

Abstract
Purpose
In the development of diabetic retinopathy, retinal mitochondria become dysfunctional, and mitochondrial DNA (mtDNA) is damaged. Because retinopathy is a progressive disease, and circulating glucose levels are high in diabetes, our aim was to investigate if peripheral blood mtDNA damage can serve as a potential biomarker of diabetic retinopathy.
Methods
Peripheral blood mtDNA damage was investigated by extended-length PCR in rats and mice, diabetic for 10 to 12 months (streptozotocin-induced, type 1 model), and in 12- and 40-week-old Zucker diabetic fatty rats (ZDF, type 2). Mitochondrial copy number (in gDNA) and transcription (in cDNA) were quantified by qPCR. Similar parameters were measured in blood from diabetic patients with/without retinopathy.
Results
Peripheral blood from diabetic rodents had significantly increased mtDNA damage and decreased copy numbers and transcription. Lipoic acid administration in diabetic rats, or Sod2 overexpression or MMP-9 knockdown in mice, the therapies that prevent diabetic retinopathy, also ameliorated blood mtDNA damage and restored copy numbers and transcription. Although blood from 40-week-old ZDF rats had significant mtDNA damage, 12-week-old rats had normal mtDNA. Diabetic patients with retinopathy had increased blood mtDNA damage, and decreased transcription and copy numbers compared with diabetic patients without retinopathy and nondiabetic individuals.
Conclusions
Type 1 diabetic rodents with oxidative stress modulated by pharmacologic/genetic means, and type 2 animal model and patients with/without diabetic retinopathy, demonstrate a strong relation between peripheral blood mtDNA damage and diabetic retinopathy, and suggest the possibility of use of peripheral blood mtDNA as a noninvasive biomarker of diabetic retinopathy.
Keywords: biomarker, diabetic retinopathy, DNA damage, mitochondria
Diabetes has become the epidemic of the 21st century, and chronic elevation of glucose in this lifelong disease results in many macro- and microvascular complications. Diabetic retinopathy is the leading cause of blindness in working adults, and it affect more than 90% of patients after 25 years of diabetes.1 It is considered one of the most feared complications of diabetes, and early detection for this progressing disease is critical in providing a good quality of life for diabetic patients. Pioneering Diabetes Control and Complication Trials (DCCT) have demonstrated that the degree and duration of elevated blood glucose are the major contributing factors in the development/progression of this blinding disease; however, many patients, particularly those with type 2 diabetes, have clinically an uncertain history of hyperglycemia. The DCCT, and the follow-up Epidemiology of Diabetes Interventions and Complications (EDIC) study, have also documented that the prior hyperglycemia leaves long-lasting detrimental effects on major diabetic complications, suggesting a “metabolic memory” phenomenon,2,3 and clinical and experimental studies have shown that diabetic retinopathy continues to progress even after hyperglycemia is reversed by normal glycemia.3,4
In the pathogenesis of diabetic retinopathy, the retina undergoes many molecular, biochemical, and functional abnormalities in diabetes, but, despite great progress in the field, the exact mechanism responsible for the pathogenesis of diabetic retinopathy remains unclear.1,5 Studies have clearly demonstrated that these metabolic abnormalities are interlinked, and generation of superoxide in the mitochondria, the energy center of the cell, is considered as the unifying molecule connecting them together.6,7 Using rodent models of diabetic retinopathy, we have shown that the retinal mitochondria are dysfunctional and their DNA (mtDNA) is damaged, the transcription of mtDNA-encoded genes, essential for the functioning of the electron transport chain, is reduced, and mtDNA copy numbers are decreased. The compromised electron transport chain system continues to fuel into the vicious cycle of free radicals. Furthermore, reversal of poor glycemic control in rodents by good glycemic control does not benefit mtDNA damage, the electron transport chain remains dysfunctional and free radicals continue to self-propagate. Accumulation of matrix metalloproteinase-9 (MMP-9), an enzyme that degrades the extracellular matrix, is increased in the mitochondria, resulting in damage of mitochondrial membrane integrity, and the activity of mitochondrial superoxide scavenging enzyme, manganese superoxide dismutase (MnSOD), is compromised. The expression of Sod2, the gene encoding for MnSOD, is also attenuated in the retina. A similar phenomenon also is seen in the retina from type 2 diabetic rodents,8 and in human donors with diabetic retinopathy.9–16
Diabetic retinopathy has a fairly long asymptomatic phase, and some patients may develop early disease complications, such as background retinopathy or microalbuminuria.17 Procuring retinal tissue from patients is not feasible, nor is vitreous sampling, which could have the potential to represent the metabolic status of the retina, as it requires an invasive procedure by an ophthalmologist with potentially sight-threatening risks. Thus, identification of biomarkers in the body fluids is essential to proper manage the disease. Because diabetes is a whole-body disease with high glucose levels in the circulation, the aim of this study was to investigate if mtDNA damage in the blood could be used as surrogate biomarker of retinal mtDNA damage and the development of diabetic retinopathy. We examined mtDNA damage in the blood obtained from animal models (rats and mice) of diabetic retinopathy, and investigated the effect of pharmacologic (administration of lipoic acid in diabetic rats) or genetic (overexpression of Sod2 or MMP-9 knockdown in mice) interventions that are shown to ameliorate retinal mtDNA damage and the development of diabetic retinopathy.10,18 Peripheral blood from Zucker diabetic fatty (ZDF) rats, type 2 diabetes model, at 12 weeks of age (no signs of retinopathy) and 40 weeks of age (detectable retinal histopathology) was also analyzed. To translate the results from experimental models to the clinical settings, mtDNA damage also was quantified in the blood obtained from diabetic patients with and without retinopathy, and in age-matched nondiabetic subjects.
