

Abstract
Destabilization refers to a memory that becomes unstable when reactivated and is susceptible to disruption by amnestic agents. Here we delineated the cellular mechanism underlying the destabilization of drug memory. Mice were conditioned with methamphetamine (MeAM) for 3 d, and drug memory was assessed with a conditioned place preference (CPP) protocol. Anisomycin (ANI) was administered 60 min after the CPP retrieval to disrupt reconsolidation. We found that destabilization of MeAM CPP after the application of ANI was blocked by the N-methyl-d-aspartate receptor (NMDAR) antagonist MK-801 and the NR2B antagonist ifenprodil (IFN) but not by the NR2A antagonist NVP-AAM077 (NVP). In addition, decrease in the phosphorylation of GluR1 at Serine845 (p-GluR1-Ser845), decrease in spine density, and a reduction in the AMPAR/NMDAR ratio in the basolateral amygdala (BLA) were reversed after the MK-801 treatment. The effect of ANI on destabilization was prevented by the protein phosphatase 2B (calcineurin, CaN) inhibitors cyclosporine A (CsA) and FK-506 and the protein phosphatase 1 (PP1) inhibitors calyculin A (CA) and okadaic acid (OA). These results suggest that memory destabilization involves the activation of NR2B-containing NMDARs, which in turn allows the influx of Ca2+. Increased intracellular Ca2+ stimulates CaN, leading to the dephosphorylation and inactivation of inhibitor 1 and the activation of PP1. PP1 then dephosphorylates p-GluR1-Ser845 to elicit AMPA receptor (AMPAR) endocytosis and destabilization of the drug memory.
Drug addiction is associated with functional changes in the central nervous system particularly at the postsynaptic glutamate receptors (Zhong et al. 2006; Kauer and Malenka 2007; Huang et al. 2008) and the brain circuits of learning and memory (Everitt et al. 2001). The changes in synapses as well as network circuitry result in impaired cognitive control of motivated behavior leading to compulsive drug-seeking behavior. Accumulative evidence indicates that drug addiction is a process of strengthened learning (Hyman 2005). In clinics, addict patients often relapse in the treatment process. One of the critical factors is the reinstatement of drug motivation when they come into contact with a drug-related environmental cue (Tang and Dani 2009; Thanos et al. 2009).
The amygdala is the brain area in which synaptic plasticity occurs after classical fear conditioning and is in charge of other negative emotions (LeDoux 2000). In addition, the amygdala plays a regulating role for positive emotional memory. Many studies have suggested that the amygdala plays an important role in the formation of stimulus–reward associations and is required for representation of the sensory-specific properties of conditioned reinforcers in drug reinforcers (Schoenbaum et al. 1998; Blundell et al. 2001; Setlow et al. 2002). It has been shown that lesions (Brown and Fibiger 1993; Meil and See 1997) or inactivation (Fuchs and See 2002; Kantak et al. 2002) of basolateral amygdala blocked cue-induced reinstatement of cocaine- and heroin-seeking behavior. The amygdala was activated in the presence of cocaine-related stimuli in imaging studies (Childress et al. 1999). Inhibition of the immediate early gene Zif268 in the BLA prior to exposure to cocaine-associated cues decreased the expression of incubation of cocaine craving (Lee et al. 2006). Cue–reward learning increased synaptic strength and enhanced the spike activity of BLA neurons in response to a conditioned stimulus (Tye et al. 2008). Furthermore, rats that acquired amphetamine CPP exhibited increased frequency of mEPSC and the number of excitatory synapses in the BLA (Rademacher et al. 2010).
Memory consolidation is thought to be a dynamic process. Memory is labile after acquisition but becomes stabilized over time, a process termed consolidation that requires new protein synthesis. However, consolidated memory becomes destabilized such that it can be disrupted if a protein synthesis inhibitor is present at the time of reactivation (Nader et al. 2000; Nader 2003). The mechanism of memory destabilization remains elusive. We used a CPP protocol to measure the preference for a context cue as an index of drug reward (Mucha et al. 1982; Mueller and Stewart 2000). In this study, we aim to delineate the mechanism of memory destabilization. The NR2B subtype of the NMDAR antagonist was first shown to prevent the destabilization of cue-dependent fear memories (Ben Mamou et al. 2006). This observation was later supported by a study showing the requirement of NR2B-containing NMDARs for memory destabilization and NR2A-containing NMDARs for memory restabilization within the basolateral amygdala (Milton et al. 2013). In contextual fear memory, Rao-Ruiz et al. (2011) suggested that retrieval-induced endocytosis of GluR2 AMPARs in the dorsal hippocampus underlied memory destabilization of contextual fear memory (Rao-Ruiz et al. 2011). Our hypothesis is that memory retrieval induces the activation of NR2B-containing NMDARs resulting in an influx of Ca2+ and the activation of CaN and PP1, leading to the dephosphorylation of p-GluR1-Ser845 and subsequently the endocytosis of synaptic AMPARs and the destabilization of drug memory.
Results
Destabilization of MeAM CPP is blocked by the NMDA receptor antagonist
We determined whether the activation of NMDARs was required for the destabilization of MeAM CPP by the application of its antagonist MK-801. On Day 1, mice were placed in the training chamber for habituations. On Day 2, mice were conditioned with MeAM (2 mg/kg, i.p.) or saline for 3 d (Days 2–4) and tested 24 h later (CPP test, Day 5). Mice were injected with MK-801 (1.0 µg/side, n = 10) or a vehicle (n = 10) bilaterally into the amygdala 30 min before the CPP test. ANI (62.5 µg/side) was administered 60 min after the CPP test to block reconsolidation of MeAM CPP. CPP was assessed 24 h after the CPP test (Day 6, retest) (Fig. 1A). A mixed two-way ANOVA revealed main effects of group (MK-801 vs. vehicle, F(1,18) = 7.896, P = 0.0116), days (F(2,36) = 27.62, P < 0.001) and a significant interaction (F(2,36) = 5.522, P = 0.0081). Post hoc Bonferroni's tests showed that both MK-801- and vehicle-treated mice spent significantly more time in the previously MeAM-paired chamber on Day 3 (CPP test) compared with Day 1 baseline level (P < 0.001), suggesting that the mice acquired CPP memory. In the vehicle-treated mice, CPP score on Day 6 (retest) was significantly different from that on Day 5 (CPP test) (P < 0.01) and is equivalent to the baseline level (P > 0.9), suggesting that ANI disrupted reconsolidation of MeAM CPP. In contrast, in the MK-801-treated mice, CPP score on Day 5 was similar to that on Day 6 (P > 0.9). These results suggest that destabilization of CPP memory was blocked by MK-801. The infusion cannula tip locations are shown in Figure 1C. Only mice with cannula tips at or within the boundaries of LA and BLA were included in the data analysis.
