

Abstract
Analogues of the anticancer natural product oximidine II were prepared and evaluated for cytotoxicity. One analogue of oximidine II that carries a C15 allylic amide side chain as well as two analogues with C15 vinyl sulfone side chains were found to lack cytotoxicity against the cancer cell line SK‐Mel‐5, thereby confirming the necessity of the C15 enamide side chain of oximidine II for cytotoxicity. Four analogues, designed by comparative molecular similarity index analysis (CoMSIA), that feature a less complex macrolactone scaffold were prepared and tested. The two analogues carrying a C15 vinyl sulfone group and the two analogues with a C15 oximidine II enamide side chain showed weak cytotoxicity against the SK‐Mel‐5 cell line and other cell lines, indicating that the designed simplified macrocycles cannot replace the oximidine II macrocycle.
Keywords: antitumor agents, benzolactone enamides, cytotoxicity, natural products, oximidines
Introduction
Salicylihalamides A (1, Figure 1) and B were discovered as the first members of a class of natural products that are characterized by the presence of an acyl enamine (enamide) side chain appended to a salicylate macrolactone core.1, 2 Subsequently, four other members of this family were discovered including the lobatamides,3 apicularens,4 CJ‐compounds,5 and oximidines6, 7, 8 (oximidine II (2) in Figure 1). Testing of the benzolactone enamide natural products by Boyd et al. revealed that these molecules are potent cytotoxic agents with nanomolar cytotoxicity across a variety of cell lines.9 By using a COMPARE analysis,10 these researchers deduced that the benzolactone enamides exert their mechanism of action by inhibition of the eukaryotic transport protein vacuolar‐(H+)‐adenosine triphosphatase (V‐ATPase). Further biological assessment showed that these molecules are selective inhibitors of only mammalian types of V‐ATPases.
Figure 1.
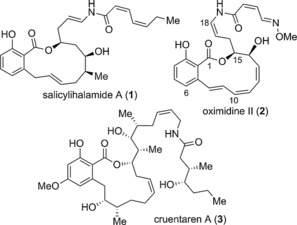
Structures of representative benzolactone natural products: salicylihalamide A (1), oximidine II (2), and cruentaren A (3).
V‐ATPases are responsible for pH homeostasis in various intracellular organelles such as the Golgi apparatus, endosomes,11 and lysosomes.12, 13 This pH control results in the regulation of cellular processes such as the degradation of proteins and other molecules, receptor‐mediated endocytosis, and coupled transport of various small molecules. More specifically, these enzymes are crucial in the processes of renal acidification,14 bone resorption and degradation,15 control of cytoplasmic pH,13 and sperm maturation.16 As V‐ATPases play a variety of cellular roles, their malfunction has been implicated in a variety of diseases including HIV, osteoporosis, and cancer.13, 17
The benzolactone class of natural products has attracted significant attention from both the synthetic and medicinal chemistry communities due to their unique architecture and their various bioactivities; particularly, their potent anticancer properties.18, 19 Analogue studies20, 21 have been aimed mainly at exploring the structure–activity relationships (SAR) for the enamide side chain, stemming from the known instability of the salicylihalamide side chain.2 The enamide could be hydrolyzed into the corresponding aldehyde and amide fragments in aqueous media and more importantly from a chemotherapeutic discovery perspective, be inactivated prior to reaching its intended target. Hydrolysis inside the acidic environment of cancer cells is also a possible concern. Work by De Brabander and co‐workers has shown that the benzolactone enamides are likely irreversible inhibitors of V‐ATPase.22 However, their efforts to incorporate isotopically labeled salicylihalamide into the V‐ATPase system and elucidate the exact binding mode have not been successful.22 Medicinal chemistry campaigns to replace the enamide with various electrophilic Michael acceptors has led to analogues that retain the V‐ATPase‐inhibitory properties of their parent natural product(s) but in general had lost cytotoxic potency.18 One notable exception is the saliphenylhalamide analogue synthesized by De Brabander. This molecule was able to retain cytotoxic properties while demonstrating enhanced aqueous stability relative to salicylihalamide.23 The necessity of an enamide side chain for cytotoxicity was brought into question with the isolation of another benzolactone natural product, cruentaren A (3, Figure 1).24, 25 Cruentaren A contains an allylic amide side chain—not a conjugated enamide—yet still possesses potent cytotoxicity. This molecule acts through a different mechanism of action, targeting mitochondrial ATP synthase (F‐ATPase), not V‐ATPase as seen with the enamide‐containing natural products.24
