

Abstract
The ability of the ventral hippocampus (VH) for long-lasting long-term potentiation (LTP) and the mechanisms underlying its lower ability for short-lasting LTP compared with the dorsal hippocampus (DH) are unknown. Using recordings of field excitatory postsynaptic potentials (EPSPs) from the CA1 field of adult rat hippocampal slices, we found that 200-Hz stimulation induced nondecremental LTP that was maintained for at least 7 h and was greater in the DH than in the VH. The interaction of NMDA receptors with L-type voltage-dependent calcium channels appeared to be more effective in the DH than in the VH. Furthermore, the LTP was significantly enhanced in the DH only, between 2 and 5 h post-tetanus. Furthermore, the mGluR5 contributed to the post-tetanic potentiation more in the VH than in the DH.
The hippocampus is a brain structure with paramount capacity for phenomena of short-term and long-term synaptic plasticity (Bliss et al. 2007). Remarkably, this ability is not uniformly distributed along the long axis of the structure. Namely, the ability for long-term potentiation (LTP) induced by high-frequency stimulation (HFS) is strikingly lower in the ventral hippocampus (VH) compared with the dorsal hippocampus (DH) (Papatheodoropoulos and Kostopoulos 2000; Maruki et al. 2001; Colgin et al. 2004; Maggio and Segal 2009; Kenney and Manahan-Vaughan 2013; Keralapurath et al. 2014). This is one of the most notable differences recently found in the intrinsic circuitry between the DH and the VH. Long-term synaptic plasticity is thought to be a fundamental mechanism that supports learning and memory (Matynia et al. 2002; Takeuchi et al. 2014). Thus, differences in the ability for LTP induction might contribute to the well-known functional segregation along the long (dorsoventral or septotemporal) axis of the hippocampus (Moser and Moser 1998; Fanselow and Dong 2010; Bast 2011; Goosens 2011; Small et al. 2011; Bannerman et al. 2014; Strange et al. 2014). Despite the established difference in the magnitude of HFS-induced LTP between the two hippocampus segments, the underlying mechanisms remain unknown. This is because, although HFS-induced LTP can be blocked by antagonists of NMDARs (Park et al. 2014), HFS might induce compound LTP in the CA1 field with the participation of L-type voltage-dependent calcium channels (L-VDCCs), (Grover and Teyler 1990; Cavus and Teyler 1996; Morgan and Teyler 2001; Bayazitov et al. 2007) and of metabotropic glutamate receptor-5 (mGluR5) (Jia et al. 1998). NMDAR-dependent and L-VDCC-dependent LTP might have distinct implication in certain types of learning and memory (Morris et al. 1986; Tsien et al. 1996; Borroni et al. 2000; Moosmang et al. 2005). In addition, NMDAR-LTP and L-VDCC-LTP might preferentially be involved in retention of information over short and long periods of time, respectively (Borroni et al. 2000).
A parameter of LTP particularly relevant for the implications of LTP in the memory function is the persistence of potentiation (Martin et al. 2000; Lynch 2004). Nondecremental LTP lasting for several weeks has been recorded in the DH of intact animal (Staubli and Lynch 1987) and for several hours in slices prepared from the DH (Alger and Teyler 1976; Reymann et al. 1985). However, it is completely unknown whether VH, with its relatively lower ability for LTP induction, can sustain long-lasting LTP. In the present study, we examined the ability of the DH and the VH to show persistent changes in synaptic transmission following HFS and the contribution of NMDARs, L-VDCCs, and mGluR5 to LTP.
