

Abstract
The eukaryotic mRNA 5′ cap structure is indispensible for pre-mRNA processing, mRNA export, translation initiation, and mRNA stability. Despite this importance, structural and biophysical studies that involve capped RNA are challenging and rare due to the lack of a general method to prepare mRNA in sufficient quantities. Here, we show that the vaccinia capping enzyme can be used to produce capped RNA in the amounts that are required for large-scale structural studies. We have therefore designed an efficient expression and purification protocol for the vaccinia capping enzyme. Using this approach, the reaction scale can be increased in a cost-efficient manner, where the yields of the capped RNA solely depend on the amount of available uncapped RNA target. Using a large number of RNA substrates, we show that the efficiency of the capping reaction is largely independent of the sequence, length, and secondary structure of the RNA, which makes our approach generally applicable. We demonstrate that the capped RNA can be directly used for quantitative biophysical studies, including fluorescence anisotropy and high-resolution NMR spectroscopy. In combination with 13C-methyl-labeled S-adenosyl methionine, the methyl groups in the RNA can be labeled for methyl TROSY NMR spectroscopy. Finally, we show that our approach can produce both cap-0 and cap-1 RNA in high amounts. In summary, we here introduce a general and straightforward method that opens new means for structural and functional studies of proteins and enzymes in complex with capped RNA.
Keywords: mRNA capping, 5′ cap, mRNA decapping, vaccinia virus capping enzyme, 7-methylguanylate cap structure
INTRODUCTION
The 5′ guanine-N7-methyl cap structure is a central architectural feature of eukaryotic mRNA (Reddy et al. 1974; Furuichi and Miura 1975; Shatkin 1976). The presence of the cap structure is essential for almost all subsequent steps in the life cycle of an mRNA, including pre-mRNA splicing (Izaurralde et al. 1994; Fresco and Buratowski 1996; Schwer and Shuman 1996), mRNA export (Hamm and Mattaj 1990), and the initiation of translation (Both et al. 1975; Filipowicz 1978). In addition, the mRNA cap structure stabilizes mRNA by protecting it against 5′-3′ exonucleolytic degradation (Furuichi et al. 1977), and removal of the cap structure irreversibly targets the transcript for decay (Parker and Song 2004). In addition, acquisition of a cap structure is a common mechanism by which viruses ensure efficient translation of their RNA and evasion of the host immune system (Daffis et al. 2010; Decroly et al. 2012).
In the basic eukaryotic cap structure (m7G or cap-0), a 5′ N7-methylguanosine is attached to the mRNA body by an unusual 5′-5′ triphosphate linkage (Fig. 1A). The capping enzymes are recruited to the RNA polymerase II through the carboxy-terminal domain (CTD) (Cho et al. 1997) and act cotranscriptionally when the transcript has reached a length of about 20 to 30 nucleotides (nt) (Salditt-Georgieff et al. 1980). Three successive enzymatic steps are required for formation of cap-0 and involve an RNA triphosphatase, an RNA guanylyltransferase, and an RNA guanine-N7-methyltransferase activity. The RNA triphosphatase removes the 5′ γ-phosphate from the nascent pre-mRNA to form an mRNA that contains a 5′ diphosphate. Subsequently the RNA guanylyltransferase transfers GMP from a GTP donor to the 5′ end of the RNA, which results in the formation of the unmethylated cap structure. Finally, the guanine-N7-methyltransferase uses S-adenosyl methionine (SAM) as a methyl donor to methylate the N7 position of the guanine base (Fig. 1A). Although the order of these catalytic steps is conserved in eukaryotes, the enzymes that perform these functions differ between species. In yeast, all three activities reside in separate proteins (Gu et al. 2010), whereas in higher eukaryotes several activities reside on a single protein chain (Takagi et al. 1997).
FIGURE 1.
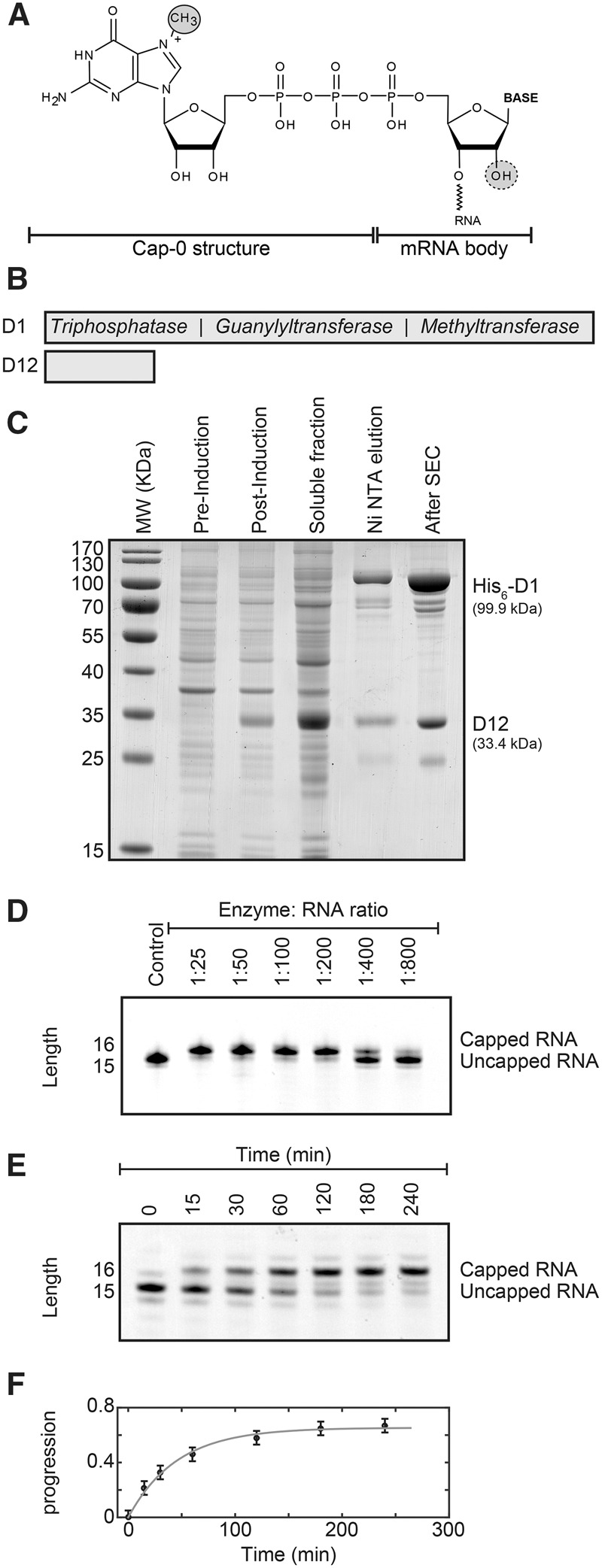
Purification and activity of the vaccinia virus capping enzyme. (A) Structure of the mRNA cap-0. The N7-methyl group in the cap structure is circled. The ribose 2′-OH group that is methylated to yield the cap-1 structure is indicated with a dashed circle. (B) Schematic representation of the vaccinia virus capping enzyme complex that contains the D1 protein with the catalytically active sites and the D12 protein that stabilizes the D1 protein. The D1 protein contains three activities: First, the triphosphatase activity hydrolyzes the RNA 5′ triphosphate into a 5′ diphosphate and inorganic phosphate. Second, the guanylyltransferase activity transfers GMP from GTP onto the 5′ end of the diphosphate RNA. In this step, the 5′-5′ triphosphate bond is formed and pyrophosphate is released. Finally, the methyltransferase activity uses SAM (S-adenosyl methionine) as a methyl donor to methylate the guanine N7 position. (See also Supplemental Fig. 1). (C) Overexpression and purification of the vaccinia virus capping enzyme. The designed plasmid contains the DNA of the D1 and D12 proteins that are codon optimized for overexpression in Escherichia coli (Supplemental Fig. 2). Shown are the E. coli cells before induction of protein expression (pre-induction) and after overexpression of the enzyme complex for 12 h (post-induction), the soluble fraction and the protein after Ni-NTA affinity purification (Ni NTA elution), and the final enzyme complex after size exclusion chromatography (After SEC). Note that the expression of the stabilizing D12 protein is higher than that of the D1 enzyme. The excess of the D12 protein is removed during Ni-NTA purification as only the D1 protein contains an affinity tag. The yield of the purification is ∼12 mg of pure enzyme complex from 5 L of E. coli culture. (D) The purified complex possesses a high mRNA capping activity. Shown are capping reactions with different enzyme:RNA ratios after 1 h incubation time. The capping reaction can be easily followed using Urea-PAGE analysis as the capping reaction adds one base to the RNA substrate. The optimal ratio varies slightly depending on the substrate RNA and should be determined using small-scale capping reactions. (E) To obtain insights into the kinetics of the capping reaction, an uncapped RNA was incubated with a low amount of capping enzyme (1:750). The RNA is capped to a larger degree after longer (>1 h) incubation times. (F) Quantification of the capping reaction shown in E. The solid gray line is a fit of the progression of the reaction [where the progression is defined as capped/(capped + uncapped) RNA] to the experimental data. From the initial slope of the progression curve we extract that the enzyme can cap 16 RNA substrates per minute. The rate of the capping reaction drops over time due to the loss of activity of the enzyme after incubation times that are longer than 1 h.