We are now reporting our efforts to discover bioactive analogues with better chemical stability by installing physiologically stable warheads onto the benzolactone framework to create analogues that would retain the cytotoxic properties observed with this natural product class. Drawing experience from our syntheses of the salicylihalamides and oximidine II,26, 27, 28 we envisioned that we could rapidly assemble the benzolactone scaffolds and affix different side chains for investigation of the anticancer properties of the analogues. To this end, we planned to install the various side chains onto either the oximidine II or structurally less complex salicylihalimide‐like cores. The design of the simplified analogues relied on an in silico analysis using the CoMSIA field for V‐ATPase inhibitors that had been generated earlier by our research group.29
Results and Discussion
The presence of the allylic side chain in cruentaren A led us to question if the electrophilic enamide functionality is necessary for cytotoxicity, or whether some other binding mode (such as hydrogen bonding) could be responsible for cytotoxicity. The synthesis of this oximidine II allylic amide homologue 10 began with vinyl iodide 4 (Scheme 1), an intermediate in previous syntheses of oximidine II.28, 30 A palladium‐mediated methoxycarbonylation reaction31 stereospecifically installed the allylic carbon with a functional handle to explore various carbon–nitrogen bond formation reactions. Ultimately, we discovered that reduction of the ester into the corresponding allylic alcohol followed by a Mitsunobu reaction with maleimide using a modified Walker protocol allowed us to install the requisite allylic nitrogen atom.32, 33 Under Luche reduction conditions, mono‐reduction of one of the amide bonds of maleimide 7 generated hemiaminal 8, which was condensed with O‐methoxyamine to generate oxime 9 as an inseparable mixture of E/Z oxime stereoisomers (3:1 ratio). Bis‐TBS removal revealed the desired allylic amide homologue 10 as only the E‐oxime stereoisomer. As shown in Table 1, analogue 10 was not cytotoxic, thereby confirming the need for an enamide side chain for the cytotoxicity of oximidine II. We next questioned whether the enamide was uniquely responsible for the cytotoxicity of oximidine II, or if other electrophilic warheads could replace the unstable enamide.
Scheme 1.

Synthesis of the allylic amide homologue 10: a) Pd(PPh3)4, DIPEA, CO (1 atm), MeOH, THF, 85 %; b) THF, HF/pyridine, RT, 89 % for 5 b, 80 % for 6 b, 69 % for 8 b, 89 % for 10; c) DIBAl‐H, CH2Cl2, −78 °C, 90 %; d) maleimide, DEAD, PPh3, THF, 0 °C, 62 %; e) CeCl3⋅7 H2O, NaBH4, MeOH, 0 °C, 86 %; f) MeONH3Cl, NaHCO3, MeOH, reflux, 34 %, 3:1 E/Z oxime ratio.
Table 1.
Cytotoxicity evaluation of synthesized analogues.
| Compound | IC50 [μm][a] | |||
|---|---|---|---|---|
| SK‐Mel‐5[b] | SK‐Mel‐28[b] | HL‐60[c] | MCF‐7[d] | |
| paclitaxel | 0.0083±0.0061, n=3 | 0.0079, n=1 | 0.0051±0.0035, n=2 | 0.0015±0.0007, n=3 |
| oximidine II (2) | 0.62±0.59, n=3 | 50, n=1 | ND | ND |
| allylic amide 10 | >60, n=2 | >60, n=2 | ND | ND |
| diene sulfone 24 | 25±1, n=2 | >60, n=1 | 41, n=1 | 41, n=1 |
| diene sulfone 25 | 32±3, n=2 | 49±7, n=2 | 52, n=1 | >60, n=2 |
| triene sulfone 36 | >60, n=1 | ND | ND | ND |
| triene sulfone 46 | 30, n=1 | ND | ND | ND |
| diene enamide 55 | 32, n=1 | 44, n=1 | 50, n=1 | 76, n=1 |
| diene enamide 56 | 24, n=1 | 57, n=1 | 25, n=1 | >100 |
[a] ND: not determined; n=number of times tested; values are the mean±SEM for n=3 values, and the mean±SD for n=2 values. [b] Human melanoma. [c] Human promyelocytic leukemia. [d] Human breast carcinoma.
