

Abstract
Multi-day tracking of cells in culture systems can provide valuable information in bioscience experiments. We report the development of a cell culture imaging system, named EmSight, which incorporates multiple compact Fourier ptychographic microscopes with a standard multiwell imaging plate. The system is housed in an incubator and presently incorporates six microscopes. By using the same low magnification objective lenses as the objective and the tube lens, the EmSight is configured as a 1:1 imaging system that, providing large field-of-view (FOV) imaging onto a low-cost CMOS imaging sensor. The EmSight improves the image resolution by capturing a series of images of the sample at varying illumination angles; the instrument reconstructs a higher-resolution image by using the iterative Fourier ptychographic algorithm. In addition to providing high-resolution brightfield and phase imaging, the EmSight is also capable of fluorescence imaging at the native resolution of the objectives. We characterized the system using a phase Siemens star target, and show four-fold improved coherent resolution (synthetic NA of 0.42) and a depth of field of 0.2 mm. To conduct live, long-term dopaminergic neuron imaging, we cultured ventral midbrain from mice driving eGFP from the tyrosine hydroxylase promoter. The EmSight system tracks movements of dopaminergic neurons over a 21 day period.
OCIS codes: (110.0180) Microscopy, (170.0110) Imaging systems, (170.3880) Medical and biological imaging
1. Introduction
Live cell imaging is widely used in various bioscience experiments for a better understanding of dynamic cellular behaviors such as migration [1], division [2,3], differentiation [4], interaction with the environment [5], and organelle-level events [6]. In the most common way to perform live cell imaging, one builds a specialized incubation and imaging chamber onto a conventional microscope, and one images the cultured cells directly on the microscope stage [7]. Recent advances with this method include robotic well plate transport systems [8] to increase throughput and the implementation of sufficiently compact and robust systems that can reside within an incubator [9]. Throughput of such systems is limited by the microscope itself. The number of resolvable pixels, characterized by the space-bandwidth product (SBP) [10], accessible through the microscope is limited at present (typically about 10 megapixels [11]). This constrains the rate of image acquisition or throughput that one can achieve.
Over the last few years, several new microscopy methods were developed with the purpose of overcoming the SBP limit of the conventional microscope. On-chip microscopes have demonstrated successful high resolution and large field-of-view (FOV) imaging of the cell cultures from within the incubator in the brightfield mode [12,13] and the fluorescence mode [14]. However, the on-chip microscopes have an inherent problem: cells need to be grown on top of the imaging sensor. This is a marked departure from the conventional cell culture workflow and therefore the technology has not found major use. Other lensless imaging methods, such as digital in-line holography [15–17] and fiber-optic lensless imaging [18,19], can work without this restriction, but they can provide high imaging SBP either in the brightfield mode or fluorescence mode, and they are not compatible with an in-vitro live cell imaging of well plate format.
Fourier ptychographic microscopy (FPM), another recently developed microscopy method, can overcome the SBP limit of conventional microscopes [20–27]. The sample is illuminated by a series of LED sources in an array, thus providing a series of illumination directions. The images are combined in the spatial frequency domain using the Fourier ptychographic reconstruction algorithm, and a higher resolution image is rendered without sacrificing the FOV. Several aspects suggest that a FPM based cell culture imaging method would be an appropriate technology fit for high throughput cell culture imaging. First, by using low magnification objective lenses and improving the image resolution through FPM processing, we can obtain large FOV imaging of cell cultures at improved resolution. Second, the digital refocusing [20] capability associated with FPM processing, can allow post-processing and correct defocus problems associated with cell culture imaging. This is a marked benefit as the user can retain and analyze images that might be rejected with conventional imaging techniques. In addition, this eliminates the need for mechanical auto-focusing solutions and simplifies the imaging system. Third, the imaging system would still be capable of collecting fluorescence images, albeit at the native resolution of the imaging system. Fourth, the configuration of the imaging system can be simple. A recent research study showed the possibility of building a compact Fourier ptychographic microscope using a cellphone lens, FPscope [28]. Fifth, we expect that a well-designed FPM-based system can be made cost-effectively and compactly to provide simultaneous imaging capability through multiple imaging units. This ability to perform parallel imaging can boost the effective system SBP by a factor equal to the number of parallel units.
This paper reports an incubator embedded cell culture imaging system, termed EmSight, which employs the FPM for high resolution bright field imaging along with mid-resolution fluorescence imaging functionality. By configuring a 1:1 imaging system using two identical inexpensive objective lenses, the EmSight reduced the size of one imaging system to allow spacing on 30 mm centers, less than the spacing of a 6-well ANSI-standard plate. This allowed us to assign a compact Fourier ptychographic microscope to each well of a 6-well plate, removing the necessity of any mechanical movement for the well plate imaging.
Recently, live cell imaging via FPM has been reported [29]. In that paper, the authors implemented a high speed FPM imaging system (greater than 1 frame per second) that is capable of tracking fast dynamic movement of cells during in-vitro culture. The system was built on a conventional inverted microscope platform and is designed for examining a single culture. This work is distinct in that the EmSight system is designed to be sufficiently compact to be housed in an incubator, cost-effective and is capable of imaging several wells in a culture well plate over a long timeline (a few weeks). Both the Emsight system and the system in [29] are designed for cell culture experiments. However, they are each designed to fit the needs of different cell culture experimental formats. EmSight is designed for streaming a lot of culture FPM data associated with multiple cultures from an incubator; while the system in [29] is focused on very fast imaging of a single cell culture.