In yeast, the most predominant RNA cap is the cap-0 structure (Sripati et al. 1976), whereas RNA in higher eukaryotes can be additionally methylated at the nucleotides that follow this cap (Adams and Cory 1975; Furuichi and Shatkin 2000). The most common modification is methylation of the 2′-O position of the ribose of the first nucleotide that follows the cap structure (cap-1), which takes place in the nucleus. The mRNA can be further methylated in the cytoplasm at the 2′-O position of the second ribose to yield a cap-2 structure. In different eukaryotes the levels of cap-0, cap-1, and cap-2 in mRNA vary. The amount of methylation has increased during evolution (Banerjee 1980) and is implicated in immunogenicity (Daffis et al. 2010; Devarkar et al. 2016).
Viruses that replicate in the eukaryotic cytoplasm rely on their own genes that code for the enzymes that provide a cap structure for the viral transcript. To that end, viruses either carry genes that code for a viral RNA capping machinery or that code for a cap-snatching machinery that transfers a cap from cellular mRNA (Reguera et al. 2016). Hence, cap structures have been observed in the majority of such viral RNAs (Shatkin 1976), although with different levels of methylation. As an example, cap-0 structures have been found on mRNA from the tobacco mosaic virus (Zimmern 1975) and cap-1 structures on mRNA from the vaccinia virus (Wei and Moss 1975). The capping enzyme of the vaccinia virus is a complex of the two viral proteins D1 and D12 (Fig. 1B; Ensinger et al. 1975; Martin et al. 1975; Shuman et al. 1980). The three activities are combined in the 97-kDa D1 protein, where the RNA 5′-triphosphatase and guanylyltransferase activities are located in the N-terminal half (Myette and Niles 1996) and the methyltransferase activity localizes to the C-terminal half of the protein (Mao and Shuman 1994). The 31-kDa D12 protein has no catalytic activity, but stimulates the methyltransferase activity of the D1 protein (Mao and Shuman 1994; De la Pena et al. 2007). A structure of the complete D1:D12 vaccinia virus complex has recently been determined and provides insights into the modulation of the catalytic activity by extensive interdomain contacts (Kyrieleis et al. 2014). The vaccinia virus also contains a 2′-O-methyltransferase enzyme (VP39) that converts the cap-0 structure into a cap-1 structure (Barbosa and Moss 1978; Schnierle et al. 1992; Hodel et al. 1998).
Despite the essential role the cap structure plays, detailed knowledge about the interactions of the 5′ end of mRNA with adapter proteins and decapping enzymes is limited. This is, to a large degree, due to challenges related to the in vitro production of pure and homogeneous capped RNA in large quantities (>500 nmol, or 15 mg for an RNA body of 100 nt) that are required for structural studies. Currently, multiple strategies have been proposed to produce capped RNA using enzymatic or chemical methods or a combination of both.
The vaccinia virus capping system has been used to enzymatically prepare limited amounts of capped RNA based on an in vitro transcribed mRNA body (Paterson and Rosenberg 1979; Green et al. 1983; Ray et al. 2006). These capped RNAs are mainly used for small-scale applications including in vitro translation experiments and assays that rely on detection of fluorescent (Gunawardana et al. 2015) or radiolabeled mRNA. Initially, enzymatic capping using the vaccinia virus capping enzyme was reported to be inefficient (Paterson and Rosenberg 1979; Contreras et al. 1982; Pelletier and Sonenberg 1985). Nowadays, the functional enzyme can be obtained commercially; however, the amount of enzyme required to produce high milligram (µmol) amounts of RNA prevents general applicability, and some of the protocols are standardized only for long (>60–100 nt) RNAs. In an alternative approach to obtain capped RNA, a cap analog (m7G-ppp-X) is added in high concentrations directly to the in vitro transcription reaction, which results in the incorporation of the cap structure at the 5′ end of the RNA (Contreras et al. 1982; Konarska et al. 1984; Pelletier and Sonenberg 1985; Nielsen and Shapiro 1986). However, due to the almost symmetric nature of the cap analog, the cap is incorporated in the reverse order in the RNA in at least a third of the transcripts (Pasquinelli et al. 1995). To prevent this reverse cap orientation, “anti-reverse” cap analogs can be used in the transcription reaction, where the 3′ OH group of the m7G nucleotide is modified (Grudzien-Nogalska et al. 2007), which results in the incorporation of a non-natural cap-like structure in the RNA. For very short RNA sequences (<9 nt), the incorporation of a reverse cap can also be prevented by the use of gene 4 primase (Matsuo et al. 2000) or a fragment thereof (Peyrane et al. 2007) that incorporates cap analogs solely in the correct orientation.
Chemically synthesized RNA is not directly suitable for enzymatic capping reactions, as the 5′ end of the RNA is not phosphorylated and thus not a substrate for guanylyltransferase enzymes. Chemically synthesized RNAs can, however, be di- or triphosphorylated at the 5′ end, although often in small scale and with yields in the range of 50% (Brownlee et al. 1995). Recently, optimizations in the solid-phase synthesis of 5′ triphosphate RNA allowed for the production of longer RNA bodies that are up to 130 nt in length (Nagata et al. 2010; Goldeck et al. 2014). The full chemical synthesis of capped RNA sequences is very challenging and inefficient due to the instability of N7-methylguanosine under acidic and basic conditions. This problem has been bypassed by synthesizing nonmethylated capped short RNAs that were N7-methylated in a subsequent enzymatic step (Thillier et al. 2012). Full chemical synthesis of small (1–3 bases) RNAs is feasible, even with very complex methylation patterns (Lewdorowicz et al. 2007); however, longer capped RNAs seem too challenging for chemical synthesis. Finally, it has been reported that 7-methylguanosine 5′-diphosphate imidazolide can be chemically coupled with 5′-monophosphorylated oligoribonucleotides to form capped RNAs, albeit with yields in the order of 40% (Sawai et al. 1999).
In summary, established in vitro methods for the production of capped RNA have severe limitations, including low yields and the limitation to short oligonucleotides. This is especially reflected in the low number of structures of proteins in complex with capped RNA (>1 nt) that have been determined to date. Currently, these structures include the human 2′-O-ribose methyltransferase CMTr1 with a capped 4-mer that was produced by chemical coupling (Smietanski et al. 2014), the 2′-O-ribose methyltransferase of vaccinia virus with a capped 6-mer that was produced by in vitro transcription in the presence of a cap analog (Hodel et al. 1998), the 2′-O-ribose methyltransferase in the NS5 protein from dengue virus with an 8-mer cap-0 viral RNA that was produced using a cap analog (Zhao et al. 2015), the dengue virus methyltransferase in complex with a 5′-capped RNA 8-mer that was chemically synthesized (Yap et al. 2010), and the innate immune receptor RIG-I that contains a chemically synthesized 24-nt-long capped hairpin RNA (Devarkar et al. 2016). The high-resolution structural data available is thus confined to a small subset of the numerous enzymes and proteins that directly interact with the mRNA cap structure. This limits our understanding of how these cap-binding factors are able to modulate mRNA metabolism and prompted us to establish a general, easy, rapid, and cost-efficient method for large-scale production of homogeneously capped RNA.