To date, only α,β‐unsaturated ketones and esters have been investigated as electrophilic enamide side chain replacements in this natural product family.34 Although these Michael acceptors showed V‐ATPase inhibition similar to their parent compounds, they displayed 10‐ to 100‐fold decreased anticancer activities in cellular assays. These mixed results led us to investigate the vinyl sulfone moiety as an electrophilic functional group. Under normal physiological conditions, vinyl sulfone groups do not undergo nucleophilic addition. Nucleophilic addition occurs only when strategic hydrogen bonding interactions, typically present in an enzyme active site, are available to activate the functionality.35 We designed these analogues assuming that the acidic environment of the V‐ATPase could activate the electrophilic vinyl sulfone moiety for nucleophilic addition, analogously to the activation of the enamide moiety.
Molecular modeling
We performed computational analysis studies to aid in the development of simplified benzolactone‐like core scaffolds in the preparation of a SAR campaign to explore various enamide replacements. In particular, we focused on a benzolactone framework with decreased rigidity, hypothesizing that these more saturated molecules would be easier to synthesize, yet still display potent V‐ATPase inhibition. This strategy is supported by the fact that the known benzolactone enamides contain different substituents and varying degrees of unsaturation within the macrocycle. In addition, studies have shown that some structural modifications to the lactone moiety can be tolerated without significant loss of activity for salicylihalamide36 and related apicularen analogues.37 Using molecular modeling data obtained from a previous comparative molecular similarity indices analysis (CoMSIA)38/3D‐QSAR study completed by our group as the training set,29 a simplified, partially saturated, oximidine‐like scaffold was discovered with predicted V‐ATPase activity similar to that of the parent oximidine II molecule. In silico predictions of oximidine analogues considered for synthesis and testing were performed via the CoMSIA38 module within the Tripos Molecular Spreadsheet in SYBYL (version 8.0; Tripos Inc., St. Louis, MO (USA), 2009). Each molecule was sketched in SYBYL and optimized according to default molecular mechanics parameters using the Tripos Molecular Force‐field39 and Gasteiger–Marsili electrostatics.40 Molecules were then aligned according to best field fit within the CoMSIA field for V‐ATPase inhibitors generated by Blackman et al.29 CoMSIA scores were then generated via the molecular spreadsheet for each field‐fit molecule, and corresponding pIC50 values were predicted by perturbing the CoMSIA score with the three‐parameter perturbation model described by Blackman et al.,29 in which the perturbation terms were computed via the Dragon molecular descriptor computation software package.41 In this manner, we would preliminarily predict low‐nanomolar IC50 values for both compound 24 (2.7 nm) and 25 (6.1 nm) as activities within the assay reported by Wu et al.34 The rationale for this potency is evident from Figure 2 in that both compound 24 and 25 align well with the CoMSIA features. In both cases the terminal phenyl group is situated at the center of a large feature with favorable van der Waals interactions. Compound 24 is able to orient its sulfone oxygen atoms favorably toward an electropositive feature, thus providing a prediction for enzymatic activation of the electrophilic moiety; the extra carbon atom on the side chain of compound 25 disrupts this electrostatic complementarity, but is able to position itself in a manner that enables the phenyl group to exploit a somewhat greater lipophilic overlay.
Figure 2.
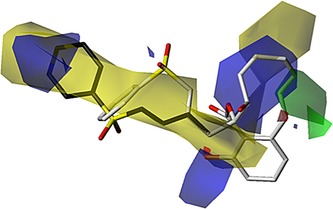
Overlay of compounds 24 and 25 onto CoMSIA fields showing isosurfaces for the following pharmacophore features: favorable van der Waals regions, steric barriers (green), and areas favorable for electropositive (red) and electronegative (blue) interactions with ligand polar groups. Both compounds are rendered according to default CPK coloring for heteroatoms; compound 24 has white carbon atoms, and compound 25 has black. CoMSIA isosurfaces correspond to default mean⋅coefficient values as implemented in the Tripos Molecular Spreadsheet.