Figure 1.

Effect of NMDAR antagonist on anisomycin-mediated the disruption of MeAM CPP. (A) Time line of the experiment (B). (B) Mice were injected with MK-801 (1.0 µg/side, n = 10) or vehicle (n = 10) bilaterally into the amygdala 30 min before the CPP test. ANI (62.5 µg/side) was administered bilaterally into the amygdala 60 min after the CPP test. CPP was assessed 24 h after the CPP test (retest). The vehicle control is saline, same as the vehicle control in Figure 2B. Therefore, the data of vehicle control are combined to make number of 10 and reused in Figure 2B. (***) P < 0.001 vs. retest of the vehicle-ANI group. (##) P < 0.01, CPP test vs. retest. (C) The distribution of cannula tips in the amygdala from mice infused with MK-801 (filled triangle) or vehicle (open circle). (D) The experimental procedure was the same as in B. Cell extracts were prepared 1 h after the retest. The MeAM-paired mice (n = 4) exhibited higher levels of p-GluR1-Ser845 when compared with the saline-paired mice (n = 4). ANI-mediated (n = 4) reversal of the increase in p-GluR1-Ser845 in the MeAM-paired mice was prevented in the MK-801-treated mice (n = 3). (*) P < 0.05, (**) P < 0.01 vs. ANI. (#) P < 0.05 vs. saline.
Phosphorylation of GluR1 at Serine-845 (p-GluR1-Ser845) is important for the trafficking of AMPARs in an activity-dependent manner. Phosphorylated GluR1-Ser845 by cAMP-dependent protein kinase (PKA) increases channel open probability and increases surface expression of AMPARs through recycling the GluR1 pool after their endocytosis (Banke et al. 2000; Man et al. 2007). One-way ANOVA showed that there were significant differences among the four groups in the level of p-GluR1-Ser845 (F(3,11) = 8.851, P = 0.0029, Fig. 1D). MeAM-paired mice (n = 4) exhibited higher levels of p-GluR1-Ser845 compared with saline-paired mice (n = 4) (Bonferroni post hoc analysis, P = 0.0441). The level of p-GluR1-Ser845 was reduced by ANI (n = 4, P = 0.0042, MeAM vs. ANI). Furthermore, the ANI-mediated decrease in p-GluR1-Ser845 was reversed after MK-801 treatment (n = 3, P = 0.0306, MK-801-ANI vs. ANI). Thus, the activation of NMDARs is required for the dephosphorylation of p-GluR1-Ser845.
We further determined which subtypes of NMDARs were responsible for MeAM CPP destabilization. The selective NR2A receptor antagonist NVP-AAM077 (NVP, 2.5 µg/side, n = 11), the NR2B receptor antagonist ifenprodil (IFN, 1 µg/side, n = 10) or the vehicle (n = 10) was infused to the amygdala 30 min before the CPP test. ANI (62.5 µg/side) was administered 60 min after the CPP test. CPP was assessed 24 h after the CPP test (retest). A mixed two-way ANOVA revealed main effects of group (IFN vs. vehicle, F(1,18) = 7.778, P = 0.0121), days (F(2,36) = 15.06, P < 0.001), and a significant interaction (F(2,36) = 6.846, P = 0.003) in the IFN treatment (Fig. 2A) but not in the NVP treatment (Fig. 2B) (NVP vs. vehicle, F(1,19) = 0.02643, P = 0.8726; days, F(2,38) = 37.77, P < 0.001; interaction, F(2,38) = 0.7959, P = 0.4586). In the IFN-treated mice, CPP score in Day 5 was similar to that on Day 6 (P > 0.9). In contrast, in the vehicle-treated mice, CPP score on Day 6 (retest) was significantly different from that on Day 5 (CPP test) (P < 0.001) and is equivalent to the baseline level (P > 0.9). These results suggest that destabilization of CPP memory was blocked by IFN but not by NVP. The infusion cannula tip locations of IFN and NVP are shown in Figure 2C and D, respectively.
Figure 2.

Anisomycin-mediated disruption of MeAM CPP is blocked by NR2B-containing but not by NR2A-containing NMDAR antagonist. (A,B) Mice were injected with the NR2B receptor antagonist ifenprodil (IFN, 1.0 µg/side, n = 10), the selective NR2A receptor antagonist NVP-AAM077 (NVP, 2.5 µg/side, n = 11), or the vehicle (n = 10) to the amygdala 30 min before the CPP test. ANI (62.5 µg/side) was administered 60 min after the CPP test. CPP was assessed 24 h after the CPP test (retest). (***) P < 0.001 vs. retest of the vehicle-ANI group. (C) The distribution of cannula tips in the amygdala from mice infused with either IFN (filled circle) or vehicle (open triangle). (D) The distribution of cannula tips in the amygdala from mice infused with NVP (asterisk) or vehicle (open square). (E,F) The experimental procedure was the same as in (A,B). Cell extracts were prepared 1 h after the retest. MeAM-paired mice (n = 4) exhibited higher level of p-GluR1-Ser845 when compared with the saline-paired mice (n = 4). The level of p-GluR1-Ser845 was reduced by ANI treatment (n = 4). Furthermore, the ANI-mediated decrease in p-GluR1-Ser845 was reversed by IFN (n = 4) treatment but not by NVP treatment (n = 3). (**) P < 0.01 vs. ANI. (#) P < 0.05 vs. saline.
In the Western blotting analysis, the one-way ANOVA showed that there were significant differences in the level of p-GluR1-Ser845 among the four groups (Fig. 2E, F(3,12) = 12.02, P = 0.0006 in the IFN treatment; Fig. 2F, F(3,12) = 16.84, P < 0.001 in the NVP treatment), and a Bonferroni post hoc analysis revealed that the level of p-GluR1-Ser845 in the MeAM-paired group (n = 4) was higher than that in the saline-paired group (n = 4) (Fig. 2E, P = 0.0403 and Fig. 2F, P = 0.0385). The level of p-GluR1-Ser845 was reduced by the ANI treatment (n = 4) (Fig. 2E, P = 0.0023 and Fig. 2F, P = 0.0013, MeAM vs. ANI). Furthermore, the decrease in p-GluR1-Ser845 was reversed by IFN (n = 4) but not by the NVP treatment (n = 3) (Fig. 2E, P = 0.0021, IFN-ANI vs. ANI; Fig. 2F, P = 0.5932, NVP-ANI vs. ANI), suggesting that NR2B mediated the dephosphorylation of p-GluR1-Ser845.