Hippocampal slices from 41 Wistar male rats, 2–4 months old, were used. All experimental treatment and procedures were conducted in accordance with the European Communities Council Directive Guidelines (86/609/EEC, JL 358, 1, December 12, 1987) for the care and use of laboratory animals, and they have been approved by the Prefectural Animal Care and Use Committee (No: EL 13BIO04). In addition, all efforts have been made to minimize the number and the suffering of animals used. Animals were decapitated after deep anesthesia with diethyl ether. The brain was removed and placed in chilled (2°C–4°C) standard artificial cerebrospinal fluid (ACSF) containing 124 mM NaCl, 4 mM KCl, 2 mM MgSO4, 2 mM CaCl2, 1.25 mM NaH2PO4, 26 mM NaHCO3 and 10 mM glucose, equilibrated with 95% O2 and 5% CO2 gas mixture at pH = 7.4. The hippocampi were excised free and 500 µm-thick transverse slices were prepared from the regions extending >1 and <4 mm from the DH and the VH using a McIlwain tissue chopper. Slices were immediately transferred and maintained to an interface type recording chamber continuously perfused with ACSF at a rate of 0.8–1.0 mL/min and humidified with a mixed gas containing 95% O2 and 5% CO2 at a constant temperature of 31 ± 0.5°C. Slices were left to equilibrate for at least 2 h before recordings were started. It should be noted that a relatively long period of preincubation is thought as essential for a stable long-term recording and the study of late-phase plasticity (Sajikumar et al. 2005). Field potentials consisting of the presynaptic fiber volley (Fv) and the excitatory postsynaptic potential (fEPSP) were recorded from the stratum radiatum of the CA1 field using carbon fibers (7 µm diameter). Test electrical pulses at the Schaffer collaterals were delivered every 30 sec using a bipolar platinum–iridium electrode (25 µm wire diameter, with an interwire distance of 100 µm) placed in stratum radiatum at a distance of ∼350 µm from the recording electrode toward the CA3. Signals were acquired, band-pass filtered at 0.5 Hz–2 kHz, digitized at 10 kHz and off-line analyzed. The maximum slope of fEPSP and the amplitude of Fv were measured. LTP was induced by HFS consisting of 10 trains of stimuli delivered at 5-sec intervals with each train composed of 40 pulses at 200 Hz (Grover and Teyler 1990; Cavus and Teyler 1996). The fEPSP/Fv ratio was used to quantify synaptic effectiveness. The following drugs were used (purchased from Tocris Cookson Ltd, UK): the antagonist of NMDAR 3-((R)-2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (CPP, 10 µM); the selective antagonist of mGluR5 3-((2-methyl-1,3-thiazol-4-yl)ethynyl)pyridine hydrochloride (MTEP, 200 µM); and the blocker of L-type voltage-dependent calcium channels nimodipine (20 µM). Stock solutions of CPP and MTEP were prepared in distilled water, whereas stock solution of nimodipine was prepared in dimethylsulfoxide (DMSO) at a concentration that when diluted for bath application the final volume of DMSO was lower than 0.05%. Nimodipine was applied for at least 45 min before the delivery of HFS and for 5 min after HFS, while CPP and MTEP were applied for 20 min before and 5 min after the delivery of HFS. The nonparametric Wilcoxon signed-rank test or the Friedman two-way ANOVA and the Mann–Whitney U test were used for comparisons between related and independent groups of values, respectively. The values of LTP throughout the text represent the means ± S.E.M and “n” indicates the number of slices and animals (slices/animals) used in the analysis. The detection of statistically significant differences was made using the number of slices.
