carbene-Catalyzed Cross-Coupling of Aldehydes with Unactivated Imines. Chem Commun. 2007:852–854. doi: 10.1039/b611646a. [DOI] [PubMed] [Google Scholar]
- 80.Enders D, Henseler A, Lowins S. N-Heterocyclic Carbene Catalyzed Nucleophilic Acylation of Trifluoromethyl Ketimines. Synthesis. 2009:4125–4128. [Google Scholar]
- 81.Sun L-H, Liang Z-Q, Jia W-Q, Ye S. Enantioselective N-Heterocyclic Carbene Catalyzed Aza-Benzoin Reaction of Enals with Activated Ketimines. Angew Chem Int Ed. 2013;52:5803–5806. doi: 10.1002/anie.201301304. [DOI] [PubMed] [Google Scholar]
- 82.DiRocco DA, Rovis T. Catalytic Asymmetric Cross-Aza-Benzoin Reactions of Aliphatic Aldehydes with N-Boc-Protected Imines. Angew Chem Int Ed. 2012;51:5904–5906. doi: 10.1002/anie.201202442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ciganek E. Esters of 2,3-Dihydro-3-oxobenzofuran-2-acetic Acid and 3,4-Dihydro-4-oxo-2H-1-benzopyran-3-acetic Acid by Intramolecular Stetter Reactions. Synthesis. 1995:1311–1314. [Google Scholar]
- 84.An intramolecular Stetter reaction was demonstrated by Trost in the context of an approach to hirsutic acid; see: Trost BM, Shuey CD, DiNinno F., Jr A Stereocontrolled Total Synthesis of (.+-.)-Hirsutic Acid C. J Am Chem Soc. 1979;101:1284–1285.
- 85.Read de Alaniz J, Rovis T. The Catalytic Asymmetric Intramolecular Stetter Reaction. Synlett. 2009:1189–1207. doi: 10.1055/s-0029-1216654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Read de Alaniz J, Kerr MS, Moore JL, Rovis T. Scope of the Asymmetric Intramolecular Stetter Reaction Catalyzed by Chiral Nucleophilic Triazolinylidene Carbenes. J Org Chem. 2008;73:2033–2040. doi: 10.1021/jo702313f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cullen SC, Rovis T. Catalytic Asymmetric Stetter Reaction Onto Vinylphosphine Oxides and Vinylphosphonates. Org Lett. 2008;10:3141–3144. doi: 10.1021/ol801047k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang Z, Yu Z, Wang Y, Shi D. N-Heterocyclic Carbene Catalyzed Intramolecular Hydroacylation of Alkynylphosphonates. Synthesis. 2012:1559–1568. [Google Scholar]
- 89.McErlean CSP, Willis AC. Application of an Intramolecular Stetter Reaction to Access trans,syn,trans-Fused Pyrans. Synlett. 2009:233–236. [Google Scholar]
- 90.Vedachalam S, Wong Q-L, Maji B, Zeng J, Ma J, Liu X-W. N-Heterocyclic Carbene Catalyzed Intramolecular Hydroacylation of Activated Alkynes: Synthesis of Chromones. Adv Synth Catal. 2011;353:219–225. [Google Scholar]
- 91.Wang Z-Q, Wang Y, Shi D-Q. N-Heterocyclic Carbene-Catalyzed Domino Hydroacylation/Stetter Reactions of Salicyl Alkynylphosphonates and Aromatic Aldehydes. Arkivoc. 2013;(iv):88–97. [Google Scholar]
- 92.Law KR, McErlean CSP. Extending the Stetter Reaction with 1,6-Acceptors. Chem Eur J. 2013;19:15852–15855. doi: 10.1002/chem.201303435. [DOI] [PubMed] [Google Scholar]
- 93.Zeitler K, Mager I. An Efficient and Versatile Approach for the Immobilization of Carbene Precursors via Copper-Catalyzed [3+2]-Cycloaddition and their Catalytic Application. Adv Synth Catal. 2007;349:1851–1857. [Google Scholar]
- 94.Hara O, Kume A, Sugiura M, Maeba I. Fluorous Thiazolium Salts for the Intramolecular Stetter Reaction. Heterocycles. 2008;76:1027–1031. [Google Scholar]
- 95.Aupoix A, Vo-Thanh G. Solvent-Free Synthesis of Alkylthiazolium-Based Ionic Liquids and their Use as Catalysts in the Intramolecular Stetter Reaction. Synlett. 2009:1915–1920. [Google Scholar]
- 96.Rong Z-Q, Li Y, Yang G-Q, You S-L. D-Camphor-Derived Triazolium Salts for Enantioselective Intramolecular Stetter Reactions. Synlett. 2011:1033–1037. [Google Scholar]
- 97.Jia M-Q, Li Y, Rong Z-Q, You S-L. Synthesis of (1R,2R)-DPEN-Derived Triazolium Salts and Their Application in Asymmetric Intramolecular Stetter Reactions. Org Biomol Chem. 2011;9:2072–2074. doi: 10.1039/c1ob00025j. [DOI] [PubMed] [Google Scholar]
- 98.Soeta T, Tabatake Y, Ukaji Y. An Asymmetric Intramolecular Stetter Reaction Catalyzed by a Chiral Triazolium Precatalyst Bearing a Pyridine Moiety. Tetrahedron. 2012;68:10188–10193. [Google Scholar]
- 99.Rafíński Z, Kozakiewicz A, Rafińska K. (−)-β-Pinene-Derived N-Heterocyclic Carbenes: Application to Highly Enantioselective Intramolecular Stetter Reaction. ACS Catal. 2014;4:1404–1408. [Google Scholar]
- 100.Li Y, Shi F-Q, He Q-L, You S-L. N-Heterocyclic Carbene-Catalyzed Cross-Coupling of Aldehydes with Arylsulfonyl Indoles. Org Lett. 2009;11:3182–3185. doi: 10.1021/ol9013238. [DOI] [PubMed] [Google Scholar]
- 101.(a) Bhunia A, Yetra SR, Bhojgude SS, Biju AT. Efficient Synthesis of γ-Keto Sulfones by NHC-Catalyzed Intermolecular Stetter Reaction. Org Lett. 2012;14:2830–2833. doi: 10.1021/ol301045x. [DOI] [PubMed] [Google Scholar]; (b) Patra A, Bhunia A, Biju AT. Facile Synthesis of γ-Ketophosphonates by an Intermolecular Stetter Reaction onto Vinylphosphonates. Org Lett. 2014;16:4798–4801. doi: 10.1021/ol502262d. [DOI] [PubMed] [Google Scholar]
- 102.Vedachalam S, Tan SM, Teo HP, Cai S, Liu X-W. N-Heterocyclic Carbene Catalyzed C-Glycosylation: A Concise Approach from Stetter Reaction. Org Lett. 2012;14:174–177. doi: 10.1021/ol202959y. [DOI] [PubMed] [Google Scholar]
- 103.Bortolini O, Fantin G, Fogagnolo M, Giovannini PP, Massi A, Pacifico S. Thiazolium-catalyzed Intermolecular Stetter Reaction of Linear and Cyclic Alkyl α-Diketones. Org Biomol Chem. 2011;9:8437–8444. doi: 10.1039/c1ob06480k. [DOI] [PubMed] [Google Scholar]
- 104.Miyashita A, Numata A, Suzuki Y, Iwamoto K-I, Higashino T. Olefin-Insertion Reaction between the Carbonyls of Benzils; Formation of 1,4-Diketones by Michael Additon Catalyzed by Cyanide Ion. Chem Lett. 1997;26:697–698. [Google Scholar]
- 105.Takaki K, Ohno A, Hino M, Shitaoka T, Komeyama K, Yoshida H. N-Heterocyclic Carbene-Catalyzed Double Acylation of Enones with Benzils. Chem Commun. 2014:12285–12288. doi: 10.1039/c4cc05436a. [DOI] [PubMed] [Google Scholar]
- 106.Zhang J, Xing C, Tiwari B, Chi YR. Catalytic Activation of Carbohydrates as Formaldehyde Equivalents for Stetter Reaction with Enones. J Am Chem Soc. 2013;135:8113–8116. doi: 10.1021/ja401511r. [DOI] [PubMed] [Google Scholar]
- 107.Liu P, Lei M, Ma L, Hu L. An Efficient Synthesis of 2-Aminofuran-3-carbonitriles via Cascade Stetter-γ-Ketonitrile Cyclization Reaction Catalyzed by N-Heterocyclic Carbene. Synlett. 2011:1133–1136. [Google Scholar]
- 108.Yu C, Lu J, Li T, Wang D, Qin B, Zhang H, Yao C. A NHC-Involved, Cascade, Metal-Free, and Three-Component Synthesis of 2,3-Diarylated Fully Substituted Furans under Solvent-Free Conditions. Synlett. 2011:2420–2424. [Google Scholar]
- 109.(a) Hong BC, Dange NS, Hsu CS, Liao JH. Sequential Organocatalytic Stetter and Michael-Aldol Condensation Reaction: Asymmetric Synthesis of Fully Substituted Cyclopentenes via a [1 + 2 + 2] Annulation Strategy. Org Lett. 2010;12:4812–4815. doi: 10.1021/ol101969t. [DOI] [PubMed] [Google Scholar]; (b) Hong BC, Dange NS, Hsu CS, Liao JH, Lee GH. Dynamic Kinetic Asymmetric Synthesis of Five Contiguous Stereogenic Centers by Sequential Organocatalytic Stetter and Michael–Aldol Reaction: Enantioselective Synthesis of Fully Substituted Cyclopentanols Bearing a Quaternary Stereocenter. Org Lett. 2011;13:1338–1341. doi: 10.1021/ol200006e. [DOI] [PubMed] [Google Scholar]
- 110.Sánchez-Larios E, Gravel M. Diastereoselective Synthesis of Indanes via a Domino Stetter–Michael Reaction. J Org Chem. 2009;74:7536–7539. doi: 10.1021/jo901468h. [DOI] [PubMed] [Google Scholar]
- 111.(a) Sánchez-Larios E, Holmes JM, Daschner CL, Gravel M. NHC-Catalyzed Spiro Bis-Indane Formation via Domino Stetter–Aldol–Michael and Stetter–Aldol–Aldol Reactions. Org Lett. 2010;12:5772–5775. doi: 10.1021/ol102685u. [DOI] [PubMed] [Google Scholar]; (b) Sánchez-Larios E, Holmes JM, Daschner CL, Gravel M. NHC-Catalyzed Spiro Bis-Indane Formation via Domino Stetter–Aldol–Michael and Stetter–Aldol–Aldol Reactions. Synthesis. 2011:1896–1904. doi: 10.1021/ol102685u. [DOI] [PubMed] [Google Scholar]