We examined whether the alteration of excitatory synaptic transmission in the amygdala of MeAM-paired mice could be blocked by ANI. Amygdala slices were made 1 h after the behavioral tests, and whole-cell recordings were made from the soma of visually identified pyramidal-like neurons located in the LA. As shown in Figure 3A, the one-way ANOVA for AMPAR/NMDAR ratio revealed a significant effect of group (F(3,26) = 8.637, P = 0.004), and a Bonferroni post hoc analysis revealed that the MeAM-paired mice (nine neurons from five mice) exhibited a significantly higher AMPAR/NMDAR ratio compared with the saline-paired mice (six neurons from four mice) (P = 0.0298, MeAM vs. saline). ANI treatment (eight neurons from six mice) significantly reduced the AMPAR/NMDAR ratio, and the effect of ANI was reversed by Tat-GluR23Y (seven neurons from four mice, P = 0.0039, GluR23Y-ANI vs. ANI), confirming that AMPARs endocytosis was involved in the reduction of the excitatory synaptic transmission. A separate group of mice after MeAM conditioning were infused with MK-801 (1.0 µg/side) or the vehicle to the amygdala 30 min before the CPP test. ANI (62.5 µg/side) was administered 60 min after the CPP test. As shown in Figure 3B, the one-way ANOVA for the AMPAR/NMDAR ratio showed a significant group effect (F(2,21) = 31.43, P < 0.001), and the Bonferroni post hoc analysis revealed that the MK-801-treated mice (eight neurons from four mice) exhibited a significantly higher level of AMPAR/NMDAR ratio when compared with the vehicle-treated mice (seven neurons from three mice, P < 0.001, MK-801-ANI vs. vehicle-ANI). In contrast, there was no difference in the AMPAR/NMDAR ratio between the NVP-treated (nine neurons from five mice) and the vehicle-treated mice (P > 0.5, NVP-ANI vs. vehicle-ANI), suggesting that the reduction in the excitatory synaptic transmission was mediated by the NR2B NMDARs.
Figure 3.
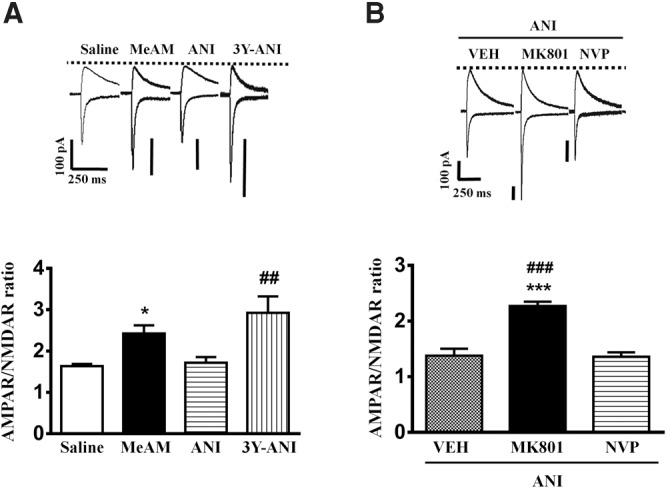
Anisomycin-mediated reversal of MeAM CPP-induced increase in the AMPAR/NMDAR ratio at the thalamo-LA synapses is blocked by MK-801. (A) Plot of AMPAR/NMDAR ratio of the saline-paired (n = 6, four mice), MeAM-paired (n = 9, five mice), ANI treatment (n = 8, six mice), and Tat-GluR23Y-treated mice (15 pmol/side, n = 7, four mice). AMPAR-mediated EPSC was evoked when the neurons were voltage-clamped at −70 mV, whereas NMDAR-mediated EPSC was determined at a holding potential of +40 mV in the presence of the AMPAR antagonist CNQX (10 µM). (*) P < 0.05 vs. saline. (##) P < 0.01 vs. ANI. (B) Plot of AMPAR/NMDAR ratio in the vehicle- (n = 7, 3 mice), MK-801- (15 pmol/side, n = 8, 4 mice), and NVP- (2.5 µg/side, n = 9, 5 mice)-treated mice. (***) P < 0.001 vs. vehicle-ANI group. (###) P < 0.001 vs. NVP-ANI group.
Experience-induced changes in dendritic spine stability serve as the mechanism for the maintenance of long-term memories. We determined whether MeAM CPP affected the number of dendritic spines in the amygdala. A one-way ANOVA revealed a significant difference in spine density among the five groups under consideration (F(4,143) = 39.93, P < 0.001). Figure 4 shows that MeAM-paired mice exhibited a higher number of dendritic spines (27.9 ± 1.2, n = 28 spines from three mice) than that of saline-paired mice (13.7 ± 0.6, n = 30 spines from three mice, Bonferroni post hoc analysis, P < 0.001, MeAM vs. saline). The increase in dendritic spines was abolished by ANI (16.3 ± 1.0, n = 30 spines from three mice, P < 0.001, MeAM vs. ANI). Furthermore, the reduction in the dendritic spines was reversed after Tat-GluR23Y (26.5 ± 1.2, n = 30 spines from three mice, P < 0.001, GluR23Y-ANI vs. ANI) or MK-801 treatment (26.5 ± 1.1, n = 30 spines from three mice, P < 0.001, MK-801-ANI vs. ANI), suggesting that the reduction in the number of dendritic spines was mediated by AMPAR endocytosis.
Figure 4.
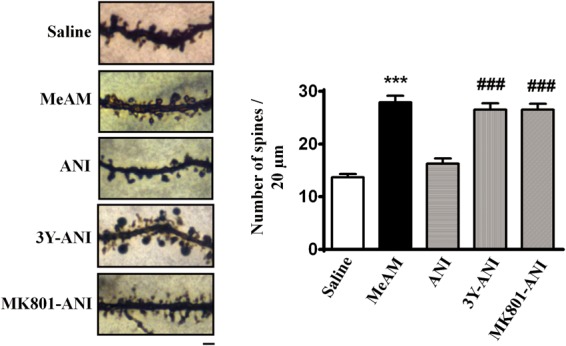
Anisomycin-mediated reversal of MeAM CPP-induced increase in spine density in LA is blocked by MK-801. Representative images and statistical analyses of Golgi staining from the lateral nucleus of amygdala of the saline-paired (n = 30, three mice), MeAM-paired (n = 30, three mice), ANI treatment- (n = 30, mice), Tat-GluR23Y- (n = 30, 3 mice), and MK-801- (n = 30, 3 mice)-treated mice. (***) P < 0.001 vs. saline. (###) P < 0.001 vs. ANI. Bar, 2 µm.