On the base of input–output curves, the baseline stimulation that was comparable between the DH and the VH (101 ± 7 µΑ vs 89 ± 5 µA, Mann–Whitney U test, P > 0.05) was set at the strength that produced an fEPSP equal to 20% of the maximum response and was similar between the DH (1.63 ± 0.84 mV/msec, n = 51/41) and VH (1.83 ± 0.1 mV/msec, n=52/41). The baseline fEPSP used to study the effects of HFS corresponded to half-maximum fEPSP/Fv (2.04 ± 0.16 and 2.57 ± 0.4 in DH and VH, respectively, Mann–Whitney U test, P > 0.05). During HFS the current intensity (176 ± 10 µA and 158 ± 9 µA in DH and VH, respectively, Mann–Whitney U test, P > 0.05) was increased to evoke an fEPSP equal to 85.2 ± 2.5% of the maximum values and corresponded to 3.0 ± 0.13 mV/msec in the DH and 3.2 ± 0.17 mV/msec in the VH. We first examined the ability of the two hippocampal segments to sustain long-lasting LTP by monitoring the synaptic effectiveness for 7 h post-HFS. HFS was applied to 12 DH slices obtained from 12 rats and from nine VH slices obtained from nine rats. The immediate effects of HFS were different between the DH and the VH. In particular, the DH showed a gradual increase in fEPSP/Fv that culminated during the first 8–10 min after the delivery of HFS, while the VH displayed an immediate onset post-tetanic potentiation (PTP), which decayed gradually over time to reach a steady-state potentiation level at 40–50 min after HFS (Fig. 1). Accordingly, the potentiation during the first 3 min post-tetanus was significantly higher in the VH than in the DH (comparison at each individual time point, Mann–Whitney U test, P < 0.05). HFS produced a significant short-term depression in Fv that lasted for 35 min in both hippocampal poles (Wilcoxon test at each time point, P < 0.05, inset in Fig. 1), suggesting that HFS produced a transient depression in the presynaptic excitability. As shown in the main graph of Figure 1, HFS induced robust nondecremental LTP in the synaptic effectiveness in both the DH and the VH that was evident at 60 min and persisted for at least 7 h post-tetanus. In particular, the increase of fEPSP/Fv measured at each of the seven consecutive hours after HFS was 85.2 ± 10.4%, 93.4 ± 9.9%, 107.6 ± 13.8%, 120.3 ± 17.2%, 127.5 ± 24.7%, 126.4 ± 27.9% and 123.8 ± 27.2% in DH (n = 12/12) and 55.6 ± 2.4%, 53.2 ± 2.7%, 52.3 ± 3.8%, 51.9 ± 4.0%, 56.3 ± 2.7%, 52.5 ± 2.3%, and 61.7 ± 4.8% in VH (n = 9/9), respectively (Wilcoxon test for each hippocampal pole and at each hour, P < 0.05). Furthermore, the amplitude of LTP was significantly higher in the DH than in the VH at all times from the 40th minute to the end of monitoring (Mann–Whitney U test performed at each of the 7 h after HFS, P < 0.05). Interestingly, in the majority of DH slices (10 of 12 slices) but in no VH slices we observed that the potentiation was gradually and significantly increased during the post-induction period of recording (Friedman two-way ANOVA, P < 0.05). Specifically, the potentiation in DH significantly increased from the 2 h post-HFS and reached a plateau at the 5 h (Wilcoxon signed-rank test between the time points of 2 or 3 h and the consequent time points, P < 0.05). These results indicate that the mechanisms underlying the persistence of LTP for several hours might differ between the DH and the VH.
Figure 1.
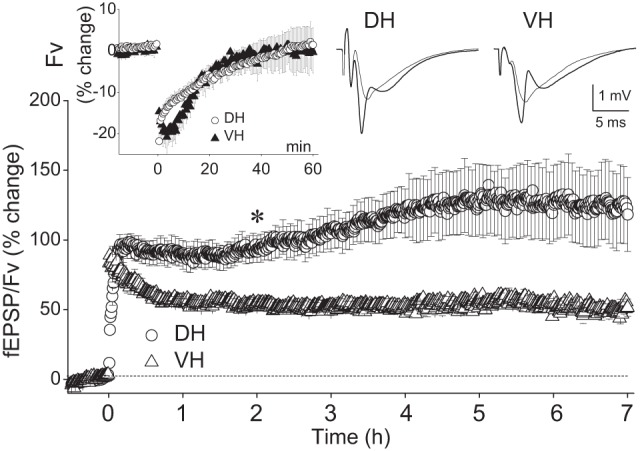
VH compared with DH displays lower LTP amplitude but similar ability to sustain nondecremental LTP. In the main diagram the fEPSP/Fv ratio is plotted as a function of time, one point per 30 sec (S.E.M. are shown in one every five points, for clarity). DH and VH data were collected from 12 and nine animals, respectively. Each animal contributed with a single slice. The magnitude of LTP of fEPSP/Fv was significantly higher in the DH than that in the VH at every hour post-tetanus (Mann–Whitney U test at each hour, P < 0.05). In addition, the LTP was enhanced in the DH but not in the VH during the recording period. The asterisk indicates the beginning of significant LTP enhancement in the DH; the potentiation reached a plateau at the 5 h (for statistics, see main text). Representative recordings before (thin line traces) and 7 h (thick line traces) after the delivery of HFS are shown at the top right of the plot. Note that the immediate post-tetanic potentiation (i.e., during the first minutes after the delivery of HFS) was higher in the VH than that in the DH. The time courses of the HFS-induced changes in the fiber volley (Fv) during the 60 min post-tetanus are shown in the inset. Fv was transiently depressed in both hippocampal poles similarly.