Methods
Animals
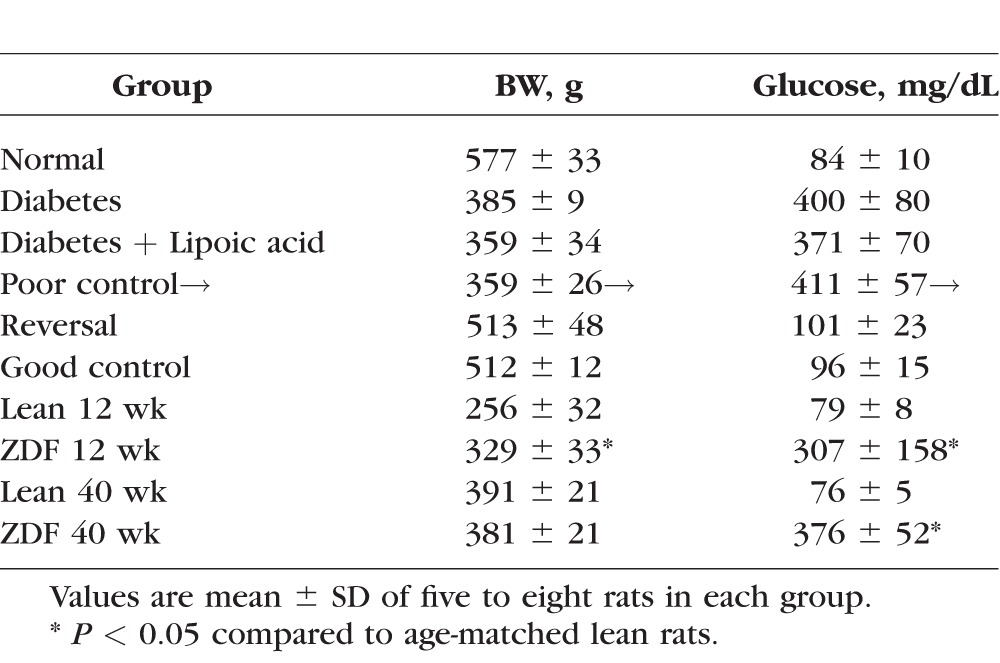
Wistar rats (male, 200 g) were made diabetic by streptozotocin (55 mg/kg body weight [BW], intraperitoneally), and soon after establishment of hyperglycemia (3–4 days after induction of diabetes), the rats were divided into four groups: rats in group 1 remained in poor glycemic control for 11 to 12 months (Diab group, glycated hemoglobin [GHb] >10%) and rats in group 2 were in poor control for 6 months followed by good control for 6 additional months (PC-Rev group). In group 3, rats were maintained in good control (GC group, GHb approximately 6.0%), and rats in group 4 received diet supplemented with lipoic acid (400 mg/kg BW) for the entire duration of the experiment (D+LA group). Normal age-matched rats served as their controls (Table 1). Rats diabetic for 11 to 12 months had slightly higher serum triglycerides compared with their age-matched normal rats (350–475 mg/dL in diabetes versus 250–325 mg/dL in normal). As a type 2 diabetes model, 12-week and 40-week-old male ZDF rats (Charles River Laboratories, Wilmington, MA, USA) were used, and age-matched lean rats served as controls. Compared with lean rats, ZDF rats (both 12 and 40 weeks old) were severely hyperglycemic (GHb values approximately 6% versus approximately 12%), and hyperlipidemic (serum triglycerides 300–400 mg/dL in lean versus 800–1600 mg/dL in ZDF rats).8 The entire animal colony was weighed two times a week, and their blood glucose was monitored once every week and GHb every 8 to 9 weeks, according to the procedures reported previously.11,16
Table 1.
Glycemic Control in Rats
Mice, hemizygous overexpressing MnSOD (MnSOD-Tg, C57BL/6 background),10 MMP-9 gene-knockout (MMP-9-KO, B6.FVB (Cg)-Mmp9tm1Tvu/J),18 and wild-type C57BL/6 (WT) were made diabetic by streptozotocin injection (55 mg/kg BW) using the methods described previously.10,18 After approximately 10 months of diabetes, blood was collected by tail puncture in tubes containing 50 μL EDTA. Treatment of animals conformed to the ARVO Resolution on the Use of Animals in Research, and was approved by the Wayne State University's Animal Care and Use Committee.