- 112.Sun F-G, Huang X-L, Ye S. Diastereoselective Synthesis of 4-Hydroxytetralones via a Cascade Stetter–Aldol Reaction Catalyzed by N-Heterocyclic Carbenes. J Org Chem. 2010;75:273–276. doi: 10.1021/jo902376t. [DOI] [PubMed] [Google Scholar]
- 113.Sun F-G, Ye S. Diastereoselective Synthesis of 3-Hydroxyindanones via N-Heterocyclic Carbene Catalyzed [4+1] Annulation of Phthalaldehyde and 1,2-Diactivated Michael Acceptors. Synlett. 2011:1005–1009. [Google Scholar]
- 114.Tiebes J. Diploma Thesis. RWTH Aachen; Aachen, Germany: 1990. Untersuchung Zur Katalytischen, Enantioselektiven C-C-Verknuepfung mit N-Chiral-Substituierten Thiazoliumsalzen. [Google Scholar]
- 115.Enders D, Han J, Henseler A. Asymmetric Intermolecular Stetter Reactions Catalyzed by a Novel Triazolium Derived N-Heterocyclic Carbene. Chem Commun. 2008:3989–3991. doi: 10.1039/b809913h. [DOI] [PubMed] [Google Scholar]
- 116.Enders D, Han J. Asymmetric Intermolecular Stetter Reactions of Aromatic Heterocyclic Aldehydes- with Arylidenemalonates. Synthesis. 2008:3864–3868. [Google Scholar]
- 117.Kim SM, Jin MY, Cui Y, Kim YS, Zhang L, Song CE, Ryu DH, Yang JW. N-Heterocyclic Carbene-Catalysed Intermolecular Stetter Reactions of Acetaldehyde. Org Biomol Chem. 2011;9:2069–2071. doi: 10.1039/c0ob01178a. [DOI] [PubMed] [Google Scholar]
- 118.(a) Liu Q, Perreault S, Rovis T. Catalytic Asymmetric Intermolecular Stetter Reaction of Glyoxamides with Alkylidenemalonates. J Am Chem Soc. 2008;130:14066–14067. doi: 10.1021/ja805680z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liu Q, Rovis T. Enantio- and Diastereoselective Intermolecular Stetter Reaction of Glyoxamide and Alkylidene Ketoamides. Org Lett. 2009;11:2856–2859. doi: 10.1021/ol901081a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.DiRocco DA, Oberg KM, Dalton DM, Rovis T. Catalytic Asymmetric Intermolecular Stetter Reaction of Heterocyclic Aldehydes with Nitroalkenes: Backbone Fluorination Improves Selectivity. J Am Chem Soc. 2009;131:10872–10874. doi: 10.1021/ja904375q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Um JM, DiRocco DA, Noey EL, Rovis T, Houk KN. Quantum Mechanical Investigation of the Effect of Catalyst Fluorination in the Intermolecular Asymmetric Stetter Reaction. J Am Chem Soc. 2011;133:11249–11254. doi: 10.1021/ja202444g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.DiRocco DA, Rovis T. Catalytic Asymmetric Intermolecular Stetter Reaction of Enals with Nitroalkenes: Enhancement of Catalytic Efficiency through Bifunctional Additives. J Am Chem Soc. 2011;133:10402–10405. doi: 10.1021/ja203810b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Moore JL, Silvestri AP, Read de Alaniz J, DiRocco DA, Rovis T. Mechanistic Investigation of the Enantioselective Intramolecular Stetter Reaction: Proton Transfer Is the First Irreversible Step. Org lett. 2011;13:1742–1745. doi: 10.1021/ol200256a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sánchez-Larios E, Thai K, Bilodeau F, Gravel M. Highly Enantioselective Intermolecular Stetter Reactions of β-Aryl Acceptors: α-Ketoester Moiety as Handle for Activation and Synthetic Manipulations. Org Lett. 2011;13:4942–4945. doi: 10.1021/ol202040b. [DOI] [PubMed] [Google Scholar]
- 124.DiRocco DA, Noey EL, Houk KN, Rovis T. Catalytic Asymmetric Intermolecular Stetter Reactions of Enolizable Aldehydes with Nitrostyrenes: Computational Study Provides Insight into the Success of the Catalyst. Angew Chem Int Ed. 2012;51:2391–2394. doi: 10.1002/anie.201107597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Fang X, Chen X, Lv H, Chi YR. Enantioselective Stetter Reactions of Enals and Modified Chalcones Catalyzed by N-Heterocyclic Carbenes. Angew Chem Int Ed. 2011;50:11782–11785. doi: 10.1002/anie.201105812. [DOI] [PubMed] [Google Scholar]
- 126.Jousseaume T, Wurz NE, Glorius F. Highly Enantioselective Synthesis of α-Amino Acid Derivatives by an NHC-Catalyzed Intermolecular Stetter Reaction. Angew Chem Int Ed. 2011;50:1410–1414. doi: 10.1002/anie.201006548. [DOI] [PubMed] [Google Scholar]
- 127.Wurz NE, Daniliuc CG, Glorius F. Highly Enantioselective Intermolecular Stetter Reaction of Simple Acrylates: Synthesis of α-Chiral γ-Ketoesters. Chem Eur J. 2012;18:16297–16301. doi: 10.1002/chem.201202432. [DOI] [PubMed] [Google Scholar]
- 128.He J, Zheng J, Liu X, She X, Pan X. N-Heterocyclic Carbene Catalyzed Nucleophilic Substitution Reaction for Construction of Benzopyrones and Benzofuranones. Org Lett. 2006;8:4637–4640. doi: 10.1021/ol061924f. [DOI] [PubMed] [Google Scholar]
- 129.He J, Tang S, Liu J, Su Y, Pan X, She X. N-Heterocyclic Carbene Catalyzed Intramolecular Nucleophilic Addition of Carbonyl Anion Equivalents to Enol Ethers. Tetrahedron. 2008;64:8797–8800. [Google Scholar]
- 130.Biju AT, Wurz NE, Glorius F. N-Heterocyclic Carbene-Catalyzed Cascade Reaction Involving the Hydroacylation of Unactivated Alkynes. J Am Chem Soc. 2010;132:5970–5971. doi: 10.1021/ja102130s. [DOI] [PubMed] [Google Scholar]
- 131.Padmanaban M, Biju AT, Glorius F. Efficient Synthesis of Benzofuranones: N-Heterocyclic Carbene (NHC)/Base-Catalyzed Hydroacylation–Stetter–Rearrangement Cascade. Org Lett. 2011;13:5624–5627. doi: 10.1021/ol2023483. [DOI] [PubMed] [Google Scholar]
- 132.Franz JF, Fuchs PJW, Zeitler K. A Versatile Combined N-Heterocyclic Carbene and Base-Catalyzed Multiple Cascade Approach for the Synthesis of Functionalized Benzofuran-3-(2H)-ones. Tetrahedron Lett. 2011;52:6952–6956. [Google Scholar]
- 133.(a) Piel I, Steinmetz M, Hirano K, Fröhlich R, Grimme S, Glorius F. Highly Asymmetric NHC-Catalyzed Hydroacylation of Unactivated Alkenes. Angew Chem Int Ed. 2011;50:4983–4987. doi: 10.1002/anie.201008081. [DOI] [PubMed] [Google Scholar]; (b) Hirano K, Biju AT, Piel I, Glorius F. N-Heterocyclic Carbene-Catalyzed Hydroacylation of Unactivated Double Bonds. J Am Chem Soc. 2009;131:14190–14191. doi: 10.1021/ja906361g. [DOI] [PubMed] [Google Scholar]
- 134.DiRocco DA, Rovis T. Organocatalytic Hydroacylation of Unactivated Alkenes. Angew Chem Int Ed. 2011;50:7982–7983. doi: 10.1002/anie.201102920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Read de Alaniz J, Rovis T. A Highly Enantio- and Diastereoselective Catalytic Intramolecular Stetter Reaction. J Am Chem Soc. 2005;127:6284–6289. doi: 10.1021/ja0425132. [DOI] [PubMed] [Google Scholar]
- 136.Domingo LR, Saéz JA, Arnó M. An ELF Analysis of the C–C Bond Formation Step in the N-Heterocyclic Carbene-Catalyzed Hydroacylation of Unactivated C–C Double Bonds. RSC Adv. 2012:7127–7134. [Google Scholar]
- 137.Biju AT, Glorius F. Intermolecular N-Heterocyclic Carbene Catalyzed Hydroacylation of Arynes. Angew Chem Int Ed. 2010;49:9761–9764. doi: 10.1002/anie.201005490. [DOI] [PubMed] [Google Scholar]
- 138.(a) Schedler M, Fröhlich R, Daniliuc CG, Glorius F. 2,6-Dimethoxyphenyl-Substituted N-Heterocyclic Carbenes (NHCs): A Family of Highly Electron-Rich Organocatalysts. Eur J Org Chem. 2012:4164–4171. [Google Scholar]; (b) Liu F, Bugaut X, Schedler M, Fröhlich R, Glorius F. Designing N-Heterocyclic Carbenes: Simultaneous Enhancement of Reactivity and Enantioselectivity in the Asymmetric Hydroacylation of Cyclopropenes. Angew Chem Int Ed. 2011;50:12626–12630. doi: 10.1002/anie.201106155. [DOI] [PubMed] [Google Scholar]
- 139.Schedler M, Wang D-S, Glorius F. NHC-Catalyzed Hydroacylation of Styrenes. Angew Chem Int Ed. 2013;52:2585–2589. doi: 10.1002/anie.201209291. [DOI] [PubMed] [Google Scholar]
- 140.(a) Walia JS, Rao DH, Singh M. Conjugate Schiff Bases. Addition of Cyanide Ion to Cinnamalmethylamine and Cinnamalaniline. Indian J Chem. 1964;2:437–439. [Google Scholar]; (b) Walia JS, Hanumantha R, Singh M, Nath R. Cyanide Ion-Catalyzed Reactions of Conjugated Azomthines. Chem Ind. 1967:583–585. [Google Scholar]