With this knowledge in hand, we developed a synthetic route toward a (5E)‐7,8‐dihydromacrolactone core, with an appended vinyl sulfone functionality (analogues 24 and 25, Figure 2). Both one‐ and two‐carbon methylene spacer analogues 24 and 25 were synthesized (Scheme 2), as we were uncertain how the distance between the electrophilic vinyl sulfone moiety and the macrocycle would affect cytotoxicity. A Brown asymmetric allylation42 reaction between butanal 11 and protected allylic alcohol 13 began our synthesis of analogue 24, producing secondary alcohol 14 as a single diastereomer in excellent yield and enantioselectivity (95:5). Transesterification of alcohol 14 with salicylate diene 31 followed by TBS protection of the resultant phenol generated triene 16 as the precursor for the ring‐closing metathesis (RCM) reaction to form the macrocycle. The salicylate diene 31 was prepared from stannylpentenol 27 43 by oxidation to aldehyde 28 followed by methenylation to form diene 29 and reaction with the known3 salicylate 30.
Scheme 2.
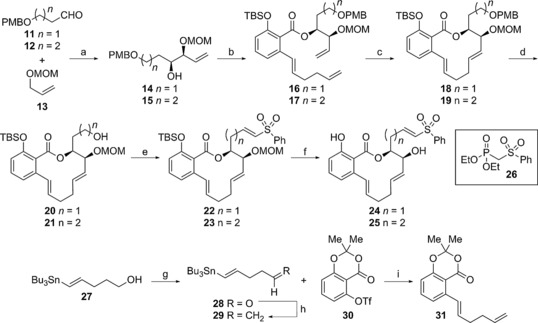
Synthesis of vinyl sulfone analogues 24 and 25: a) 13, sBuLi, THF, −78 °C, then (+)‐β‐methoxydiisopinocamphenylborane, THF, −78 °C to −90 °C, then BF3⋅OEt2, then 11/12, 80 % for 14, 88 % for 15; b) NaHMDS, THF, 0 °C, then 31, THF, 0 °C, then TBSCl, imidazole; c) Grubbs 2nd‐gen. catalyst, PhMe, reflux, 60 %, two steps for 18, 61 %, two steps for 19; d) DDQ, CH2Cl2, 0 °C, 50 % for 20, 63 % for 21; e) SO3⋅Py, Et3N, DMSO, CH2Cl2, 0 °C; f) phosphonate 26, NaH, THF, 0 °C, then aldehyde, 48 %, two steps for 22, 65 %, two steps for 23; f) 4 m HCl, MeOH, 82 % for 24, 85 % for 25; g) SO3⋅Py, Et3N, DMSO, CH2Cl2, 0 °C; h) CH3PPh3Br, tBuOK, 0 °C to RT; i) Pd(PPh3)4, LiCl, THF, 70 °C, 93 %, three steps.
In agreement with previous trends26, 27 and our design rationale, reaction of seco‐cycle 16 using the Grubbs second‐generation ruthenium catalyst produced diene macrocycle 18 in 60 % yield over two steps (from 14). Oxidative cleavage of the para‐methoxybenzyl (PMB) ether, oxidation of the resultant primary alcohol to the corresponding aldehyde, followed by a Horner–Wadsworth–Emmons (HWE) olefination with phosphonate 26 installed the desired vinyl sulfone unit. Global deprotection revealed the vinyl sulfone analogue 24. Two‐carbon vinyl sulfone analogue 25 was synthesized in an analogous manner following these established protocols. Using this sequence, we were able to access the (5E)‐7,8‐dihydromacrolactone vinyl sulfone analogues in only eight linear steps from known starting materials.
When biological testing revealed that derivatives 24 and 25 showed only weak cytotoxic effects (Table 1), we decided to synthesize the vinyl sulfone side chain analogue 36 (Scheme 3) containing the oximidine II triene macrocyclic core and its C15 vinyl sulfone homologue 46 (Scheme 4). In the oximidine II system (and in other benzolactone enamide molecules), the electrophilic position of the enamide side chain is located at a position three carbon atoms away from the macrolactone core. Both analogues were readily available using chemistry previously developed in our laboratories.
Scheme 3.