Human Samples
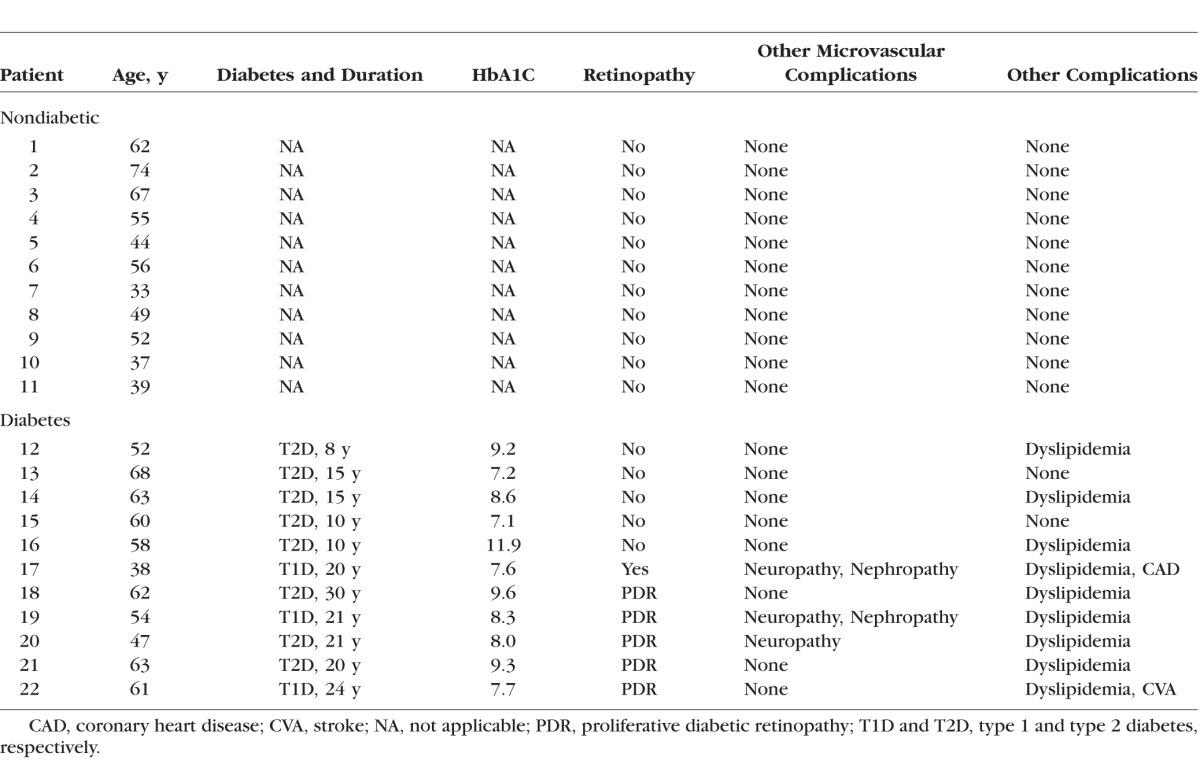
Diabetic and nondiabetic patients at the Kresge Eye Institute, or Department of Medicine, Wayne State University, Detroit, MI, USA, were recruited either before scheduled retinal surgery or during an appointment for routine eye care or their routine appointment. Diabetic patients with retinal changes were graded using the International Clinical Disease Severity Scale for Diabetic Retinopathy.19 The study was approved by the Wayne State University Institutional Review Board, and written informed consent was received from the patients before obtaining their blood sample. The protocol complied with all aspects of the Health Insurance Portability and Accountability Act, and was conducted in accordance with the tenets of the Declaration of Helsinki. Blood was collected in tubes containing EDTA as an anticoagulant, and was used for DNA and RNA isolation. Clinical characteristics of human subjects (diabetic and nondiabetic) are included in Table 2.
Table 2.
Patient Characteristics
DNA Damage
DNA from blood was isolated using the protocol specified in Qiagen kit for blood DNA isolation (Qiagen, Valencia, CA, USA), with minor modifications. Briefly, blood was digested in lysis buffer for 1 hour at 56°C, and this was followed by addition of 100% ethanol. The samples were loaded onto Qiagen columns, and the DNA was eluted in AE buffer. Mitochondrial DNA damage was performed in 10 ng DNA by quantitative extended length PCR using Elongase Enzyme Mix PCR kit (Invitrogen, Carlsbad, CA, USA) and sequence-specific primers, following the method routinely used in our laboratory11–14; Table 3 shows the primer sequences. Long region of mtDNA (9–13 kb) and also the D-loop region (800–1400 bp) were amplified in a reaction mixture containing 1× PCR buffer A and B, 200 mM dNTP, 1 unit Elongase, and 0.1 mM primers using an initial incubation cycle of 1 minute at 75°C, 1 minute at 94°C, and 24 cycles of 15 seconds at 94°C and 12 minutes at 65°C. The final extension was performed for 10 minutes at 72°C. The short region of mtDNA (115–220 bp) and that of the D-loop (90–150 bp) were amplified using specific PCR primers. The thermal cycling conditions included 94°C for 5 minutes, 28 cycles of 1 minute at 94°C, 30 seconds at 60°C, 1 minute at 72°C, and the final extension was for 10 minutes at 72°C. The PCR products were separated on 1.2% and 2.0% agarose gel for long and short amplification products, respectively. The intensity of PCR products was quantified using Carestream digital software (Rochester, NY, USA), and relative amplification was calculated by normalizing the intensity of the long PCR product to the short PCR product; decrease in the amplification ratio was indicative of an increase in DNA damage.11,20
Table 3.