Involvement of protein phosphatases in destabilization of MeAM memory
Phosphorylated GluR1-Ser845 induces AMPAR insertion into plasma membrane and thereby strengthens synaptic transmission. In contrast, dephosphorylation of p-GluR1-Ser845 triggers AMPAR internalization and weakens transmission (Man et al. 2007). Because calcineurin (CaN) is stimulated in response to activation of NMDAR, leading to long-term depression (LTD) (Mulkey et al. 1994; Hodgkiss and Kelly 1995; Lee et al. 1998; Lu et al. 2000; Zeng et al. 2001), we examined whether CaN was involved in the destabilization of MeAM memory. CaN inhibitor cyclosporin A (CsA, 2.5 µg/side, n = 14), FK-506 (5 µg/side, n = 13), or vehicle (n = 10) was infused to the amygdala 30 min before the CPP test. ANI (62.5 µg/side) was administered 60 min after the CPP test. CPP was assessed 24 h after the CPP test (retest). As shown in Figure 5A, a mixed two-way ANOVA revealed a significant day × treatment interaction (F(4,68) = 3.031, P = 0.0232). ANI disrupted MeAM CPP in the vehicle-treated mice (Bonferroni post hoc analysis, P = 0.001) but not in the CsA- or FK-506-treated mice (P > 0.1 and P > 0.1, respectively). Moreover, CsA- or FK-506-treated mice exhibited significantly higher levels of MeAM CPP in the retest compared with the vehicle-treated mice (Bonferroni post hoc analysis, P = 0.0003, CsA-ANI vs. vehicle-ANI; P = 0.0109, FK-506-ANI vs. vehicle-ANI), indicating that CsA and FK-506 blocked the destabilization of MeAM CPP. The data suggest that the destabilization of MeAM CPP is mediated by CaN. The infusion cannula tip locations of CsA and FK-506 are shown in Figure 5B.
Figure 5.

Anisomycin-mediated disruption of MeAM CPP is blocked by calcineurin inhibitors cyclosporin A and FK-506. (A) Mice were injected with calcineurin inhibitor cyclosporin A (CsA, 2.5 µg/side, n = 14), FK-506 (5 µg/side, n = 13), or vehicle (n = 10) to the amygdala 30 min before the CPP test. ANI (62.5 µg/side) was administered 60 min after the CPP test. CPP was assessed 24 h after the CPP test (retest). (*) P < 0.05, (***) P < 0.001 vs. retest of vehicle-ANI group. (##) P < 0.01, CPP test vs. retest. (B) The distribution of cannula tips in the amygdala from mice infused with CsA (hash) or FK-506 (filled diamond). (C) The experimental procedure was same as in A. Cell extracts were prepared 1 h after the retest. ANI-mediated (n = 3) reduction of p-GluR1-Ser845 in MeAM-paired mice (n = 4) was reversed in the FK-506-treated mice (n = 3). (*) P < 0.05 vs. FK-506-ANI. (D) Representative images and statistical analyses of Golgi staining from the lateral nucleus of amygdala of the vehicle- (n = 30, three mice), FK506- (n = 30, three mice), and okadaic acid- (n = 30, three mice) treated mice. (***) P < 0.001 vs. vehicle-ANI. Bar, 2 µm.
In the Western blotting analysis, the reduction of p-GluR1-Ser845 was blocked by FK-506 treatment (one-way ANOVA, F(2,7) = 15.76, P = 0.0026; Bonferroni post hoc analysis, P = 0.0105, ANI vs. FK-506; n = 4, 3 and 3 in MeAM, ANI, and FK-506-ANI, respectively) (Fig. 5C). The FK-506-treated mice (27.5 ± 1.1, n = 30 spines from three mice) had a higher number of dendritic spines compared with the vehicle-treated mice (19.5 ± 0.7, n = 30 spines from three mice) (one-way ANOVA, F(2,87) = 26.29, P < 0.001; Bonferroni post hoc analysis, P < 0.001, FK-506-ANI vs. vehicle-ANI) (Fig. 5D). In addition, in electrophysiological recordings, the FK-506-treated mice (six neurons from four mice) exhibited significantly higher AMPAR/NMDAR ratio than those of the vehicle-treated mice (seven neurons from three mice, P = 0.02, FK-506-ANI vs. vehicle-ANI) (Fig. 6).
Figure 6.
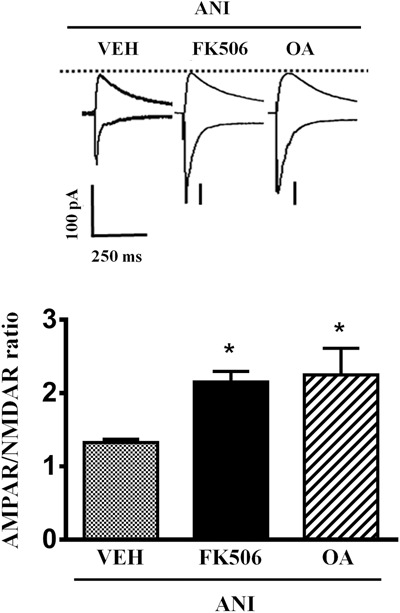
Anisomycin-mediated reversal of MeAM CPP-induced increase in the AMPAR/NMDAR ratio at the thalamo-LA synapses is blocked by calcineurin and PP1 inhibitors. Plot of AMPAR/NMDAR ratio in the vehicle- (n = 7, three mice), FK-506- (5 µg/side, n = 6, four mice), and OA- (10 ng/side, n = 5, three mice)-treated mice. (*) P < 0.05, vs. vehicle-ANI group.