Here, we designed an optimized expression system for the vaccinia virus capping complex that allows for the straightforward purification of large quantities of the highly active enzyme. We then show that the enzyme we produce is able to cap a very wide variety of RNA species, where capping efficiency is virtually independent of the length, sequence, and structure of the RNA body. We establish that milligram amounts of capped RNA can be obtained in biological laboratories in very short times and that the capped RNAs are amenable for a wide range of biophysical and structural studies.
RESULTS
Cloning and purification of the vaccinia virus capping complex
To be able to overexpress the D1:D12 vaccinia virus capping complex (Fig. 1B, bottom) in E. coli, we designed an expression vector (Supplemental Fig. S1, see Materials and Methods). We wished to have a system at hand that ensures high levels of overexpression and that allows for a straightforward and reproducible purification of the complex. To that end, we combined coexpression of both proteins for optimal stability of the complex and with a single purification tag on the D1 enzyme for convenient purification (De la Pena et al. 2007; Kyrieleis et al. 2014) with codon optimization of the D1 and D12 genes for E. coli expression. Using this expression system, we could overexpress the viral capping complex and isolate it using two standard purification steps (Ni affinity chromatography followed by size exclusion chromatography; Fig. 1C; see Materials and Methods). The purification of the enzyme takes 1.5 d and typically yields 12 mg pure enzyme from 5 L of E. coli culture, which is sufficient to prepare 5–10 µmol capped RNA.
RNA body preparation
The RNA body that we use for the capping reaction was prepared using established in vitro transcription using the T7 polymerase. In combination with the standard T7 ϕ6.5 promoter, this enzyme prefers a guanine as the first base, whereas in combination with the T7 ϕ2.5 promoter, transcription starts with an adenine (Coleman et al. 2004; Ray et al. 2006). As templates for the in vitro transcription reaction, we used plasmid DNA that contains an HDV-ribozyme at the 3′ end and DNA primers in a nonmethylated or methylated form (Helmling et al. 2015).
Capping reaction
To test the activity of the vaccinia virus capping complex on the RNA body, we performed capping reactions using different enzyme:RNA ratios. The capping reaction was then analyzed using Urea-PAGE, where the RNA was visualized using methylene blue (Fig. 1D). During the capping reaction a methylated guanidine is added to the RNA body, which results in a shift of the mobility of the transcript. Interestingly, we found that we could cap an RNA body with close to 100% efficiency within 1 h in all cases. The efficiency depends, however, on the enzyme:RNA ratio and in all subsequent experiments we used small-scale tests to optimize this ratio before large-scale capping reactions were performed. We found that optimal enzyme:substrate ratios varied between 1:10 and 1:500, depending on the RNA body. Longer incubation times can increase the yield of the capping reaction (Fig. 1E); however, the enzyme loses activity over the course of hours (Fig. 1F). Complete capping is thus most reliably assured in reaction times that do not exceed 1–2 h.
After the capping reaction, the capped RNA was separated from the enzyme using a heat precipitation step, which quantitatively removed all protein without loss of the target RNA (Supplemental Fig. S2). Finally, the RNA could be separated from nucleotides and salts by isopropanol precipitation and desalting steps. The yield of the final capped RNA product depends mainly on the efficiency of the transcription reaction, as the RNA body was capped close to 100% in all cases. Routinely, we obtained 500 nmol (15 mg for a 100-mer RNA body) from a 10-mL transcription reaction. From the transcribed RNA to the pure capped product the total experimental time is ∼3.5 h (1 h capping reaction, 2 h precipitation, and 30 min desalting).
To confirm that the capping reaction indeed added a cap-0 structure to the RNA body, we performed native mass spectrometry on an RNA body of 31 nt before and after the capping reaction (Supplemental Fig. S3). As starting material, we used a mixture of 5′ mono -, di-, and triphosphorylated RNA and the identity of all species could be unambiguously identified in the native mass spectrum. After the capping reaction, we detected a mixture of 5′ monophosphorylated RNA and of the cap-0 RNA. These data confirm that the RNA body requires a di- or triphosphate at the 5′ end in order to be a substrate for the vaccinia capping enzyme and, more importantly, this shows that the capping enzyme properly adds a cap-0 structure to the RNA.
Dependence of the capping on the RNA body
To test the dependence of the efficiency of the vaccinia capping enzyme on length, sequence, and secondary structure of the RNA body, we performed a large set of capping reactions, where we varied single parameters. To test how the substrate length modulates capping efficiency, we performed capping reactions using substrates with RNA bodies between 2 and 100 nt. If the RNA body is over 40 nt, it is not possible to directly follow the capping reaction using a standard Urea-PAGE analysis, as a difference of one base cannot be clearly resolved anymore. To follow the capping reaction we therefore designed a substrate that contains a unique RNase A cleavage site that is located 15 bases downstream from the 5′ end of the RNA body (Fig. 2A). In that manner, we can perform the capping reaction on the full-length intact RNA and observe the progress of the capping reaction after cleaving with RNase A. Importantly, from this set of experiments we can conclude that RNAs between 40 and 100 nt are fully and efficiently capped. To test whether very short RNAs are also a substrate for the vaccinia virus capping enzyme, we used RNAs between 2 and 5 nt. Interestingly, we observe that the capping enzyme efficiently and fully caps even dinucleotide sequences (Fig. 2B). Taken together, the vaccinia capping enzyme is able to efficiently cap RNA fully independent of substrate length.
FIGURE 2.

The RNA capping reaction is robust and independent of substrate RNA length, sequence, or secondary structure. (A) Capping reactions using RNA substrates of different lengths (between 100 and 40 nt). To visualize the capping efficiency a single U was introduced at position 15. RNAse A digestion of the RNA (before and after capping) released the 5′ end of the substrate that only contained 15 nt. The mass shift due to the capping reaction can be readily observed using Urea-PAGE analysis. The capping efficiency of all RNAs was close to 100%. (B) Shorter RNA sequences (2–5 bases) are also efficiently capped using the purified capping complex. (C) The capping reaction is independent of sequence because both GA and CU rich sequences can be capped with efficiencies close to 100%. Note that the GA and CU RNA migrate slightly differently due to the base composition. (D) The capping reaction is efficient when the first base of the RNA body is an adenine. (E) RNA secondary structure only interferes with the capping reaction when there is no 5′ overhang. A stable RNA hairpin that contains no 5′ overhang is not capped, whereas a stable hairpin with a 5′ overhang of 2 or 4 bases is efficiently capped.
To evaluate whether the sequence of the RNA plays a role in the capping process, we used RNA bodies that are either GA rich or CU rich. Both substrates are fully and efficiently capped, indicating that the primary sequence of the RNA body does not influence the capping efficiency (Fig. 2C). In addition, we established that the RNA capping reaction also functions when the first base of the transcript is an adenine, as transcripts produced with the T7 ϕ2.5 promoter are also fully capped (Fig. 2D).
Finally, to test whether RNA secondary structure has an influence on the capping efficiency, we used a very stable GC hairpin structure with either no, two, or four bases overhang at the 5′ end. Of these RNA bodies only the RNA hairpin that contains a fully base-paired 5′ end is not a substrate for the capping enzyme (Fig. 2E). The other RNA bodies are efficiently and fully capped, indicating that stable secondary structure elements do not interfere with the capping reaction unless the 5′ end is part of a stable double strand.
In summary, we here show that the vaccinia capping enzyme is able to cap RNA in an efficient manner that is practically independent of the length, sequence, and structure of the RNA body.
Preparative preparation of short capped RNAs
For a number of structural and functional studies it might be advantageous to prepare capped RNA with a minimal possible length. Above, we showed that RNA as short as 2 nt can be efficiently capped. In vitro transcription reactions of very short RNAs (<10 nt) are, however, often very inefficient. We therefore established an alternative approach to prepare large quantities of short and capped RNA based on the vaccinia capping and the T7 polymerase enzymes. In brief, we demonstrate that it is convenient to transcribe and cap a longer RNA body, followed by the preparative digestion of this product using a specific RNase (Fig. 3A). Indeed, we show that a 30-nt-long RNA is efficiently capped and quantitatively cleaved using RNase A (Fig. 3B). In an alternative approach the longer RNA can be digested using site-specific RNase H cleavage (Duss et al. 2010). The cleaved fragments can be conveniently separated using anion-exchange chromatography (Fig. 3C), which yield highly pure and capped short RNA fragments.
FIGURE 3.