In section 2, we describe the system configuration of the EmSight and discuss its imaging principle. In section 3, we characterize the system using a phase Siemens star target and fluorescent microbeads, and report our culture imaging demonstration. In section 4, we show experimental results from longitudinal live cell imaging experiments of dopaminergic and other neurons in mouse ventral midbrain cultures. Finally, we summarize the results and discuss future directions and possible applications of the system.
2. System configuration
EmSight is a compact imaging system designed for live-cell imaging within standard biological incubators, usually within a volume of 200 liters. Incubators have several horizontal shelves which can hold plates and flasks. The system utilizes two adjacent shelves, 250 mm apart. We attach the LED matrix (32 × 32 LEDs, 4 mm pitch) for FPM illumination to the bottom surface of the upper shelf, as shown in Fig. 1(A). With this configuration, each LED in the matrix can be shared for FPM illumination by several imaging units, which reduces the effective footprint of the imaging unit, making more parallel experiments possible in the limited culture volume inside the incubator. This configuration also frees up the space between the well plate and the LED matrix, which gives us enough space for easy access to the well plate on the EmSight and eliminates the possibility of unnecessary light reflections from additional surfaces. As shown in Fig. 1(B), at present the EmSight is designed for live cell imaging in the 6-well plate format. Six identical imaging units are arranged in an EmSight body in order to image all wells in a 6-well plate which is loaded on top of the EmSight body. For the fluorescence illumination, high-power LEDs and excitation filters are at the horizontal edge of the plate, illuminating the imaged region directly from the side of the wells without blocking the light path from the LED matrix. In order to eliminate the possible background noise such as light reflection from surfaces and unexpected excitation of green LEDs in the LED matrix, the high power blue LEDs have openings only to the sample positions blocking other straying light paths. The assembled EmSight prototype is shown in Fig. 1(C). The size of the EmSight is 125 mm (W) x 133 mm (L) x 170 mm (H). The casing is 3D printed using a Makerbot 3D printer.
Fig. 1.

System configuration and imaging method of the EmSight. (A) Conceptual design of the EmSight system, attached to two adjacent shelves in an incubator. An LED matrix is attached to the bottom surface of the upper shelf for the FPM illumination, and EmSight modules are attached on the next shelf for the cell culture imaging. (B) The EmSight is designed for the 6-well plate format. Six units of the same 1:1 imaging system shown in (D) are built into an EmSight and a 6-well ANSI standard plate is loaded on top of the EmSight. Six high power LEDs and excitation filters are installed into the side of the plate holders, providing fluorescence illumination. (C) The EmSight prototype. The size of the EmSight is 125 mm (W) × 133 mm (L) × 170 mm(H) including a 6-well plate and a high power LED module. (D) EmSight uses the identical objective lenses for the objective and the tube lens, configuring a 1:1 imaging system. The LED matrix turns on one by one sequentially for the FPM illumination; and high power LEDs provide the fluorescence excitation.
The imaging unit inside the EmSight consists of a pair of the same objectives which are configured to perform 1:1 imaging. The configuration is shown in Fig. 1(D). We use Olympus 4X/NA0.1 objective lenses in our prototype. The EmSight system uses 5 MP CMOS cameras (Imaging Source, DMK 23UP031, Pixel size: 2.2 µm) as detectors and the sensor size is matched to the field size of the imaging system. Furthermore, we match the raw resolution of the objective lens with the pixel size limited resolution from the camera in the 1:1 imaging system, which enables full usage of native NA of objective lens both in FPM imaging and fluorescence imaging. During operation, we capture 169 images of the sample per well, each with a different LED illumination angle, and stitch the data together in the spatial frequency domain using the Fourier ptychographic algorithm [20] to render a higher-resolution brightfield image. For each well, we first determine the LED that is centered on the well by checking the brightness of images from different LED positions without samples. Then we calibrate the positional error between the LEDs and EmSight using the vignette image from the center LED (Appendix). After calibration of the positional error, a sample well plate is loaded and 13 × 13 LEDs around each center LED are chosen for imaging each well sequentially. To perform fluorescence imaging, we insert an emission filter (540 nm/50 nm) between each objective and its tube lens and illuminate the sample using a high power LED (470 nm) and an excitation filter (475 nm/50 nm). These filter sets and high power LED can be changed accordingly for each fluorophore in use. By choosing the brightfield illumination wavelength to fall within the passband of the emission filter (> 93% transmission), we can collect both brightfield and fluorescence images without losing photons while the filters remain in place. Custom built Matlab scripts control the operation of LED matrix and high power LEDs, and synchronously capture images from the cameras. After a 6-well plate is loaded on the EmSight, the incubator is closed during the experiment and data from the camera is saved at the pre-specified time points into a desktop computer which is located outside of the incubator. Current prototype of EmSight uses 14 cables (2 for a LED matrix, 6 for high power LEDs, and 6 for cameras) for power and communication with a computer. The exposure time for bright field and fluorescence imaging depends on the sample in the experiment. In our neuron imaging experiment, we imaged each sample with three different exposure times (8 ms, 64 ms, and 500 ms) and combined them into a HDR (High Dynamic Range) raw image, and we captured fluorescence signal from the sample with 1 sec exposure time. The total imaging time for a single frame data set (3 × 169 raw FPM images + 1 fluorescence image) is 5 min, and an additional 1 min is required for transmitting and saving 700 MB of the raw data.