- 141.Singh G, Mandal AK. Dimerisation of Cinnamalaniline with Potassium Cyanide: A Novel Synthesis of 4,5-trans-Diphenyl-1-phenyl-2-phenylimino-2,3,4,5-tetrahydro-1H –azepine. Heterocycles. 1982;18:291–294. [Google Scholar]
- 142.Sohn SS, Rosen EL, Bode JW. N-Heterocyclic Carbene-Catalyzed Generation of Homoenolates: γ-Butyrolactones by Direct Annulations of Enals and Aldehydes. J Am Chem Soc. 2004;126:14370–14371. doi: 10.1021/ja044714b. [DOI] [PubMed] [Google Scholar]
- 143.(a) Burstein C, Glorius F. Organocatalyzed Conjugate Umpolung of α,β-Unsaturated Aldehydes for the Synthesis of γ-Butyrolactones. Angew Chem Int Ed. 2004;43:6205–6208. doi: 10.1002/anie.200461572. [DOI] [PubMed] [Google Scholar]; (b) Burstein C, Tschan S, Xie X, Glorius F. N-Heterocyclic Carbene-Catalyzed Conjugate Umpolung for the Synthesis of γ-Butyrolactones. Synthesis. 2006:2418–2439. [Google Scholar]
- 144.Cohen DT, Scheidt KA. Cooperative Lewis acid/N-Heterocyclic Carbene Catalysis. Chem Sci. 2012;3:53–57. doi: 10.1039/C1SC00621E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Jang KP, Hutson GE, Johnston RC, McCusker EO, Cheong PH–Y, Scheidt KA. Asymmetric Homoenolate Additions to Acyl Phosphonates through Rational Design of a Tailored N-Heterocyclic Carbene Catalyst. J Am Chem Soc. 2014;136:76–79. doi: 10.1021/ja410932t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Nair V, Vellalath S, Poonoth M, Mohan R, Suresh E. N-Heterocyclic Carbene Catalyzed Reaction of Enals and 1,2-Dicarbonyl Compounds: Stereoselective Synthesis of Spiro γ-Butyrolactones. Org Lett. 2006;8:507–509. doi: 10.1021/ol052926n. [DOI] [PubMed] [Google Scholar]
- 147.Nair V, Vellalath S, Poonoth M, Suresh E, Viji S. N-Heterocyclic Carbene Catalyzed Reaction of Enals and Diaryl-1,2 diones via Homoenolate: Synthesis of 4,5,5-Trisubstituted γ-Butyrolactones. Synthesis. 2007:3195–3200. [Google Scholar]
- 148.(a) Sun LH, Shen LT, Ye S. Highly Diastereo- and Enantioselective NHC-Catalyzed [3+2] Annulation of Enals and Isatins. Chem Commun. 2011;47:10136–10138. doi: 10.1039/c1cc13860j. [DOI] [PubMed] [Google Scholar]; (b) Nawaz F, Zaghouani M, Bonne D, Chuzel O, Rodriguez J, Coquerel Y. Design, Synthesis, and Organocatalytic Activity of N-Heterocyclic Carbenes Functionalized with Hydrogen-Bond Donors in Enantioselective Reactions of Homoenolates. Eur J Org Chem. 2013:8253–8264. [Google Scholar]
- 149.Dugal-Tessier J, O’Bryan EA, Schroeder TBH, Cohen DT, Scheidt KA. An N-Heterocyclic Carbene/Lewis Acid Strategy for the Stereoselective Synthesis of Spirooxindole Lactones. Angew Chem Int Ed. 2012;51:4963–4967. doi: 10.1002/anie.201201643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Li Y, Zhao Z-A, He H, You S-L. Stereoselective Synthesis of γ-Butyrolactones via Organocatalytic Annulations of Enals and Keto Esters. Adv Synth Catal. 2008;350:1885–1890. [Google Scholar]
- 151.Li J-L, Sahoo B, Daniliuc C-G, Glorius F. Conjugate Umpolung of β,β-Disubstituted Enals by Dual Catalysis with an N-Heterocyclic Carbene and a Brønsted Acid: Facile Construction of Contiguous Quaternary Stereocenters. Angew Chem Int Ed. 2014;53:10515–10519. doi: 10.1002/anie.201405178. [DOI] [PubMed] [Google Scholar]
- 152.Zhao X, DiRocco DA, Rovis T. N-Heterocyclic Carbene and Brønsted Acid Cooperative Catalysis: Asymmetric Synthesis of trans-γ-Lactams. J Am Chem Soc. 2011;133:12466–12469. doi: 10.1021/ja205714g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lv H, Jia W-Q, Sun L-H, Ye S. N-Heterocyclic Carbene Catalyzed [4+3] Annulation of Enals and o-Quinone Methides: Highly Enantioselective Synthesis of Benzo-ε-Lactones. Angew Chem Int Ed. 2013;52:8607–8610. doi: 10.1002/anie.201303903. [DOI] [PubMed] [Google Scholar]
- 154.Nair V, Poonoth M, Vellalath S, Suresh E, Thirumalai R. An N-Heterocyclic Carbene-Catalyzed [8 + 3] Annulation of Tropone and Enals via Homoenolate. J Org Chem. 2006;71:8964–8965. doi: 10.1021/jo0615706. [DOI] [PubMed] [Google Scholar]
- 155.He M, Bode JW. Catalytic Synthesis of γ-Lactams via Direct Annulations of Enals and N-Sulfonylimines. Org Lett. 2005;7:3131–3134. doi: 10.1021/ol051234w. [DOI] [PubMed] [Google Scholar]
- 156.Rommel M, Fukuzumi T, Bode JW. Cyclic Ketimines as Superior Electrophiles for NHC-Catalyzed Homoenolate Additions with Broad Scope and Low Catalyst Loadings. J Am Chem Soc. 2008;130:17266–17267. doi: 10.1021/ja807937m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.He M, Bode JW. Enantioselective, NHC-Catalyzed Bicyclo-β-Lactam Formation via Direct Annulations of Enals and Unsaturated N-Sulfonyl Ketimines. J Am Chem Soc. 2008;130:418–419. doi: 10.1021/ja0778592. [DOI] [PubMed] [Google Scholar]
- 158.Chan A, Scheidt KA. Direct Amination of Homoenolates Catalyzed by N-Heterocyclic Carbenes. J Am Chem Soc. 2008;130:2740–2741. doi: 10.1021/ja711130p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Raup DEA, Cardinal-David B, Holte D, Scheidt KA. Cooperative catalysis by carbenes and Lewis acids in a highly stereoselective route to γ-lactams. Nat Chem. 2010;2:766–771. doi: 10.1038/nchem.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Chan A, Scheidt KA. Highly Stereoselective Formal [3 + 3] Cycloaddition of Enals and Azomethine Imines Catalyzed by N-Heterocyclic Carbenes. J Am Chem Soc. 2007;129:5334–5335. doi: 10.1021/ja0709167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Phillips EM, Reynolds TE, Scheidt KA. Highly Diastereo- and Enantioselective Additions of Homoenolates to Nitrones Catalyzed by N-Heterocyclic Carbenes. J Am Chem Soc. 2008;130:2416–2417. doi: 10.1021/ja710521m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Seayad J, Patra PK, Zhang Y, Ying JY. Organocatalytic Synthesis of N-Phenylisoxazolidin-5-ones and a One-Pot Synthesis of β-Amino Acid Esters. Org Lett. 2008;10:953–956. doi: 10.1021/ol800003n. [DOI] [PubMed] [Google Scholar]
- 163.Ikota H, Ishida T, Tsukano C, Takemoto Y. Synthesis of 3,3-Disubstituted Indoline-2-Thiones Catalyzed by an N-Heterocyclic Carbene. Chem Commun. 2014;50:8871–8874. doi: 10.1039/c4cc04047c. [DOI] [PubMed] [Google Scholar]
- 164.Yang L, Tan B, Wang F, Zhong G. An Unexpected N-Heterocyclic Carbene-Catalyzed Annulation of Enals and Nitroso Compounds. J Org Chem. 2009;74:1744–1746. doi: 10.1021/jo802515g. [DOI] [PubMed] [Google Scholar]
- 165.Siddiqui IR, Srivastava A, Shamim S, Srivastava A, Shireen, Waseem MA, Singh RKP. Highly Diastereoselective NHC-Catalyzed [4+3] Annulation of Enals, Alde-hydes and N-Phenyl Urea/Thiourea for the Synthesis of Monocyclic trans-1,3-Diazepanes. Synlett. 2013;24:2586–2590. [Google Scholar]
- 166.Nair V, Vellalath S, Poonoth M, Suresh E. N-Heterocyclic Carbene-Catalyzed Reaction of Chalcones and Enals via Homoenolate: an Efficient Synthesis of 1,3,4-Trisubstituted Cyclopentenes. J Am Chem Soc. 2006;128:8736–8737. doi: 10.1021/ja0625677. [DOI] [PubMed] [Google Scholar]
- 167.Nair V, Babu BP, Vellalath S, Varghese V, Raveendran AE, Suresh E. Nucleophilic Heterocyclic Carbene Catalyzed Annulation of Enals to Chalcones in Methanol: A Stereoselective Synthesis of Highly Functionalized Cyclopentanes. Org Lett. 2009;11:2507–2510. doi: 10.1021/ol900571x. [DOI] [PubMed] [Google Scholar]
- 168.(a) Nair V, Babu BP, Vellalath S, Suresh E. Stereoselective Synthesis of Spirocyclopentanones via N-Heterocyclic Carbene-Catalyzed Reactions of Enals and Dienones. Chem Commun. 2008:747–749. doi: 10.1039/b715733a. [DOI] [PubMed] [Google Scholar]; (b) Verma P, Patni PA, Sunoj RB. Mechanistic Insights on N-Heterocyclic Carbene-Catalyzed Annulations: The Role of Base-Assisted Proton Transfers. J Org Chem. 2011;76:5606–5613. doi: 10.1021/jo200560t. [DOI] [PubMed] [Google Scholar]