Large-scale preparation of short, capped RNAs. (A) A longer RNA (30 nt) is prepared using in vitro transcription and capped as described above. The RNA contains a unique cleavage site for an RNase, and we here used RNase A to cleave the RNA after a single uracil. (B) Urea-PAGE analysis of the capping reaction and RNase digestion (lanes 1–3) and of the subsequent purification of the digested RNA (lanes 4,5). (C) The two RNA fragments are separated using a Dionex DNAPac column. The drawn line corresponds to the absorbance at 260 nm, whereas the dashed line corresponds to the salt gradient that is used to elute the RNA.
Separation of capped and uncapped RNA
In all our experiments we observed a capping efficiency of close to 100%. In case the capping reaction turns out less efficient, it might, however, be necessary to separate uncapped substrate from capped product. To that end we made use of the eIF4E binding protein that specifically interacts with capped RNA and not with RNA that has a 5′ end devoid of m7 methylated guanine. We incubated a mixture of 50% capped and 50% uncapped RNA with purified His6-tagged eIF4E protein and Ni-NTA resin (Fig. 4A). The uncapped RNA did not interact with the eIF4E protein on the resin and could thus be removed in a single wash step. The capped RNA, on the other hand, formed a tight complex with the eIF4E on the resin. This eIF4E:capped RNA complex was then eluted from the Ni-NTA resin using higher imidazole concentrations. Finally, the capped RNA could be separated from eIF4E protein using a phenol–chloroform extraction step. In summary, we show that it is possible and straightforward to quantitatively separate capped and uncapped RNA.
FIGURE 4.

(A) In case the capping reaction is incomplete, the uncapped RNA can be separated from the capped RNA using an eIF4E pulldown. N-terminally His6-tagged eIF4E was attached to Ni-NTA resin and a mixture of 50:50 capped and uncapped RNA was added (a + b). The uncapped RNA did not interact with the eIF4E and could be washed away. Capped RNA interacted tightly with the eIF4E protein and was thus retained on the beads. The protein:RNA complex was eluted using imidazole. The capped RNA was separated from the eIF4E protein using a phenol–chloroform extraction. (B) The capped RNA is a substrate for the Dcp1:Dcp2 decapping enzyme. An RNA body of 15 nucleotides was capped using the capping enzyme complex and subsequently decapped using the Dcp1:Dcp2 decapping complex.
Translation and decapping assay using the capped RNA
To demonstrate the functionality of the capped RNA, we prepared a capped transcript that codes for the GFP protein and used this in in vitro translation assays (Supplemental Fig. S4). As expected, the translation efficiency is significantly increased after the capping reaction, proving that the modification occurred correctly.
In addition, capped RNA is a substrate for a number of decapping enzymes, including DcpS and the Dcp1:Dcp2 complex (Parker and Song 2004). To confirm that the capped RNA that we prepared here is indeed a substrate for the decapping enzymes we incubated a capped RNA of 15 nt with the purified Dcp1:Dcp2 complex. Urea-PAGE analysis of the RNA shows that the cap structure is, as expected, fully removed (Fig. 4B). We hereby demonstrate the suitability of the capped RNA for activity assays that circumvent radioactive labeling.
Binding studies using the capped RNA
To demonstrate general applicability in biophysical studies, we investigated the complex between capped RNA and eIF4E. To that end, we used fluorescence anisotropy and high-resolution NMR methods as examples. In a first set of experiments, we quantified the interaction strength between the eIF4E protein and the capped RNA using fluorescence anisotropy experiments with a fluorescently labeled capped RNA of 31 nt. To probe the strength of the capped-RNA:eIF4E interaction, we added the Drosophila melanogaster (Dm.) protein in increasing amounts to the RNA and observed changes in the fluorescence anisotropy (Fig. 5A). From the binding curve, we extracted a kD for the RNA:protein interaction of 294 (42) nM. Previously, the affinity between the Dm. eIF4E and m7GDP (an RNA cap analog that lacks the complete RNA body and that contains two instead of three phosphate groups) was determined to be ∼700 nM, using ITC (Kinkelin et al. 2012). The slightly better affinity of the protein for capped RNA in comparison to that for m7GDP suggests that there are a small number of contacts between the cap binding protein and the mRNA that involve parts of the RNA that are outside the methylated guanine and first two phosphates in the cap linker. This is in agreement with biophysical and structural studies on the murine eIF4E protein that shows contributions of the γ-phosphate in the eIF4E:cap interaction (Niedzwiecka et al. 2002). In summary, our and previous studies show that the interaction between the mRNA cap structure and eIF4E is mainly mediated through the cap structure and the 5′-5′ triphosphate linker and that there are no extensive contacts between the RNA body and the cap binding protein. The fact that the mRNA body does not interact with the eIF4E protein is also confirmed in structural studies with cap analogs, as there the first base of the RNA body is not oriented in a fixed position on the eIF4E structure (Fig. 5B).
FIGURE 5.

Affinity measurements using capped RNA. (A) Fluorescence anisotropy binding studies to probe the interaction strength between eIF4E and a capped RNA (kD = 294 ± 42 nM). (B) Known structures of eIF4E in complex with the m7GpppG or m7GpppA cap analog (Tomoo et al. 2005; Liu et al. 2009; Peter et al. 2015) show that the position of the first nucleotide of the RNA body (pink) is not well defined. This indicates that the interaction between eIF4e and the mRNA is mediated almost exclusively through the m7GTP part of the cap-0 structure.
NMR studies of the capped RNA
To show the potential of our RNA capping method for structural biology, we performed high-resolution NMR experiments. In those experiments, the interaction between the capped RNA and the eIF4E protein was probed using two complementary approaches. First, we prepared 15N-labeled eIF4E and recorded NMR spectra in the absence and presence of a capped RNA with an RNA body of 5 nt (Fig. 6A). Upon addition of the RNA, we observed clear chemical shift perturbations (CSPs) in the eIF4E resonances that directly report on the specific interaction between the cap binding protein and the capped RNA. The large number of residues that undergo CSPs is in agreement with the enclosure of the cap structure into the core of the eIF4E protein (Matsuo et al. 1997; Kinkelin et al. 2012). In agreement with the limited role the RNA body plays in the cap recognition process, the NMR spectrum of eIF4E in complex with the capped RNA and that of eIF4E in complex with m7-GDP are very similar (Supplemental Fig. S5).
FIGURE 6.

NMR studies using capped RNA. (A) 2D 1H-15N TROSY spectrum of the eIF4E cap binding protein in the absence (black) and presence (red) of a twofold excess of a 5-mer RNA that contains a 5′ cap-0 structure. (B) 1H-13C methyl TROSY spectrum of a 5-mer RNA that contains a m7-[13CH3]/2′-O-[13CH3] cap-1 structure in the absence (black) and presence (red) of eIF4E.
In a second set of NMR experiments, we labeled the capped RNA with NMR active methyl groups. To achieve that, we used 13C-labeled SAM (see Materials and Methods) during the RNA capping reaction. In a second step, we modified this 13C-labeled cap-0 RNA further using the vaccinia virus VP39 enzyme that modifies a cap-0 RNA into a cap-1 RNA. This reaction was also performed in the presence of 13C-labeled SAM. The resulting cap-1 RNA thus contained two NMR active methyl groups, at the N7 position of the guanine in the cap structure and at the 2′-O position of the first nucleotide of the RNA body. The resulting NMR spectrum clearly displays two resonances (Fig. 6B). The two resonances were assigned based on NMR spectra of a methyl labeled cap-0 RNA (data not shown) that only displayed a single resonance. Upon addition of eIF4E to the cap-1 RNA, the guanine N7-methyl group was broadened beyond detection, whereas the ribose 2′-O methyl group was unaffected (Fig. 6B). These data are also in agreement with the lack of significant interactions between the eIF4E protein and the first nucleotide of the RNA body. In addition, these experiments show that the cap-0 RNA can be entirely converted into a cap-1 RNA with the use of the VP39 enzyme from the vaccinia virus.