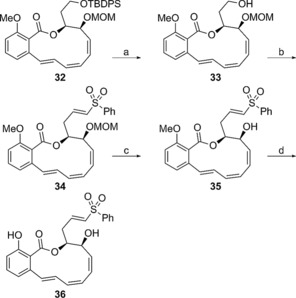
Synthesis of vinyl sulfone analogue 36: a) TBAF, THF, 88 %; b) 1. PDC, 4 Å MS, CH2Cl2, 0 °C, 2. 26, NaH, THF, then aldehyde, THF, 0 °C, 47 %, two steps; c) CBr4, iPrOH, 75 °C, 78 %; d) BCl3, CH2Cl2, 0 °C, 75 %.
Scheme 4.
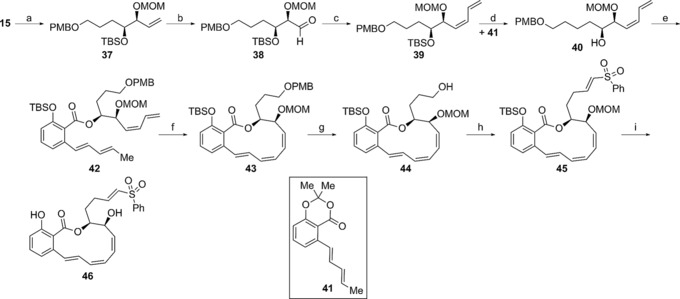
Synthesis of vinyl sulfone analogue 46: a) TBSCl, imidazole, DMF, 75 °C, 71 %; b) 1. OsO4, NMO, H2O, THF, 2. Pb(OAc)4, K2CO3, MeOH, 0 °C; c) Ti(OiPr)4, allyldiphenylphosphine, tBuLi, THF, −78 °C to 0 °C, then MeI; d) TBAF, THF, 19 %, four steps; e) NaHMDS, THF, 0 °C, then 41, THF, 0 °C, then TBSCl, imidazole, 91 %; f) Grubbs 2nd‐gen. catalyst, PhMe, reflux, 22 %; g) DDQ, CH2Cl2, 0 °C to RT, 98 %; h) 1. PDC, 4 Å MS, CH2Cl2, 2. 26, NaH, THF, 0 °C, then aldehyde, 47 %, two steps; i) 4 m HCl, MeOH, 68 %.
The synthesis of the vinyl sulfone side chain analogue 36 began with triene macrocycle 32, an intermediate in our recent synthesis of oximidine II.28 Silyl group removal, oxidation of the primary alcohol to the corresponding aldehyde, and HWE olefination using phosphonate 26 43 yielded vinyl phenylsulfone intermediate 34. Subjecting this compound to a two‐step alkyl ether deprotection sequence generated the desired vinyl sulfone 36. Assays (Table 1) revealed that this compound is devoid of cytotoxicity in the cell lines tested.
RCM reactions are well‐established strategies in the synthesis of various macrocyclic benzolactone enamides, including oximidine II.30, 44 The usefulness of this methodology is demonstrated here in the synthesis of vinyl sulfone analogue 46 (Scheme 4). Our synthesis of analogue 46 began with the above‐mentioned alcohol 15 (see Scheme 2). Silylation, oxidative cleavage of the terminal olefin, Wittig olefination, and desilylation furnished diene 40, ready for a transesterification reaction with salicylate 41.28 Transesterification reactions are well‐established protocols for the generation of salicylate esters.45 This transesterification followed by silylation of the resultant phenol gave the RCM precursor 42 in excellent yield over two steps. The RCM reaction, using the Grubbs second‐generation catalyst, gave 43 in low yields but allowed for material throughput toward the final desired analogue. Dimerization produced the major by‐product in this macrocyclization reaction. Removal of the PMB ether from macrocycle 43 under oxidative conditions, followed by oxidation to the corresponding aldehyde, HWE olefination with phosphonate 26, and global protecting group removal provided the target vinyl sulfone 46.
Unfortunately, biological testing revealed that oximidine sulfone analogue 46 does not possess cytotoxic activity. To verify the validity of our in silico modeling and design of the simplified oximidine II macrolactone core, we synthesized and tested analogues 55 and 56 (Scheme 5) possessing the enamide side chain.
Scheme 5.