- 169.Chiang P-C, Kaeobamrung J, Bode JW. Enantioselective, Cyclopentene-Forming Annulations via NHC-Catalyzed Benzoin–Oxy-Cope Reactions. J Am Chem Soc. 2007;129:3520–3521. doi: 10.1021/ja0705543. [DOI] [PubMed] [Google Scholar]
- 170.Cardinal-David B, Raup DEA, Scheidt KA. Cooperative N-Heterocyclic Carbene/Lewis Acid Catalysis for Highly Stereoselective Annulation Reactions with Homoenolates. J Am Chem Soc. 2010;132:5345–5347. doi: 10.1021/ja910666n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Cohen DT, Cardinal-David B, Scheidt KA. Lewis Acid Activated Synthesis of Highly Substituted Cyclopentanes by the N-Heterocyclic Carbene Catalyzed Addition of Homoenolate Equivalents to Unsaturated Ketoesters. Angew Chem Int Ed. 2011;50:1678–1682. doi: 10.1002/anie.201005908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Seetha Lakshmi KC, Sinu CR, Padmaja DVM, Gopinathan A, Suresh E, Nair V. A Novel Intramolecular Homoenolate Annulation Leading to the Formation of Cyclopentene-Fused Macrocycles. Org Lett. 2014;16:5532–5335. doi: 10.1021/ol5023239. [DOI] [PubMed] [Google Scholar]
- 173.Guo C, Schedler M, Daniliuc CG, Glorius F. N-Heterocyclic Carbene Catalyzed Formal [3+2] Annulation Reaction of Enals: An Efficient Enantioselective Access to Spiro-Heterocycles. Angew Chem Int Ed. 2014;53:10232–10236. doi: 10.1002/anie.201405381. [DOI] [PubMed] [Google Scholar]
- 174.Chan A, Scheidt KA. Conversion of α,β-Unsaturated Aldehydes into Saturated Esters: An Umpolung Reaction Catalyzed by Nucleophilic Carbenes. Org Lett. 2005;7:905–908. doi: 10.1021/ol050100f. [DOI] [PubMed] [Google Scholar]
- 175.Sohn SS, Bode JW. Catalytic Generation of Activated Carboxylates from Enals: A Product-Determining Role for the Base. Org Lett. 2005;7:3873–3876. doi: 10.1021/ol051269w. [DOI] [PubMed] [Google Scholar]
- 176.Nair V, Sinu CR, Babu BP, Varghese V, Jose A, Suresh E. Novel Nucleophilic Heterocyclic Carbene Mediated Stereoselective Conjugate Addition of Enals to Nitrostyrenes via Homoenolate. Org Lett. 2009;11:5570–5573. doi: 10.1021/ol901918x. [DOI] [PubMed] [Google Scholar]
- 177.(a) Maji B, Ji L, Wang S, Vedachalam S, Ganguly R, Liu XW. N-Heterocyclic Carbene Catalyzed Homoenolate-Addition Reaction of Enals and Nitroalkenes: Asymmetric Synthesis of 5-Carbon-Synthon δ-Nitroesters. Angew Chem Int Ed. 2012;51:8276–8280. doi: 10.1002/anie.201203449. [DOI] [PubMed] [Google Scholar]; (b) White NA, DiRocco DA, Rovis T. Asymmetric N-Heterocyclic Carbene Catalyzed Addition of Enals to Nitroalkenes: Controlling Stereochemistry via the Homoenolate Reactivity Pathway To Access δ-Lactams. J Am Chem Soc. 2013;135:8504–8507. doi: 10.1021/ja403847e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Chiang P-C, Rommel M, Bode JW. α-Hydroxyenones as Mechanistic Probes and Scope-Expanding Surrogates for α,β-Unsaturated Aldehydes in N-Heterocyclic Carbene-Catalyzed Reactions. J Am Chem Soc. 2009;131:8714–8718. doi: 10.1021/ja902143w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Fu Z, Xu J, Zhu T, Leong WWY, Chi YR. β-Carbon Activation of Saturated Carboxylic Esters Through N-Heterocyclic Carbene Organocatalysis. Nat Chem. 2013;5:835–839. doi: 10.1038/nchem.1710. [DOI] [PubMed] [Google Scholar]
- 180.Guin J, De Sarkar S, Grimme S, Studer A. Biomimetic Carbene-Catalyzed Oxidations of Aldehydes Using TEMPO. Angew Chem Int Ed. 2008;47:8727–8730. doi: 10.1002/anie.200802735. [DOI] [PubMed] [Google Scholar]
- 181.White NA, Rovis T. Enantioselective N-Heterocyclic Carbene-Catalyzed β-Hydroxylation of Enals Using Nitroarenes: An Atom Transfer Reaction That Proceeds via Single Electron Transfer. J Am Chem Soc. 2014;136:14674–14677. doi: 10.1021/ja5080739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Du Y, Wang Y, Li X, Shao G, Webster RD, Chi YR. N-Heterocyclic Carbene Organocatalytic Reductive β,β-Coupling Reactions of Nitroalkenes via Radical Intermediates. Org Lett. 2014;16:5678–5681. doi: 10.1021/ol5027415. [DOI] [PubMed] [Google Scholar]
- 183.(a) Vora HU, Rovis T. Asymmetric N-Heterocyclic Carbene (NHC) Catalyzed Acyl Anion Reactions. Aldrichimica Acta. 2011;44:3–10. [PMC free article] [PubMed] [Google Scholar]; (b) Vora HU, Wheeler P, Rovis T. Exploiting Acyl and Enol Azolium Intermediates via N-Hetero- cyclic Carbene-Catalyzed Reactions of α-Reducible Aldehydes. Adv Synth Cat. 2012;354:1617–1639. doi: 10.1002/adsc.201200031. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ryan SJ, Candish L, Lupton DW. Acyl Anion Free N-Heterocyclic Carbene Organocatalysis. Chem Soc Rev. 2013;42:4906–4917. doi: 10.1039/c3cs35522e. [DOI] [PubMed] [Google Scholar]; (d) Fraile A, Parra A, Tortosa M, Alemán J. Organocatalytic Transformations of Alkynals, Alkynones, Propriolates, and Related Electron-Deficient Alkynes. Tetrahedron. 2014;70:9145–9173. [Google Scholar]
- 184.(a) Khaleeli N, Li R, Townsend CA. Origin of the β-Lactam Carbons in Clavulanic Acid from an Unusual Thiamine Pyrophosphate-Mediated Reaction. J Am Chem Soc. 1999;121:9223–9224. [Google Scholar]; (b) Merski M, Townsend CA. Observation of an Acryloyl–Thiamin Diphosphate Adduct in the First Step of Clavulanic Acid Biosynthesis. J Am Chem Soc. 2007;129:15750–15751. doi: 10.1021/ja076704r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.For ynals as α,β-unsaturated acylazolium precursors, see: Zeitler K. Stereoselective Synthesis of (E)-α,β-Unsaturated Esters via Carbene-Catalyzed Redox Esterification. Org Lett. 2006;8:637–640. doi: 10.1021/ol052826h.Maki BE, Chan A, Phillips EM, Scheidt KA. Tandem Oxidation of Allylic and Benzylic Alcohols to Esters Catalyzed by N-Heterocyclic Carbenes. Org Lett. 2007;9:371–374. doi: 10.1021/ol062940f.Maki BE, Chan A, Scheidt KA. Protonation of Homoenolate Equivalents Generated by N-Heterocyclic Carbenes. Synthesis. 2008:1306–1315. doi: 10.1055/s-2008-1072516.Kaeobamrung J, Mahatthananchai J, Zheng P, Bode JW. An Enantioselective Claisen Rearrangement Catalyzed by N-Heterocyclic Carbenes. J Am Chem Soc. 2010;132:8810–8812. doi: 10.1021/ja103631u.Mahatthananchai J, Zheng P, Bode JW. α,β-Unsaturated Acyl Azoliums from N-Heterocyclic Carbene Catalyzed Reactions: Observation and Mechanistic Investigation. Angew Chem Int Ed. 2011;50:1673–1677. doi: 10.1002/anie.201005352.Mahatthananchai J, Kaeobamrung J, Bode JW. Chiral N-Heterocyclic Carbene-Catalyzed Annulations of Enals and Ynals with Stable Enols: A Highly Enantioselective Coates–Claisen Rearrangement. ACS Catal. 2012;2:494–503. doi: 10.1021/cs300020t.Zhu ZQ, Zheng XL, Jiang NF, Wan X, Xiao JC. Chiral N-Heterocyclic Carbene Catalyzed Annulation of α,β-Unsaturated Aldehydes with 1,3-Dicarbonyls. Chem Commun. 2011;47:8670–8672. doi: 10.1039/c1cc12778k.Zhu ZQ, Xiao JC. N-Heterocyclic Carbene-Catalyzed Reaction of Alkynyl Aldehydes with 1,3-Keto Esters or 1,3-Diketones. Adv Synth Catal. 2010;352:2455–2458.
- 186.For α-bromoenals as α,β-unsaturated acylazolium precursors, see: Sun FG, Sun LH, Ye S. N-Heterocyclic Carbene-Catalyzed Enantioselective Annulation of Bromoenal and 1,3-Dicarbonyl Compounds. Adv Synth Catal. 2011;353:3134–3138.Yetra SR, Bhunia A, Patra A, Mane MV, Vanka K, Biju AT. Enantioselective N-Heterocyclic Carbene-Catalyzed Annulations of 2-Bromoenals with 1,3-Dicarbonyl Compounds and Enamines via Chiral α,β-Unsaturated Acylazoliums. Adv Synth Catal. 2013;355:1089–1097.Yetra SR, Kaicharla T, Kunte SS, Gonnade RG, Biju AT. Asymmetric N-Heterocyclic Carbene (NHC)-Catalyzed Annulation of Modified Enals with Enolizable Aldehydes. Org Lett. 2013;15:5202–5205. doi: 10.1021/ol4026155.Yao C, Wang D, Lu J, Li T, Jiao W, Yu C. N-Heterocyclic Carbene Catalyzed Reactions of α-Bromo-α,β-Unsaturated Aldehydes/α,β-Dibromoaldehydes with 1,3-Dinucleophilic Reagents. Chem Eur J. 2012;18:1914–1917. doi: 10.1002/chem.201103358.