Then, we examined the contribution of NMDARs, L-VDCCs, and mGluR5 in the induction of LTP in DH and VH. The effects of HFS delivered under blockade of NMDARs, L-VDCCs, or mGluR5 are shown in Figure 2. In the DH, immediately after the application of HFS in the presence of 10 µM CPP, we observed a transient depression in the synaptic effectiveness that lasted for 2–3 min. This post-tetanic depression was followed by a gradual recovery of the response over the next 2–3 min that was replaced by potentiation at 5 min post-tetanus. A steady-state level of potentiation was reached at ∼15 min post-tetanus. On the contrary, the VH displayed an immediate PTP that lasted only 1–2 min, while afterward the response reached a stable potentiation level. Thus, under blockade of NMDARs, the ventral synapses displayed short but evident PTP while the dorsal ones showed post-tetanic depression instead of potentiation. Interestingly, the Fv was transiently depressed after HFS in both hippocampal poles, and the time course of this depression was similar to that observed in ACSF. Sixty minutes after HFS the fEPSP/Fv showed significant potentiation of similar amplitude in DH (22.1 ± 3.9%, n = 20/10, Wilcoxon test, P < 0.001) and VH (15.2 ± 2.3%, n = 19/10, Wilcoxon test, P < 0.001), (Mann–Whitney U test between DH and VH, P > 0.05), (Fig. 2A,B). However, the magnitude of this NMDAR-independent LTP was strongly reduced compared with that observed in ACSF (72.7 ± 6.7% in DH, n = 16/12 and 42.7 ± 4.1% in VH, n = 16/11; Mann–Whitney U test between standard ACSF and CPP in either pole, P < 0.001), demonstrating that NMDARs significantly contributed to induction of LTP in both hippocampal poles. We observed similar results also under blockade of L-VDCCs. Specifically, HFS applied to slices bathed with 20 µM nimodipine effectively induced LTP in both DH (28.0 ± 7.4%, n = 6/3, Wilcoxon test, P < 0.005), and VH (14.2 ± 5.5%, n = 6/3, Wilcoxon test, P < 0.005) (Fig. 2A,B). Furthermore, the magnitude of LTP was similar between the DH and the VH but significantly smaller compared with that of LTP induced in ACSF (Mann–Whitney U test between standard ACSF and nimodipine, P < 0.05 in DH and P < 0.05 in VH), demonstrating that L-VDCCs significantly contributed to LTP in both poles. Interestingly, the magnitude of LTP under blockade of NMDARs was similar to that observed under blockade of L-VDCCs, in both the DH and VH. Then, we examined the possible involvement of mGluR5, by applying HFS in the presence of 200 µM MTEP. As shown in Figure 2C,D, the potentiation induced under MTEP and measured at 60 min post-induction in DH (59.5 ± 6.7%, n = 5/3) and VH (33.7 ± 7.0%, n = 5/3) was similar to that observed under standard conditions. Therefore, the difference in the magnitude of LTP remained significantly different between the DH and the VH (Mann–Whitney U test, P < 0.05). However, the PTP induced under MTEP and measured at 10, 20, and 30 min was significantly smaller compared with ACSF in both DH and VH (Mann–Whitney U test for each group and time point, P < 0.05). These experiments demonstrated that 200 Hz HFS, in either hippocampal pole, induced a compound LTP with robust NMDAR- and L-VDCC-dependent components. Hence, the LTP observed under CPP and nimodipine can be attributed to L-VDCCs and NMDARs, respectively. Then, the similar magnitudes of L-VDCC- and NMDAR-dependent LTP implied that both mechanisms, when activated independently of each other, have similar contribution to LTP between the DH and the VH. Notably, however, the magnitude of compound LTP was larger than the algebraic sum of NMDAR- and L-VDCC-dependent LTP (∼50% and ∼30% in DH and VH, respectively), suggesting that NMDARs and L-VDCCs interact between each other to facilitate LTP in both hippocampal poles. Importantly, this facilitation was considerably more effective in the DH than in the VH, although the LTP that was induced by each of the two mechanisms independently was similar between the DH and the VH. To illustrate the contribution of each of the two mechanisms to the compound LTP induced under ACSF, we subtracted the magnitude of potentiation induced under blockade of NMDARs or L-VDCCs from the magnitude of compound LTP. As shown in Figure 3, NMDARs and L-VDCCs similarly contributed to LTP in DH and VH, yet the contribution of NMDARs to the short-term potentiation was higher in the DH than that in the VH. In addition, the contribution of mGluR5 to short-term potentiation was larger in the VH than that in the DH (Mann–Whitney U test, P < 0.05) (Fig. 3). It is known that