DISCUSSION
Functional and structural studies of proteins and enzymes that specifically recognize the 5′ end of mRNA are technically very challenging due to the lack of cost-effective and efficient ways to prepare larger quantities of capped RNA. Here, we show that the vaccinia capping enzyme is capable of capping RNA in quantities that by far exceed those that can be obtained using alternative (chemical or commercial enzymatic) methods. Importantly, we used a number of systematic tests (Figs. 2, 3) to show that the capping efficiency is almost independent of the length, sequence, and structure of the substrate RNA. The only RNA that we were not able to cap using the vaccinia enzyme is a substrate where the most 5′ nucleotide is part of a very stable hairpin. Due to the efficiency of the capping reaction, the obtained quantities of capped RNA are solely limited by the amounts of substrate. In our hands, we were able to obtain up to 500 nmol (15 mg for a 100-mer RNA body) of capped RNA from a 10-mL T7 transcription reaction. It is important to note that these quantities are more than sufficient for NMR applications and crystallization screening.
To have easy access to the vaccinia capping enzyme complex, we designed a highly optimized expression and purification setup (Fig. 1C; Supplemental Fig. S1). Importantly, the methods that are required for the enzyme preparation are generally available in biological laboratories, which makes the introduced system easily accessible. In our laboratory we produce large quantities of the enzyme and store this at −20°C without loss of activity for at least several months. Based on an appropriate mRNA body, capped RNA for biophysical studies (Figs. 5, 6) can then be produced within 3.5 h. The expression vector for the enzyme complex can be obtained from the authors upon request to facilitate future studies.
The capping method can be adapted for the production of various modified RNAs that serve specific purposes. First, we show that thio-uridine containing RNA can be capped efficiently. This enables, e.g., the addition of a fluorescent or spin label into the capped RNA, which we use here for fluorescence anisotropy measurements (Fig. 5). Secondly, NMR active nuclei can be introduced at specific positions in the capped RNA. We show that the use of 13C-SAM results in the site-directed labeling of the methyl groups in the cap structure. Equivalently, the use of NMR active GTP would result in the specific labeling of the cap base. Thirdly, we show that the cap-0 structure that the vaccinia virus produces can be used as a substrate for enzymes that modify the cap-0 structure into a cap-1 structure (Fig. 6). Finally, the capped RNA can potentially be used as a building block to produce complex mRNAs in a strategy where capped RNA fragments and uncapped RNA fragments can be enzymatically ligated (Duss et al. 2010). In brief, we show that the modular nature of the capping reaction system can be exploited to produce various modified capped RNAs in large scales.
In summary, we here introduce an easy, fast, and cost-efficient protocol to prepare large quantities of capped RNA. We envision that this will spark future structural and biophysical studies of mRNA and its associated proteins and enzymes.
MATERIALS AND METHODS
Cloning of vaccinia capping enzyme
The sequences of the vaccinia capping enzyme subunits (UniProt identifiers P04298 and P04318, respectively) were optimized for expression in E. coli and synthesized by GenScript. Synthetic D1 and D12 sequences were inserted into the first and second multiple cloning sites of a modified pRSF-duet vector (Novagen), respectively (Supplemental Fig. S1). This vector carries an N-terminal TEV protease cleavable His-tag on D1 in the first multiple cloning site and an untagged D12 expressed from the second multiple cloning site, where both proteins have their own T7 promoter. In order to obtain a plasmid with reduced copy number, both multiple cloning sites encompassing both promoters and ORFs were then transferred to a pET-based (Novagen) vector backbone (Supplemental Fig. S1).
Purification of vaccinia capping enzyme
The vaccinia capping enzyme complex is coexpressed in E. coli BL21 Gold (DE3) pLysS cells. Cells were grown at 37°C to a cell density of OD600 = 0.6. Temperature was decreased to 20°C and cells were induced by addition of 0.2 mM IPTG. Twelve hours after induction, pelleted cells were resuspended in buffer A (50 mM Tris pH 8, 100 mM NaCl, 5 mM imidazole, 10% glycerol, 1 mM DTT) supplemented with 0.1% Triton X-100 and 1 mM PMSF and incubated on ice for 15 min with lysozyme. After lysis by sonification, the cell lysate was centrifuged at 50,000g for 30 min and the supernatant was applied to Ni-NTA resin (QIAGEN) equilibrated in buffer A. To remove unbound proteins, the column was washed with 10 column volumes buffer A. The protein complex was eluted from the resin by buffer A containing 200 mM imidazole. The elution fraction was dialyzed against buffer A without imidazole overnight. Size exclusion chromatography was performed in buffer A without imidazole using a Superdex S200 column. The pure capping enzyme complex was concentrated, aliquoted, and stored in buffer that contains 50% glycerol at −20°C.
In vitro transcription of RNA
RNA was produced by in vitro transcription (IVT) using DNA primers as templates for transcription by T7 RNA polymerase, which requires a duplex template in the promoter (Supplemental Table S1). Therefore, the reaction contained an equimolar amount of antisense strand primers bearing the T7 promoter sequence and sense strand primers encompassing 5′-target RNA sequence (in reverse complement)-CC–T7 promoter (in reverse compliment)-3′. Some of the antisense primers were methylated at the 2′-O position of the last 2 nt (Helmling et al. 2015) in order to reduce 3′ inhomogeneity. The use of methylated primers is indicated in the RNA construct list below.
IVT reactions were set up with a final concentration for both primers of 1 µM, 40 mM Tris pH 8, 5 mM DTT, 1 mM spermidine, 0.01% Triton X-100, 4 mM each of NTP in the RNA sequence, 0.2 µM T7 RNA polymerase (purified in the laboratory as described previously [Audin et al. 2013]), and 20–60 mM MgCl2. The optimal Mg2+ concentration was determined in small-scale reactions. Transcription reaction mixtures were incubated at 37°C for at least 4 h. Precipitated pyrophosphate was cleared by addition of EDTA and the product RNA was precipitated as described below.
RNA precipitation
RNA was precipitated with 0.3 M NaOAc pH 5.2 and 0.7 volumes of isopropanol at −20°C for at least 2 h. The reaction was then centrifuged at 9000g and −5°C for 1 h and the pellet was washed with cold 75% ethanol and centrifuged again for 30 min. The resulting pellet was air dried and resuspended in the desired buffer for further applications. For short RNAs (2–10 nt), precipitation was carried out with 0.2 M NaCl and 3.5 volumes of EtOH (replacing the NaOAc and isopropanol) and a precipitation time of 12 h.
RNA purification using anion-exchange chromatography
RNA from IVT was purified using anion exchange chromatography on a 22 × 250 mm DNAPac PA100 column (Dionex) operated at 80°C. The column was run in buffers containing 20 mM Tris pH 8.0 and 5 M Urea (pH adjusted to 8.0 at room temperature). RNA of different lengths was eluted with a NaCl gradient. Salt gradients were designed according to the respective RNA lengths to be separated. Short RNAs (2–10 nt) typically elute between 50 and 200 mM NaCl while longer constructs (60–80 nt) elute at 300–400 mM NaCl. RNA containing fractions were analyzed using urea-PAGE, pooled and precipitated as described above. After precipitation, RNAs were desalted using a prepacked PD-10 Desalting column (GE Healthcare) and subsequently concentrated using a speedvac device.
Urea-PAGE
Analysis of RNA length was performed using Urea-PAGE. Polyacrylamide gels containing 8 M urea in TBE buffer were prepared with an acrylamide content suitable for the desired resolution (8%–21%). Gels were run with 10 mA until the sample had penetrated the gel completely and were further run with 15–20 mA. Gels were then rinsed with water and stained with a solution of 2g/L methylene blue in 0.4 M NaAc, pH 4.7 and destained in water.
RNA capping protocol
Capping reactions were performed in buffer containing 50 mM Tris, pH 8, 5 mM KCl, 1 mM MgCl2, and 1 mM DTT with 20 µM RNA. 0.5 mM GTP, 0.1 mM SAM, and capping enzyme according to the previously determined substrate to enzyme ratio were added. The reaction was incubated at 37°C and prepared for analysis by Urea-PAGE by adding an equal volume of sample buffer containing 8 M Urea, 20 mM EDTA, 2 mM Tris pH 8, and Bromophenol blue and Xylene Cyanol.
As capping efficiency varies between different RNA constructs, the most efficient ratio of capping enzyme to RNA molecules was determined in small-scale test reactions of 20 µL. Enzyme to RNA ratios typically varied between 1:10 and 1:100. Preparative scale reactions were scaled up accordingly. After capping, the reaction was heated to 70°C for 10 min and centrifuged for 10 min at 4500g to remove the capping enzyme from the reaction. Capped RNA was then precipitated and resuspended in an appropriate buffer and stored at −20°C.