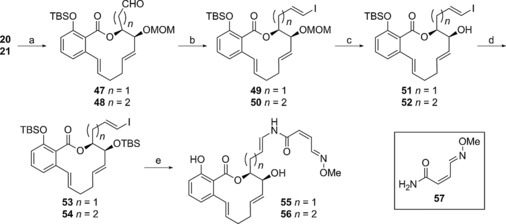
Synthesis of simplified benzolactones with an oximidine enamide side chain: a) DMP, CH2Cl2, 88 % for 47, 72 % for 48; b) CrCl2, CHI3, THF/dioxane, 86 %, 9:1 E/Z ratio for 49, 84 %, 9:1 E/Z ratio for 50; c) CBr4, iPrOH, 75 °C, 86 % for 51, 96 % for 52; d) TBSOTf, 2,6‐lutidine, CH2Cl2, −78 °C to 0 °C, 78 % for 53, 84 % for 54; e) 1. 57, CuI, DMEDA, Cs2CO3, DMA, 2. HF⋅Py, Py, THF, 40 %, two steps for 55, 30 %, two steps for 56.
Analogue 55 (Scheme 5) was prepared from the previously described intermediate 20 (Scheme 2), which underwent alcohol oxidation followed by Takai olefination of the resultant aldehyde to provide vinyl iodide 49. Protecting group manipulation prepared iodide 53 for installation of the enamide side chain. The side chain was attached via a copper‐mediated C−N coupling reaction with amide 57, and global desilylation finalized the synthesis of analogue 55. Two‐carbon enamide analogue 56 was synthesized in a similar manner by following the established procedure.
Cytotoxicity testing
The NIH cytotoxicity screen showed that the benzolactone enamides display their most significant cytotoxicity toward melanoma and leukemia cell lines.9 These natural products are especially potent against SK‐Mel‐5, SK‐Mel‐28, and HL‐60 cells. Based on this precedent, our analogues were tested against these cell lines in addition to the breast cancer cell line MCF‐7. Each of our analogues displayed significant lower cytotoxicity than synthetic oximidine II. Additionally, the respective deprotected versions of intermediates 5 a, 6 a, and 8 a, compounds 5 b, 6 b, and 8 b, toward the synthesis of the allylic amide analogue were also inactive, including the Michael acceptor Z‐enoate compound 5 b (data not shown). Some weak cytotoxic activity was retained for diene sulfone 25 and the enamides 55 and 56. These results provide further support for previous analogue studies that demonstrated the necessity of an acyl enamine side chain for bioactivity and that the designed macrolactone scaffold investigated herein cannot replace the rigid macrocycle of the natural products.
To rationalize a structural basis for the observed differences in activity, a molecular overlay was generated for compounds 2 and 55. Plausible low‐energy conformations for both compounds were obtained by structural optimization using the VEGA ZZ software package46 and were aligned via the PyMOL program.47 Using pairwise alignment among conserved atoms, the macrocycles of the two compounds overlay very closely; however, the polar side chains exhibit much greater variation. To enhance the comparison, a survey of side chain conformational variations were explored for compound 55, with Figure 3 illustrating the structure producing the closest alignment with compound 2.
Figure 3.
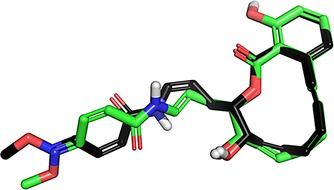
Graphic showing the alignment of compound 2 (black carbon atoms) with compound 55. Non‐carbon atoms (oxygen, nitrogen and polar hydrogens) are rendered by CPK coloring.
It is unlikely that the very minor structural differences within the macrocycles of the two compounds can explain the variation in bioactivity; however, the one structurally unconserved double bond within the side chains does produce a key difference in the electrostatic profiles of the two molecules. In contrasting a cis configuration adjacent to the amide in compound 2 with a trans structure in compound 55, one concludes that the two side chains are unlikely to avail themselves of the same set of receptor hydrogen bonding features, even if a broad range of side chain conformational space is explored. The superior activity of compound 2 may thus be likely ascribed to an ability to form side chain hydrogen bonds that compound 55 is unable to access.
Conclusions
In summary, side chain analogues of oximidine II were synthesized to probe the hypothesis that the enamide functionality of the parent compound could be replaced with physiologically stable functional groups bearing an allylic amide or an electrophilic vinyl sulfone functionality, and that the analogues would retain bioactivity. However, because the allylic amide and sulfone side chain analogues lack cytotoxicity, we have added further evidence that an enamide moiety is required for the biological activity of the benzolactone enamide family of natural products. Analogues were also designed and synthesized following computational studies which predicted that flexible and less complex macrolactones would fit well into the V‐ATPase binding pocket. These analogues showed weak cytotoxic activities, demonstrating that this simplified scaffold cannot replace the macrocycle of the natural products.