3. System characterization
In order to characterize the resolution of the EmSight, we used Siemens star targets and fluorescence beads as samples following a new coherent imaging resolution standard described in [30]. Figure 2(A1) shows a microscope image of a Siemens star intensity target (Edmund optics, #58833, smallest periodicity: 4.37 µm) taken with a 4X/NA 0.1 objective with planar illumination, and Fig. 2(B1) shows a raw image of the same sample from the EmSight with the center LED illumination. We define the resolution of the imaging system as the smallest periodicity of unambiguously resolved for all spokes in the Siemens star target. Figure 1(A2) and (B2) show the profile of amplitude values along a red line indicated in Fig. 1(A1) and (B1), respectively. The smallest resolvable periodicity of the objective lens is 4.57 µm and the smallest resolvable periodicity of the EmSight native imaging capability (without applying FPM processing) is 4.80 µm, which indicates that the EmSight is almost fully using the NA provided by the objective lens in the 1:1 imaging configuration. Because the main sample of the EmSight is mostly transparent live cells, we fabricated a Siemens star phase sample by etching a slide glass using FIB (Focused Ion Beam). The largest and smallest periodicity is 4 µm and 1 µm, respectively, and the depth of the etched surface is about 230 nm. The raw intensity image and the FPM reconstructed phase image of the sample are shown in Fig. 2(C1) and (C2), respectively. In Fig. 2(C3), fully resolved line profile (blue line) and the smallest resolved line profile (red line) are shown. By imaging the sample from 169 different LED directions and combining the low resolution images using Fourier ptychographic algorithm (synthetic NA of 0.42), the resolution of the system is increased by about 4 times, resolving all the spokes of periodicity of 1.26 µm.
Fig. 2.

Resolution of the EmSight. (A and B) Siemens star intensity target. (A1) Conventional microscope image using a 4X/NA 0.1 objective lens. (B1) Raw image from EmSight. (A2 and B2) Profile of the smallest resolved lines through a red line indicated in (A1) and (B1), respectively. (C) Siemens star phase target. (C1) Raw image from EmSight. (C2) FPM reconstructed phase image from EmSight. (C3) Profile of the resolved lines through red and blue lines indicated in (C2). (D) Mixture of 4.5 µm green fluorescence beads and non-fluorescent beads. (D1) Overlay image of the raw bright field image and the fluorescence image (Green color). (D2) FPM reconstructed intensity image from EmSight. (D3) FPM reconstructed phase image. (D4) Thickness profile of a bead, based on the reconstructed phase through a line indicated in (D3).
Next we used a mixture of green fluorescence beads and non-fluorescent beads (4.5 µm diameter) to check the fluorescence and FPM phase imaging performance. A raw bright field image overlaid with a fluorescence image (green color) is shown in Fig. 2(D1). We expect EmSight users to use the fluorescence images to identify targets labeled by, or expressing specific fluorophores, and FPM reconstructed higher-resolution images to compensate for the diffraction-limited fluorescence image of the targets. For simplicity, we did not apply resolution improving methods to the fluorescence imaging in the current EmSight design, however, we note that the resolution of the fluorescence images may be enhanced by adopting special illumination methods such as patterned illumination or scattering illumination [31] or aberration correcting methods [32] for those applications requiring higher-resolution fluorescence imaging. As shown in Fig. 2(D1), the fluorescence beads are clearly distinguishable from the non-fluorescent beads by the fluorescence signal. Two beads that are attached to each other (labeled with red arrow) are resolved from the FPM reconstructed intensity (Fig. 2(D2)) and phase (Fig. 2(D3)) images, and the fluorescence image (Fig. 2(D1)) gives us information on which bead is fluorescent amongst them.
A complex field that contains intensity and phase information of the sample can be rendered by the iterative Fourier ptychographic algorithm [21]. In order to verify the quantitative accuracy of the phase information, we converted the reconstructed phase information into the thickness of the bead using Eq. (1).
| (1) |
where T is the thickness of the bead, λ is the wavelength of the light, Δϕ is the phase relative to the background phase, and Δn is the refractive index difference between the sample and the background. In Fig. 2(B), the polystyrene beads (n = 1.58) are immersed in oil (n = 1.515) and green LEDs (530 nm) are used for illumination. The converted line profile indicated in Fig. 2(D3) is shown in Fig. 2(D4). The measured curve closely matched the expected profile of an ideal sphere, which indicates the FPM reconstructed phase information of the EmSight is quantitatively accurate. We note that there are small asymmetries in the phase of some beads in Fig. 2(D3). These small asymmetries can be attributed to the abrupt phase change of boundary between the bead and surrounding oil.
