

Abstract
Luc7 is an essential 261-amino acid protein subunit of the Saccharomyces cerevisiae U1 snRNP. To establish structure–function relations for yeast Luc7, we conducted an in vivo mutational analysis entailing N- and C-terminal truncations and alanine scanning of phylogenetically conserved amino acids, including two putative zinc finger motifs, ZnF1 and ZnF2, and charged amino acids within the ZnF2 module. We identify Luc7-(31–246) as a minimal functional protein and demonstrate that whereas mutations of the CCHH ZnF2 motif are lethal, mutations of the ZnF1 CCCH motif and the charged residues of the ZnF2 modules are not. Though dispensable for vegetative growth in an otherwise wild-type background, the N-terminal 18-amino acid segment of Luc7 plays an important role in U1 snRNP function, evinced by our findings that its deletion (i) impaired the splicing of SUS1 pre-mRNA; (ii) was synthetically lethal absent other U1 snRNP constituents (Mud1, Nam8, the TMG cap, the C terminus of Snp1), absent the Mud2 subunit of the Msl5•Mud2 branchpoint binding complex, and when the m7G cap-binding site of Cbc2 was debilitated; and (iii) bypassed the need for the essential DEAD-box ATPase Prp28. Similar phenotypes were noted for ZnF1 mutations C45A, C53A, and C68A and ZnF2 domain mutations D214A, R215A, R216A, and D219A. These findings highlight the contributions of the Luc7 N-terminal peptide, the ZnF1 motif, and the ZnF2 module in stabilizing the interactions of the U1 snRNP with the pre-mRNA 5′ splice site and promoting the splicing of a yeast pre-mRNA, SUS1, that has a nonconsensus 5′ splice site.
Keywords: Luc7, Prp28, U1 snRNP, spliceosome assembly
INTRODUCTION
pre-mRNA splicing initiates when the U1 snRNP engages the intron 5′ splice site (5′SS). The Saccharomyces cerevisiae U1 snRNP consists of a trimethylguanosine (TMG) capped 568-nt U1 snRNA, a seven-subunit Sm protein ring (present also in the U2, U4, and U5 snRNPs), and ten U1-specific protein subunits: Prp39, Prp40, Snu71, Snu56, Snp1, Mud1, Luc7, Prp42, Nam8, and Yhc1 (Gottschalk et al. 1998; Fortes et al. 1999a; Schwer et al. 2011). Base-pairing of the U1 snRNA leader motif 5′-ACUUAC sequence with the consensus yeast 5′SS element 5′-GUAUGU nucleates an initial U1•pre-mRNA complex. Cross-intron bridging interactions between the yeast U1 snRNP at the 5′SS and the Msl5•Mud2 heterodimer at the branchpoint sequence 5′-UACUAAC then stabilize a commitment complex, which provides a scaffold for recruitment of the U2 snRNP to the branchpoint (Abovich and Rosbash 1997). The U1 snRNP is eventually ejected from the spliceosome at the step when the U5•U4•U6 tri-snRNP complex joins en route to forming a pre-mRNA•U2•U5•U6 spliceosome. Dissociation of U1 snRNP is triggered by the DEAD-box ATPase Prp28 (Staley and Guthrie 1999; Chen et al. 2001; Jacewicz et al. 2014), acting to disrupt the short U1:5′SS RNA duplex or remodel protein–RNA contacts at the 5′SS (or both).
Traditional and systems genetic approaches, as well as structure-guided mutagenesis, have highlighted a rich network of genetically buffered functions during early spliceosome assembly in budding yeast, embracing the U1-specific snRNP proteins Mud1, Nam8, Yhc1 and Snp1, the U1 snRNA, the TMG cap, the Cbc2•Sto1 nuclear m7G cap-binding complex (CBC), the DEAD-box ATPase Prp28, and the Msl5•Mud2 branchpoint-binding complex (Liao et al. 1991; Abovich et al. 1994; Colot et al. 1996; Gottschalk et al. 1998; Hausmann et al. 2008; Wilmes et al. 2008; Costanzo et al. 2010; Chang et al. 2012; Qiu et al. 2012; Schwer et al. 2013; Schwer and Shuman 2014, 2015; Jacewicz et al. 2015). This network is defined by the numerous instances in which null alleles of inessential players (e.g., Mud1, Nam8, Mud2) or benign mutations in essential factors (e.g., Yhc1, Snp1, Msl5) elicit synthetic lethal and sick phenotypes when combined with other benign mutations in the splicing machinery.
Several of the essential subunits of the S. cerevisiae U1 snRNP are still comparatively uncharted with respect to their structure–activity relations and genetic interactions. In the present study, we focus on Luc7, a 261-aa polypeptide encoded by the YDL087C ORF on chromosome 4. The LUC7 gene was identified initially in a genetic screen for synthetic lethality with cbc2Δ sto1Δ double-deletion of the nuclear CBC subunits (Fortes et al. 1999b). The “LUC” screen (Lethal Unless CBC is produced) yielded 14 complementation groups of LUC mutants, many of which could be assigned to known spliceosome components, including (i) U1 snRNP subunits Mud1, Nam8, Snu56, and Snu71; (ii) the SmD3 subunit of the Sm ring; and (iii) the Mud2 subunit of the branchpoint binding protein (Fortes et al. 1999b). Subsequent studies showed that Luc7 is a stoichiometric subunit of the yeast U1 snRNP and that a luc7-ts allele was associated with reduced efficiency of splicing of a reporter pre-mRNA with a nonconsensus 5′SS sequence GUAUAU (Fortes et al. 1999a). U1 snRNP isolated from a luc7-ts strain grown at restrictive temperature was deficient in both Luc7 and Snu71 subunits (Fortes et al. 1999a). Two-hybrid experiments identified a physical interaction between Luc7 and the essential FF1 domain of Prp40 (Ester and Uetz 2008). It was suggested that the tandem FF domains of Prp40 nucleate a Prp40•Luc7•Snu71 subcomplex with the yeast U1 snRNP (Ester and Uetz 2008). In vitro crosslinking of Luc7 to the 5′ exon of a 32P-labeled 4-thio-U substituted pre-mRNA substrate that was incubated in a yeast extract, in a manner dependent on the 5′SS consensus sequence in the pre-mRNA, led to the suggestion that Luc7 stabilizes the U1 snRNP•pre-mRNA complex (Puig et al. 2007).