- 187.For esters and acyl fluorides as α,β-unsaturated acylazolium precursors, see: Ryan SJ, Candish L, Lupton DW. N-Heterocyclic Carbene-Catalyzed Generation of α,β-Unsaturated Acyl Imidazoliums: Synthesis of Dihydropyranones by their Reaction with Enolates. J Am Chem Soc. 2009;131:14176–14177. doi: 10.1021/ja905501z.Candish L, Lupton DW. The Total Synthesis of (−)-7-Deoxyloganin via N-Heterocyclic Carbene Catalyzed Rearrangement of α,β-Unsaturated Enol Esters. Org Lett. 2010;12:4836–4839. doi: 10.1021/ol101983h.Cheng J, Huang Z, Chi YR. NHC Organocatalytic Formal LUMO Activation of α,β-Unsaturated Esters for Reaction with Enamides. Angew Chem Int Ed. 2013;52:8592–8596. doi: 10.1002/anie.201303247.Chen XY, Gao ZH, Song CY, Zhang CL, Wang ZX, Ye S. N-Heterocyclic Carbene Catalyzed Cyclocondensation of α,β-Unsaturated Carboxylic Acids: Enantioselective Synthesis of Pyrrolidinone and Dihydropyridinone Derivatives. Angew Chem Int Ed. 2014;53:11611–11615. doi: 10.1002/anie.201407469.
- 188.For oxidative methods for α,β-unsaturated acylazolium generation, see: De Sarkar S, Studer A. NHC-Catalyzed Michael Addition to α,β-Unsaturated Aldehydes by Redox Activation. Angew Chem Int Ed. 2010;49:9266–9269. doi: 10.1002/anie.201004593.Rong ZQ, Jia MQ, You SL. Enantioselective N-Heterocyclic Carbene-Catalyzed Michael Addition to α,β-Unsaturated Aldehydes by Redox Oxidation. Org Lett. 2011;13:4080–4083. doi: 10.1021/ol201595f.Wanner B, Mahatthananchai J, Bode JW. Enantioselective Synthesis of Dihydropyridinones via NHC-Catalyzed Aza-Claisen Reaction. Org Lett. 2011;13:5378–5381. doi: 10.1021/ol202272t.Kravina AG, Mahatthananchai J, Bode JW. Enantioselective, NHC-Catalyzed Annulations of Trisubstituted Enals and Cyclic N-Sulfonylimines via α,β-Unsaturated Acyl Azoliums. Angew Chem Int Ed. 2012;51:9433–9436. doi: 10.1002/anie.201204145.De Sarkar S, Grimme S, Studer A. NHC Catalyzed Oxidations of Aldehydes to Esters: Chemoselective Acylation of Alcohols in Presence of Amines. J Am Chem Soc. 2010;132:1190–1191. doi: 10.1021/ja910540j.Maji B, Vedachalan S, Ge X, Cai S, Liu XW. N-Heterocyclic Carbene-Mediated Oxidative Esterification of Aldehydes: Ester Formation and Mechanistic Studies. J Org Chem. 2011;76:3016–3023. doi: 10.1021/jo200275c.Corey EJ, Gilman NW, Ganem BE. New Methods for the Oxidation of Aldehydes to Carboxylic Acids and Esters. J Am Chem Soc. 1968;90:5616–5617.Corey EJ, Katzenellenbogen JA, Gilman NW, Roman SA, Erickson BW. Stereospecific Total Synthesis of the dl-C18 Cecropia Juvenile Hormone. J Am Chem Soc. 1968;90:5618–5620.Gilman NW. The Preparation of Carboxylic Amides from Aldehydes by Oxidation. J Chem Soc D. 1971:733–734.
- 189.(a) Ryan SJ, Candish L, Lupton DW. N-Heterocyclic Carbene-Catalyzed (4 + 2) Cycloaddition/Decarboxylation of Silyl Dienol Ethers with α,β-Unsaturated Acid Fluorides. J Am Chem Soc. 2011;133:4694–4697. doi: 10.1021/ja111067j. [DOI] [PubMed] [Google Scholar]; (b) Candish L, Lupton DW. Concise Formal Synthesis of (−)-7-Deoxyloganinvia N-Heterocyclic Carbene Catalysed Rearrangement of α,β-Unsaturated Enol Esters. Org Biomol Chem. 2011;9:8182–8189. doi: 10.1039/c1ob05953j. [DOI] [PubMed] [Google Scholar]
- 190.(a) Candish L, Lupton DW. N-Heterocyclic Carbene-Catalyzed Ireland–Coates Claisen Rearrangement: Synthesis of Functionalized β-Lactones. J Am Chem Soc. 2013;135:58–61. doi: 10.1021/ja310449k. [DOI] [PubMed] [Google Scholar]; (b) Candish L, Forsyth CM, Lupton DW. N-tert-Butyl Triazolylidenes: Catalysts for the Enantioselective (3+2) Annulation of α,β-Unsaturated Acyl Azoliums. Angew Chem Int Ed. 2013;52:9149–9152. doi: 10.1002/anie.201304081. [DOI] [PubMed] [Google Scholar]
- 191.Samanta RC, Maji B, De Sarkar S, Bergander K, Fröhlich R, Mück-Lichtenfeld C, Mayr H, Studer A. Nucleophilic Addition of Enols and Enamines to α,β-Unsaturated Acyl Azoliums: Mechanistic Studies. Angew Chem Int Ed. 2012;51:5234–5238. doi: 10.1002/anie.201109042. [DOI] [PubMed] [Google Scholar]
- 192.Lyngvi E, Bode JW, Schoenebeck F. A Computational Study of the Origin of Stereoinduction in NHC-Catalyzed Annulation Reactions of α,β-Unsaturated Acyl Azoliums. Chem Sci. 2012;3:2346–2350. doi: 10.1039/C2SC20331F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Du D, Hu Z, Jin J, Lu Y, Tang W, Lu T. N-Heterocyclic Carbene-Catalyzed Three-Component Domino Reaction of Alkynyl Aldehydes with Oxindoles. Org Lett. 2012;14:1274–1277. doi: 10.1021/ol300148f. [DOI] [PubMed] [Google Scholar]
- 194.Lu Y, Tang W, Zhang Y, Du D, Lu T. N-Heterocyclic Carbene-Catalyzed Annulations of Enals and Ynals with Indolin-3-ones: Synthesis of 3,4-Dihydropyrano[3,2-b]indol-2-ones. Adv Synth Catal. 2013;355:321–326. [Google Scholar]
- 195.Romanov-Michailidis F, Besnard C, Alexakis A. N-Heterocyclic Carbene-Catalyzed Annulation of α-Cyano-1,4-diketones with Ynals. Org Lett. 2012;14:4906–4909. doi: 10.1021/ol3022287. [DOI] [PubMed] [Google Scholar]
- 196.Ni Q, Song X, Raabe G, Enders D. N-Heterocyclic Carbene-Catalyzed Enantioselective Annulation of Indolin-3-ones with Bromoenals. Chem Asian J. 2014;9:1535–1538. doi: 10.1002/asia.201402052. [DOI] [PubMed] [Google Scholar]
- 197.Zhang H-R, Dong Z-W, Yang Y-J, Wang P-L, Hui X-P. N-Heterocyclic Carbene-Catalyzed Stereoselective Cascade Reaction: Synthesis of Functionalized Tetrahydroquinolines. Org Lett. 2013;15:4750–4753. doi: 10.1021/ol4024985. [DOI] [PubMed] [Google Scholar]
- 198.De Sarkar S, Biswas A, Samanta RC, Studer A. Catalysis with N-Heterocyclic Carbenes Under Oxidative Conditions. Chem Eur J. 2013;19:4664–4678. doi: 10.1002/chem.201203707. [DOI] [PubMed] [Google Scholar]
- 199.Ragsdale SW. Pyruvate Ferredoxin Oxidoreductase and Its Radical Intermediate. Chem Rev. 2003;103:2333. doi: 10.1021/cr020423e. [DOI] [PubMed] [Google Scholar]
- 200.(a) Wallach O. Ueber die Einwirkung von Cyankalium auf Chloral; Eine Neue Darstellungsweise der Dichloressigsäure. Ber Dtsch Chem Ges. 1873;6:114–119. [Google Scholar]; (b) Christmann M. Otto Wallach: Founder of Terpene Chemistry and Nobel Laureate 1910. Angew Chem Int Ed. 2010;49:9580–9586. doi: 10.1002/anie.201003155. [DOI] [PubMed] [Google Scholar]
- 201.Kötz A, Otto K. Gleichzeitige Reduktion und Oxydation. (Erste Abhandlung.) Dichlorbrenztraubensäure, -Nitril und -Ester aus Trichlormilchsäure, -Nitril und -Ester. J Prakt Chem. 1913;88:531–552. [Google Scholar]
- 202.Castells J, Llitjos H, Moreno-Mañas M. Nitrobenzene Aldehyde Oxidations Catalyzed by Conjugate Bases of Thiazolium Ions. Tetrahedron Lett. 1977;18:205–206. [Google Scholar]
- 203.Biswas A, De Sarkar S, Tebben L, Studer A. Enantioselective Cyclopropanation of Enals by Oxidative N-Heterocyclic Carbene Catalysis. Chem Commun. 2012;48:5190–5192. doi: 10.1039/c2cc31501g. [DOI] [PubMed] [Google Scholar]
- 204.Biswas A, De Sarkar S, Fröhlich R, Studer A. Highly Stereoselective Synthesis of 1,2,3-Trisubstituted Indanes via Oxidative N-Heterocyclic Carbene-Catalyzed Cascades. Org Lett. 2011;13:4966–4969. doi: 10.1021/ol202108a. [DOI] [PubMed] [Google Scholar]