Primer Sequences
Copy Numbers
Mitochondrial copy numbers were quantified in genomic DNA using a SYBR green–based quantitative PCR (qPCR) and primers for cytochrome b (cytb) as mtDNA marker and β-actin as nuclear DNA marker.20
Transcription
Mitochondrial DNA transcription was performed in RNA extracted from blood by Trizol reagent (Invitrogen). Complementary DNA (cDNA) was synthesized by High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA, USA), and transcripts of mtDNA-encoded cytb of complex III and NADPH dehydrogenase 6 (ND6) of complex I of the electron transport chain were quantified by SYBR green–based qPCR using β-ACTIN as a housekeeping gene.11,13
Retinal Histopathology
The retina from formalin-fixed eyes of ZDF and lean rats was rinsed overnight with running water, digested with 3% crude trypsin (Invitrogen-Gibco, Grand Island, NY, USA) containing 200 mM sodium fluoride. Trypsin-digested microvasculature was mounted on a glass slide, and stained with periodic acid Schiff-hematoxylin to count the degenerative capillaries.18
Statistics Analysis
The data obtained from rodents were statistically analyzed using Sigma Stat software (SPSS, Chicago, IL, USA). For multiple group comparison, 1-way ANOVA followed by Student-Newman-Keuls test was used for data with normal distribution, whereas Kruskal-Wallis 1-way analysis followed by Dunn's test was performed for data that did not present normal distribution. A 2-tailed t-test was used for comparison between the data obtained from two groups of patients. Data are expressed as mean ± SD, and P < 0.05 was considered statistically significant.
Results
Type 1 Diabetes
Damage of retinal mtDNA in diabetes is well documented11–16; to evaluate if mtDNA damage is also observed in the peripheral blood, extended-length PCR was performed. As shown in Figure 1a, blood from diabetic rats had significantly reduced ratio of long to short amplicons of mtDNA, compared with the blood from age-matched normal rats, suggesting higher mtDNA damage in diabetes. Consistent with mtDNA damage as a whole, its D-loop region in the same diabetic rats was also significantly damaged (Fig. 1b).
Figure 1.

Mitochondrial DNA damage in peripheral blood of diabetic rats. DNA damage was determined by amplifying the (a) 13.4-kb and 210-bp amplicons of the mtDNA, and (b) 819-bp and 148-bp amplicons in the D-loop region of the mtDNA. The relative amplification was quantified by normalizing the intensity of the long PCR product to the short PCR product. Decrease in the amplification ratio indicated an increase in the DNA damage. (c) Copy numbers of mtDNA were determined in the genomic DNA by qPCR using cytb as mtDNA marker and β-actin as a nuclear DNA marker. Transcripts of mtDNA-encoded (d) cytb and (e) ND6 were quantified by SYBR green–based qPCR using β-actin as a housekeeping gene. Each measurement was made in duplicate in five to seven rats in each group, and the values are represented as mean ± SD. Norm, normal; Diab, diabetes; PC-Rev, poor control for 6 months followed by good control for 6 months; GC, good glycemic control. *P versus normal and #P < 0.05 versus diabetes.
Damaged mtDNA results in decreased mtDNA copy numbers; to investigate if decrease in mtDNA copy numbers is also seen in the peripheral blood, genomic DNA from blood was analyzed. Figure 1c clearly shows a 30% to 40% decrease in the ratio of cytb to β-actin in blood genomic DNA in diabetic rats compared with age-matched normal rats. In the same animals, the transcription of mtDNA, as determined by the gene transcripts of mtDNA-encoded cytb or ND6, were also decreased by more than 50% (Figs. 1d–e), further suggesting that the blood could mimic similar to diabetes-induced mtDNA damage as seen in the retina.
Amelioration of Blood mtDNA Damage by Regulation of Oxidative Stress
Administration of lipoic acid prevents retinal oxidative stress, mtDNA damage, and the development of retinopathy in diabetic rats.21 To investigate if lipoic acid also ameliorates blood mtDNA damage, damage to mtDNA and its D-loop region was determined in the peripheral blood from the diabetic rats treated with lipoic acid. As shown in Figures 1a and 1b, administration of lipoic acid ameliorated diabetes-induced damage to the peripheral blood mtDNA and the D-loop region.
Reversal of Hyperglycemia and Blood mtDNA Damage
Clinical studies have shown that the re-institution of good glycemic control in the early stages of diabetes prevents the development of diabetic retinopathy, but if this glycemic control is instituted after a period of hyperglycemia, retinopathy continues to progress and this metabolic memory phenomenon is also duplicated in diabetic rodents.2–4 Consistent with this, peripheral blood from the rats that were maintained in good control for the entire duration (GC group) did not show significant damage to the mtDNA or D-loop region, but that from the rats maintained in poor glycemic control for 6 months followed by 6 months of good control (PC-Rev group), had significantly higher damage in both mtDNA and D-loop region compared with the rats that remained normal. Similarly, diabetes-induced decrease in mtDNA copy numbers and transcription was also not reversed by 6 months of good control that had followed 6 months of poor control. The values obtained from the rats in PC-Rev group were similar to those obtained from the rats in the diabetes group (Fig. 1).