CaN, which is activated by the Ca2+ influx through the NMDARs, dephosphorylates inhibitor-1 and subsequently activates protein phosphatase 1 (PP1) activity. We examined the involvement of PP1 in the destabilization of MeAM memory by infusing PP1 inhibitors calyculin A (CA, 5 pmol/side, n = 8), okadaic acid (OA, 10 ng/side, n = 8), or vehicle (n = 9) to the amygdala 30 min before the CPP test. ANI (62.5 µg/side) was administered 60 min after the CPP test. CPP was assessed 24 h after the CPP test (retest). As shown in Figure 7A, a mixed two-way ANOVA revealed a significant day × treatment interaction (F(4,42) = 2.649, P = 0.0464). ANI disrupted MeAM CPP in the vehicle-treated mice (Bonferroni post hoc analysis, P = 0.0004) but not in the CA- or OA-treated mice (P > 0.1 and P > 0.1, respectively). Moreover, the CA- or OA-treated mice exhibited significantly higher levels of MeAM CPP in the retest compared with the vehicle-treated mice (Bonferroni post hoc analysis, P = 0.0107, CA-ANI vs. vehicle-ANI; P = 0.0120, OA-ANI vs. vehicle-ANI), indicating that CA and OA blocked the destabilization of MeAM CPP. The infusion cannula tip locations of CA and OA are shown in Figure 7B. Moreover, Western blotting analysis showed that CA (n = 5, Fig. 7C) and OA (n = 4, Fig. 7D) reversed ANI-mediated dephosphorylation of p-GluR1-Ser845 (one-way ANOVA, F(2,12) = 6.670, P = 0.0113 and F(2,11) = 17.29, P = 0.0004, respectively; Bonferroni post hoc analysis, P = 0.0305, ANI vs. CA-ANI and P = 0.0314, ANI vs. OA-ANI). Furthermore, there was a significantly higher AMPAR/NMDAR ratio in the OA-treated mice (five neurons from three mice) when compared with that of the vehicle-treated mice (P = 0.013, OA-ANI vs. vehicle-ANI) (Fig. 6) and a significant increase in the number of dendritic spines in the amygdala of the OA-treated mice (27.4 ± 0.9, n = 30 spines from three mice) when compared with that found in the vehicle-treated mice (19.5 ± 0.7, n = 30 spines from three mice, P < 0.001, OA-ANI vs. vehicle-ANI) (Fig. 5D). These data suggest that destabilization of MeAM CPP is mediated by PP1.
Figure 7.

Anisomycin-mediated disruption of MeAM CPP is blocked by PP1 inhibitors calyculin A and okadaic acid. (A) Mice were injected with either PP1 inhibitors calyculin A (CA, 5 pmol/side, n = 8), okadaic acid (OA, 10 ng/side, n = 8), or the vehicle (n = 9) to the amygdala 30 min before the CPP test. ANI (62.5 µg/side) was administered 60 min after the CPP test. CPP was assessed 24 h after the CPP test (retest). (*) P < 0.05 vs. Retest of the vehicle-ANI group. (###) P < 0.001, CPP test vs. retest. (B) The distribution of cannula tips in the amygdala from mice infused with CA (fx1) or OA (filled heart). (C,D) The experimental procedure was the same as in A. Cell extracts were prepared 1 h after the retest. ANI (n = 5) reduction of p-GluR1-Ser845 in the MeAM-paired mice (n = 5) was reversed in both the CA- (n = 5) and OA- (n = 4)-treated mice. (*) P < 0.05 vs. CA-ANI or OA-ANI.
Discussion
We have previously shown that destabilization of MeAM CPP after the application of ANI could be blocked after bilateral injection of Tat-GluR23Y into the BLA, suggesting that AMPA receptor endocytosis in the BLA is critical for destabilization (Yu et al. 2013). However, little is known about signal transduction leading to AMPA receptor endocytosis in the BLA during destabilization of drug memory. In the present study, we found that MK-801 and ifenprodil blocked the destabilization of MeAM CPP, suggesting the involvement of NR2B-containing NMDARs. Memory retrieval initiated two consecutive but opposite processes: the previously established memory became destabilized and then required a de novo protein synthesis-dependent reconsolidation to persist. Both memory destabilization and reconsolidation processes were blocked by nonselective NMDAR antagonists. Recently, using a fear conditioning procedure, Milton et al. (2013) showed memory destabilization and reconsolidation to be mediated by different subtypes of NMDARs; NR2B-containing NMDARs-mediated memory destabilization, whereas NR2A-containing NMDARs were required for memory reconsolidation. Consistent with these results, we found that memory destabilization was blocked by MK-801 and selective NR2B-containing NMDARs ifenprodil, whereas selective NR2A-containing NMDARs NVP-AAM077 was ineffective on destabilization.
NR2A- and NR2B-containing NMDARs are linked to different intracellular signaling pathways. For example, the carboxy-terminal domain of NR2B suppresses CREB, while NR2A-containing NMDAR activation promotes CREB phosphorylation (Hardingham et al. 2002; Martel et al. 2012). Thus, it is likely that memory retrieval induces destabilization of that memory by the activation of NR2B-containing NMDARs, leading to the stimulation of calcineurin and de-phosphorylation of p-GluR1-Ser845. This is followed by reconsolidation that requires the activation of NR2A-containing NMDARs and the activation of protein kinases. It is worth noting that memory retrieval induces a brief destabilization process and a longer process of restabilization to reconsolidate the memory. Thus, nonselective NMDAR antagonists will predominantly act on NR2A-containing NMDARs and block reconsolidation (Milton et al. 2013).
How is activation of NR2B-containing NMDARs required for destabilization of MeAM CPP? CaN, a protein Ser/Thr phosphatase, has high affinity for Ca2+ and can be activated by nanomolar concentrations of Ca2+ (Cohen and Klee 1988). CaN has been shown to be involved in long-term depression (LTD) (Mulkey et al. 1994; Hodgkiss and Kelly 1995), a form of synaptic weakening, and depotentiation of previously potentiated synapses (Zhuo et al. 1999; Jouvenceau et al. 2003; Kang-Park et al. 2003; Lin et al. 2003). In the present study, we found that destabilization of MeAM CPP after application of ANI and dephosphorylation of p-GluR1-Ser845 were blocked by calcineurin inhibitors, cyclosporine A, and FK-506. Ser845 on GluR1 is one of the most important for regulation of AMPAR trafficking. Phosphorylated Ser845 increases AMPAR open probability (Banke et al. 2000) and insertion into the membrane (Man et al. 2007) and thereby strengthens synaptic transmission. In contrast, Ser845 dephosphorylation triggers AMPAR internalization and weakens transmission (Man et al. 2007). It is likely that memory retrieval initiates glutamate release and the activation of NMDARs leading to Ca2+ influx. Increased intracellular Ca2+ stimulated calcineurin activity could directly and/or indirectly dephosphorylate p-GluR1-Ser845. We found that destabilization of MeAM CPP was blocked by PP1 inhibitors, suggesting that calcineurin stimulated PP1 by dephosphorylation of Inhibitor 1. Activation of PP1 catalyzed dephosphorylation of p-GluR-Ser845, resulting in AMPAR endocytosis that was crucial for memory deconsolidation. This could explain why memory became labile when retrieved. Conversely, in the absence of anisomycin, memory underwent protein synthesis-dependent reconsolidation for long-term retention.