- 205.Walsh PJ, Kozlowski MC. Fundamentals of Asymmetric Catalysis; University Science Books; Sausalito: 2009. p. 563. [Google Scholar]
- 206.Chiang P-C, Kim Y, Bode JW. Catalytic Amide Formation with α′-Hydroxyenones as Acylating Reagents. Chem Commun. 2009:4566–4568. doi: 10.1039/b909360e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Mo J, Shen L, Chi YR. Direct β-Activation of Saturated Aldehydes to Formal Michael Acceptors through Oxidative NHC Catalysis. Angew Chem Int Ed. 2013;52:8588–8591. doi: 10.1002/anie.201302152. [DOI] [PubMed] [Google Scholar]
- 208.Zhao Y-M, Tam Y, Wang Y-J, Li Z, Sun J. N-Heterocyclic Carbene-Catalyzed Internal Redox Reaction of Alkynals: An Efficient Synthesis of Allenoates. Org Lett. 2012;14:1398–1401. doi: 10.1021/ol300111m. [DOI] [PubMed] [Google Scholar]
- 209.Qi J, Xie X, Han R, Ma D, Yang J, She X. N-Heterocyclic Carbene (NHC)-Catalyzed/Lewis Acid Mediated Conjugate Umpolung of Alkynyl Aldehydes for the Synthesis of Butenolides: A Formal [3+2] Annulation. Chem Eur J. 2013;19:4146–4150. doi: 10.1002/chem.201204386. [DOI] [PubMed] [Google Scholar]
- 210.ElSohly AM, Wespe DA, Poore TJ, Snyder SA. An Efficient Approach to the Securinega Alkaloids Empowered by Cooperative N-Heterocyclic Carbene/Lewis Acid Catalysis. Angew Chem Int Ed. 2013;52:5789–5794. doi: 10.1002/anie.201301849. [DOI] [PubMed] [Google Scholar]
- 211.Lee A, Scheidt KA. A Cooperative N-Heterocyclic Carbene/Chiral Phosphate Catalysis System for Allenolate Annulations. Angew Chem Int Ed. 2014;53:7594–7598. doi: 10.1002/anie.201403446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.For selected examples, see: Van Veldhuizen JJ, Garber SB, Kingsbury JS, Hoveyda AH. A Recyclable Chiral Ru Catalyst for Enantioselective Olefin Metathesis. Efficient Catalytic Asymmetric Ring-Opening/Cross Metathesis in Air. J Am Chem Soc. 2002;124:4954–4955. doi: 10.1021/ja020259c.Van Veldhuizen JJ, Gillingham DG, Barber SB, Kataoka O, Hoveyda AH. Chiral Ru-Based Complexes for Asymmetric Olefin Metathesis: Enhancement of Catalyst Activity through Steric and Electronic Modifications. J Am Chem Soc. 2003;125:12502–12508. doi: 10.1021/ja0302228.Hoveyda AH, Gillingham DG, Van Veldhuizen JJ, Kataoka O, Barber SB, Kingsbury JS, Harrity JPA. Ru Complexes Bearing Bidentate Carbenes: From Innocent Curiosity to Uniquely Effective Catalysts for Olefin Metathesis. Org Biomol Chem. 2004;2:8–23. doi: 10.1039/b311496c.Lee KS, Brown MK, Hird AW, Hoveyda AH. A Practical Method for Enantioselective Synthesis of All-Carbon Quaternary Stereogenic Centers through NHC-Cu-Catalyzed Conjugate Additions of Alkyl- and Arylzinc Reagents to β-Substituted Cyclic Enones. J Am Chem Soc. 2006;128:7182–7184. doi: 10.1021/ja062061o.Brown MK, May TL, Baxter CA, Hoveyda AH. All-Carbon Quaternary Stereogenic Centers by Enantioselective Cu-Catalyzed Conjugate Additions Promoted by a Chiral N-Heterocyclic Carbene. Angew Chem Int Ed. 2007;46:1097–1100. doi: 10.1002/anie.200604511.Lee Y, Akiyama K, Gillingham DG, Brown MK, Hoveyda AH. Highly Site- and Enantioselective Cu-Catalyzed Allylic Alkylation Reactions with Easily Accessible Vinylaluminum Reagents. J Am Chem Soc. 2008;130:446–447. doi: 10.1021/ja0782192.Wu H, Radomkit S, O’Brien JM, Hoveyda AH. Metal-Free Catalytic Enantioselective C–B Bond Formation: (Pinacolato)boron Conjugate Additions to α,β-Unsaturated Ketones, Esters, Weinreb Amides, and Aldehydes Promoted by Chiral N-Heterocyclic Carbenes. J Am Chem Soc. 2012;134:8277–8285. doi: 10.1021/ja302929d.
- 213.(a) Wang G, Chen X, Miao G, Yao W, Ma C. Divergent NHC-Catalyzed Oxidative Transformations of 3-Bromoenal: Selective Synthesis of 2H-Pyran-2-Ones and Chiral Dihydropyranones. J Org Chem. 2013;78:6223–6232. doi: 10.1021/jo400950j. [DOI] [PubMed] [Google Scholar]; (b) Wu Y, Yao W, Pan L, Zhang Y, Ma C. N-Heterocyclic Carbene Catalyzed Transformations of 3-Halopropenals to the Equivalents of β-Acylvinyl Anions. Org Lett. 2010;12:640–643. doi: 10.1021/ol902961y. [DOI] [PubMed] [Google Scholar]
- 214.Zheng C, Yao W, Zhang Y, Ma C. Chiral Spirooxindole–Butenolide Synthesis through Asymmetric N-Heterocyclic Carbene-Catalyzed Formal (3 + 2) Annulation of 3-Bromoenals and Isatins. Org Lett. 2014;16:5028–5031. doi: 10.1021/ol502365r. [DOI] [PubMed] [Google Scholar]
- 215.Douglas J, Churchill G, Smith AD. NHCs in Asymmetric Organocatalysis: Recent Advances in Azolium Enolate Generation and Reactivity. Synthesis. 2012;44:2295–2309. [Google Scholar]
- 216.Connor EF, Nyce GW, Myers M, Möck A, Hedrick JL. First Example of N-Heterocyclic Carbenes as Catalysts for Living Polymerization: Organocatalytic Ring-Opening Polymerization of Cyclic Esters. J Am Chem Soc. 2002;124:914–915. doi: 10.1021/ja0173324. [DOI] [PubMed] [Google Scholar]
- 217.(a) Grasa GA, Kissling RM, Nolan SP. N-Heterocyclic Carbenes as Versatile Nucleophilic Catalysts for Transesterification/Acylation Reactions. Org Lett. 2002;4:3583–3586. doi: 10.1021/ol0264760. [DOI] [PubMed] [Google Scholar]; (b) Nyce GW, Lamboy JA, Connor EF, Waymouth RM, Hedrick JL. Expanding the Catalytic Activity of Nucleophilic N-Heterocyclic Carbenes for Transesterification Reactions. Org Lett. 2002;4:3587–3590. doi: 10.1021/ol0267228. [DOI] [PubMed] [Google Scholar]; (c) Grasa GA, Güveli T, Singh R, Nolan SP. Efficient Transesterification/Acylation Reactions Mediated by N-Heterocyclic Carbene Catalysts. J Org Chem. 2003;68:2812–2819. doi: 10.1021/jo0267551. [DOI] [PubMed] [Google Scholar]; (d) Movassaghi M, Schmidt MA. N-Heterocyclic Carbene-Catalyzed Amidation of Unactivated Esters with Amino Alcohols. Org Lett. 2005;7:2453–2456. doi: 10.1021/ol050773y. [DOI] [PubMed] [Google Scholar]; (e) Lai CL, Lee HM, Hu CH. Theoretical Study on the Mechanism of N-Heterocyclic Carbene Catalyzed Transesterification Reactions. Tetrahedron Lett. 2005;46:6265–6270. [Google Scholar]
- 218.(a) Suzuki Y, Yamauchi K, Muramatsu K, Sato M. First Example of Chiral N-Heterocyclic Carbenes as Catalysts for Kinetic Resolution. Chem Commun. 2004:2770–2771. doi: 10.1039/b411855c. [DOI] [PubMed] [Google Scholar]; (b) Suzuki Y, Muramatsu K, Yamauchi K, Morie Y, Sato M. Chiral N-Heterocyclic Carbenes as Asymmetric Acylation Catalysts. Tetrahedron. 2006;62:302–310. [Google Scholar]
- 219.Kano T, Sasaki K, Maruoka K. Enantioselective Acylation of Secondary Alcohols Catalyzed by Chiral N-Heterocyclic Carbenes. Org Lett. 2005;7:1347–1349. doi: 10.1021/ol050174r. [DOI] [PubMed] [Google Scholar]
- 220.Chow KY-K, Bode JW. Catalytic Generation of Activated Carboxylates: Direct, Stereoselective Synthesis of β-Hydroxyesters from Epoxyaldehydes. J Am Chem Soc. 2004;126:8126–8127. doi: 10.1021/ja047407e. [DOI] [PubMed] [Google Scholar]
- 221.Reynolds NT, Read de Alaniz J, Rovis T. Conversion of α-Haloaldehydes into Acylating Agents by an Internal Redox Reaction Catalyzed by Nucleophilic Carbenes. J Am Chem Soc. 2004;126:9518–9519. doi: 10.1021/ja046991o. [DOI] [PubMed] [Google Scholar]