Digital refocusing is one of the most important features of the FPM and it is especially useful in live cell imaging, where multi-day or multi-week experiments can be vitiated by image defocus caused by system drifts and well plate misalignments. By introducing a quadratic defocus phase term into the support constraint in the spatial frequency domain, we can digitally refocus the defocused raw images during the FPM reconstruction [20]. In order to characterize the refocusing performance of the EmSight, we intentionally image a defocused Siemens star phase target and digitally refocused the image during the FPM reconstruction (see Fig. 3). For the depth-of-field (DOF) test, we built the equivalent imaging system on the optics table and used a mechanical stage for making displacement of the sample from the detection system. For the refocusing of the defocused sample, we use the angular spectrum wave propagation method [33,34]. By the discrete sampling of the camera and the Nyquist sampling theorem, there is a maximum distance that the angular spectrum method can be applied as in Eq. (2).
| (2) |
where z is the distance of wave propagation, λ is wavelength of light, u and v are spatial frequencies in x and y directions, and Δu and Δv are sampling intervals in x and y directions. In our system, we matched the NA of objective lens with camera pixel size, and we used 128 pixel x 128 pixel for each tile of image reconstruction, therefore . However, this is the theoretical maximum distance which the angular spectrum method can be applied without violating the Nyquist sampling theorem in the noise-free condition, and is not representing actual limit which resolution starts to deteriorate. In order to consider actual experimental condition, we experimentally measured DOF by finding the maximum sample displacement where the resolution of the image starts to deteriorate. As shown in Fig. 3(A) and 3(B), the EmSight refocused the sample successfully without deteriorating resolution within the defocusing range of ± 100 µm, which indicates that the DOF of the EmSight is about 200 µm. As an example of digital refocusing on a biological sample, one of the defocused mouse neuron culture images and its refocused FPM reconstruction phase image are shown in Fig. 3(C). Blurry neurites of the neurons in the defocused image are clearly refocused in the FPM reconstructed image.
Fig. 3.

Digital refocusing of the EmSight. (A) Z = + 100 µm, and (B) Z = −100 µm defocused Siemens star phase target. (A1 and B1) Raw image. (A2 and B2) Digitally refocused FPM phase image. (A3 and B3) Line profile of the smallest resolved lines along a red line indicated in (A2) and (B2), respectively. (C1) A defocused mouse midbrain culture. (C2) Digitally refocused FPM phase image.
We also used the EmSight to image a mouse ventral midbrain culture sample in order to test the FOV of the system and to assess the fluorescence imaging capability of the system. The sample was fixed with paraformaldehyde (PFA) and immersed in phosphate buffered saline (PBS) for the imaging. The full FOV of one imaging unit of the EmSight is 5.7 mm × 4.3 mm, as shown in Fig. 4(A). This sample was originally seeded with dopaminergic (DA) neurons that were engineered to express eGFP. We were able to identify 15 DA neurons through the observed eGFP signals within the FOV shown in Fig. 4(A). Images from three representative DA neurons are shown in Fig. 4(C)-4(E). The eGFP fluorescence signal intensity captured from the EmSight system is strong enough to be used to identify the target DA neurons, as shown in Fig. 4(C1)-4(E1). Figure 4(C2)-4(E2) show the FPM reconstructed phase images at distances of 1.8 mm, 2.2 mm, and 3.4 mm from the center of the FOV, respectively. Neurites of the neuron which are not distinguished in the raw images are clearly visible in the reconstructed phase images as indicated in red arrows. For comparison, fluorescence image and Zernike phase contrast image of a dopaminergic neuron (DA neuron) are shown in Fig. 4(B) using 20X/NA0.4 objective lenses. We note that the neuron sample in our experiment is a primary mixed culture and the surface of the well plate is covered with glial cells. The unevenness of the background in Fig. 4(C2)-4(E2) is the image of the glial cells. These glial cells can be seen in the Zernike Phase contrast image of Fig. 4(B2). Also dark halos surrounding some cell bodies in the phase images are attributable to the physical changes the DH cells made to its immediate environment. The same halos (white in phase contrast) are observed in the phase contrast image as well.
Fig. 4.

Large FOV imaging of the EmSight. The sample is mouse ventral midbrain cultures. (A) Full FOV FPM reconstructed phase image. The size of FOV is 5.7 mm × 4.3 mm. (B) Images of a dopaminergic neuron (DA neuron) from conventional microscope (20X/NA0.4). (B1) Fluorescence image, and (B2) phase contrast image. (C-E) Enlarged images at a distance of 1.8 mm, 2.2 mm, and 3.4 mm away from the center of the FOV, respectively. (C1-E1) Raw images overlaid with fluorescence images (Green color). In this GENSAT strain, dopaminergic neurons produce the eGFP signal. (C2-E2) The reconstructed phase images. Neurites of the neuron which are not distinguished in the raw images are clearly visible in the reconstructed phase images (red arrows).
4. Live cell culture imaging
To conduct live cell imaging using the EmSight system, we cultured ventral midbrain from the GENSAT tyrosine hydroxylase (TH)-eGFP strain [35], a BAC transgenic mice line driving eGFP from the TH promoter. The cultures were grown based on the procedure we have recently established for the long-term primary culture of mouse ventral midbrain neurons [36,37]. Each of the neuron-glia ventral midbrain cultures consisted of a glial cell monolayer, DA neurons generating TH-eGFP, and other midbrain neurons. We employed the EmSight system to image and monitor the cultures over a 3-week period.