Genetic Manipulations Inhibit the Development of Diabetic Retinopathy and Blood mtDNA Damage
To further confirm the utility of peripheral blood mtDNA damage as a possible biomarker of diabetic retinopathy, blood from mice overexpressing Sod2 or from mice with the MMP-9 gene suppressed was analyzed; these genetically modulated mice are protected from the development of diabetic retinopathy.10,18 As with the rats, blood from WT diabetic mice had significantly higher degree of mtDNA and D-loop damage than the blood from age-matched normal mice (Fig. 2); however, overexpression of Sod2, or suppression of MMP-9, also prevented diabetes-induced mtDNA and D-loop damage in the peripheral blood of these mice (Fig. 2).
Figure 2.

Peripheral blood mtDNA damage in diabetic mice, and effect of genetic manipulation of Sod2 or MMP-9. Blood from WT mice, or mice overexpressing Sod2 or knocked out for MMP-9, and diabetic for 10 months was analyzed for damage by amplifying (a) 10.0 kb and 116-bp amplicons of the mtDNA, and (b) 830-bp and 103-bp amplicons of the D-loop region. The intensity of the long PCR product was normalized to that of the short PCR product. Each measurement was made in duplicate in six to eight mice in each group. The values are presented as mean ± SD. WT-N, wild-type normal mice; WT-D, wild-type mice diabetic for approximately 10 months; Sod2-D and MMP9-D, diabetic mice overexpressing Sod2 or being knocked out for MMP-9 respectively. *P and #P < 0.05 compared with WT-N and WT-D, respectively.
Type 2 Diabetes
More than 90% of diabetic patients have type 2 diabetes, and they also have high circulating lipids. In addition to hyperglycemia, hyperlipidemia is also now considered as a major component associated with the development of diabetic retinopathy.22 Peripheral blood from 12- and 40-week-old ZDF rats was analyzed. Despite severe hyperglycemic/hyperlipidemia in both 12- and 40-week-old ZDF rats, 40-week-old ZDF rats had significant damage to mtDNA and its D-loop in the peripheral blood, but 12-week-old rats showed no change compared with their age-matched lean rats (Figs. 3a, 3b). Furthermore, in the same peripheral blood samples, mtDNA copy numbers and transcription were also significantly decreased in 40-week-old ZDF rats compared with no change in 12-week-old rats (Figs. 3c, 3d). Retinal vasculature from 12-week-old ZDF rats did not show any pathology associated with diabetic retinopathy; ZDF rats and their age-matched lean rats had three to six degenerative capillaries, but these numbers were approximately 4-fold higher in 40-week-old ZDF rats (Fig. 4a). Consistent with this, although retina from 12-week-old ZDF rats did not present any increase in mtDNA damage, 40-week-old ZDF rats showed significant increase in mtDNA damage (Fig. 4b).
Figure 3.

Damage of mtDNA and its transcription in the peripheral blood from type 2 diabetic rats. Peripheral blood from ZDF and age-matched lean rats (LN), 12 weeks and 40 weeks of age, was analyzed for (a) mtDNA and (b) D-loop damage by extended-length PCR. The relative amplification was quantified by normalizing the intensity of the long PCR product to the short PCR product. Decrease in the amplification ratio indicated an increase in the DNA damage. (c) Copy numbers of mtDNA were determined in the genomic DNA by qPCR using cytb as mtDNA marker and β-actin as a nuclear DNA marker. (d) Transcripts of mtDNA-encoded cytb and ND6 were quantified by SYBR green–based qPCR using β-actin as a housekeeping gene. Each measurement was made in duplicate in five to seven rats in each group, and the values are represented as mean ± SD. *P versus age-matched lean rat.
Figure 4.

Retinal histopathology and mtDNA damage in ZDF rats: (a) trypsin-digested retinal microvessels from 12- and 40-week-old ZDF rats were stained with periodic acid Schiff-hematoxylin for acellular capillaries and the arrow indicates an acellular capillary; the picture shows microvasculature from 12- and 40-week-old ZDF rats and 40-week-old lean rats; (b) mtDNA damage in the retina was determined by extended-length PCR. Values are represented as mean ± SD from five to seven rats per group; *P < 0.05 compared with age-matched lean rats. Measurements were performed on five to six rats in each group.
Human
Concomitant with increased peripheral blood mtDNA damage in the animal models at a stage when histopathology associated with diabetic retinopathy can be observed, mtDNA damage was significantly higher in the blood from patients with documented diabetic retinopathy compared with their age-matched nondiabetic counterparts. However, diabetic patients without retinopathy did not present mtDNA damage, and the values obtained from diabetic patients with and without documented retinopathy were significantly different from each other (P < 0.05; Fig. 5a). Similar differences among these two groups of diabetic patients were observed in the D-loop damage (Fig. 5b). Furthermore, although increased mtDNA/D-loop damage in the peripheral blood from patients with diabetic retinopathy was accompanied by decreased mtDNA copy numbers (ratio of CYTB and β-ACTIN expression in the blood genomic DNA; Fig. 6a) and subnormal mtDNA transcription (mRNA levels of mtDNA-encoded CYTB; Fig. 6b), such impairments were not detectable in the peripheral blood from diabetic patients without retinopathy; the values obtained from nondiabetic patients and diabetic patients without retinopathy were not significantly different from each other.
Figure 5.

Mitochondrial DNA damage in the blood from patients with diabetic retinopathy. Damage was determined by amplifying (a) 8.8-kb and 223-bp amplicons in the mtDNA, and (b) 1.4-kb and 90-bp amplicons in the D-loop region. Each measurement was made in duplicate in blood from five to six diabetic patients each with retinopathy (Diab-Ret) or without retinopathy (Diab-No Ret), and 7 to 10 nondiabetic subjects (Norm), and the values are represented as mean ± SD. *P < 0.05 versus Normal.