interaction between NMDARs and L-VDCCs during HFS can produce compound LTP which has higher magnitude compared with LTP induced by activation of only NMDARs or L-VDCCs (Cavus and Teyler 1996; Bayazitov et al. 2007). Hence, the higher magnitude of HFS-induced compound LTP in the DH compared with the VH suggests that the interaction between NMDARs and L-VDCCs is more effective in the dorsal than in the ventral segment of the hippocampus. Results from previous studies have shown that NMDARs differ between the two hippocampal poles regarding their subunit composition and the functionality under conditions of endogenous or exogenous activation. Specifically, the different subunit composition of NMDARs between the DH and the VH (Pandis et al. 2006) suggested that NMDARs permit larger currents in VH than that in DH. Furthermore, strong presynaptic activity (Papatheodoropoulos 2015b) or exogenous activation of NMDARs (Kouvaros and Papatheodoropoulos 2016) produces greater NMDAR-dependent responses in the VH than that in the DH. In addition, under conditions of induced population discharges, the synaptic contribution of NMDARs is higher in the VH than that in the DH (Papatheodoropoulos et al. 2005; Papatheodoropoulos 2015a). These observations indicate that a higher activation readiness of NMDARs could permit a higher entry of calcium ions and greater magnitude of LTP in the VH compared with that in the DH. However, we observed that the compound LTP was larger in the DH pointing to the likely case of more effective recruitment of L-VDCCs following NMDAR-mediated depolarization in the DH than that in the VH. However, there is no comparative data concerning the status of L-VDCCs between the DH and the VH. In addition, other types of VDCCs (Sabatini and Svoboda 2000; Yasuda et al. 2003; Cavazzini et al. 2005) and/or calcium release from internal stores following neuromodulatory transmission (McKay et al. 2007; Raymond 2007) could also participate to enhance HFS-induced synaptic potentiation and thus contribute to the difference in LTP between the DH and the VH. Finally, the LTP-enhancing calcium permeable AMPA (Jia et al. 1996; Plant et al. 2006) could also contribute to the observed differences.
Figure 2.

Effects of blockade of NMDARs, L-VDCCs, and mGluR5 on LTP in DH and VH. Time course of HFS-induced changes in fEPSP/Fv observed under standard conditions and in the presence of antagonists of NMDARs CPP (10 µM), L-VDCCs nimodipine (Nimo, 20 µM), and mGluR5 MTEP (200 µM) in DH (A,C) and VH (B,D). Representative examples of responses before and 60 min after HFS (thin and thick line traces, respectively) are shown on the top of each graph. Calibration bars: 0.5 mV and 2 msec. The numbers of slices/animals used in each condition were as follows: DH, 16/13 (ASCF), 20/10 (CPP), 6/3 (Nimo) and 5/3 (MTEP); VH, 16/11 (ACSF), 19/10 (CPP), 6/3 (Nimo), and 5/3 (MTEP). Note that the independent contribution of NMDARs and L-VDCCs was similar in the two hippocampal poles. However, the interaction between the two mechanisms (which occurred under standard conditions) produced LTP of greater magnitude in DH than in VH.
Figure 3.

Contribution of NMDARs, L-VDCCs, and mGluR5 to synaptic plasticity in DH and VH. The graphs show the percentage of the three mechanisms contribution to compound synaptic potentiation, comparatively between the DH and the VH. (A,B). In each diagram are shown the compound (total) changes in fEPSP/Fv observed under standard conditions (presented in Fig. 2) and the changes that resulted after subtracting the percentage values obtained under blockade of NMDARs, L-VDCCs, or mGluR5 from the total ones, for DH (A) and VH (B). To facilitate comparisons between the DH and the VH, the contribution of each of the three mechanisms to synaptic plasticity in the two hippocampal poles are shown in separate plots (C–E). The contribution of each mechanism to compound LTP is expressed as the proportion of reduction of LTP in the presence of the corresponding blocker (i.e., CPP, nimodipine, and MTEP for NMDARs, L-VDCCs, and mGluR5, respectively). Note that the proportional contribution of NMDARs and L-VDCCs to the LTP was rather comparable between the DH and the VH; however, the contribution of NMDARs to the potentiation during the first 8 min post-tetanus was higher in the DH than in VH (Mann–Whitney U test, P < 0.05). In addition, the contribution of mGluR5 to the synaptic potentiation during the first 30 min post-tetanus was larger in the VH than in DH (Mann–Whitney U test between DH and VH at 10, 20, and 30 min, P < 0.05).