The primary structure of S. cerevisiae Luc7 is shown in Figure 1, aligned to the Luc7 homologs from Aspergillus fumigatus (Genbank accession XP_750789), Trichoderma atroviride (Genbank accession EHK44709), and Homo sapiens (Luc7-like 1 isoform a; Genbank accession NP_060502). As noted previously (Puig et al. 2007), Luc7 proteins are distinguished by two putative zinc finger (ZnF) motifs, which we refer to henceforth as ZnF1 (comprising Cys45, Cys53, Cys68, and His72 in S. cerevisiae Luc7) and ZnF2 (Cys201, Cys204, His220, and His226 in S. cerevisiae Luc7) (Fig. 1). To our inspection, there are no other instructive amino acid sequence motifs in S. cerevisiae Luc7. The longer 325-amino acid human Luc7-like polypeptide has a unique C-terminal segment that is rich in runs of arginines, with interspersed acidic residues, and Ser-Arg dipeptide repeats (Fig. 1). The A. fumigatus and T. atroviride Luc7 proteins have C-terminal extensions peppered with runs of glycines and Arg-Gly repeats, respectively. S. cerevisiae Luc7 has no such low-complexity C-terminal segment. A key distinction between the S. cerevisiae and human Luc7 proteins is that the former is an integral subunit of the yeast U1 snRNP, whereas the latter is not a subunit of the human U1 snRNP, which contains only three U1-specific subunits: U1-70K, U1-A, and U1-C, which are the human homologs of yeast Snp1, Mud1, and Yhc1, respectively (Kondo et al. 2015).
FIGURE 1.

Phylogeny-guided mutational analysis of Luc7. The primary structures of the Luc7 polypeptides of Saccharomyces cerevisiae (Sce), Aspergillus fumigatus (Afu), Trichoderma atroviride (Tat), and Homo sapiens (Hsa) are aligned. Positions of side chain identity/similarity are indicated by (•). Gaps in the alignment are denoted by dashes. Forward and reverse arrows indicate the boundaries of S. cerevisiae Luc7 N-terminal and C-terminal truncations, respectively. Black arrows denote biologically active truncations; red arrows indicate lethal truncations. The conserved amino acids of the N-terminal CCCH ZnF1 module are shaded gold; the amino acids of the conserved C-terminal CCHH ZnF2 module are shaded green. Eight other conserved positions subjected to mutagenesis in the present study are shaded magenta.
Here we present a phylogenetically guided in vivo mutational analysis of S. cerevisiae Luc7, entailing N- and C-terminal truncations and alanine scanning of conserved amino acids. We test a series of 23 alleles for phenotypes per se and for synthetic genetic interactions with a broad spectrum of other early-stage spliceosome components or splicing factors. We define Luc7-(31–246) as a minimal functional unit and demonstrate that whereas mutations of the CCHH ZnF2 motif are lethal, mutations of the ZnF1 CCCH motif are not. A hierarchy of mutational synergies of otherwise benign Luc7 alleles highlights the importance of the N-terminal 18-aa segment and amino acids surrounding ZnF2. By identifying LUC7 alleles that bypass the essentiality of Prp28, we implicate specific constituents of Luc7 in stabilizing the U1•5′SS interaction.
RESULTS
Mapping the proximal and distal margins of the functional Luc7 protein
A series of N-terminal and C-terminal truncation alleles were placed on CEN LEU2 plasmids under the control of the native LUC7 promoter and tested by plasmid shuffle for complementation of a luc7Δ p[CEN URA3 LUC7] strain. We found that deleting 18 or 30 amino acids from the N terminus (black arrows in Fig. 1) had no apparent impact on yeast growth at 25°C–37°C, as gauged by colony size (Fig. 2). (The LUC7-[31–261] strain formed smaller colonies at 20°C.) In contrast, an N-terminal truncation of 90 amino acids (red arrow in Fig. 1), which eliminated the ZnF1 module, was lethal (i.e., no FOA-resistant colonies were recovered after plasmid shuffle performed at 20°C, 30°C, and 37°C). Whereas deleting the C-terminal segment from amino acids 247–261 had no effect on yeast growth, further deletion of amino acids 234–246 elicited a temperature-sensitive (ts) growth defect, i.e., the LUC7-(1–233) strain grew as well as wild-type LUC7 at 20°C–25°C but grew slowly at 30°C and failed to grow at 34°C–37°C (Fig. 2). The more extensive C-terminal deletion LUC7-(1–198), which eliminated the ZnF2 module, was lethal. To query whether the dispensable N-terminal and C-terminal segments make genetically redundant contributions to Luc7 activity in vivo, we combined the deletions and found that the LUC7-(31–246) strain was viable, albeit slightly cold-sensitive (cs) (Fig. 2).
FIGURE 2.

Deletion and alanine-scanning mutagenesis of yeast Luc7. Truncated LUC7 alleles (top panel), alanine mutants of the cysteines and histidines of the ZnF1 and ZnF2 motifs (middle panels), and additional alanine mutants in the ZnF2 domain (bottom panel) were tested for activity by plasmid shuffle. The viable FOA-resistant luc7Δ strains bearing the indicated LUC7 alleles were spot-tested for growth on YPD agar at the temperatures specified. LUC7 alleles listed at the bottom of each panel failed to complement luc7Δ in the plasmid shuffle assay and were deemed lethal.
Alanine scanning mutagenesis of the ZnF1 and ZnF2 cysteines and histidines
Cys45, Cys53, Cys68, and His72 comprising the ZnF1 motif were mutated individually to alanine. The LUC7 C68A and H72A strains thrived at all temperatures from 20°C–37°C. The C45A and C53A strains grew well at 20°C–25°C but grew very slowly at 30°C–34°C (as gauged by colony size) and failed to grow at 37°C (Fig. 2). Thus, whereas a deletion of the segment from amino acids 36–90 spanning the ZnF1 motif was lethal, elimination of any one of the four putative metal-binding side chains of ZnF1 was not. These results are consistent with scenarios in which (i) the imputed metal-binding function of ZnF1 is not essential per se for Luc7 bioactivity; (ii) any three of the four ZnF1 amino acids suffices for metal-binding and bioactivity; or (iii) ZnF1 function is genetically redundant with that of ZnF2.
Entirely different results were obtained when we introduced alanines in lieu of the Cys201, Cys204, His220, and His226 residues of the ZnF2 motif. The C201A, C204A, and H226A mutations were lethal. The H220A allele gave rise to FOA-resistant colonies at 20°C during the plasmid shuffle procedure; however, the H220A strain was barely viable on YPD agar at 20°C and failed to grow at higher temperatures (Fig. 2). The fact that each of the imputed metal ligands of ZnF2 is critical for Luc7 bioactivity, whereas those of ZnF1 are not, vitiates the idea that ZnF1 and ZnF2 are functionally redundant.