Figure 6.

Copy number of mtDNA and its transcription in the peripheral blood from diabetic subjects. (a) Copy number was quantified in the genomic DNA by qPCR using CYTB as mtDNA-encoded and β-ACTIN as a nuclear DNA-encoded gene. (b) Transcripts of mtDNA-encoded CYTB were quantified in the blood cDNA by qPCR using β-ACTIN as a housekeeping gene. The values are represented as mean ± SD obtained from five to six diabetic patients each with retinopathy (Diab-Ret) or without retinopathy (Diab-No Ret), and 7 to 10 nondiabetic subjects (Norm). *P < 0.05 versus Normal.
Discussion
Duration of diabetes is considered as one of the strongest predictors of the development and progression of diabetic retinopathy. During the initial stages of this progressing disease, patients often do not present any noticeable symptoms, but by the time some vascular lesions are detected in the retina, their vision loss could become irreversible.1,23 Our previous work has shown that mtDNA damage in the retina and its vasculature has an important role in the development of diabetic retinopathy, and this damage is observed before the pathology associated with diabetic retinopathy can be noticed in diabetic rodents.9,11,14,15,20 Here, we present novel data from animal models of type 1 diabetes showing that mtDNA is damaged in the peripheral blood of diabetic rodents, this damage is not detectable during the duration of hyperglycemia when retinal histopathology is not observed, and pharmacologic/genetic manipulations that prevent the development of diabetic retinopathy prevent blood mtDNA damage. Re-institution of good control in diabetic rats, after a period of poor glycemic control, which fails to reverse retinal mtDNA damage and prevent the progression of diabetic retinopathy, also fails to prevent blood mtDNA damage. In the type 2 diabetes model, peripheral blood mtDNA damage is not observed during the age when retinopathy is not detectable. These results clearly suggest that the damage in peripheral blood mtDNA could serve as a noninvasive biomarker of the development of diabetic retinopathy. As a proof of principle, we have provided data from diabetic patients with and without retinopathy documenting increased blood mtDNA damage and decreased copy numbers only in the patients with retinopathy compared with their age-matched nondiabetic individuals.
Mammalian mitochondria have their own double-stranded small DNA (∼16 kb), which encodes genes for proteins that are critical in normal mitochondrial homeostasis.24 Due to close proximity to the free radical producing electron transport system, and lack of protective histones, mtDNA is highly susceptible to oxidative damage.25 In the pathogenesis of diabetic retinopathy, mtDNA is damaged, and the damage is more extensive at the noncoding region, the D-loop region with essential transcription/replication elements15; here we show that the diabetic rodents present similar D-loop damage in their peripheral blood.
The number of mitochondria vary depending on the energetic requirements, the environment, and the redox balance, and brain/retina cell with high metabolic rate may have more than 2000 mitochondria, compared with less than 100 mitochondria in a white blood cell.26,27 Although initial oxidative stress could increase mitochondria numbers, chronic oxidative stress decreases their numbers and increases mtDNA damage. Our previous study showed decreased mitochondria numbers in the retina and its endothelium in diabetes,14 and here we show the similar phenomenon in the blood. Blood from diabetic rodents, at a stage of diabetes when retinopathy can be detected, have lower mtDNA copy numbers, and the therapies that prevent the development of diabetic retinopathy in these rodents also prevent increase in mtDNA damage and decrease in mtDNA copy numbers in their blood. In support, depletion in mtDNA content in lymphocytes is associated with poor glycemic control, and other chronic diseases.27,28
Diabetes also compromises the repair system and mtDNA transcription. Consistent with the decreased retinal mtDNA transcription,11,12 the levels of mtDNA-encoded genes cytb and ND6 are also decreased in the blood of diabetic rodents, further supporting the validity of blood mtDNA.
In diabetes, regulation of oxidative stress via pharmacologic or genetic manipulations protects retinal mitochondria damage and the development of diabetic retinopathy.5,7,9,10,18 Here, our results demonstrate that the same manipulations also prevent mtDNA damage in peripheral blood and ameliorate decrease in copy numbers and transcription. The two genetic manipulations associated with the prevention of the development of retinopathy in diabetic mice, the suppression of MMP-9, an enzyme that is implicated in increasing mitochondrial membrane permeability and activating the pro-apoptotic machinery, and overexpression of Sod2,10,18 also prevent increased mtDNA damage in the blood. In addition, administration of lipoic acid, an antioxidant that ameliorates retinal mtDNA damage and the development of diabetic retinopathy in rats,21 also attenuates diabetes-induced mtDNA damage in the blood.
Pioneering DCCT-EDIC studies have clearly shown that the progression of retinopathy continues to benefit from the prior intensive control, and the same metabolic memory phenomenon is also observed in experimental models of diabetic retinopathy.2,3 Rodent models have shown that the retinal mitochondrial structure, function, and its DNA remain damaged, which generates a vicious cycle of superoxide, and the retinopathy continues to progress.11,12,21 Convincing data presented here demonstrate that peripheral blood mtDNA damage also embodies the same phenomenon, further strengthening an association between the blood mtDNA damage and the progression of diabetic retinopathy.