In a consolidated fear memory, gradual increase in the number of conditioned stimuli (CSs) shifts fear reconsolidation to extinction and results in the suppression of that memory. Interestingly, the transition from reconsolidation to extinction is accompanied by a gradual increase in amygdala calcineurin levels (Merlo et al. 2014). Here, we found that the destabilization of MeAM CPP after the application of ANI was prevented by the calcineurin inhibitors cyclosporine A and FK-506. Increased intracellular Ca2+-activated protein kinases that subsequently translocated to the nucleus. Activated protein kinases phosphorylated transcriptional factors such as CREB to initiate transcription and translation resulting in the reconsolidation of that memory. Increased intracellular Ca2+ could also stimulate calcineurin activity. When reconsolidation was disrupted by ANI, calcineurin could directly and/or indirectly dephosphorylate p-GluR1-Ser845.
In conclusion, we have demonstrated that the activation of NR2B containing NMDARs is required for destabilization of MeAM CPP after the application of ANI. MeAM CPP-induced increases in p-GluR1-Ser845, AMPAR/NMDAR ratio and spine density were shown to be abolished after the application of ANI. These effects as well as the destabilization of MeAM CPP were reversed by calcineurin and PP1 inhibitors. It is suggested that dephosphorylation of p-GluR1-Ser845 and the resultant AMPAR endocytosis underlies the mechanism of drug memory destabilization.
Materials and Methods
Surgery and intracranial microinjections
All procedures were approved by the Institutional Animal Care and Use Committee of the College of Medicine, National Cheng Kung University. Male C57BL/6 mice (4 to 6 wk old) were anesthetized with chloral hydrate (400 mg/kg, i.p.) and were subsequently mounted on a stereotaxic apparatus, and a cannula made of 26 gauge stainless steel tubing was implanted bilaterally into the amygdala (anteroposterior, −1.6 mm; mediolateral, ±3.5 mm; dorsoventral, −4.6 mm). The mice were handled daily and were given 7 d to recover. The drug was administered bilaterally to the amygdala in a volume of 1 µL at a rate of 0.1 µL/min. The infusion cannulas were left in place for 2 min before being withdrawn.
Drugs
Anisomycin (62.5 µg/side dissolved in saline-containing 10% DMSO, the pH value was adjusted to 7.0) was obtained from Sigma-Aldrich. A Tat-conjugated peptide designed to block the regulated clathrin-coated endocytosis of AMPARs was used with a sequence of 869-YKEGYNVYG-877 (GluR23Y, 15 pmol/side). The scrambled control peptide in which the tyrosine residues were replaced by alanine had the AKEGANVAG sequence (GluR23A, 15 pmol/side). These peptides were purchased from Kelowna International Scientific Inc. (Sanchong Dist.) and were dissolved in saline. One microgram per side (1)-MK-801 maleate (Sigma-Aldrich) and 2.5 µg/side NVP-AAM077 (Sigma-Aldrich) were dissolved in 0.9% saline. One microgram per side ifenprodil (Abcam), 5 µg/side FK-506 (Cayman), 2.5 µg/side cyclosporine A (Sigma-Aldrich), 5 pmol/side calyculin A (Alomone Labs), and 10 ng/side okadaic acid (Sigma-Aldrich) were dissolved in 10% DMSO.
Conditioned place preference paradigm
The CPP paradigm consisted of three phases: preconditioning, conditioning, and post-conditioning. For the pre- and post-conditioning phases, animals were placed in a neutral gray center compartment, and the sliding doors were removed to allow equal access to the entire apparatus for 15 min. The amount of time each mouse spent in each compartment was monitored to determine initial preferences. During the conditioning phase, the animals received either a saline or methamphetamine (MeAM) solution for six conditioning sessions. Specifically, each animal received saline injections in the morning while in their initially preferred side (saline-paired side) and MeAM injections in the afternoon in their initially nonpreferred side (drug-paired side) for 3 d. The animals were confined to the appropriate compartment for the entire conditioning session (30 min), and access to the neutral compartment was blocked by the sliding door. The data reported herein shows that the mice initially expressed a bias for a particular side, with most preferring the dark black compartment. For the post-conditioning tests, the animals were allowed to access the entire apparatus in a drug-free-state. The CPP apparatus had a biased design (i.e., the drug was administered on the initially nonpreferred side), and thus, the relative amount of time spent in each compartment before vs. after conditioning was used to assess the CPP. The CPP data reported here are shown as the “time difference,” which was calculated by subtracting the time spent in the initially nonpreferred side of the apparatus (drug-paired) and the time spent in the preferred side during the pre- and post-conditioning test. It is important to mention that overall, the MeAM-treated mice spent significantly more time in the initially nonpreferred compartment than in the initially preferred compartment following drug conditioning, suggesting a true reward measurement.
Western blotting analysis
The minislices containing basolateral amygdala were dissected from the slices and sonicated briefly in a homogenizing buffer (1% Triton X-100, 0.1% SDS, 50 mM Tris-HCl, pH 7.5, 0.3 M sucrose, 5 mM EDTA, 2 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride and 20 µg/mL leupeptin, and 4 µg/mL aprotinin). After sonication, the samples were centrifuged at 14,000 r.p.m. for 30 min at 4°C, and the supernatant was obtained. The protein concentration in the soluble fraction was then measured using a Bradford assay, with bovine serum albumin as the standard. Bound protein was resuspended in 4 µL of an SDS sample loading buffer at 95°C. The total slice lysates was resolved in 8.5% SDS–PAGE gels, blotted electrophoretically to the PVDF membrane, and blocked overnight in a TBS buffer containing 5% nonfatty milk. In the Western blot analysis, glutamate receptor 1 phosphoSer845 (1:2500; Millipore) or actin antibody (1:100,000; Chemicon) was used followed by an HRP-conjugated secondary antibody for 1 h. An enhanced chemiluminescence kit was used for detection. The Western blots were developed in the linear range used for densitometry.
Golgi staining
The experimental mice were obtained from the Animal Center of National Cheng-Kung University. After the behavioral tests, the entire brains were removed after perfusion with phosphate-buffered saline (PBS) and then processed for Golgi staining (Rapid GolgiStain Kit, FD NeuroTechnologies) to examine the histology of the dendritic spines. Coronal sections with a thickness of 150-μm were cut on a vibratome and plated on gelatin-coated microscopy slides. Images were collected using a Leica DM 2500 microscope, and MetaMorph image software was used to count the number of spines.