The persistence of synaptic plasticity is one of its fundamental properties that might support the information storage required in long-term memory (Martin et al. 2000; Lynch 2004). Accordingly, cellular processes that support long-lasting synaptic potentiation such as protein synthesis (Stanton and Sarvey 1984; Frey et al. 1988; Lee et al. 1992; Scharf et al. 2002) or the persistent activity of protein kinase Mzeta (Sacktor 2008) are required for both LTP maintenance and long-term memory. In the hippocampus, the duration of synaptic potentiation has been correlated with the occurrence of hippocampus-dependent learning and memory (Doyere and Laroche 1992). Thus, the finding of equal ability of DH and VH to sustain synaptic potentiation for several hours may be relevant for the participation of DH and VH to at least short-term memory.
PTP is a form of short-lasting synaptic plasticity (Zucker and Regehr 2002) that might have significant implications in the memory process, especially in forms of short-lasting memory (Roberto et al. 2002; Kusakari et al. 2015). Then, it could be reasonable to hypothesize that the more prominent 200 Hz-induced immediate PTP in the VH compared with the DH is suggestive of the higher ability of the VH to temporarily handle high-frequency incoming information compared with the DH.
This study shows for the first time that the VH can sustain robust, particularly long-lasting nondecremental LTP of synaptic effectiveness, as has been shown for the DH. Furthermore, the present results suggest that a more effective interaction between NMDARs and L-VDCCs can explain the higher magnitude of LTP in the DH compared with the VH.
Acknowledgments
This research has been co-financed by the European Union (European Social Fund–ESF) and Greek national funds through the Operational Program “Education-and-Lifelong-Learning” of the National Strategic Reference Framework (NSRF)–Research Funding Program: Thales. Investing in knowledge society through the European Social Fund; (# MIS: 380342).
Footnotes
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.042531.116.
References
- Alger BE, Teyler TJ. 1976. Long-term and short-term plasticity in the CA1, CA3, and dentate regions of the rat hippocampal slice. Brain Res 110: 463–480. [DOI] [PubMed] [Google Scholar]
- Bannerman DM, Sprengel R, Sanderson DJ, McHugh SB, Rawlins JN, Monyer H, Seeburg PH. 2014. Hippocampal synaptic plasticity, spatial memory and anxiety. Nat Rev Neurosci 15: 181–192. [DOI] [PubMed] [Google Scholar]
- Bast T. 2011. The hippocampal learning-behavior translation and the functional significance of hippocampal dysfunction in schizophrenia. Curr Opin Neurobiol 21: 492–501. [DOI] [PubMed] [Google Scholar]
- Bayazitov IT, Richardson RJ, Fricke RG, Zakharenko SS. 2007. Slow presynaptic and fast postsynaptic components of compound long-term potentiation. J Neurosci 27: 11510–11521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL, Morris R. 2007. Synaptic plasticity in the hippocampus. In The hippocampus book (ed. Andersen P, Morris R, Amaral D, Bliss T, O'Keefe J), pp. 343–474. [Google Scholar]
- Borroni AM, Fichtenholtz H, Woodside BL, Teyler TJ. 2000. Role of voltage-dependent calcium channel long-term potentiation (LTP) and NMDA LTP in spatial memory. J Neurosci 20: 9272–9276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavazzini M, Bliss T, Emptage N. 2005. Ca2+ and synaptic plasticity. Cell Calcium 38: 355–367. [DOI] [PubMed] [Google Scholar]
- Cavus I, Teyler T. 1996. Two forms of long-term potentiation in area CA1 activate different signal transduction cascades. J Neurophysiol 76: 3038–3047. [DOI] [PubMed] [Google Scholar]
- Colgin LL, Kubota D, Jia Y, Rex CS, Lynch G. 2004. Long-term potentiation is impaired in rat hippocampal slices that produce spontaneous sharp waves. J Physiol 558: 953–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyere V, Laroche S. 1992. Linear relationship between the maintenance of hippocampal long-term potentiation and retention of an associative memory. Hippocampus 2: 39–48. [DOI] [PubMed] [Google Scholar]
- Fanselow MS, Dong HW. 2010. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron 65: 7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey U, Krug M, Reymann KG, Matthies H. 1988. Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res 452: 57–65. [DOI] [PubMed] [Google Scholar]