Alanine scanning of conserved amino acids in the ZnF2 module
Primary structure conservation among the Luc7 proteins aligned in Figure 1 is greatest within the segment of S. cerevisiae Luc7 from amino acids 196–233 that embraces ZnF2, i.e., 25/38 positions of side chain identity in all four Luc7 homologs. We extended the alanine scan to eight invariant basic or acidic residues in this ZnF2 module: Lys197, Asp212, Asp214, Arg215, Arg216, Asp219, Lys224, and Arg233 (shaded magenta in Fig. 1). All of the eight LUC7-Ala strains were viable and grew as well as wild-type LUC7 on YPD agar at 25°C–37°C (Fig. 2). Only R216A cells were slightly slow-growing at 20°C, as gauged by colony size (Fig. 2).
Synthetic genetic interactions of Luc7 truncation mutants
To survey genetic interactions of the Luc7 truncation mutants, we tested by plasmid shuffle for complementation of strains in which the genes encoding U1 snRNP subunits Nam8 or Mud1 or the branchpoint binding protein subunit Mud2 were deleted in the luc7Δ p[CEN URA3 LUC7] background. Yeast nam8Δ, mud1Δ, and mud2Δ single mutants grow as well as wild-type yeast at all temperatures. We also tested for complementation of a luc7Δ shuffle strain lacking the gene encoding the TMG capping enzyme Tgs1. Yeast tgs1Δ cells grow well at 30°C–37°C, but they fail to grow at 20°C. In addition, we tested complementation of a luc7Δ shuffle strain in which the Cbc2 subunit of the nuclear m7G cap-binding complex has a Y24A mutation in the cap-binding pocket. Although yeast cbc2-Y24A cells grow as well as wild-type yeast at all temperatures, the Y24A mutation elicits synthetic lethality or sickness with nam8Δ, mud1Δ, and mud2Δ (Qiu et al. 2012). The results are summarized in Figure 3.
FIGURE 3.
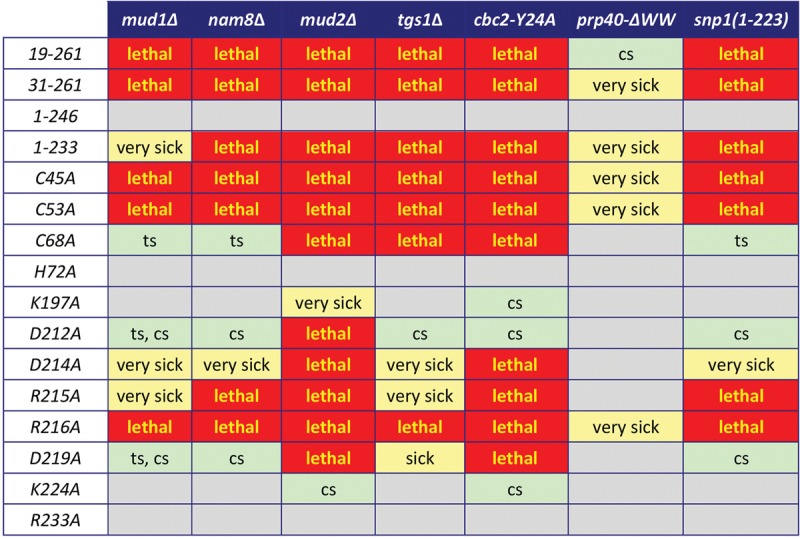
Synthetic interactions of Luc7 mutants. Synthetically lethal pairs of alleles are highlighted in red boxes. Other negative pairwise interactions are classified as sick or very sick (yellow boxes) or temperature-sensitive (ts) or cold sensitive (cs) (light green boxes). Gray boxes denote lack of mutational synergy.
The genetics highlight the importance of the N-terminal segment for Luc7 activity in vivo when other components of the U1 snRNP and the early spliceosome are perturbed. To wit, the LUC7-(19–261) allele was lethal in combination with mud2Δ, nam8Δ, mud1Δ, tgs1Δ, and cbc2-Y24A. The same synthetic lethalities were observed for the LUC7-(31–261) allele (Fig. 3). In contrast, deletion of the C-terminal segment in LUC7-(1–246) elicited no growth defects in the mud2Δ, nam8Δ, mud1Δ, tgs1Δ, or cbc2-Y24A backgrounds (Fig. 3). However, incremental deletion of the segment from amino acids 234–246 in LUC7-(1–233), which resulted in a ts growth defect by itself, was synthetically lethal with mud2Δ, nam8Δ, tgs1Δ, and cbc2-Y24A and synthetically sick with mud1Δ (Fig. 3).
Unlike the “optional” splicing factors surveyed above, the U1 snRNP subunits Prp40 and Snp1 are essential for yeast vegetative growth. Prp40 is a 583-aa protein containing two tandem WW modules at the N terminus (amino acids 1–70) and four FF domains dispersed downstream (Ester and Uetz 2008). We previously constructed a truncated chromosomal allele, prp40-(77–583) (henceforth named prp40-ΔWW), that cleanly subtracts the tandem WW modules and found that the prp40-ΔWW strain grew as well as the PRP40 control strain on YPD agar at all temperatures (Schwer et al. 2013). Here we tested the Luc7 truncation mutants for complementation of a prp40-ΔWW luc7Δ shuffle strain. LUC7-(19–261) displayed a cs growth defect in the prp40-ΔWW background. LUC7-(31–261) was synthetically sick at 34°C–37°C in combination with prp40-ΔWW; the LUC7-(31–261) prp40-ΔWW strain failed to grow at 20°C–30°C (classified as “very sick” in Fig. 3). LUC7-(1–246) elicited no growth phenotype in the prp40-ΔWW background. The LUC7-(1–233) prp40-ΔWW strain was sick at 20°C–25°C and failed to thrive at 30°C–37°C (Fig. 3, very sick).
Snp1, a 300-aa polypeptide, is the yeast homolog of human U1-70K (Kao and Siliciano 1992). We reported previously that yeast snp1Δ cells bearing a C-terminal truncation allele, snp1(1–223), grow as well as wild-type SNP1 cells on YPD agar at all temperatures (Qiu et al. 2015). snp1(1–223) displays a distinctive spectrum of mutational synergies whereby it is (i) synthetically lethal with mud2Δ; (ii) severely synthetically sick and cs with nam8Δ; (iii) benign in combination with mud1Δ; and (iv) suppresses the cs defect of tgs1Δ (Qiu et al. 2015). Testing the Luc7 truncation mutants for complementation of a snp1(1-223) luc7Δ shuffle strain revealed that the N-terminal Luc7 truncations LUC7-(19–261) and LUC7-(31–261) and the C-terminal truncation LUC7-(1–233) were lethal in combination with snp1(1–223). In contrast, the C-terminal truncation LUC7-(1–246) was benign in the snp1(1-223) background (Fig. 3).