Our ventral midbrain cultures were obtained from embryonic day 14 mouse embryos which were extracted from timed pregnant mice using a standard method [38]. The glial cells and midbrain neurons were grown in a 6-well plate and the culture medium was exchanged at three-day intervals over the 3-week imaging period. The EmSight system captured images from each well at one hr intervals for the FPM imaging. Fluorescence imaging was conducted once per day for each well. Figure 5(A) shows representative FPM phase images from the time-lapse imaging of a DA neuron and midbrain neurons. The tracked DA neuron is successfully identified using the eGFP fluorescence signal and marked with a yellow circle. By comparing each subsequent image, we choose a cell having minimum position and size change with a target cell of previous time frame as a target cell of current time frame. In our one hr period time-lapse imaging, this tracking method worked well for most of cells. For the time sequences cells were moving fast, we manually corrected the tracking error. We successfully tracked the target cell for the duration of the culture experiment (see Visualization 1 (15.3MB, AVI) ), and Fig. 5(B) shows the positional trace of the tracked target cell. Images of the entire field of view and the reconstructed FPM phase images for each of the 6 wells at culture day 7 are shown in Fig. 6.
Fig. 5.

Time-lapse imaging of the mouse ventral midbrain culture. (A) FPM phase images overlaid with fluorescence image (Green color) of a tracked dopaminergic neuron (see Visualization 1 (15.3MB, AVI) ). (B) Positional trace of the dopaminergic neuron marked in (A).
Fig. 6.
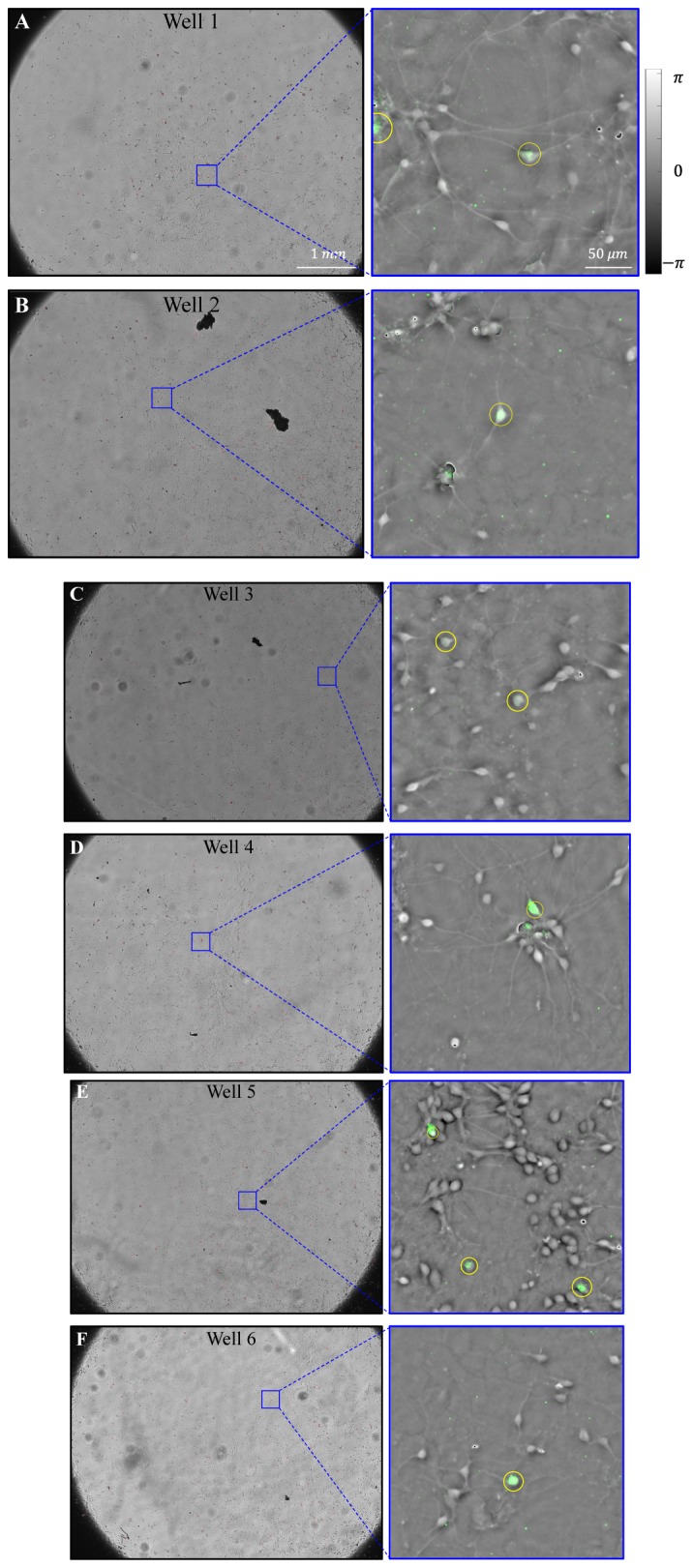
6-well plate imaging of the mouse ventral midbrain culture. (A-F) Full FOV images and FPM phase images overlaid with fluorescence image (Green color) from Well 1 to Well 6, respectively (Culture day 7).
The mouse ventral midbrain cultures used in the experiment were mixed primary cultures. During the 21-day culture experiment, actively dividing cells were found from the time-lapse images as shown in Fig. 7 (see Visualization 2 (2.4MB, AVI) ). Two putative mother cells as indicated by the red and blue arrows divided into daughter cells. These cells began dividing on approximately day 14 of culture. Such cell divisions were observed in each of the 6 wells in the plate. We think that these dividing cells may be neural stem cells or progenitor cells.
Fig. 7.

Time-lapse imaging of the dividing cells. Two mother cells marked in red and blue arrows divided into daughter cells (see Visualization 2 (2.4MB, AVI) ).