For generation of cap-1 RNA, Vaccinia MTase (NEB) was added to the capping reaction according to the manufacturer's recommendations.
RNase A cleavage
RNase A is an endoribonuclease that cleaves specifically the phosphodiester bond between a pyrimidine nucleotide and the following nucleotide, resulting in a 3′ pyrimidine nucleoside phosphate. RNAse A was used for analytic and preparative specific cleavage of GA containing RNA sequences at a single uracil nucleotide. After capping of the RNA, it was cleaved with RNase A by adding 50 ng RNase A per nmol of RNA and incubating the reaction for 10 min at 37°C. Capping efficiency was then analyzed by Urea-PAGE. After preparative RNase A cleavage, the reaction was phenol–chloroform extracted once to remove all RNase A activity from the mixture before proceeding with RNA purification using anion-exchange chromatography.
Decapping with Dcp1:2
For decapping of capped RNA, a construct of the Dcp1:Dcp2 decapping complex from S. pombe was used, which encompasses the regulatory and catalytic domain of Dcp2 (1-266) and full-length Dcp1. The enzyme was purified as described previously (Fromm et al. 2012).
Capped RNA was incubated in capping buffer supplemented with an additional 5 mM MgCl2 and Dcp1:Dcp2 in a molar ratio of enzyme to RNA of 1:20 for 30 min at 30°C. Sample buffer was added to stop the reaction and the samples were analyzed using Urea-PAGE.
Purification of eIF4E
The pEK-vH vector expressing D.m. eIF4E isoform c (UniProt identifier P48598-2) as an N-terminal His-tag fusion was a kind gift of Dr. Fulvia Bono (MPI Tübingen). His-eIF4E was expressed in E. coli DE3 BL21 cells carrying a plasmid for rare RIL codons. Cells were cultured at 37°C to an OD600 of 0.6 in LB or M9 medium supplemented with 15N NH4Cl. Expression was induced by addition of 0.5 mM IPTG and cells were shifted to 25°C overnight. Pelleted cells were resuspended and lysed in buffer E (25 mM Tris pH 8.0, 150 mM NaCl, 5 mM imidazole, 1 mM DTT) supplemented with 0.1% Triton X-100 and 100 µg/mL lysozyme. Cellular debris was removed by centrifugation for 30 min at 50,000g. The supernatant was applied to a Ni-NTA column, washed with buffer E, and protein was eluted with buffer E supplemented with an additional 200 mM imidazole. Untagged protein, used for NMR and fluorescence polarization experiments, was cleaved using TEV protease and dialyzed into buffer E before applying it to a second Ni-NTA column. Hexa-His-tagged protein was diluted into 25 mM Tris pH 8.0, 10 mM NaCl and applied to a HiTrap Q-column, washed with 25 mM Tris pH 8.0, 100 mM NaCl, and eluted with buffer containing 250 mM NaCl. Both tagged and untagged protein were concentrated and purified on a Superdex 200 column. Fractions containing monomeric protein were pooled and used for separation and titration experiments.
Separation of capped and uncapped RNAs
The mixture of capped and uncapped RNA was prepared in buffer E (25 mM Tris, pH 8.0, 150 mM NaCl, 1 mM DTT, 5 mM imidazole). A twofold molar excess of RNase-free His-eIF4E over total RNA concentration was added. Also, 0.2 mL Ni-NTA beads per used mg of His-eIF4E were equilibrated in buffer E and added to the reaction. After incubation for 10 min on ice, the mixture was applied to a spin column and centrifuged for 30 sec at 100g. The beads were washed three times with 5 volumes buffer E with the uncapped RNA residing in the flow-through. The purified complex of His-eIF4E and capped RNA was released from the Ni-NTA beads with buffer E supplemented with 200 mM imidazole. To remove the eIF4E protein from the capped RNA, the elution fraction was phenol–chloroform extracted.
Fluorescein-labeling of RNA
Fluorescein-labeled RNA was designed to contain only one uracil nucleotide and was transcribed substituting UTP with thio-UTP and purified and capped as described above. RNA was then fluorescein labeled by incubating the RNA at a concentration between 30 and 100 µM in the presence of 10 mM fluorescein in 100 mM sodium phosphate buffer (pH 8.0) for 24 h at room temperature in the dark (Ramos and Varani 1998; Audin et al. 2016). Afterward, the RNA was purified by three subsequent rounds of RNA precipitation.
Fluorescence anisotropy experiments
Fluorescence polarization experiments were carried out in buffer containing 25 mM Tris, pH 8.0, 150 mM NaCl, 1 mM DTT, and 0.005% Triton X-100 at room temperature in a Tecan infinite f200 plate reader. Excitation and emission wavelength were 485 and 535 nm, respectively. Concentration of fluorescein-labeled 31-mer RNA was 20 nM, and eIF4E concentrations ranged between 0 and 2.5 µM. Samples were prepared in triplicate and blanked against buffer only and anisotropy was averaged over ten measurements. To obtain a binding constant, the binding curves were fitted to a one-site binding model using a least-squares routine. The binding process did not influence the fluorescence intensity, which is consistent with the 15-nt distance between the label and the eIF4E binding site.
Generation of 13C-methyl-labeled S-adenosyl methionine
The purification of MetK and the synthesis of S-adenosyl methionine were carried out as described previously (Ottink et al. 2010). The plasmid carrying an N-terminally His-tagged MetK was a kind gift of Professor Jens Wöhnert (University of Frankfurt am Main).
In brief, 13C-methyl-labeled S-adenosyl methionine is synthesized from methionine and ATP by the E. coli MetK enzyme. The synthesis was carried out in 50 mM Tris, pH 8.2, 100 mM KCl, 10 mM MgCl2, 1 mM L-methionine methyl-13C, 1 mM ATP in the presence of 0.6 mg/mL MetK. The reaction was incubated for 3 h at 30°C. An SP sepharose column (HiTrap SP-FF; GE healthcare) of 30% of the reaction volume was washed with two column volumes of 500 mM HCl and then equilibrated with 20 column volumes of 50 mM HCl. The reaction mixture was adjusted to 50 mM HCl with concentrated HCl and applied to the column. After a wash step with 6 column volumes of 50 mM HCl, pure SAM was eluted with 500 mM HCl. Before storage, SAM was concentrated and the pH was adjusted to 3 with NaOH before storage at −20°C.
NMR spectroscopy
NMR spectra were recorded at 25°C on a Bruker AVIII-600 spectrometer with a room temperature probe head. Samples were prepared in 25 mM Tris, pH 8.0 and 150 mM NaCl, which was based on 100% D2O or 90% H2O/10% D2O for 13C-labeled RNA samples and 15N-labeled protein samples, respectively. The concentration of both the eIF4E sample and the cap-1 RNA sample was 100 µM and for the titration steps a molar excess of 1.5 of interacting RNA or protein was added. NMR data were processed using the NMRPipe-NMRDraw software suite and figures displaying NMR spectra were produced using NMRView (www.onemoonscientific.com).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank all laboratory members for discussions and support. We especially thank Philip Wurm for providing the fluorescently labeled capped RNA and for help in recording the fluorescence anisotropy data. We are grateful to Jens Wöhnert (University of Frankfurt am Main) for sharing the S-adenosyl methionine synthetase plasmid and to Fulvia Bono (MPI Tübignen) for sharing the eIF4E plasmid. This work was supported by the Max Planck Society and the European Research Council under the European Union's Seventh Framework Programme (FP7/2007–2013), ERC grant agreement no. 616052.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.056614.116.
Freely available online through the RNA Open Access option.