Slice preparation
The mice were decapitated, and their brains were rapidly removed and placed in a cold oxygenated artificial cerebrospinal fluid (ACSF) solution. Subsequently, the brain was hemisected and cut transversely posterior to the first branch and anterior to the last branch of the superior cerebral vein. The resulting section was glued to the chuck of a Vibroslice tissue slicer. Three hundred micrometer-thick transverse slices were cut, and the appropriate slices were placed in a beaker of oxygenated ACSF at room temperature for at least 1 h before recording. The ACSF solution had the following composition (in mM): NaCl 117, KCl 4.7, CaCl2 2.5, MgCl2 1.2, NaHCO3 25, NaH2PO4 1.2, and glucose 11. The ACSF was bubbled continuously with 95% O2–5% CO2 and had a pH of 7.4.
Whole-cell patch-clamp recordings
Whole-cell patch-clamp recordings from LA projection neurons were performed at approximately room temperature (22°C–24°C) in a superfusing chamber. Neurons were visualized with an infrared video microscope with a 40× water immersion objective on an upright microscope.
Whole-cell patch-clamp recordings were made from the LA neurons. The excitatory postsynaptic currents (EPSCs) were evoked at 0.05 Hz by extracellular stimulation of fibers emerging from the internal capsule, which originated in the medial geniculate nucleus of the thalamus, and were projected monosynaptically to the LA using a bipolar electrode. Patch electrodes were pulled from a thick-walled glass capillary (0.75 mm internal diameter and 1.5 mm outer diameter) to a tip resistance of 3–5 MΩ. The composition of the internal solution was 115 mM cesium gluconate, 5 mM NaCl, 1 mM EGTA, 0.3 mM CaCl2, 2 mM MgCl2, 5 mM Na-ATP, 0.4 mM Na-GTP, and 10 mM HEPES. The final pH of the internal solution was adjusted to 7.3 by adding either 1 M KOH or CsOH; the final osmolarity was adjusted to 290–300 mOsm by adding sucrose. Recordings were low-pass-filtered at 2.5–20 kHz and digitized at 5–50 kHz. Membrane potentials and currents were monitored and recorded using an Axopatch 700B amplifier (Molecular Devices), acquired via a Digidata 1440 series analog-to-digital interface on a Pentium computer with Clampex 10.3 software (Molecular Devices). AMPAR-mediated EPSC was evoked when the neurons were voltage-clamped at −70 mV, whereas NMDAR-mediated synaptic responses were isolated in the presence of the AMPA receptor antagonist CNQX (10 µM) and the GABA A receptor antagonist bicuculline (10 µM).
Data analysis
All values represent mean ± standard error of the mean (SEM). One-way ANOVA and Bonferroni post hoc comparisons were used to analyze the differences in the number of dendritic spines, protein levels, and electrophysiologic data. Student's t-tests were used to analyze the differences in the behavioral tests among the vehicle and drug samples. The level of significance was P < 0.05.
Acknowledgments
We thank Dr. Min-Der Lai for proofreading the manuscript. This study was supported by grants NHRI-EX101-10117NI from the National Health Research Institute, MOST103-2321-B-006-008 from the National Science Council, Aim for the Top University Project of the National Cheng-Kung University.
Author contributions: Y.-J.Y. and P.-W.G. were responsible for the study concept and design. C.-H.H., Y.-J.Y., and C.-H.C. contributed to the acquisition of animal data. C.-H.H. and Y.-J.Y. performed the electrophysiological recordings. C.-H.H. and C.-H.C. assisted with data analysis and with the interpretation of the findings. P.-W.G. drafted the manuscript. Y.-J.Y. and C.-H.C. provided critical revision of the manuscript for important intellectual content. All authors critically reviewed content and approved the final version for publication.
Footnotes
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.039941.115.
References
- Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis SF. 2000. Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J Neurosci 20: 89–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Mamou C, Gamache K, Nader K. 2006. NMDA receptors are critical for unleashing consolidated auditory fear memories. Nat Neurosci 9: 1237–1239. [DOI] [PubMed] [Google Scholar]
- Blundell P, Hall G, Killcross S. 2001. Lesions of the basolateral amygdala disrupt selective aspects of reinforcer representation in rats. J Neurosci 21: 9018–9026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EE, Fibiger HC. 1993. Differential effects of excitotoxic lesions of the amygdala on cocaine-induced conditioned locomotion and conditioned place preference. Psychopharmacology (Berl) 113: 123–130. [DOI] [PubMed] [Google Scholar]
- Childress AR, Mozley PD, McElgin W, Fitzgerald J, Reivich M, O'Brien CP. 1999. Limbic activation during cue-induced cocaine craving. Am J Psychiatry 156: 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Klee CB. 1988. Molecular aspects of cellular regulation: calmodulin, Vol. 5, pp. 225–248. Elsevier, Amsterdam. [Google Scholar]
- Everitt BJ, Dickinson A, Robbins TW. 2001. The neuropsychological basis of addictive behaviour. Brain Res Brain Res Rev 36: 129–138. [DOI] [PubMed] [Google Scholar]
- Fuchs RA, See RE. 2002. Basolateral amygdala inactivation abolishes conditioned stimulus- and heroin-induced reinstatement of extinguished heroin-seeking behavior in rats. Psychopharmacology (Berl) 160: 425–433. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. 2002. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci 5: 405–414. [DOI] [PubMed] [Google Scholar]
- Hodgkiss JP, Kelly JS. 1995. Only ‘de novo’ long-term depression (LTD) in the rat hippocampus in vitro is blocked by the same low concentration of FK506 that blocks LTD in the visual cortex. Brain Res 705: 241–246. [DOI] [PubMed] [Google Scholar]
- Huang YY, Kandel ER, Levine A. 2008. Chronic nicotine exposure induces a long-lasting and pathway-specific facilitation of LTP in the amygdala. Learn Mem 15: 603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman SE. 2005. Addiction: a disease of learning and memory. Am J Psychiatry 162: 1414–1422. [DOI] [PubMed] [Google Scholar]
- Jouvenceau A, Billard JM, Haditsch U, Mansuy IM, Dutar P. 2003. Different phosphatase-dependent mechanisms mediate long-term depression and depotentiation of long-term potentiation in mouse hippocampal CA1 area. Eur J Neurosci 18: 1279–1285. [DOI] [PubMed] [Google Scholar]