Synthetic genetic interactions of Luc7-Ala mutants
All of the viable Luc7-Ala mutants were evaluated for mutational synergies in the seven genetic backgrounds listed in Figure 3. At the severest end of the synergy spectrum was ZnF2 mutant R216A, which was benign on its own but synthetically lethal with mud1Δ, nam8Δ, mud2Δ, tgs1Δ, cbc2-Y24A, and snp1(1–223) and synthetically very sick with prp40-ΔWW (Fig. 3). Mutations of ZnF2 residues flanking Arg216 also synergized strongly. The neighboring upstream ZnF2 mutation R215A was lethal with nam8Δ, mud2Δ, cbc2-Y24A, and snp1(1–223) and very sick with mud1Δ and tgs1Δ. At the next amino acid upstream, D214A was lethal with mud2Δ and cbc2-Y24A and very sick with mud1Δ, nam8Δ, tgs1Δ, and snp1(1–223). Moving two residues further upstream, D212A was lethal with mud2Δ, elicited cs defects in the nam8Δ, cbc2-Y24A, and snp1(1–223) backgrounds, caused ts and cs defects in mud1Δ cells, and exacerbated the cold-sensitive growth defect of tgs1Δ cells (i.e., raising the restrictive temperature) (Fig. 3). The neighboring downstream mutation D219A was lethal with mud2Δ and cbc2-Y24A, sick with tgs1Δ, cs with nam8Δ and snp1(1-223), and both cs and ts with mud1Δ (Fig. 3). The mutations at the outer margins of the conserved ZnF2 module displayed milder synergies that were confined to mud2Δ and cbc2-Y24A (e.g., K197A and K224A) or elicited no synthetic phenotype in any of the backgrounds surveyed (e.g., R233A).
Synergies of ZnF1 alanine mutations ranged from the entirely benign in the case of H72A to the pan-catastrophic for C45A and C53A. Whereas C45A and C53A caused ts growth defects by themselves, these alleles were lethal with mud1Δ, nam8Δ, mud2Δ, tgs1Δ, cbc2-Y24A, and snp1(1–223) and very sick with prp40-ΔWW. The C68A mutation, which caused no growth phenotype per se, was lethal with mud2Δ, tgs1Δ, and cbc2-Y24A and ts with mud1Δ, nam8Δ, and snp1(1–223) (Fig. 3).
Luc7 mutations bypass the essentiality of Prp28
Yeast Prp28 is an essential DEAD-box ATPase implicated in displacing the U1 snRNP from the 5′SS during the transition from a pre-mRNA•U1•U2 spliceosome to a pre-mRNA•U2•U5•U6 spliceosome. The requirement for Prp28 for vegetative growth can be bypassed by mutations in the essential U1 snRNP subunits Yhc1, Prp42, and Snu71, and by specific U1 snRNA mutations located within and flanking the segment that base-pairs with the intron 5′SS (Chen et al. 2001; Hage et al. 2009; Schwer et al. 2013; Schwer and Shuman 2014, 2015), the common theme being that such mutations are thought to weaken the U1•5′SS contacts and thereby allay the need for Prp28 during U1 snRNP ejection from the early spliceosome.
Here we queried whether any of the Luc7 mutations might bypass Prp28, by transforming a prp28Δ luc7Δ p[CEN URA3 PRP28 LUC7] strain with CEN LEU2 LUC7 plasmids and selecting for growth on medium containing FOA. Control experiments affirmed that plasmid shuffle with a wild-type LUC7 plasmid yielded no FOA-resistant survivors, whereas cotransformation with a wild-type LUC7 plasmid and a CEN HIS3 PRP28 plasmid did. We found that the N-terminal truncations LUC7-(19–261) and LUC7-(31–261) were able to sustain growth of prp28Δ luc7Δ cells on FOA, as were several of the LUC7-Ala mutants. The viable luc7Δ prp28Δ cells bearing these bypass alleles were tested for growth on YPD agar (Fig. 4). LUC7-(19–261) prp28Δ cells grew as well as wild-type LUC7 PRP28 cells at 37°C, 34°C, and 30°C, as gauged by colony size, but they grew slowly at 25°C and did not thrive at 20°C. LUC7-(31–261) prp28Δ cells grew as well as wild-type at 37°C and 34°C, were slower growing at 30°C and 25°C, and did not thrive at 20°C.
FIGURE 4.

Luc7 mutations that bypass Prp28. Yeast prp28Δ luc7Δ cells bearing the indicated LUC7 mutant allele on a CEN LEU2 plasmid were spot-tested for growth on YPD agar at the temperatures specified. Control strains were those that had been cotransformed with plasmids bearing the wild-type PRP28 and LUC7 genes.
Among the Ala mutants, the ZnF2 module allele R215A evinced the strongest Prp28 bypass phenotype, i.e., LUC7-R215A prp28Δ cells grew well at 37°C, 34°C, and 30°C, but did not thrive at 25°C and 20°C. Flanking mutations D214A and R216A also bypassed Prp28 to permit wild-type growth at 37°C and 34°C, while sustaining slow or no growth at lower temperatures. ZnF2 alleles D212A and D219A were comparatively weak Prp28 bypass suppressors, in that they sustained slow growth of prp28Δ cells and only at 34°C–37°C. We surmise that higher temperatures destabilize the weakened U1•5′SS interface in cells bearing the aforementioned Luc7 mutants and thereby permit U1 snRNP ejection without the assistance of Prp28. However, when the U1:5′SS duplex pairing is more stable at lower temperatures, Prp28 is still required. Cold-sensitivity is a characteristic shared with most of the other prp28 bypass mutations described to date (Chen et al. 2001; Hage et al. 2009; Schwer et al. 2013; Schwer and Shuman 2014, 2015).
Exceptions to the cold-sensitivity of Prp28 bypass were evident for ZnF1 alleles C45A and C53A, which supported slow growth of prp28Δ cells at 25°C but did not allow growth at 37°C (Fig. 4). The lack of bypass at 37°C reflects the fact that the C45A and C53A mutations by themselves elicit a ts growth defect (Fig. 2). In contrast, ZnF1 mutant C68A, which has no growth phenotype on its own, affords a relatively weak Prp28 bypass, but it does so in the typical cs fashion (Fig. 4).