- 222.Burns NZ, Baran PS, Hoffmann RW. Redox Economy in Organic Synthesis. Angew Chem Int Ed. 2009;48:2854–2867. doi: 10.1002/anie.200806086. [DOI] [PubMed] [Google Scholar]
- 223.(a) Enders D, Grossmann A, Van Craen D. N-Heterocyclic Carbene Catalyzed Synthesis of Oxime Esters. Org Biomol Chem. 2013;11:138–141. doi: 10.1039/c2ob26974k. [DOI] [PubMed] [Google Scholar]; (b) Song X, Ni Q, Grossmann A, Enders D. N-Heterocyclic Carbene-Catalyzed One-Pot Synthesis of Hydroxamic Esters. Chem Asian J. 2013;8:2965–2969. doi: 10.1002/asia.201300938. [DOI] [PubMed] [Google Scholar]
- 224.Reynolds NT, Rovis T. Enantioselective Protonation of Catalytically Generated Chiral Enolates as an Approach to the Synthesis of α-Chloroesters. J Am Chem Soc. 2005;127:16406–16407. doi: 10.1021/ja055918a. [DOI] [PubMed] [Google Scholar]
- 225.Vora HU, Rovis T. N-Heterocyclic Carbene Catalyzed Asymmetric Hydration: Direct Synthesis of α-Protio and α-Deuterio α-Chloro and α-Fluoro Carboxylic Acids. J Am Chem Soc. 2010;132:2860–2861. doi: 10.1021/ja910281s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Vora HU, Rovis T. Nucleophilic Carbene and HOAt Relay Catalysis in an Amide Bond Coupling: An Orthogonal Peptide Bond Forming Reaction. J Am Chem Soc. 2007;129:13796–13797. doi: 10.1021/ja0764052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Bode JW, Sohn SS. N-Heterocyclic Carbene-Catalyzed Redox Amidations of α-Functionalized Aldehydes with Amines. J Am Chem Soc. 2007;129:13798–13799. doi: 10.1021/ja0768136. [DOI] [PubMed] [Google Scholar]
- 228.Binanzer M, Hsieh S-Y, Bode JW. Catalytic Kinetic Resolution of Cyclic Secondary Amines. J Am Chem Soc. 2011;133:19698–19701. doi: 10.1021/ja209472h. [DOI] [PubMed] [Google Scholar]
- 229.Zhao X, Ruhl KE, Rovis T. N-Heterocyclic-Carbene-Catalyzed Asymmetric Oxidative Hetero-Diels–Alder Reactions with Simple Aliphatic Aldehydes. Angew Chem Int Ed. 2012;51:12330–12333. doi: 10.1002/anie.201206490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Song X, Ni Q, Zhu C, Raabe G, Enders D. Asymmetric N-Heterocyclic Carbene Catalyzed Annulation of 2-Alkenylbenzothiazoles with α-Chloro Aldehydes. Synthesis. 2015:421–428. doi: 10.1055/s-0034-1379369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Zhang Y-R, Lv H, Zhou D, Ye S. Chiral N-Heterocyclic Carbene-Catalyzed Formal [4+2] Cycloaddition of Ketenes with Enones: Highly Enantioselective Synthesis of trans- and cis-δ-Lactones. Chem Eur J. 2008;14:8473–8476. doi: 10.1002/chem.200801165. [DOI] [PubMed] [Google Scholar]
- 232.Huang X-L, He L, Shao P-L, Ye S. [4+2] Cycloaddition of Ketenes with N-Benzoyldiazenes Catalyzed by N-Heterocyclic Carbenes. Angew Chem Int Ed. 2009;48:192–195. doi: 10.1002/anie.200804487. [DOI] [PubMed] [Google Scholar]
- 233.Wurz RP. Chiral Dialkylaminopyridine Catalysts in Asymmetric Synthesis. Chem Rev. 2007;107:5570–5595. doi: 10.1021/cr068370e. [DOI] [PubMed] [Google Scholar]
- 234.Song X, Ni Q, Shu C, Raabe G, Enders D. Asymmetric N-Heterocyclic Carbene Catalyzed Annulation of 2-Alkenylbenzothiazoles with α-Chloro Aldehydes. Synthesis. 2015;47:421–428. doi: 10.1055/s-0034-1379369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Ni Q, Song X, Xiong J, Raabe G, Enders D. Regio- and Stereoselective Synthesis of Benzothiazolo-pyrimidinones via an NHC-Catalyzed Mannich/Lactamization Domino Reaction. Chem Commun. 2015;51:1263–1266. doi: 10.1039/c4cc08594a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Zhang Y-R, He L, Wu X, Shao P-L, Ye S. Chiral N-Heterocyclic Carbene Catalyzed Staudinger Reaction of Ketenes with Imines: Highly Enantioselective Synthesis of N-Boc β-Lactams. Org Lett. 2008;10:277–280. doi: 10.1021/ol702759b. [DOI] [PubMed] [Google Scholar]
- 237.Duguet N, Campbell CD, Slawin AMZ, Smith AD. N-Heterocyclic Carbene Catalysed β-Lactam Synthesis. Org Biomol Chem. 2008;6:1108–1113. doi: 10.1039/b800857b. [DOI] [PubMed] [Google Scholar]
- 238.Jian T-Y, He L, Tang C, Ye S. N-Heterocyclic Carbene Catalysis: Enantioselective Formal [2+2] Cycloaddition of Ketenes and N-Sulfinylanilines. Angew Chem Int Ed. 2011;50:9104–9107. doi: 10.1002/anie.201102488. [DOI] [PubMed] [Google Scholar]
- 239.Ni Q, Zhang H, Grossmann A, Loh CCJ, Merkens C, Enders D. Asymmetric Synthesis of Pyrroloindolones by N-Heterocyclic Carbene Catalyzed [2+3] Annulation of α-Chloroaldehydes with Nitrovinylindoles. Angew Chem Int Ed. 2013;52:13562–13566. doi: 10.1002/anie.201305957. [DOI] [PubMed] [Google Scholar]
- 240.Duong HA, Cross MJ, Louie J. N-Heterocyclic Carbenes as Highly Efficient Catalysts for the Cyclotrimerization of Isocyanates. Org Lett. 2004;6:4679–4681. doi: 10.1021/ol048211m. [DOI] [PubMed] [Google Scholar]
- 241.(a) Wang XN, Shen LT, Ye S. NHC-Catalyzed Enantioselective [2 + 2] and [2 + 2 + 2] Cycloadditions of Ketenes with Isothiocyanates. Org Lett. 2011;13:6382–6385. doi: 10.1021/ol202688h. [DOI] [PubMed] [Google Scholar]; (b) Wang XN, Shen LT, Ye S. Enantioselective [2+2+2] Cycloaddition of Ketenes and Carbon Disulfide Catalyzed by N-Heterocyclic Carbenes. Chem Commun. 2011;47:8388–8390. doi: 10.1039/c1cc12316e. [DOI] [PubMed] [Google Scholar]
- 242.(a) He M, Struble JR, Bode JW. Highly Enantioselective Azadiene Diels–Alder Reactions Catalyzed by Chiral N-Heterocyclic Carbenes. J Am Chem Soc. 2006;128:8418–8420. doi: 10.1021/ja062707c. [DOI] [PubMed] [Google Scholar]; (b) He M, Uc GJ, Bode JW. Chiral N-Heterocyclic Carbene Catalyzed, Enantioselective Oxodiene Diels–Alder Reactions with Low Catalyst Loadings. J Am Chem Soc. 2006;128:15088–15089. doi: 10.1021/ja066380r. [DOI] [PubMed] [Google Scholar]
- 243.Wang X-N, Lv H, Huang X-L, Ye S. Asymmetric Esterification of Ketenes Catalyzed by an N-Heterocyclic Carbene. Org Biomol Chem. 2009;7:346–350. doi: 10.1039/b815139c. [DOI] [PubMed] [Google Scholar]
- 244.Concellón C, Duguet N, Smith AD. N-Heterocyclic Carbene-Mediated Enantioselective Addition of Phenols to Unsymmetrical Alkylarylketenes. Adv Synth Catal. 2009;351:3001–3009. [Google Scholar]
- 245.Douglas J, Ling KB, Concellón C, Churchill G, Slawin AMZ, Smith AD. NHC-Mediated Chlorination of Unsymmetrical Ketenes: Catalysis and Asymmetry. Eur J Org Chem. 2010:5863–5869. [Google Scholar]
- 246.Li L, Du D, Ren J, Wang Z. N-Heterocyclic Carbene Catalyzed Domino Reactions of Formylcyclopropane 1,1-Diesters: Construction of a 6-5-5 Tricyclic Pyrrolo[1,2-a]indole. Eur J Org Chem. 2011:614–618. doi: 10.1021/jo900650h. [DOI] [PubMed] [Google Scholar]
- 247.For recent reviews on NHC catalyzed domino reactions, see: Pellissier H. Recent Developments in Asymmetric Organocatalytic Domino Reactions. Adv Synth Catal. 2012;354:237–294.Grossmann A, Enders D. N-Heterocyclic Carbene Catalyzed Domino Reactions. Angew Chem Int Ed. 2012;51:314–325. doi: 10.1002/anie.201105415.Chauhan P, Enders D. N-Heterocyclic Carbene Catalyzed Activation of Esters: A New Option for Asymmetric Domino Reactions. Angew Chem Int Ed. 2014;53:1485–1487. doi: 10.1002/anie.201309952.