5. Discussion and conclusion
We have successfully developed an incubator embedded cell culture imaging microscope (EmSight) system based on the Fourier ptychographic microscopy (FPM) method. We used two inexpensive low magnification objectives to perform 1:1 imaging of the target culture onto the imaging sensor chip. By using the FPM method, a single imaging unit of the EmSight makes a synthetic NA of 0.42 and the FOV of 5.7 mm × 4.3 mm within the footprint of 3 cm × 3 cm. This EmSight prototype contained six sets of the imaging units for a 6-well plate cell culture imaging. We think the EmSight system can be a useful imaging solution for parallel culture experiments which have different manipulations for each well, as drug testing. As shown in Fig. 7, EmSight can also be used for tracking cell lines or stem cell differentiation.
We expect that the present configuration of the EmSight system can be further expanded to 12-well ANSI plates if the imaging sensor board is redesigned for higher density. We note that we captured wells sequentially in our neuron imaging experiment because the time between each imaging sequence (1 hr) was long enough to capture each well one at a time. For a simultaneous imaging from 6 wells, we can upgrade imaging cameras to cameras with high dynamic range and apply the multiple LED illumination method from [29,39] in order to boost capturing speed for a frame data set.
We conducted a set of 21-day mouse ventral midbrain culture experiments to demonstrate the long-term cell culture imaging capabilities of the EmSight system. With its naturally long DOF of 0.2 mm, defocused images acquired during the culture process can be digitally refocused using the iterative Fourier ptychographic reconstruction algorithm. The fluorescence imaging functionality built in the EmSight enabled the specific identification of DA (eGFP-positive) neurons from mixed primary cultures. From the time-lapse imaging, eGFP positive DA neurons were successfully tracked, i.e. the distance and the direction the cell moved was determined.
This type of analysis can be applied to the study of certain brain diseases, such as Parkinson’s disease (PD). PD involves a widespread neuronal malfunction, especially pronounced degeneration of DA neurons of the substantia nigra pars compacta (SNc) [40–43]. Tracking of DA neurons in a culture can provide useful insights into the characteristics of DA neurons during degeneration. By the wide FOV imaging functionality combined with fluorescence imaging, EmSight is well suited for identifying and tracking of such a rare event in fully populated cells over long periods of time. We plan to apply improved image processing methods to the time-lapse images in future research. Such methods will enable one to obtain data on neurite length and neurite branching. These are key aspects of neuronal development, degeneration, and repair.
Acknowledgments
We would like to thank Xiaoze Ou and Jaebum Chung for helpful discussions, and Daniel Martin for fabrication of a Siemens star phase target. Jinho Kim’s work was supported by the Kwanjeong Educational Foundation.
Appendix. LED position calibration
Fourier ptychographic algorithm requires accurate illumination direction information from different LEDs in order to stitch the raw images into correct locations in the spatial frequency domain. Because the main body of the EmSight is separated from the LED matrix, we need to calibrate the LED position relative to the sensor camera for the accurate reconstruction. We use a vignette image from the center LED illumination as shown in Fig. 8(A)
Fig. 8.

LED position calibration. (A) Vignette image from the center LED. (B) BW image of well aligned vignette image. Center of BW image (blue dot) is located at the center of imaging sensor (red dot) (C) Black and white image of misaligned vignette image. Center of vignette image (blue dot) is shifted from the center of imaging sensor (red dot) (D) Lookup table for the LED displacement to the camera.
for this purpose. In order to find the center of the vignette image, we convert the vignette image into a black and white (BW) image. When the LED position is well aligned with the camera, the center of BW image (blue dot) is located at the center of imaging sensor (red dot) as shown in Fig. 8(B). When there is a misalignment between the LED and the camera, the center of BW image (blue dot) is shifted from the center of the imaging sensor (red dot) as shown in Fig. 8(C). We use the shift of the center of vignette image as an indicator of LED displacement and make a lookup table by moving LED matrix relative to the camera as shown in Fig. 8(D). Blue closed circle is the number of pixel shift when LED moved x direction, and red closed circle is the number of pixel shift when LED moved y direction. In order to consider the shift in both directions, the number of pixel shifts in x and y are measured as the LED is translated in diagonal direction (open circles). After loading an EmSight into the incubator, we measure the number of pixel shifts of the vignette in x and y directions and find the LED displacement using the lookup table. The curve of open circles in lookup table is used only when the both of the resultant displacement in x and y directions are more than 1 mm.