Taken together, the mutational synergies and Prp28 bypass experiments underscore the significant contributions of the Luc7 N terminus and the ZnF1 and ZnF2 modules to early spliceosome assembly and stability.
Luc7 mutations affect splicing of SUS1 pre-mRNA
We surveyed our collection of Luc7 mutations that had no effect per se on yeast growth for their effects on the efficacy of in vivo splicing of two specific yeast pre-mRNAs: GLC7, which has a single intron and is efficiently spliced in wild-type cells (Fig. 5A); and SUS1, a two-intron-containing transcript (Fig. 5B). We chose SUS1 in light of our prior findings that SUS1 splicing is adversely affected by the Cbc2-Y24A mutation (Qiu et al. 2012) and by Msl5 mutations that perturb the Msl5•branchpoint RNA interface (Jacewicz et al. 2015). SUS1 is one of the few yeast genes that contain two introns and it is the splicing of the first intron (which has a nonconsensus 5′ splice site GUAUGA and a nonconsensus branchpoint sequence UACUGAC) that is selectively impaired in the absence of CBC (Hossain et al. 2009).
FIGURE 5.
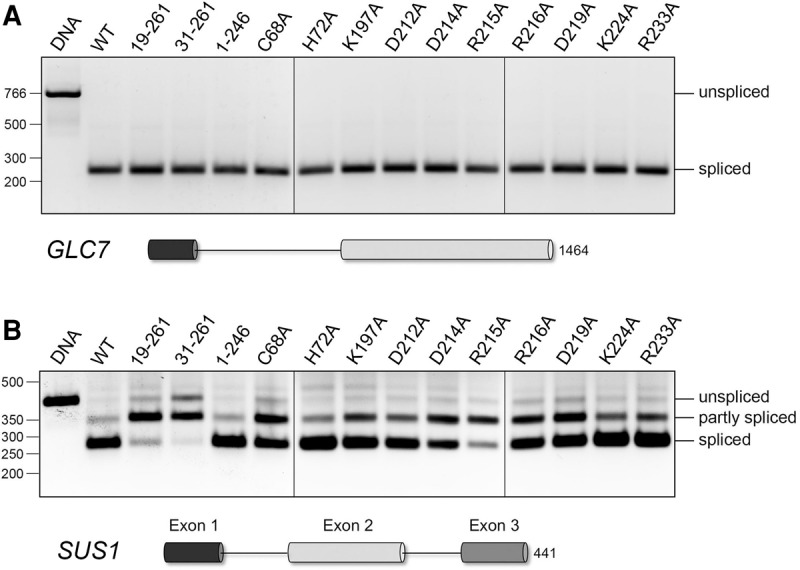
Luc7 mutations affect splicing of SUS1 pre-mRNA in vivo. RNA isolated from the indicated LUC7 strains was reverse transcribed with an oligo(dT) primer. cDNAs were PCR-amplified with gene-specific sense and antisense primer pairs derived from the first and last exons of the GLC7 (panel A) and SUS1 (panel B) genes. The exon–intron organization of the pre-mRNAs is shown, with exons depicted as horizontal cylinders. The PCR products were resolved by native agarose gel electrophoresis and visualized by staining with ethidium bromide. The products of PCR amplification of genomic DNA with the same primer pairs are shown in lanes labeled “DNA.” The positions and sizes (bp) of linear duplex DNA markers are indicated on the left. The RT-PCR products of unspliced, partly spliced, and fully spliced transcripts are specified on the right.
cDNAs synthesized by oligo(dT)-primed reverse transcription of total RNA from yeast strains with wild-type and mutant LUC7 alleles were PCR-amplified using gene-specific primer pairs flanking the first and last introns, such that agarose gel electrophoresis would resolve the longer PCR fragments derived from unspliced pre-mRNA (which comigrates with the control fragment amplified from a genomic DNA template) and the shorter PCR fragments derived from mature spliced mRNA or, in the case of SUS1, partially spliced intermediates. None of the Luc7 mutations affected GLC7 splicing (Fig. 5A).
As reported previously (Hossain et al. 2009; Qiu et al. 2012; Jacewicz et al. 2015), the SUS1 transcripts in wild-type cells consisted predominantly of doubly spliced mature mRNA plus a minor component of partly spliced intermediate species (Fig. 5B). In LUC7-(19–261), -(31–261), and R215A cells, the mature mRNA was diminished and the singly spliced intermediate comprised the majority species (Fig. 5B). These three LUC7 alleles that exerted the greatest impact on SUS1 splicing efficiency were also ones that exhibited the widest spectrum of synthetic lethalities and that were most effective in bypassing Prp28. LUC7 alleles C68A, D214A, R216A, and D219A also affected SUS1 splicing efficiency, manifest by a shift in the transcript distribution toward the partly spliced intermediate, albeit not to the point that it was the majority species detected by RT-PCR (Fig. 5B). C68A, D214A, R216A, and D219A all displayed multiple synthetic genetic interactions and were able to bypass Prp28. In contrast, the LUC7-(1–246), H72A, R233A, and K224A alleles had little or no impact on SUS1 splicing (as gauged by the relative amounts of fully spliced versus partly spliced species); these alleles were the ones that (i) displayed either no synthetic phenotypes—in the case of LUC7-(1–246), H72A, and R233A—or narrowly mild mutation synergies (K224A); and (ii) were unable to bypass Prp28.
We surveyed the same wild-type and mutant LUC7 strains for splicing of the MATa1 pre-mRNA, which contains two introns, albeit both with consensus splicing signals. PCR amplification of the MATa1 cDNA with primers in exon 1 and exon 3 revealed efficient removal of both introns in wild-type cells, where the mature spliced mRNA was the predominant species, with a minority fraction of partially spliced intermediate, and hardly any unspliced precursor (Supplemental Fig. S1A). Whereas the LUC7-(31–261) strain had a diminished level of mature MATa1 transcript, the other LUC7 mutants maintain the wild-type distribution of mature and partially spliced species.