- 248.Dong X, Yang W, Hu W, Sun J. N-Heterocyclic Carbene Catalyzed Enantioselective α-Fluorination of Aliphatic Aldehydes and α-Chloro Aldehydes: Synthesis of α-Fluoro Esters, Amides, and Thioesters. Angew Chem Int Ed. 2014;54:660–663. doi: 10.1002/anie.201409961. [DOI] [PubMed] [Google Scholar]
- 249.Hao L, Du Y, Lv H, Chen X, Jiang H, Shao Y, Chi YR. Enantioselective Activation of Stable Carboxylate Esters as Enolate Equivalents via N-Heterocyclic Carbene Catalysts. Org Lett. 2012;14:2154–2157. doi: 10.1021/ol300676w. [DOI] [PubMed] [Google Scholar]
- 250.Shen L-T, Shao P-L, Ye S. N-Heterocyclic Carbene-Catalyzed Cyclization of Unsaturated Acyl Chlorides and Ketones. Adv Synth Catal. 2011;353:1943–1948. [Google Scholar]
- 251.Wang X-N, Zhang Y-Y, Ye S. Enantioselective Synthesis of Spirocyclic Oxindole-β-Lactones via N-Heterocyclic Carbene-Catalyzed Cycloaddition of Ketenes and Isatins. Adv Synth Catal. 2010;352:1892–1895. [Google Scholar]
- 252.Mo J, Chen X, Chi YR. Oxidative γ-Addition of Enals to Trifluoromethyl Ketones: Enantioselectivity Control via Lewis Acid/N-Heterocyclic Carbene Cooperative Catalysis. J Am Chem Soc. 2012;134:8810–8813. doi: 10.1021/ja303618z. [DOI] [PubMed] [Google Scholar]
- 253.Yao C, Xiao Z, Liu R, Li T, Jiao W, Yu C. N-Heterocyclic-Carbene-Catalyzed Reaction of α-Bromo-α,β-Unsaturated Aldehyde or α,β-Dibromoaldehyde with Isatins: An Efficient Synthesis of Spirocyclic Oxindole–Dihydropyranones. Chem Eur J. 2013;19:456–459. doi: 10.1002/chem.201202655. [DOI] [PubMed] [Google Scholar]
- 254.Zhao Y-M, Cheung MS, Lin Z, Sun J. Enantioselective Synthesis of β,γ-Unsaturated α-Fluoroesters Catalyzed by N-Heterocyclic Carbenes. Angew Chem Int Ed. 2012;51:10359–10363. doi: 10.1002/anie.201204521. [DOI] [PubMed] [Google Scholar]
- 255.Chen X-Y, Xia F, Cheng J-T, Ye S. Highly Enantioselective γ-Amination by N-Heterocyclic Carbene Catalyzed [4+2] Annulation of Oxidized Enals and Azodicarboxylates. Angew Chem Int Ed. 2013;52:10644–10647. doi: 10.1002/anie.201305571. [DOI] [PubMed] [Google Scholar]
- 256.He L, Jian T-Y, Ye S. N-Heterocyclic Carbene Catalyzed Aza-Morita–Baylis–Hillman Reaction of Cyclic Enones with N-Tosylarylimines. J Org Chem. 2007;72:7466–7468. doi: 10.1021/jo071247i. [DOI] [PubMed] [Google Scholar]
- 257.He L, Zhang Y-R, Huang X-L, Ye S. Chiral Bifunctional N-Heterocyclic Carbenes: Synthesis and Application in the Aza-Morita-Baylis-Hillman Reaction. Synthesis. 2008:2825–2829. [Google Scholar]
- 258.Chen X-Y, Xia F, Ye S. Catalytic MBH Reaction of β-Substituted Nitroalkenes with Azodicarboxylates. Org Biomol Chem. 2013;11:5722–5726. doi: 10.1039/c3ob40804c. [DOI] [PubMed] [Google Scholar]
- 259.Chen X-Y, Sun L-H, Ye S. N-Heterocyclic Carbene Catalyzed [4+2] Cycloaddition of Nitroalkenes with Oxodienes. Chem Eur J. 2013;19:4441–4445. doi: 10.1002/chem.201204539. [DOI] [PubMed] [Google Scholar]
- 260.Atienza RL, Scheidt KA. N-Heterocyclic Carbene-Promoted Rauhut–Currier Reactions between Vinyl Sulfones and α,β-Unsaturated Aldehydes. Aust J Chem. 2011;64:1158–1164. doi: 10.1071/ch11264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Matsuoka S-I, Shimakawa S, Takagi K, Suzuki M. Organocatalytic Head-to-Tail Dimerization of Methacrolein via Conjugate Addition of Methanol: An Alcohol Activation Mechanism Proved by Electrospray Ionization Mass Spectrometry. Tetrahedron Lett. 2011;52:6835–6838. [Google Scholar]
- 262.Fischer C, Smith SW, Powell DA, Fu GC. Umpolung of Michael Acceptors Catalyzed by N-Heterocyclic Carbenes. J Am Chem Soc. 2006;128:1472–1473. doi: 10.1021/ja058222q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Atienza RL, Roth HS, Scheidt KA. N-Heterocyclic Carbene-Catalyzed Rearrangements of Vinyl Sulfones. Chem Sci. 2011:1772–1776. doi: 10.1039/C1SC00194A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.(a) Matsuoka SI, Ota Y, Washio A, Katada A, Ichioka K, Takagi K, Suzuki M. Organocatalytic Tail-to-Tail Dimerization of Olefin: Umpolung of Methyl Methacrylate Mediated by N-Heterocyclic Carbene. Org Lett. 2011;13:3722–3725. doi: 10.1021/ol201380m. [DOI] [PubMed] [Google Scholar]; (b) Biju AT, Padmanaban M, Wurz NE, Glorius F. N-Heterocyclic Carbene Catalyzed Umpolung of Michael Acceptors for Intermolecular Reactions. Angew Chem Int Ed. 2011;50:8412–8415. doi: 10.1002/anie.201103555. [DOI] [PubMed] [Google Scholar]
- 265.Zhang Y, Schmitt M, Falivene L, Caporaso L, Cavallo L, Chen EYX. Organocatalytic Conjugate-Addition Polymerization of Linear and Cyclic Acrylic Monomers by N-Heterocyclic Carbenes: Mechanisms of Chain Initiation, Propagation, and Termination. J Am Chem Soc. 2013;135:17925–17942. doi: 10.1021/ja4088677.He J, Zhang Y, Chen EYX. Synthesis of Pyridine- and 2-Oxazoline-Functionalized Vinyl Polymers by Alane-Based Frustrated Lewis Pairs. Synlett. 2014:1534–1538.Hong M, Chen EYX. Proton-Transfer Polymerization (HTP): Converting Methacrylates to Polyesters by an N-Heterocyclic Carbene. Angew Chem Int Ed. 2014;53:11900–11906. doi: 10.1002/anie.201406630.For a review, see: Chen X-Y, Ye S. N-Heterocyclic Carbene-Catalyzed Reactions of C–C Unsaturated Bonds. Org Biomol Chem. 2013;11:7991–7998. doi: 10.1039/c3ob41469h.
- 266.Kato T, Matsuoka S-I, Suzuki M. Cooperative N-Heterocyclic Carbene/Brønsted Acid Catalysis for the Tail-to-Tail (Co)dimerization of Methacrylonitrile. J Org Chem. 2014;79:4484–4491. doi: 10.1021/jo500514e. [DOI] [PubMed] [Google Scholar]
- 267.Matsuoka S-I, Tochigi Y, Takagi K, Suzuki M. Sequential One-Pot and Three-Component Reactions of an N-Heterocyclic Carbene to Form 4-(1,2,4-triazol-5-ylidene)pyrrolidine-2,5-diones: A Tandem Umpolung/Annulation Sequence via Deoxy-Breslow Intermediates. Tetrahedron. 2012;68:9836–9841. [Google Scholar]
- 268.Schedler M, Wurz NE, Daniliuc CG, Glorius F. N-Heterocyclic Carbene Catalyzed Umpolung of Styrenes: Mechanistic Elucidation and Selective Tail-to-Tail Dimerization. Org Lett. 2014;16:3134–3137. doi: 10.1021/ol501256d. [DOI] [PubMed] [Google Scholar]
- 269.Sinu CR, Suresh E, Nair V. N-Heterocyclic Carbene Catalyzed Reaction of Cinnamils Leading to the Formation of 2,3,8-Triaryl Vinyl Fulvenes: An Uncommon Transformation. Org Lett. 2013;15:6230–6233. doi: 10.1021/ol4030748. [DOI] [PubMed] [Google Scholar]
- 270.Maji B, Horn M, Mayr H. Nucleophilic Reactivities of Deoxy Breslow Intermediates: How Does Aromaticity Affect the Catalytic Activities of N-Heterocyclic Carbenes? Angew Chem Int Ed. 2012;51:6231–6235. doi: 10.1002/anie.201202327. [DOI] [PubMed] [Google Scholar]
- 271.Lee K-S, Zhugralin AR, Hoveyda AH. Efficient C–B Bond Formation Promoted by N-Heterocyclic Carbenes: Synthesis of Tertiary and Quaternary B-Substituted Carbons through Metal-Free Catalytic Boron Conjugate Additions to Cyclic and Acyclic α,β-Unsaturated Carbonyls. J Am Chem Soc. 2009;131:7253–7255. doi: 10.1021/ja902889s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Kleeberg C, Crawford AG, Batsanov AS, Hodgkinson P, Apperley DC, Cheung MS, Marder TB. Spectroscopic and Structural Characterization of the CyNHC Adduct of B2pin2 in Solution and in the Solid State. J Org Chem. 2012;77:785–789. doi: 10.1021/jo202127c. [DOI] [PubMed] [Google Scholar]
- 273.O’Brien JM, Hoveyda AM. Metal-Free Catalytic C–Si Bond Formation in an Aqueous Medium. Enantioselective NHC-Catalyzed Silyl Conjugate Additions to Cyclic and Acyclic α,β-Unsaturated Carbonyls. J Am Chem Soc. 2011;133:7712–7715. doi: 10.1021/ja203031a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Blanc R, Commeiras L, Parrain J-L. N-Heterocyclic Carbene-Mediated Organocatalytic Transfer of Tin onto Aldehydes: New Access to α-Silyloxyalkylstannanes and γ-Silyloxyallylstannanes. Adv Synth Catal. 2010;352:661–666. [Google Scholar]
- 275.Phillips EM, Riedrich M, Scheidt KA. N-Heterocyclic Carbene-Catalyzed Conjugate Additions of Alcohols. J Am Chem Soc. 2010;132:13179–13181. doi: 10.1021/ja1061196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Kang Q, Zhang Y. N-Heterocyclic Carbene-Catalyzed aza-Michael Addition. Org Biomol Chem. 2011;9:6715–6720. doi: 10.1039/c1ob05429e. [DOI] [PubMed] [Google Scholar]
- 277.Hans M, Delaude L, Rodriguez J, Coquerel Y. N-Heterocyclic Carbene Catalyzed Carba-, Sulfa-, and Phospha-Michael Additions with NHC CO2 Adducts as Precatalysts. J Org Chem. 2014;79:2758–2764. doi: 10.1021/jo500108a. [DOI] [PubMed] [Google Scholar]
- 278.Perez F, Ren Y, Boddaert T, Rodriguez J, Coquerel Y. A Stable N-Heterocyclic Carbene Organocatalyst for Hydrogen/Deuterium Exchange Reactions Between Pseudoacids and Deuterated Chloroform. J Org Chem. 2014;80:1092–1097. doi: 10.1021/jo502578x. [DOI] [PubMed] [Google Scholar]
- 279.(a) Boddaert T, Coquerel Y, Rodriguez J. Organocatalytic Activity of N-Heterocyclic Carbenes in the Michael Addition of 1,3-Dicarbonyl Compounds: Application to a Stereoselective Spirocyclization Sequence. Adv Synth Catal. 2009;351:1744–1748. [Google Scholar]; (b) Boddaert T, Coquerel Y, Rodriguez J. Combination of Rearrangement with Metallic and Organo Catalyses – a Step- and Atom-Economical Approach to a-Spirolactones and –lactams. Eur J Org Chem. 2011:5061–5070. [Google Scholar]
- 280.Boddaert T, Coquerel Y, Rodriguez J. N-Heterocyclic Carbene-Catalyzed Michael Additions of 1,3-Dicarbonyl Compounds. Chem Eur J. 2011;17:2266–2271. doi: 10.1002/chem.201002538. [DOI] [PubMed] [Google Scholar]
- 281.Chen J, Huang Y. Asymmetric Catalysis with N-Heterocyclic Carbenes as Non-Covalent Chiral Templates. Nat Commun. 2014;5:3437. doi: 10.1038/ncomms4437. [DOI] [PubMed] [Google Scholar]
- 282.Izquierdo J, Hutson GE, Cohen DT, Scheidt KA. A Continuum of Progress: Applications of N-Hetereocyclic Carbene Catalysis in Total Synthesis. Angew Chem Int Ed. 2012;51:11686–11698. doi: 10.1002/anie.201203704. [DOI] [PMC free article] [PubMed] [Google Scholar]