Funding
National Institute of Health (NIH) (R01AI6226-01); Caltech Agency Award (AMGEN.96EYES)
References and links
- 1.Angelini T. E., Hannezo E., Trepat X., Marquez M., Fredberg J. J., Weitz D. A., “Glass-like dynamics of collective cell migration,” Proc. Natl. Acad. Sci. U.S.A. 108(12), 4714–4719 (2011). 10.1073/pnas.1010059108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dey-Guha I., Wolfer A., Yeh A. C., G Albeck J., Darp R., Leon E., Wulfkuhle J., Petricoin E. F., 3rd, Wittner B. S., Ramaswamy S., “Asymmetric cancer cell division regulated by AKT,” Proc. Natl. Acad. Sci. U.S.A. 108(31), 12845–12850 (2011). 10.1073/pnas.1109632108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Costa M. R., Ortega F., Brill M. S., Beckervordersandforth R., Petrone C., Schroeder T., Götz M., Berninger B., “Continuous live imaging of adult neural stem cell division and lineage progression in vitro,” Development 138(6), 1057–1068 (2011). 10.1242/dev.061663 [DOI] [PubMed] [Google Scholar]
- 4.Matsui T., Takano M., Yoshida K., Ono S., Fujisaki C., Matsuzaki Y., Toyama Y., Nakamura M., Okano H., Akamatsu W., “Neural stem cells directly differentiated from partially reprogrammed fibroblasts rapidly acquire gliogenic competency,” Stem Cells 30(6), 1109–1119 (2012). 10.1002/stem.1091 [DOI] [PubMed] [Google Scholar]
- 5.Lin S., Fonteno S., Weng J. H., Talbot P., “Comparison of the toxicity of smoke from conventional and harm reduction cigarettes using human embryonic stem cells,” Toxicol. Sci. 118(1), 202–212 (2010). 10.1093/toxsci/kfq241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Srinivasan R., Richards C. I., Xiao C., Rhee D., Pantoja R., Dougherty D. A., Miwa J. M., Lester H. A., “Pharmacological chaperoning of nicotinic acetylcholine receptors reduces the endoplasmic reticulum stress response,” Mol. Pharmacol. 81(6), 759–769 (2012). 10.1124/mol.112.077792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.http://www.zeiss.com/microscopy/en_us/products/microscope-components/incubation.html.
- 8.http://www.nikoninstruments.com/Products/Live-Cell-Screening-Systems/BioStation-CT.
- 9.http://www.essenbioscience.com/essen-products/incucyte/.
- 10.Lohmann A. W., Dorsch R. G., Mendlovic D., Zalevsky Z., Ferreira C., “Space-bandwidth product of optical signals and systems,” J. Opt. Soc. Am. A 13(3), 470–473 (1996). 10.1364/JOSAA.13.000470 [DOI] [Google Scholar]
- 11.Zheng G., Ou X., Yang C., “0.5 gigapixel microscopy using a flatbed scanner,” Biomed. Opt. Express 5(1), 1–8 (2014). 10.1364/BOE.5.000001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng G., Lee S. A., Antebi Y., Elowitz M. B., Yang C., “The ePetri dish, an on-chip cell imaging platform based on subpixel perspective sweeping microscopy (SPSM),” Proc. Natl. Acad. Sci. U.S.A. 108(41), 16889–16894 (2011). 10.1073/pnas.1110681108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jung J. H., Han C., Lee S. A., Kim J., Yang C., “Microfluidic-integrated laser-controlled microactuators with on-chip microscopy imaging functionality,” Lab Chip 14(19), 3781–3789 (2014). 10.1039/C4LC00790E [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han C., Pang S., Bower D. V., Yiu P., Yang C., “Wide field-of-view on-chip Talbot fluorescence microscopy for longitudinal cell culture monitoring from within the incubator,” Anal. Chem. 85(4), 2356–2360 (2013). 10.1021/ac303356v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bishara W., Su T. W., Coskun A. F., Ozcan A., “Lensfree on-chip microscopy over a wide field-of-view using pixel super-resolution,” Opt. Express 18(11), 11181–11191 (2010). 10.1364/OE.18.011181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bishara W., Sikora U., Mudanyali O., Su T. W., Yaglidere O., Luckhart S., Ozcan A., “Holographic pixel super-resolution in portable lensless on-chip microscopy using a fiber-optic array,” Lab Chip 11(7), 1276–1279 (2011). 10.1039/c0lc00684j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greenbaum A., Sikora U., Ozcan A., “Field-portable wide-field microscopy of dense samples using multi-height pixel super-resolution based lensfree imaging,” Lab Chip 12(7), 1242–1245 (2012). 10.1039/c2lc21072j [DOI] [PubMed] [Google Scholar]
- 18.Coskun A. F., Sencan I., Su T. W., Ozcan A., “Wide-field lensless fluorescent microscopy using a tapered fiber-optic faceplate on a chip,” Analyst (Lond.) 136(17), 3512–3518 (2011). 10.1039/c0an00926a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coskun A. F., Sencan I., Su T. W., Ozcan A., “Lensless wide-field fluorescent imaging on a chip using compressive decoding of sparse objects,” Opt. Express 18(10), 10510–10523 (2010). 10.1364/OE.18.010510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng G., Horstmeyer R., Yang C., “Wide-field, high-resolution Fourier ptychographic microscopy,” Nat. Photonics 7(9), 739–745 (2013). 10.1038/nphoton.2013.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ou X., Horstmeyer R., Yang C., Zheng G., “Quantitative phase imaging via Fourier ptychographic microscopy,” Opt. Lett. 38(22), 4845–4848 (2013). 10.1364/OL.38.004845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horstmeyer R., Yang C., “A phase space model of Fourier ptychographic microscopy,” Opt. Express 22(1), 338–358 (2014). 