Finally, we evaluated the effects of the Luc7 N-terminal truncations on the splicing of the single-intron YFR045w and PMI40 pre-mRNAs. YFR045w, which has a nonconsensus 5′ splice site GUAAGU and a nonconsensus branchpoint sequence UAUUAAC, was relatively inefficiently spliced in wild-type cells (e.g., compared to GLC7); unspliced YFR045w precursor was evident by RT-PCR, but was less abundant than the spliced mature transcript (Supplemental Fig. S1B). We found that YFR045w splicing was impeded in LUC7-(19-261) and LUC7-(31-261) cells, where the mature YFR045w mRNA was diminished and the unspliced pre-mRNA was now the majority species (Supplemental Fig. S1B). (Note that YFR045w splicing in LUC7-[1–246] cells was no different from the wild-type strain [Supplemental Fig. S1B].)
In contrast, the PMI40 pre-mRNA, which has a nonconsensus branchpoint sequence (AACUAAC) but a consensus 5′SS, was spliced very efficiently in wild-type LUC7 cells and was unaffected by the LUC7 N-terminal truncations (Supplemental Fig. S1C).
DISCUSSION
The present study affords new genetic insights into Luc7 function and structure–activity relations. We defined Luc7-(31–246) as a minimal functional unit for complementation of vegetative growth of a luc7Δ null strain. Though dispensable for growth in an otherwise wild-type background, the N-terminal peptide of Luc7 (1MSTMSTPAAEQRKLVEQL18) plays an important role in U1 snRNP function, which was evinced by our findings that the LUC7-(19-261) allele (i) impaired the splicing of SUS1 and YFR045w pre-mRNAs in vivo; (ii) was synthetically lethal absent other U1 snRNP constituents (Mud1, Nam8, the TMG cap, the C terminus of Snp1), absent Mud2, and when the cap-binding site of nuclear CBC was debilitated; and (iii) bypassed the need for the essential DEAD-box ATPase Prp28. This constellation of effects implicates the N-terminal peptide in fulfilling two aspects of Luc7 function: in stability of the U1•pre-mRNA complex and in aiding the splicing of yeast pre-mRNAs that have a nonconsensus 5′SS element. The N-terminal peptide includes eight positions of side chain conservation in all four Luc7 proteins aligned in Figure 1.
Alanine scanning revealed that whereas mutations of the CCHH ZnF2 motif are lethal, mutations of the ZnF1 CCCH motif are not, signifying that the ZnF1 and ZnF2 motifs are not functionally redundant. Although the ZnF2 module bracketed by the essential putative metal-binding cysteines and histidines is conserved among the Luc7 homologs, none of the eight alanine changes introduced in lieu of conserved basic and acidic residues affected yeast growth. Yet, the alanine scan highlighted the functional importance of individual ZnF2 module constituents Asp214, Arg215, Arg216, and Asp219, as gauged by their effects on SUS1 splicing, synergies with other splicing mutations, and Prp28 bypass suppression. The results suggest a scenario in which a putative metal-coordination complex formed by the CCHH ZnF2 motif establishes a specific tertiary structure of the 201CEVCGAYLSRLDTDRRLADHFLGKIH226 segment that is required for Luc7 activity, likely via contacts made by the conserved charged amino acids that our mutagenesis identified as relevant. It is noteworthy that Ester and Uetz (2008) had previously identified a 15-aa peptide within the ZnF2 module—214DRRLADHFLGKIHLG228—in their screen of an overlapping Luc7 peptide library for interaction with the tandem FF1-2 domains of yeast Prp40. Focusing further on binding of this peptide to the Prp40 FF1 domain, they conducted an alanine scan of every position in the Luc7 peptide. Most pertinent to our results, they reported that alanine substitutions for Asp214, Arg215, Arg216, Asp219, and His220 did not diminish Luc7 peptide interaction with the Prp40 FF1 domain (Ester and Uetz 2008). We infer that the in vivo phenotypes we see for the D214A, R215A, R216A, D219A, and H220A alleles do not reflect a simple defect in the imputed Luc7•Prp40-FF1 interaction. Because Ester and Uetz (2008) did note that a K224A mutation of the Luc7 peptide nearly effaced its interaction with Prp40-FF1, it is conceivable that the mild in vivo mutational synergies noted here for the LUC7-K224A allele might arise via an effect on Luc7•Prp40-FF1 interaction.
Three of the candidate metal-ligands of the ZnF1 motif (Cys45, Cys53, and Cys68), though not individually essential for yeast survival, are functionally relevant by the following criteria: inefficient SUS1 splicing (in C68A cells); temperature-sensitive growth defects (C45A and C53A); and severe synthetic phenotypes and Prp28 bypass (C45A, C53A, and C68A). His72 was unique in that its mutation to alanine elicited no effects in any of the tests we applied. Assuming that the ZnF1 motif really does coordinate a metal, it is possible that the place of the His72 ligand in the metal coordination complex can be taken either by another (albeit nonconserved) histidine in the vicinity of the ZnF1 motif (e.g., His40 or His76) or by a water molecule.
The genetic analysis here establishes a foundation for understanding Luc7 function that will ultimately hinge on obtaining a crystal structure of yeast Luc7 per se and/or a cryo-EM structure of the complete yeast U1 snRNP.
MATERIALS AND METHODS
Luc7 expression plasmids and mutants
A 1.8-kb DNA segment bearing the LUC7 gene was amplified from S. cerevisiae genomic DNA by PCR using a forward primer that introduced a SacI site 500-bp upstream of the Luc7 translation initiation site and a reverse primer to introduce a XhoI site 500-bp downstream from the stop codon. The PCR product was digested with SacI and XhoI and inserted between the SacI and XhoI sites of pRS316 (CEN URA3). A second yeast expression plasmid, p415-LUC7 (CEN LEU2), containing the LUC7 ORF plus 500 bp of upstream and 500 bp of downstream genomic DNA, was constructed so as to introduce a 5′ BamHI site and a 3′ HindIII site immediately flanking the ORF. N-terminal truncation Luc7-(19–261) was generated by PCR using a forward primer that introduced a BamHI site preceding the Met19 codon. Luc7-(31–261) was generated with a forward primer that introduced a 5′ BamHI site and a Met codon in lieu of the Tyr30 codon. C-terminal truncations were generated by PCR using reverse primers that introduced stop codons and flanking HindIII sites in lieu of Asn106, Gln199, Glu234 or Asn247. The truncated PCR products were inserted into pRS415-LUC7 in lieu of the wild-type ORF to generate plasmids p415-LUC7-(19–261), p415-LUC7-(31–261), p415-LUC7-(1–198), p415-LUC7-(1–233), p415-LUC7-(1–246), and p415-LUC7-(31–246). Single alanine mutations were introduced into the LUC7 gene by two-stage PCR overlap extension with mutagenic primers. The PCR products were digested with BamHI and HindIII and inserted into the pRS415-based expression vector to generate a series of p415-LUC7-Ala plasmids. The LUC7 genes were sequenced completely to confirm that no unwanted changes were acquired during amplification and cloning.