The validity of peripheral mtDNA damage as a possible biomarker of diabetic retinopathy is further supported by our results from type 2 diabetic animal models; although mtDNA damage and its transcription are not observed in peripheral blood of ZDF rats at 12 weeks of age, an age when these animals show no signs of retinopathy and retinal mtDNA damage,8 at 40 weeks, an age when retinopathy and mtDNA damage are detectable in the retina and its vasculature, mtDNA damage is also observed in the blood. These results clearly suggest that despite severe hyperglycemia/hyperlipidemia, during the earlier stages of the disease, peripheral blood mtDNA is not altered, but with the extending duration of hyperglycemia/hyperlipidemia (40 weeks of age), blood also shows mtDNA damage. These results also document that type 1 and type 2 diabetes share similar phenomena. As with experimental models, mtDNA damage and decrease in its transcription and copy numbers have been observed in the retinal microvasculature from human donors with diabetic retinopathy.12,13 The results presented here clearly demonstrate that the peripheral blood from patients with diabetic retinopathy also has increased mtDNA damage and decreased copy numbers compared with their age-matched nondiabetic counterparts; however, such damage is not detected in the blood from diabetic patients without retinopathy. These results support a close association between peripheral blood mtDNA damage and diabetic retinopathy, and confirm the validity of peripheral blood mtDNA damage as a possible biomarker of diabetic retinopathy. Consistent with our results, others have shown a decrease in mtDNA copy numbers and increase in mtDNA damage in the peripheral blood from diabetic patients with proliferative retinopathy and neuropathy.29,30 We recognize that the number of patients used in the present study is limited, and we do not have details about the severity of hyperglycemia; additional blood samples will be required to confirm relationship between mtDNA damage in the blood and the severity of retinopathy, blood sugar control, and other systemic factors. In addition, we cannot rule out the role of other systemic conditions, including blood pressure and other diabetic complications, in mtDNA damage in the blood of these patients. However, data from diabetic patients with/without retinopathy is consistent with our results from animal models of type 1 diabetes with pharmacologic/genetic therapies to inhibit the development of diabetic retinopathy,10,18,21 and ZDF rats with/without retinopathy, and support the possibility of blood mtDNA damage as a potential biomarker of diabetic retinopathy. Furthermore, a similar metabolic phenomenon is observed in the progression of diabetic retinopathy,4 and, here our results showing the failure of mtDNA damage in blood to benefit from reversal of hyperglycemia imply a strong relationship between blood mtDNA damage and diabetic retinopathy.
In conclusion, we show a direct relation between diabetic retinopathy and mtDNA damage in the peripheral blood. This is supported by our convincing results from type 1 diabetic rodent models with and without modulating their metabolic abnormalities by either a pharmacologic or by a genetic approach, type 2 diabetic model with severe hyperlipidemia/hyperglycemia at an age when retinopathy is not detectable to the age of animals when retinal mtDNA and retinopathy are present, and peripheral blood from patients with and without diabetic retinopathy. Damaged mtDNA in peripheral blood is now being considered as one of the biomarkers of the obstructive sleep apnea syndrome31 and also for traumatic brain injury32; the results presented here clearly suggest its use as a possible biomarker of diabetic retinopathy.
Acknowledgments
The authors thank Shikha Tewari, PhD, for help with some initial measurements, and Doug Putt and Mangayarkarasi Thandampallayam Ajjeya for their help in the maintenance of diabetic animal colony. The authors alone are responsible for the text and the writing of the paper.
Supported in part by grants from the National Institutes of Health (EY014370, EY017313, EY022230), the Thomas Foundation, and an unrestricted grant to the Department of Ophthalmology from the Research to Prevent Blindness.