- Goosens KA. 2011. Hippocampal regulation of aversive memories. Curr Opin Neurobiol 21: 460–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover LM, Teyler TJ. 1990. Two components of long-term potentiation induced by different patterns of afferent activation. Nature 347: 477–479. [DOI] [PubMed] [Google Scholar]
- Jia Z, Agopyan N, Miu P, Xiong Z, Henderson J, Gerlai R, Taverna FA, Velumian A, MacDonald J, Carlen P, et al. 1996. Enhanced LTP in mice deficient in the AMPA receptor GluR2. Neuron 17: 945–956. [DOI] [PubMed] [Google Scholar]
- Jia Z, Lu Y, Henderson J, Taverna F, Romano C, Abramow-Newerly W, Wojtowicz JM, Roder J. 1998. Selective abolition of the NMDA component of long-term potentiation in mice lacking mGluR5. Learn Mem 5: 331–343. [PMC free article] [PubMed] [Google Scholar]
- Kenney J, Manahan-Vaughan D. 2013. NMDA receptor-dependent synaptic plasticity in dorsal and intermediate hippocampus exhibits distinct frequency-dependent profiles. Neuropharmacology 74: 108–118. [DOI] [PubMed] [Google Scholar]
- Keralapurath MM, Clark JK, Hammond S, Wagner JJ. 2014. Cocaine- or stress-induced metaplasticity of LTP in the dorsal and ventral hippocampus. Hippocampus 24: 577–590. [DOI] [PubMed] [Google Scholar]
- Kouvaros S, Papatheodoropoulos C. 2016. Major dorsoventral differences in the modulation of the local CA1 hippocampal network by NMDA, mGlu5, adenosine A2A and cannabinoid CB1 receptors. Neuroscience 317: 47–64. [DOI] [PubMed] [Google Scholar]
- Kusakari S, Saitow F, Ago Y, Shibasaki K, Sato-Hashimoto M, Matsuzaki Y, Kotani T, Murata Y, Hirai H, Matsuda T, et al. 2015. Shp2 in forebrain neurons regulates synaptic plasticity, locomotion, and memory formation in mice. Mol Cell Biol 35: 1557–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EH, Hung HC, Lu KT, Chen WH, Chen HY. 1992. Protein synthesis in the hippocampus associated with memory facilitation by corticotropin-releasing factor in rats. Peptides 13: 927–937. [DOI] [PubMed] [Google Scholar]
- Lynch MA. 2004. Long-term potentiation and memory. Physiol Rev 84: 87–136. [DOI] [PubMed] [Google Scholar]
- Maggio N, Segal M. 2009. Differential modulation of long-term depression by acute stress in the rat dorsal and ventral hippocampus. J Neurosci 29: 8633–8638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SJ, Grimwood PD, Morris RG. 2000. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci 23: 649–711. [DOI] [PubMed] [Google Scholar]
- Maruki K, Izaki Y, Nomura M, Yamauchi T. 2001. Differences in paired-pulse facilitation and long-term potentiation between dorsal and ventral CA1 regions in anesthetized rats. Hippocampus 11: 655–661. [DOI] [PubMed] [Google Scholar]
- Matynia A, Kushner SA, Silva AJ. 2002. Genetic approaches to molecular and cellular cognition: a focus on LTP and learning and memory. Annu Rev Genet 36: 687–720. [DOI] [PubMed] [Google Scholar]
- McKay BE, Placzek AN, Dani JA. 2007. Regulation of synaptic transmission and plasticity by neuronal nicotinic acetylcholine receptors. Biochem Pharmacol 74: 1120–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moosmang S, Haider N, Klugbauer N, Adelsberger H, Langwieser N, Muller J, Stiess M, Marais E, Schulla V, Lacinova L, et al. 2005. Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J Neurosci 25: 9883–9892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan SL, Teyler TJ. 2001. Electrical stimuli patterned after the theta-rhythm induce multiple forms of LTP. J Neurophysiol 86: 1289–1296. [DOI] [PubMed] [Google Scholar]
- Morris RG, Anderson E, Lynch GS, Baudry M. 1986. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-d-aspartate receptor antagonist, AP5. Nature 319: 774–776. [DOI] [PubMed] [Google Scholar]
- Moser MB, Moser EI. 1998. Functional differentiation in the hippocampus. Hippocampus 8: 608–619. [DOI] [PubMed] [Google Scholar]
- Pandis C, Sotiriou E, Kouvaras E, Asprodini E, Papatheodoropoulos C, Angelatou F. 2006. Differential expression of NMDA and AMPA receptor subunits in rat dorsal and ventral hippocampus. Neuroscience 140: 163–175. [DOI] [PubMed] [Google Scholar]