Yeast strains and tests of Luc7 function in vivo
To develop a plasmid shuffle assay for gauging mutational effects on Luc7 function, we generated a luc7Δ strain that relies for viability on maintenance of a CEN URA3 LUC7 plasmid (p316-LUC7). A BY4743 diploid strain in which one chromosomal LUC7 locus from ORF positions +1 to +787 was replaced with a kanMX cassette was transformed with p316-LUC7. The diploid was sporulated, asci were dissected, and haploid luc7Δ Ura+ progeny were recovered. The luc7Δ [p316-LUC7] cells were resistant to kanamycin and unable to grow on medium containing 0.75 mg/mL FOA (5-fluoroorotic acid). The luc7Δ [p316-LUC7] cells were transfected with CEN LEU2 LUC7 plasmids expressing wild-type or mutated Luc7 proteins. Individual Leu2+ transformants were selected and streaked on agar medium containing FOA. LUC7 mutant alleles that failed to give rise to FOA-resistant colonies at any temperature after incubation for 8 d at 20°C, 30°C, or 37°C were deemed lethal. Individual FOA-resistant colonies with viable LUC7 alleles were grown to mid-log phase in YPD broth and adjusted to A600 of 0.1. Aliquots (3 µL) of serial 10-fold dilutions were spotted to YPD agar plates, which were incubated at 20°C, 25°C, 30°C, 34°C, and 37°C.
Tests of mutational synergy
We developed plasmid shuffle assays to test the effects of Luc7 mutations in mud2Δ, nam8Δ, mud1Δ, tgs1Δ, cbc2-Y24A, prp40-ΔWW, and snp1-(1–223) genetic backgrounds. Via standard genetic manipulations of mating, sporulation, and dissection, we generated mud2Δ luc7Δ [p316-LUC7], nam8Δ luc7Δ [p316-LUC7], mud1Δ luc7Δ [p316-LUC7], tgs1Δ luc7Δ [p316-LUC7], cbc2-Y24A luc7Δ [p316-LUC7], prp40-ΔWW luc7Δ [p316-LUC7], and snp1Δ luc7Δ [p316-SNP1-LUC7] haploid strains, which were unable to grow on FOA-containing medium unless they had been previously transformed with wild-type LUC7 or a functional LUC7 mutant allele on a CEN LEU2 plasmid or, in the case of snp1Δ luc7Δ [p316-SNP1-LUC7], a CEN LEU2 LUC7 plasmid plus a CEN HIS3 plasmid bearing the snp1-(1–223) allele.
Test of Prp28 bypass
Heterozygous prp28Δ/PRP28 luc7Δ/LUC7 diploids were obtained by crossing prp28Δ::natMX p316-PRP28 (CEN URA3 PRP28) with luc7Δ::kanMX p316-LUC7 cells of the opposite mating type, selecting diploids on YPD medium containing clonNat plus G418, and plating them to FOA-containing medium to select against the two URA3 plasmids. The heterozygous diploid was then transfected with a CEN URA3 LUC7 PRP28 plasmid (p316-LUC7-PRP28) in which the LUC7 gene (nucleotides −500 to +500) is arranged in a head-to-tail configuration with the PRP28 gene (nucleotides −520 to +560). Ura+ heterozygous diploids were subjected to sporulation and tetrad dissection, after which haploid luc7Δ prp28Δ [p316-LUC7-PRP28] progeny were recovered. The luc7Δ prp28Δ [p316-LUC7-PRP28] strain was unable to grow on FOA medium unless it had been subjected to prior cotransformation with CEN LEU2 LUC7 and CEN HIS3 PRP28 plasmids bearing the wild-type LUC7 and PRP28 genes. Transformation with the CEN LEU2 LUC7 plasmid alone yielded no FOA-resistant survivors. To test for bypass suppression of prp28Δ by LUC7 mutants, luc7Δ prp28Δ [p316-LUC7-PRP28] cells were transfected with CEN LEU2 plasmids expressing truncated or alanine-substituted Luc7 proteins. Individual Leu2+ transformants were selected and streaked on agar medium containing FOA. LUC7 mutant alleles that gave rise to FOA-resistant colonies after incubation for up to 8 d at 20°C, 30°C, or 37°C were deemed Prp28 bypass suppressors. Viable prp28Δ colonies with bypass LUC7 alleles were grown to mid-log phase in YPD broth and adjusted to A600 of 0.1. Aliquots (3 µL) of serial 10-fold dilutions were spotted to YPD agar plates, which were incubated at 20°C, 25°C, 30°C, 34°C, and 37°C.
RT-PCR assays of pre-mRNA splicing in vivo
Yeast luc7Δ p316-LUC7 haploid strains with wild-type LUC7 or a “benign” LUC7 mutant allele (i.e., no growth defect on YPD agar) on a CEN LEU2 plasmid were grown in liquid YPD medium at 30°C until A600 reached 0.6–0.8. Cells (20 A600 units) were harvested by centrifugation and total cellular RNA was isolated by the hot phenol method. The RNA preparations were treated with DNase I. First-strand cDNA synthesis was carried out by using the ProtoScript First Strand cDNA synthesis kit (NEB) with 1 µg of total RNA template and an oligo(dT)23 primer. Aliquots of the mixtures were then used for 35 cycles of PCR amplification by Taq polymerase (NEB) of the SUS1 cDNAs with 0.2 µM oligonucleotide primers 5′-TGGATACTGCGCAATTAAAGAGTC and 5′-TCATTGTGTATCTACAATCTCTTCAAG, complementary to the first and third exons of the SUS1 transcript, and of the GLC7 cDNAs with 0.2 µM primers 5′-CCTGGTCAACAAGTTGATCTAGAAG and 5′-CTAAGGATTGTTTACCACGGTCGAC, complementary to the two GLC7 exons. The PCR products were resolved by electrophoresis through a native 2% agarose gel and visualized by staining with ethidium bromide.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health–National Institute of General Medical Sciences grants GM52470 (S.S. and B.S.) and GM102961 (B.S.).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.056911.116.