- Kang-Park MH, Sarda MA, Jones KH, Moore SD, Shenolikar S, Clark S, Wilson WA. 2003. Protein phosphatases mediate depotentiation induced by high-intensity theta-burst stimulation. J Neurophysiol 89: 684–690. [DOI] [PubMed] [Google Scholar]
- Kantak KM, Black Y, Valencia E, Green-Jordan K, Eichenbaum HB. 2002. Dissociable effects of lidocaine inactivation of the rostral and caudal basolateral amygdala on the maintenance and reinstatement of cocaine-seeking behavior in rats. J Neurosci 22: 1126–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC. 2007. Synaptic plasticity and addiction. Nat Rev Neurosci 8: 844–858. [DOI] [PubMed] [Google Scholar]
- LeDoux JE. 2000. Emotion circuits in the brain. Annu Rev Neurosci 23: 155–184. [DOI] [PubMed] [Google Scholar]
- Lee HK, Kameyama K, Huganir RL, Bear MF. 1998. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron 21: 1151–1162. [DOI] [PubMed] [Google Scholar]
- Lee JL, Milton AL, Everitt BJ. 2006. Cue-induced cocaine seeking and relapse are reduced by disruption of drug memory reconsolidation. J Neurosci 26: 5881–5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Yeh SH, Leu TH, Chang WC, Wang ST, Gean PW. 2003. Identification of calcineurin as a key signal in the extinction of fear memory. J Neurosci 23: 1574–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YM, Mansuy IM, Kandel ER, Roder J. 2000. Calcineurin-mediated LTD of GABAergic inhibition underlies the increased excitability of CA1 neurons associated with LTP. Neuron 26: 197–205. [DOI] [PubMed] [Google Scholar]
- Man HY, Sekine-Aizawa Y, Huganir RL. 2007. Regulation of {α}-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor trafficking through PKA phosphorylation of the Glu receptor 1 subunit. Proc Natl Acad Sci 104: 3579–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martel MA, Ryan TJ, Bell KF, Fowler JH, McMahon A, Al-Mubarak B, Komiyama NH, Horsburgh K, Kind PC, Grant SG, et al. 2012. The subtype of GluN2 C-terminal domain determines the response to excitotoxic insults. Neuron 74: 543–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meil WM, See RE. 1997. Lesions of the basolateral amygdala abolish the ability of drug associated cues to reinstate responding during withdrawal from self-administered cocaine. Behav Brain Res 87: 139–148. [DOI] [PubMed] [Google Scholar]
- Merlo E, Milton AL, Goozee ZY, Theobald DE, Everitt BJ. 2014. Reconsolidation and extinction are dissociable and mutually exclusive processes: behavioral and molecular evidence. J Neurosci 34: 2422–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton AL, Merlo E, Ratano P, Gregory BL, Dumbreck JK, Everitt BJ. 2013. Double dissociation of the requirement for GluN2B- and GluN2A-containing NMDA receptors in the destabilization and restabilization of a reconsolidating memory. J Neurosci 33: 1109–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucha RF, van der Kooy D, O'Shaughnessy M, Bucenieks P. 1982. Drug reinforcement studied by the use of place conditioning in rat. Brain Res 243: 91–105. [DOI] [PubMed] [Google Scholar]
- Mueller D, Stewart J. 2000. Cocaine-induced conditioned place preference: reinstatement by priming injections of cocaine after extinction. Behav Brain Res 115: 39–47. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Endo S, Shenolikar S, Malenka RC. 1994. Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature 369: 486–488. [DOI] [PubMed] [Google Scholar]
- Nader K. 2003. Memory traces unbound. Trends Neurosci 26: 65–72. [DOI] [PubMed] [Google Scholar]
- Nader K, Schafe GE, Le Doux JE. 2000. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature 406: 722–726. [DOI] [PubMed] [Google Scholar]
- Rademacher DJ, Rosenkranz JA, Morshedi MM, Sullivan EM, Meredith GE. 2010. Amphetamine-associated contextual learning is accompanied by structural and functional plasticity in the basolateral amygdala. J Neurosci 30: 4676–4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao-Ruiz P, Rotaru DC, van der Loo RJ, Mansvelder HD, Stiedl O, Smit AB, Spijker S. 2011. Retrieval-specific endocytosis of GluA2-AMPARs underlies adaptive reconsolidation of contextual fear. Nat Neurosci 14: 1302–1308. [DOI] [PubMed] [Google Scholar]
- Schoenbaum G, Chiba AA, Gallagher M. 1998. Orbitofrontal cortex and basolateral amygdala encode expected outcomes during learning. Nat Neurosci 1: 155–159. [DOI] [PubMed] [Google Scholar]
- Setlow B, Holland PC, Gallagher M. 2002. Disconnection of the basolateral amygdala complex and nucleus accumbens impairs appetitive pavlovian second-order conditioned responses. Behav Neurosci 116: 267–275. [DOI] [PubMed] [Google Scholar]
- Tang J, Dani JA. 2009. Dopamine enables in vivo synaptic plasticity associated with the addictive drug nicotine. Neuron 63: 673–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanos PK, Bermeo C, Wang GJ, Volkow ND. 2009. D-cycloserine accelerates the extinction of cocaine-induced conditioned place preference in C57bL/c mice. Behav Brain Res 199: 345–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tye KM, Stuber GD, de Ridder B, Bonci A, Janak PH. 2008. Rapid strengthening of thalamo-amygdala synapses mediates cue-reward learning. Nature 453: 1253–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YJ, Chang CH, Gean PW. 2013. AMPA receptor endocytosis in the amygdala is involved in the disrupted reconsolidation of Methamphetamine-associated contextual memory. Neurobiol Learn Mem 103: 72–81. [DOI] [PubMed] [Google Scholar]
- Zeng H, Chattarji S, Barbarosie M, Rondi-Reig L, Philpot BD, Miyakawa T, Bear MF, Tonegawa S. 2001. Forebrain-specific calcineurin knockout selectively impairs bidirectional synaptic plasticity and working/episodic-like memory. Cell 107: 617–629. [DOI] [PubMed] [Google Scholar]
- Zhong W, Dong Z, Tian M, Cao J, Xu T, Xu L, Luo J. 2006. Opiate withdrawal induces dynamic expressions of AMPA receptors and its regulatory molecule CaMKIIalpha in hippocampal synapses. Life Sci 79: 861–869. [DOI] [PubMed] [Google Scholar]
- Zhuo M, Zhang W, Son H, Mansuy I, Sobel RA, Seidman J, Kandel ER. 1999. A selective role of calcineurin α in synaptic depotentiation in hippocampus. Proc Natl Acad Sci 96: 4650–4655. [DOI] [PMC free article] [PubMed] [Google Scholar]