REFERENCES
- Adams JM, Cory S. 1975. Modified nucleosides and bizarre 5′-termini in mouse myeloma mRNA. Nature 255: 28–33. [DOI] [PubMed] [Google Scholar]
- Audin MJ, Dorn G, Fromm SA, Reiss K, Schutz S, Vorlander MK, Sprangers R. 2013. The archaeal exosome: identification and quantification of site-specific motions that correlate with cap and RNA binding. Angew Chem 52: 8312–8316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audin MJ, Wurm JP, Cvetkovic MA, Sprangers R. 2016. The oligomeric architecture of the archaeal exosome is important for processive and efficient RNA degradation. Nucleic Acids Res 44: 2962–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee AK. 1980. 5′-terminal cap structure in eucaryotic messenger ribonucleic acids. Microbiol Rev 44: 175–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa E, Moss B. 1978. mRNA(nucleoside-2′-)-methyltransferase from vaccinia virus. Purification and physical properties. J Biol Chem 253: 7692–7697. [PubMed] [Google Scholar]
- Both GW, Banerjee AK, Shatkin AJ. 1975. Methylation-dependent translation of viral messenger RNAs in vitro. Proc Natl Acad Sci 72: 1189–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownlee GG, Fodor E, Pritlove DC, Gould KG, Dalluge JJ. 1995. Solid phase synthesis of 5′-diphosphorylated oligoribonucleotides and their conversion to capped m7Gppp-oligoribonucleotides for use as primers for influenza A virus RNA polymerase in vitro. Nucleic Acids Res 23: 2641–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho EJ, Takagi T, Moore CR, Buratowski S. 1997. mRNA capping enzyme is recruited to the transcription complex by phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev 11: 3319–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman TM, Wang G, Huang F. 2004. Superior 5′ homogeneity of RNA from ATP-initiated transcription under the T7 ϕ2.5 promoter. Nucleic Acids Res 32: e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras R, Cheroutre H, Degrave W, Fiers W. 1982. Simple, efficient in vitro synthesis of capped RNA useful for direct expression of cloned eukaryotic genes. Nucleic Acids Res 10: 6353–6362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daffis S, Szretter KJ, Schriewer J, Li J, Youn S, Errett J, Lin TY, Schneller S, Zust R, Dong H, et al. 2010. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 468: 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De la Pena M, Kyrieleis OJ, Cusack S. 2007. Structural insights into the mechanism and evolution of the vaccinia virus mRNA cap N7 methyl-transferase. EMBO J 26: 4913–4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decroly E, Ferron F, Lescar J, Canard B. 2012. Conventional and unconventional mechanisms for capping viral mRNA. Nat Rev Microbiol 10: 51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devarkar SC, Wang C, Miller MT, Ramanathan A, Jiang F, Khan AG, Patel SS, Marcotrigiano J. 2016. Structural basis for m7G recognition and 2′-O-methyl discrimination in capped RNAs by the innate immune receptor RIG-I. Proc Natl Acad Sci 113: 596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duss O, Maris C, von Schroetter C, Allain FH. 2010. A fast, efficient and sequence-independent method for flexible multiple segmental isotope labeling of RNA using ribozyme and RNase H cleavage. Nucleic Acids Res 38: e188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ensinger MJ, Martin SA, Paoletti E, Moss B. 1975. Modification of the 5′-terminus of mRNA by soluble guanylyl and methyl transferases from vaccinia virus. Proc Natl Acad Sci 72: 2525–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipowicz W. 1978. Functions of the 5,-terminal m7G cap in eukaryotic mRNA. FEBS Lett 96: 1–11. [DOI] [PubMed] [Google Scholar]
- Fresco LD, Buratowski S. 1996. Conditional mutants of the yeast mRNA capping enzyme show that the cap enhances, but is not required for, mRNA splicing. RNA 2: 584–596. [PMC free article] [PubMed] [Google Scholar]
- Fromm SA, Truffault V, Kamenz J, Braun JE, Hoffmann NA, Izaurralde E, Sprangers R. 2012. The structural basis of Edc3- and Scd6-mediated activation of the Dcp1:Dcp2 mRNA decapping complex. EMBO J 31: 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuichi Y, Miura K. 1975. A blocked structure at the 5′ terminus of mRNA from cytoplasmic polyhedrosis virus. Nature 253: 374–375. [DOI] [PubMed] [Google Scholar]
- Furuichi Y, Shatkin AJ. 2000. Viral and cellular mRNA capping: past and prospects. Adv Virus Res 55: 135–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuichi Y, LaFiandra A, Shatkin AJ. 1977. 5′-Terminal structure and mRNA stability. Nature 266: 235–239. [DOI] [PubMed] [Google Scholar]
- Goldeck M, Tuschl T, Hartmann G, Ludwig J. 2014. Efficient solid-phase synthesis of pppRNA by using product-specific labeling. Angew Chem 53: 4694–4698. [DOI] [PubMed] [Google Scholar]
- Green MR, Maniatis T, Melton DA. 1983. Human β-globin pre-mRNA synthesized in vitro is accurately spliced in Xenopus oocyte nuclei. Cell 32: 681–694. [DOI] [PubMed] [Google Scholar]
- Grudzien-Nogalska E, Stepinski J, Jemielity J, Zuberek J, Stolarski R, Rhoads RE, Darzynkiewicz E. 2007. Synthesis of anti-reverse cap analogs (ARCAs) and their applications in mRNA translation and stability. Methods Enzymol 431: 203–227. [DOI] [PubMed] [Google Scholar]
- Gu M, Rajashankar KR, Lima CD. 2010. Structure of the Saccharomyces cerevisiae Cet1-Ceg1 mRNA capping apparatus. Structure 18: 216–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardana D, Domashevskiy AV, Gayler KR, Goss DJ. 2015. Efficient preparation and properties of mRNAs containing a fluorescent cap analog: Anthraniloyl-m7GpppG. Translation (Austin) 3: e988538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamm J, Mattaj IW. 1990. Monomethylated cap structures facilitate RNA export from the nucleus. Cell 63: 109–118. [DOI] [PubMed] [Google Scholar]
- Helmling C, Keyhani S, Sochor F, Furtig B, Hengesbach M, Schwalbe H. 2015. Rapid NMR screening of RNA secondary structure and binding. J Biomol NMR 63: 67–76. [DOI] [PubMed] [Google Scholar]
- Hodel AE, Gershon PD, Quiocho FA. 1998. Structural basis for sequence-nonspecific recognition of 5′-capped mRNA by a cap-modifying enzyme. Mol Cell 1: 443–447. [DOI] [PubMed] [Google Scholar]
- Izaurralde E, Lewis J, McGuigan C, Jankowska M, Darzynkiewicz E, Mattaj IW. 1994. A nuclear cap binding protein complex involved in pre-mRNA splicing. Cell 78: 657–668. [DOI] [PubMed] [Google Scholar]
- Kinkelin K, Veith K, Grunwald M, Bono F. 2012. Crystal structure of a minimal eIF4E-Cup complex reveals a general mechanism of eIF4E regulation in translational repression. RNA 18: 1624–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarska MM, Padgett RA, Sharp PA. 1984. Recognition of cap structure in splicing in vitro of mRNA precursors. Cell 38: 731–736. [DOI] [PubMed] [Google Scholar]
- Kyrieleis OJ, Chang J, de la Pena M, Shuman S, Cusack S. 2014. Crystal structure of vaccinia virus mRNA capping enzyme provides insights into the mechanism and evolution of the capping apparatus. Structure 22: 452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewdorowicz M, Jemielity J, Kierzek R, Shapira M, Stepinski J, Darzynkiewicz E. 2007. Solid-supported synthesis of 5′-mRNA CAP-4 from Trypanosomatids. Nucleosides Nucleotides Nucleic Acids 26: 1329–1333. [DOI] [PubMed] [Google Scholar]
- Liu W, Zhao R, McFarland C, Kieft J, Niedzwiecka A, Jankowska-Anyszka M, Stepinski J, Darzynkiewicz E, Jones DN, Davis RE. 2009. Structural insights into parasite eIF4E binding specificity for m7G and m2,2,7G mRNA caps. J Biol Chem 284: 31336–31349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, Shuman S. 1994. Intrinsic RNA (guanine-7) methyltransferase activity of the vaccinia virus capping enzyme D1 subunit is stimulated by the D12 subunit. Identification of amino acid residues in the D1 protein required for subunit association and methyl group transfer. J Biol Chem 269: 24472–24479. [PubMed] [Google Scholar]