10.1364/OE.22.000338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ou X., Zheng G., Yang C., “Embedded pupil function recovery for Fourier ptychographic microscopy,” Opt. Express 22(5), 4960–4972 (2014). 10.1364/OE.22.004960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ou X., Horstmeyer R., Zheng G., Yang C., “High numerical aperture Fourier ptychography: principle, implementation and characterization,” Opt. Express 23(3), 3472–3491 (2015). 10.1364/OE.23.003472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chung J., Ou X., Kulkarni R. P., Yang C., “Counting White Blood Cells from a Blood Smear Using Fourier Ptychographic Microscopy,” PLoS One 10(7), e0133489 (2015). 10.1371/journal.pone.0133489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams A., Chung J., Ou X., Zheng G., Rawal S., Ao Z., Datar R., Yang C., Cote R., “Fourier ptychographic microscopy for filtration-based circulating tumor cell enumeration and analysis,” J. Biomed. Opt. 19(6), 066007 (2014). 10.1117/1.JBO.19.6.066007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horstmeyer R., Ou X., Zheng G., Willems P., Yang C., “Digital pathology with Fourier ptychography,” Comput. Med. Imaging Graph. 42, 38–43 (2015). 10.1016/j.compmedimag.2014.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dong S., Guo K., Nanda P., Shiradkar R., Zheng G., “FPscope: a field-portable high-resolution microscope using a cellphone lens,” Biomed. Opt. Express 5(10), 3305–3310 (2014). 10.1364/BOE.5.003305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tian L., Liu Z., Yeh L.-H., Chen M., Zhong J., Waller L., “Computational illumination for high-speed in vitro Fourier ptychographic microscopy,” Optica 2(10), 904–911 (2015). 10.1364/OPTICA.2.000904 [DOI] [Google Scholar]
- 30.Horstmeyer R., Heintzmann R., Popescu G., Waller L., Yang C., “Standardizing the resolution claims for coherent microscopy,” Nat. Photonics 10(2), 68–71 (2016). 10.1038/nphoton.2015.279 [DOI] [Google Scholar]
- 31.Dong S., Nanda P., Shiradkar R., Guo K., Zheng G., “High-resolution fluorescence imaging via pattern-illuminated Fourier ptychography,” Opt. Express 22(17), 20856–20870 (2014). 10.1364/OE.22.020856 [DOI] [PubMed] [Google Scholar]
- 32.Chung J., Kim J., Ou X., Horstmeyer R., Yang C., “Wide field-of-view fluorescence image deconvolution with aberration-estimation from Fourier ptychography,” Biomed. Opt. Express 7(2), 352–368 (2016). 10.1364/BOE.7.000352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsushima K., Shimobaba T., “Band-Limited Angular Spectrum Method for Numerical Simulation of Free-Space Propagation in Far and Near Fields,” Opt. Express 17(22), 19662–19673 (2009). 10.1364/OE.17.019662 [DOI] [PubMed] [Google Scholar]
- 34.T. M. Kreis, M. Adms, and W. P. O. Jüptner, “Methods of Digital Holography: A comparison,” SPIE Proc. 3098, 224–233 (1997). [Google Scholar]
- 35.Gong S., Zheng C., Doughty M. L., Losos K., Didkovsky N., Schambra U. B., Nowak N. J., Joyner A., Leblanc G., Hatten M. E., Heintz N., “A gene expression atlas of the central nervous system based on bacterial artificial chromosomes,” Nature 425(6961), 917–925 (2003). 10.1038/nature02033 [DOI] [PubMed] [Google Scholar]
- 36.Henley B. M., Williams B. A., Srinivasan R., Cohen B. N., Xiao C., Mackey E. D., Wold B. J., Lester H. A., “Transcriptional regulation by nicotine in dopaminergic neurons,” Biochem. Pharmacol. 86(8), 1074–1083 (2013). 10.1016/j.bcp.2013.07.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Srinivasan R., Henley B. M., Henderson B. J., Indersmitten T., Cohen B. N., Kim C. H., McKinney S., Deshpande P., Xiao C., Lester H. A., “Smoking-relevant nicotine concentration attenuates the unfolded protein response in dopaminergic neurons,” J. Neurosci. 36(1), 65–79 (2016). 10.1523/JNEUROSCI.2126-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pruszak J., Just L., Isacson O., Nikkhah G., “Isolation and Culture of Ventral Mesencephalic Precursor Cells and Dopaminergic Neurons from Rodent Brains,” in Current Protocols in Stem Cell Biology. (Unit 2D.5)(2009) Chap. 2. [DOI] [PubMed] [Google Scholar]
- 39.Tian L., Li X., Ramchandran K., Waller L., “Multiplexed coded illumination for Fourier Ptychography with an LED array microscope,” Biomed. Opt. Express 5(7), 2376–2389 (2014). 10.1364/BOE.5.002376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hirsch E., Graybiel A. M., Agid Y. A., “Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson’s disease,” Nature 334(6180), 345–348 (1988). 10.1038/334345a0 [DOI] [PubMed] [Google Scholar]
- 41.German D. C., Manaye K., Smith W. K., Woodward D. J., Saper C. B., “Midbrain dopaminergic cell loss in Parkinson’s disease: Computer visualization,” Ann. Neurol. 26(4), 507–514 (1989). 10.1002/ana.410260403 [DOI] [PubMed] [Google Scholar]
- 42.Goto S., Hirano A., Matsumoto S., “Subdivisional involvement of nigrostriatal loop in idiopathic Parkinson’s disease and striatonigral degeneration,” Ann. Neurol. 26(6), 766–770 (1989). 10.1002/ana.410260613 [DOI] [PubMed] [Google Scholar]
- 43.Rinne J. O., Rummukainen J., Paljärvi L., Rinne U. K., “Dementia in Parkinson’s disease is related to neuronal loss in the medial substantia nigra,” Ann. Neurol. 26(1), 47–50 (1989). 10.1002/ana.410260107 [DOI] [PubMed] [Google Scholar]