REFERENCES
- Abovich N, Rosbash M. 1997. Cross-intron bridging interactions in the yeast commitment complex are conserved in mammals. Cell 89: 403–412. [DOI] [PubMed] [Google Scholar]
- Abovich N, Liao XC, Rosbash M. 1994. The yeast MUD2 protein: an interaction with PRP11 defines a bridge between commitment complexes and U2 snRNP addition. Genes Dev 8: 843–854. [DOI] [PubMed] [Google Scholar]
- Chang J, Schwer B, Shuman S. 2012. Structure-function analysis and genetic interactions of the yeast branchpoint binding protein Msl5. Nucleic Acids Res 40: 4539–4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JY, Stands L, Staley JP, Jackups RR Jr, Latus LJ, Chang TH. 2001. Specific alterations of U1-C protein or U1 small nuclear RNA can eliminate the requirement of Prp28p, an essential DEAD box splicing factor. Mol Cell 7: 227–232. [DOI] [PubMed] [Google Scholar]
- Colot HV, Stutz F, Rosbash M. 1996. The yeast splicing factor Mud13p is a commitment complex component and corresponds to CBP20, the small subunit of the nuclear cap-binding complex. Genes Dev 10: 1699–1708. [DOI] [PubMed] [Google Scholar]
- Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JL, Toufighi K, Mostafavi S, et al. 2010. The genetic landscape of a cell. Science 327: 425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ester C, Uetz P. 2008. The FF domains of yeast U1 snRNP protein Prp40 mediate interactions with Luc7 and Snu71. BMC Biochem 9: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortes P, Bilbao-Cortés D, Fornerod M, Rigaut G, Raymond W, Séraphin B, Mattaj IW. 1999a. Luc7p, a novel yeast U1 snRNP protein with a role in 5′ splice site recognition. Genes Dev 13: 2425–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortes P, Kufel J, Fornerod M, Polycarpou-Schwarz M, Lafontaine D, Tollervey D, Mattaj IW. 1999b. Genetic and physical interaction involving the yeast nuclear cap-binding complex. Mol Cell Biol 19: 6543–6553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschalk A, Tang J, Puig O, Salgado J, Neubauer G, Colot HV, Mann M, Séraphin B, Rosbash M, Lührmann R, et al. 1998. A comprehensive biochemical and genetic analysis of the yeast U1 snRNP reveals five novel proteins. RNA 4: 374–393. [PMC free article] [PubMed] [Google Scholar]
- Hage R, Tung L, Du H, Stands L, Rosbash M, Chang TH. 2009. A targeted bypass screen identifies Ynl187p, Prp42p, Snu71p, and Cbp80p for stable U1 snRNP/pre-mRNA interaction. Mol Cell Biol 29: 3941–3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausmann S, Zheng S, Costanzo M, Brost RL, Garcin D, Boone C, Shuman S, Schwer B. 2008. Genetic and biochemical analysis of yeast and human cap trimethylguanosine synthase: functional overlap of TMG caps, snRNP components, pre-mRNA splicing factors, and RNA decay pathways. J Biol Chem 283: 31706–31718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain MA, Claggett JM, Nguyen T, Johnson TL. 2009. The cap binding complex influences H2B ubiquitination by facilitating splicing of the SUS1 pre-mRNA. RNA 15: 1515–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacewicz A, Schwer B, Smith P, Shuman S. 2014. Crystal structure, mutational analysis, and RNA-dependent ATPase activity of the yeast DEAD-box pre-mRNA splicing factor Prp28. Nucleic Acids Res 42: 12885–12898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacewicz A, Chico L, Smith P, Schwer B, Shuman S. 2015. Structural basis for recognition of intron branchpoint RNA by yeast Msl5 and selective effects of interfacial mutations on splicing of yeast pre-mRNAs. RNA 21: 401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao HY, Siliciano PG. 1992. The yeast homolog of the U1 snRNP protein 70K is encoded by the SNP1 gene. Nucleic Acids Res 20: 4009–4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo Y, Oubridge C, van Roon AM, Nagai K. 2015. Crystal structure of human U1 snRNP, a small nuclear ribonucleoprotein particle, reveals the mechanism of 5′ splice site recognition. eLife 4: e04986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao XC, Tang J, Rosbash M. 1991. An enhancer screen identifies a gene that encodes the yeast U1 snRNP A protein: implications for snRNP protein function in pre-mRNA splicing. Genes Dev 7: 419–428. [DOI] [PubMed] [Google Scholar]
- Puig O, Bragado-Nilsson E, Koski T, Séraphin B. 2007. The U1 snRNP-associated factor Luc7p affects 5′ splice site selection in yeast and human. Nucleic Acids Res 35: 5874–5885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu ZR, Chico L, Chang J, Shuman S, Schwer B. 2012. Genetic interactions of hypomorphic mutations in the m7G cap binding pocket of yeast nuclear cap binding complex: an essential role for Cbc2 in meiosis via splicing of MER3 pre-mRNA. RNA 18: 1996–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu ZR, Schwer B, Shuman S. 2015. Two routes to genetic suppression of RNA trimethylguanosine cap deficiency, via C-terminal truncation of U1 snRNP subunit Snp1 or overexpression of RNA polymerase subunit Rpo26. G3 (Bethesda) 5: 1361–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Shuman S. 2014. Structure-function analysis of the Yhc1 subunit of yeast U1 snRNP and genetic interactions of Yhc1 with Mud2, Nam8, Mud1, Tgs1, U1 snRNA, SmD3 and Prp28. Nucleic Acids Res 42: 4697–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Shuman S. 2015. Structure-function analysis and genetic interactions of the Yhc1, SmD3, SmB, and Snp1 subunits of yeast U1 snRNP and genetic interactions of SmD3 with U2 snRNP subunit Lea1. RNA 21: 1173–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Erdjument-Bromage H, Shuman S. 2011. Composition of yeast snRNPs and snoRNPs in the absence of trimethylguanosine caps reveals nuclear cap binding protein as a gained U1 component implicated in the cold-sensitivity of tgs1Δ cells. Nucleic Acids Res 39: 6715–6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Chang J, Shuman S. 2013. Structure-function analysis of the 5′ end of yeast U1 snRNA highlights genetic interactions with the Msl5•Mud2 branchpoint binding complex and other spliceosome assembly factors. Nucleic Acids Res 41: 7485–7500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley JP, Guthrie C. 1999. An RNA switch at the 5′ splice site requires ATP and the DEAD box protein Prp28p. Mol Cell 3: 55–64. [DOI] [PubMed] [Google Scholar]
- Wilmes GM, Bergkessel M, Bandyopadhyay S, Shales M, Braberg H, Cagney G, Collins SR, Whitworth GB, Kress TL, Weissman JS, et al. 2008. A genetic interaction map of RNA-processing factors reveals links between Sem1/Dss1-containing complexes and mRNA export and splicing. Mol Cell 32: 735–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.