- Martin SA, Paoletti E, Moss B. 1975. Purification of mRNA guanylyltransferase and mRNA (guanine-7-) methyltransferase from vaccinia virions. J Biol Chem 250: 9322–9329. [PubMed] [Google Scholar]
- Matsuo H, Li H, McGuire AM, Fletcher CM, Gingras AC, Sonenberg N, Wagner G. 1997. Structure of translation factor eIF4E bound to m7GDP and interaction with 4E-binding protein. Nat Struct Biol 4: 717–724. [DOI] [PubMed] [Google Scholar]
- Matsuo H, Moriguchi T, Takagi T, Kusakabe T, Buratowski S, Sekine M, Kyogoku Y, Wagner G. 2000. Efficient synthesis of 13C, 15N-labeled RNA containing the cap structure m7GpppA. J Am Chem Soc 122: 2417–2421. [Google Scholar]
- Myette JR, Niles EG. 1996. Domain structure of the vaccinia virus mRNA capping enzyme. Expression in Escherichia coli of a subdomain possessing the RNA 5′-triphosphatase and guanylyltransferase activities and a kinetic comparison to the full-size enzyme. J Biol Chem 271: 11936–11944. [DOI] [PubMed] [Google Scholar]
- Nagata S, Hamasaki T, Uetake K, Masuda H, Takagaki K, Oka N, Wada T, Ohgi T, Yano J. 2010. Synthesis and biological activity of artificial mRNA prepared with novel phosphorylating reagents. Nucleic Acids Res 38: 7845–7857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedzwiecka A, Marcotrigiano J, Stepinski J, Jankowska-Anyszka M, Wyslouch-Cieszynska A, Dadlez M, Gingras AC, Mak P, Darzynkiewicz E, Sonenberg N, et al. 2002. Biophysical studies of eIF4E cap-binding protein: recognition of mRNA 5′ cap structure and synthetic fragments of eIF4G and 4E-BP1 proteins. J Mol Biol 319: 615–635. [DOI] [PubMed] [Google Scholar]
- Nielsen DA, Shapiro DJ. 1986. Preparation of capped RNA transcripts using T7 RNA polymerase. Nucleic Acids Res 14: 5936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottink OM, Nelissen FH, Derks Y, Wijmenga SS, Heus HA. 2010. Enzymatic stereospecific preparation of fluorescent S-adenosyl-L-methionine analogs. Anal Biochem 396: 280–283. [DOI] [PubMed] [Google Scholar]
- Parker R, Song H. 2004. The enzymes and control of eukaryotic mRNA turnover. Nat Struct Mol Biol 11: 121–127. [DOI] [PubMed] [Google Scholar]
- Pasquinelli AE, Dahlberg JE, Lund E. 1995. Reverse 5′ caps in RNAs made in vitro by phage RNA polymerases. RNA 1: 957–967. [PMC free article] [PubMed] [Google Scholar]
- Paterson BM, Rosenberg M. 1979. Efficient translation of prokaryotic mRNAs in a eukaryotic cell-free system requires addition of a cap structure. Nature 279: 692–696. [DOI] [PubMed] [Google Scholar]
- Pelletier J, Sonenberg N. 1985. Insertion mutagenesis to increase secondary structure within the 5′ noncoding region of a eukaryotic mRNA reduces translational efficiency. Cell 40: 515–526. [DOI] [PubMed] [Google Scholar]
- Peter D, Weber R, Kone C, Chung MY, Ebertsch L, Truffault V, Weichenrieder O, Igreja C, Izaurralde E. 2015. Mextli proteins use both canonical bipartite and novel tripartite binding modes to form eIF4E complexes that display differential sensitivity to 4E-BP regulation. Genes Dev 29: 1835–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyrane F, Selisko B, Decroly E, Vasseur JJ, Benarroch D, Canard B, Alvarez K. 2007. High-yield production of short GpppA- and 7MeGpppA-capped RNAs and HPLC-monitoring of methyltransfer reactions at the guanine-N7 and adenosine-2′O positions. Nucleic Acids Res 35: e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos A, Varani G. 1998. A new method to detect long-range protein-RNA contacts: NMR detection of electron-proton relaxation induced by nitroxide spin-labeled RNA. J Am Chem Soc 120: 10992–10993. [Google Scholar]
- Ray D, Shah A, Tilgner M, Guo Y, Zhao Y, Dong H, Deas TS, Zhou Y, Li H, Shi PY. 2006. West Nile virus 5′-cap structure is formed by sequential guanine N-7 and ribose 2′-O methylations by nonstructural protein 5. J Virol 80: 8362–8370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy R, Ro-Choi TS, Henning D, Busch H. 1974. Primary sequence of U-1 nuclear ribonucleic acid of Novikoff hepatoma ascites cells. J Biol Chem 249: 6486–6494. [PubMed] [Google Scholar]
- Reguera J, Gerlach P, Cusack S. 2016. Towards a structural understanding of RNA synthesis by negative strand RNA viral polymerases. Curr Opin Struct Biol 36: 75–84. [DOI] [PubMed] [Google Scholar]
- Salditt-Georgieff M, Harpold M, Chen-Kiang S, Darnell JE Jr. 1980. The addition of 5′ cap structures occurs early in hnRNA synthesis and prematurely terminated molecules are capped. Cell 19: 69–78. [DOI] [PubMed] [Google Scholar]
- Sawai H, Wakai H, Nakamura-Ozaki A. 1999. Synthesis and reactions of nucleoside 5′-diphosphate imidazolide. A nonenzymatic capping agent for 5′-monophosphorylated oligoribonucleotides in aqueous solution. J Org Chem 64: 5836–5840. [Google Scholar]
- Schnierle BS, Gershon PD, Moss B. 1992. Cap-specific mRNA (nucleoside-O2′-)-methyltransferase and poly(A) polymerase stimulatory activities of vaccinia virus are mediated by a single protein. Proc Natl Acad Sci 89: 2897–2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Shuman S. 1996. Conditional inactivation of mRNA capping enzyme affects yeast pre-mRNA splicing in vivo. RNA 2: 574–583. [PMC free article] [PubMed] [Google Scholar]
- Shatkin AJ. 1976. Capping of eucaryotic mRNAs. Cell 9: 645–653. [DOI] [PubMed] [Google Scholar]
- Shuman S, Surks M, Furneaux H, Hurwitz J. 1980. Purification and characterization of a GTP-pyrophosphate exchange activity from vaccinia virions. Association of the GTP-pyrophosphate exchange activity with vaccinia mRNA guanylyltransferase. RNA (guanine-7-)methyltransferase complex (capping enzyme). J Biol Chem 255: 11588–11598. [PubMed] [Google Scholar]
- Smietanski M, Werner M, Purta E, Kaminska KH, Stepinski J, Darzynkiewicz E, Nowotny M, Bujnicki JM. 2014. Structural analysis of human 2′-O-ribose methyltransferases involved in mRNA cap structure formation. Nat Commun 5: 3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sripati CE, Groner Y, Warner JR. 1976. Methylated, blocked 5′ termini of yeast mRNA. J Biol Chem 251: 2898–2904. [PubMed] [Google Scholar]
- Takagi T, Moore CR, Diehn F, Buratowski S. 1997. An RNA 5′-triphosphatase related to the protein tyrosine phosphatases. Cell 89: 867–873. [DOI] [PubMed] [Google Scholar]
- Thillier Y, Decroly E, Morvan F, Canard B, Vasseur JJ, Debart F. 2012. Synthesis of 5′ cap-0 and cap-1 RNAs using solid-phase chemistry coupled with enzymatic methylation by human (guanine-N7)-methyl transferase. RNA 18: 856–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomoo K, Matsushita Y, Fujisaki H, Abiko F, Shen X, Taniguchi T, Miyagawa H, Kitamura K, Miura K, Ishida T. 2005. Structural basis for mRNA Cap-Binding regulation of eukaryotic initiation factor 4E by 4E-binding protein, studied by spectroscopic, X-ray crystal structural, and molecular dynamics simulation methods. Biochim Biophys Acta 1753: 191–208. [DOI] [PubMed] [Google Scholar]
- Wei CM, Moss B. 1975. Methylated nucleotides block 5′-terminus of vaccinia virus messenger RNA. Proc Natl Acad Sci 72: 318–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap LJ, Luo D, Chung KY, Lim SP, Bodenreider C, Noble C, Shi PY, Lescar J. 2010. Crystal structure of the dengue virus methyltransferase bound to a 5′-capped octameric RNA. PLoS One 5: e12836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Soh TS, Lim SP, Chung KY, Swaminathan K, Vasudevan SG, Shi PY, Lescar J, Luo D. 2015. Molecular basis for specific viral RNA recognition and 2′-O-ribose methylation by the dengue virus nonstructural protein 5 (NS5). Proc Natl Acad Sci 112: 14834–14839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmern D. 1975. The 5′ end group of tobacco mosaic virus RNA is m7G5′ ppp5′ Gp. Nucleic Acids Res 2: 1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.