- Papatheodoropoulos C. 2015a. Higher intrinsic network excitability in ventral compared with the dorsal hippocampus is controlled less effectively by GABAB receptors. BMC Neurosci 16: 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papatheodoropoulos C. 2015b. Striking differences in synaptic facilitation along the dorsoventral axis of the hippocampus. Neuroscience 301: 454–470. [DOI] [PubMed] [Google Scholar]
- Papatheodoropoulos C, Kostopoulos G. 2000. Decreased ability of rat temporal hippocampal CA1 region to produce long-term potentiation. Neurosci Lett 279: 177–180. [DOI] [PubMed] [Google Scholar]
- Papatheodoropoulos C, Moschovos C, Kostopoulos G. 2005. Greater contribution of N-methyl-d-aspartic acid receptors in ventral compared to dorsal hippocampal slices in the expression and long-term maintenance of epileptiform activity. Neuroscience 135: 765–779. [DOI] [PubMed] [Google Scholar]
- Park P, Volianskis A, Sanderson TM, Bortolotto ZA, Jane DE, Zhuo M, Kaang BK, Collingridge GL. 2014. NMDA receptor-dependent long-term potentiation comprises a family of temporally overlapping forms of synaptic plasticity that are induced by different patterns of stimulation. Philos Trans R Soc Lond B Biol Sci 369: 20130131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant K, Pelkey KA, Bortolotto ZA, Morita D, Terashima A, McBain CJ, Collingridge GL, Isaac JT. 2006. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat Neurosci 9: 602–604. [DOI] [PubMed] [Google Scholar]
- Raymond CR. 2007. LTP forms 1, 2 and 3: different mechanisms for the “long” in long-term potentiation. Trends Neurosci 30: 167–175. [DOI] [PubMed] [Google Scholar]
- Reymann KG, Malisch R, Schulzeck K, Brodemann R, Ott T, Matthies H. 1985. The duration of long-term potentiation in the CA1 region of the hippocampal slice preparation. Brain Res Bull 15: 249–255. [DOI] [PubMed] [Google Scholar]
- Roberto M, Nelson TE, Ur CL, Gruol DL. 2002. Long-term potentiation in the rat hippocampus is reversibly depressed by chronic intermittent ethanol exposure. J Neurophysiol 87: 2385–2397. [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Svoboda K. 2000. Analysis of calcium channels in single spines using optical fluctuation analysis. Nature 408: 589–593. [DOI] [PubMed] [Google Scholar]
- Sacktor TC. 2008. PKMzeta, LTP maintenance, and the dynamic molecular biology of memory storage. Prog Brain Res 169: 27–40. [DOI] [PubMed] [Google Scholar]
- Sajikumar S, Navakkode S, Frey JU. 2005. Protein synthesis-dependent long-term functional plasticity: methods and techniques. Curr Opin Neurobiol 15: 607–613. [DOI] [PubMed] [Google Scholar]
- Scharf MT, Woo NH, Lattal KM, Young JZ, Nguyen PV, Abel T. 2002. Protein synthesis is required for the enhancement of long-term potentiation and long-term memory by spaced training. J Neurophysiol 87: 2770–2777. [DOI] [PubMed] [Google Scholar]
- Small SA, Schobel SA, Buxton RB, Witter MP, Barnes CA. 2011. A pathophysiological framework of hippocampal dysfunction in ageing and disease. Nat Rev Neurosci 12: 585–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton PK, Sarvey JM. 1984. Blockade of long-term potentiation in rat hippocampal CA1 region by inhibitors of protein synthesis. J Neurosci 4: 3080–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staubli U, Lynch G. 1987. Stable hippocampal long-term potentiation elicited by ‘theta’ pattern stimulation. Brain Res 435: 227–234. [DOI] [PubMed] [Google Scholar]
- Strange BA, Witter MP, Lein ES, Moser EI. 2014. Functional organization of the hippocampal longitudinal axis. Nat Rev Neurosci 15: 655–669. [DOI] [PubMed] [Google Scholar]
- Takeuchi T, Duszkiewicz AJ, Morris RG. 2014. The synaptic plasticity and memory hypothesis: encoding, storage and persistence. Philos Trans R Soc Lond B Biol Sci 369: 20130288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien JZ, Huerta PT, Tonegawa S. 1996. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell 87: 1327–1338. [DOI] [PubMed] [Google Scholar]
- Yasuda R, Sabatini BL, Svoboda K. 2003. Plasticity of calcium channels in dendritic spines. Nat Neurosci 6: 948–955. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. 2002. Short-term synaptic plasticity. Annu Rev Physiol 64: 355–405. [DOI] [PubMed] [Google Scholar]