Disclosure: M. Mishra, None; J. Lillvis, None; B. Seyoum, None; R.A. Kowluru, None
References
- 1. Frank RN. Diabetic retinopathy. N Engl J Med. 2004; 350: 48–58. [DOI] [PubMed] [Google Scholar]
- 2. Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993; 329: 977–986. [DOI] [PubMed] [Google Scholar]
- 3. Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Group. Effect of intensive diabetes therapy on the progression of diabetic retinopathy in patients with type 1 diabetes: 18 years of follow-up in the DCCT/EDIC. Diabetes. 2015; 64: 631–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kowluru RA,, Chan PS. Metabolic memory in diabetes—from in vitro oddity to in vivo problem: role of apoptosis. Brain Res Bull. 2010; 81: 297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kowluru RA,, Kowluru A,, Mishra M,, Kumar B. Oxidative stress and epigenetic modifications in the pathogenesis of diabetic retinopathy. Prog Retin Eye Res. 2015; 48: 40–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005; 54: 1615–1625. [DOI] [PubMed] [Google Scholar]
- 7. Kowluru RA. Mitochondria damage in the pathogenesis of diabetic retinopathy and in the metabolic memory associated with its continued progression. Curr Med Chem. 2013; 20: 3226–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mishra M,, Kumar B,, Kowluru A,, Kowluru RA. Hyperlipidemia and the development of diabetic retinopathy. Lab Invest. In press. [DOI] [PMC free article] [PubMed]
- 9. Kowluru RA,, Abbas SN. Diabetes-induced mitochondrial dysfunction in the retina. Invest Ophthalmol Vis Sci. 2003; 44: 5327–5334. [DOI] [PubMed] [Google Scholar]
- 10. Kanwar M,, Chan PS,, Kern TS,, Kowluru RA. Oxidative damage in the retinal mitochondria of diabetic mice: possible protection by superoxide dismutase. Invest Ophthalmol Vis Sci. 2007; 48: 3805–3811. [DOI] [PubMed] [Google Scholar]
- 11. Madsen-Bouterse SA,, Mohammad G,, Kanwar M,, Kowluru RA. Role of mitochondrial DNA damage in the development of diabetic retinopathy, and the metabolic memory phenomenon associated with its progression. Antioxid Redox Signal. 2010; 13: 797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mishra M,, Kowluru RA. Retinal mitochondrial DNA mismatch repair in the development of diabetic retinopathy, and its continued progression after termination of hyperglycemia. Invest Ophthalmol Vis Sci. 2014; 55: 6960–6967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mishra M,, Kowluru RA. Epigenetic modification of mitochondrial DNA in the development of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2015; 56: 5133–5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Santos JM,, Tewari S,, Goldberg AFX,, Kowluru RA. Mitochondria biogenesis and the development of diabetic retinopathy. Free Radic Biol Med. 2011; 51: 1849–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tewari S,, Santos JM,, Kowluru RA. Damaged mitochondrial DNA replication system and the development of diabetic retinopathy. Antioxid Redox Signal. 2012; 17: 492–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhong Q,, Kowluru RA. Epigenetic changes in mitochondrial superoxide dismutase in the retina and the development of diabetic retinopathy. Diabetes. 2011; 60: 1304–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vijan S. In the clinic. Type 2 diabetes. Ann Intern Med. 2015; 162:ITC1–ITC16. [DOI] [PubMed]
- 18. Kowluru RA,, Mohammad G,, dos Santos JM,, Zhong Q. Abrogation of MMP-9 gene protects against the development of retinopathy in diabetic mice by preventing mitochondrial damage. Diabetes. 2011; 60: 3023–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wilkinson CP,, Ferris FL,, III,, Klein RE,, et al. Proposed international clinical diabetic retinopathy and diabetic macular edema disease severity scales. Ophthalmology. 2003; 110: 1677–1682. [DOI] [PubMed] [Google Scholar]
- 20. Santos JM,, Tewari S,, Kowluru RA. A compensatory mechanism protects retinal mitochondria from initial insult in diabetic retinopathy. Free Radic Biol Med. 2012; 53: 1729–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Santos JM,, Kowluru RA. Role of mitochondria biogenesis in the metabolic memory associated with the continued progression of diabetic retinopathy and its regulation by lipoic acid. Invest Ophthalmol Vis Sci. 2011; 52: 8791–8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chew EY,, Davis MD,, Danis RP,, et al. The effects of medical management on the progression of diabetic retinopathy in persons with type 2 diabetes: the Action to Control Cardiovascular Risk in Diabetes (ACCORD) Eye Study. Ophthalmology. 2014; 121: 2443–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Aiello LP. Diabetic retinopathy and other ocular findings in the diabetes control and complications trial/epidemiology of diabetes interventions and complications study. Diabetes Care. 2014; 37: 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Scarpulla RC. Nucleus-encoded regulators of mitochondrial function: integration of respiratory chain expression nutrient sensing and metabolic stress. Biochim Biophys Acta. 2012; 1819: 1088–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stuart JA,, Brown MF. Mitochondrial DNA maintenance and bioenergetics. Biochim Biophys Acta. 2006; 1757: 79–89. [DOI] [PubMed] [Google Scholar]
- 26. Selak MA,, Lyver E,, Micklow E,, et al. Blood cells from Friedreich ataxia patients harbor frataxin deficiency without a loss of mitochondrial function. Mitochondrion. 2011; 11: 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Malik AN,, Czajka A. Is mitochondrial DNA content a potential biomarker of mitochondrial dysfunction? Mitochondrion. 2013; 13: 481–492. [DOI] [PubMed] [Google Scholar]
- 28. Xia P,, An HX,, Dang CX,, et al. Decreased mitochondrial DNA content in blood samples of patients with stage I breast cancer. BMC Cancer. 2009; 9: 454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Malik AN,, Parsade CK,, Ajaz S,, et al. Altered circulating mitochondrial DNA and increased inflammation in patients with diabetic retinopathy. Diabetes Res Clin Pract. 2015; 110: 257–265. [DOI] [PubMed] [Google Scholar]
- 30. Czajka A,, Ajaz S,, Gnudi L,, et al. Altered mitochondrial function, mitochondrial DNA and reduced metabolic flexibility in patients with diabetic nephropathy. EBioMedicine. 2015; 2: 499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim YS,, Kwak JW,, Lee KE,, et al. Can mitochondrial dysfunction be a predictive factor for oxidative stress in patients with obstructive sleep apnea? Antioxid Redox Signal. 2014; 21: 1285–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kilbaugh TJ,, Lvova M,, Karlsson M,, et al. Peripheral blood mitochondrial DNA as a biomarker of cerebral mitochondrial dysfunction following traumatic brain Injury in a porcine model. PLoS One. 2015; 10: e0130927. [DOI] [PMC free article] [PubMed] [Google Scholar]