Experimental Section
Chemistry
General: Infrared (IR) spectra were recorded on an FT‐IR instrument from thin films on NaCl plates. All NMR experiments were recorded on either 400 MHz (100 MHz) or 500 MHz (125 MHz) spectrometers as noted. Chemical shifts are reported as parts per million (ppm) using the solvent residual peak as the internal standard. Samples obtained in CDCl3 were referenced to 7.27 ppm for 1H NMR and 77.0 ppm for 13C NMR spectra. Samples obtained in [D6]DMSO were referenced to 2.50 ppm for 1H NMR and 39.5 ppm for 13C NMR spectra. Samples obtained in [D6]acetone were referenced to 2.05 ppm for 1H NMR and 29.9 ppm for 13C NMR spectra. Coupling constants (J) are reported in Hz. The multiplicities of the signals are assigned using the following abbreviations: s=singlet, d=doublet, t=triplet, q=quartet, qu=quintet, se=sextet, br=broad. Optical rotations were recorded using a polarimeter at room temperature (RT). LRMS and HRMS were obtained using the electrospray ionization (ESI) technique.
Moisture‐sensitive reactions were run in flame‐dried glassware under an atmosphere of nitrogen unless otherwise specified. THF, CH2Cl2, Et2O, and toluene were dried and deoxygenated by passing the nitrogen‐purged solvents through activated alumina columns on a solvent purification system. DMF was similarly dried by passing through a column of activated 4 Å molecular sieves on a solvent purification system. All other reagents and solvents were used as received from commercial sources unless otherwise noted. Reaction progress was monitored by thin‐layer chromatography (TLC, silica gel, 10×20 cm, 250 μm), visualizing with UV light (λ 254 nm) and developing the plates with Hanessian′s stain (blue stain). All compounds were purified by column chromatography with 230–400 mesh silica gel as described. All compounds were concentrated using standard rotary evaporator and high‐vacuum techniques.
Methyl (Z)‐4‐((3S,4S,5Z,7Z,9E)‐4,14‐bis(tert‐butyldimethylsilyloxy)‐1‐oxo‐3,4‐dihydro‐1H‐benzo[c][1]oxacyclododecin‐3‐yl)but‐2‐enoate (5 a). A flask containing Pd(PPh3)4 (71 mg, 0.661 mmol) was flushed with CO (1 atm). To this flask was added N,N′‐diisopropylethylamine (DIPEA; 0.044 mL, 0.245 mmol) and anhydrous MeOH (4.90 mL) at RT and the flask was flushed with CO again. A THF solution of vinyl iodide 4 28 (160 mg, 0.245 mmol in 1.23 mL THF) was added and the reaction was stirred under CO (1 atm) for 45 min. The reaction mixture was filtered through a pad of Celite, and the Celite layer was eluted with Et2O until the eluent was colorless. The crude material was purified by flash column chromatography (hexanes/Et2O, 100:0–95:5) to collect 122 mg of the desired ester 5 a as a yellow oil (85 %): TLC (hexanes/Et2O, 80:20): R f=0.80; =−148 (c=0.600, CHCl3); 1H NMR (400 MHz, CDCl3): δ=0.01 (s, 3 H), 0.09 (s, 3 H), 0.20 (s, 3 H), 0.22 (s, 3 H), 0.88 (s, 9 H), 0.98 (s, 9 H), 3.00–3.09 (m, 1 H), 3.55–3.63 (m, 1 H), 3.74 (s, 3 H), 4.64 (dd, J=10, 3.2 Hz, 1 H), 5.24 (td, J=9.1, 4.0 Hz, 1 H), 5.82 (t, J=12 Hz, 1 H), 5.85–5.89 (m, 2 H), 5.97 (dd, J=12, 4.1 Hz, 1 H), 6.34 (t, J=11 Hz, 1 H), 6.46 (ddd, J=12, 7.7, 5.6 Hz, 1 H), 6.64 (d, J=16 Hz, 1 H), 6.75 (d, J=8.1 Hz, 1 H), 6.79 (d, J=7.4 Hz, 1 H), 7.06 (ddd, J=16, 10, 0.5 Hz, 1 H), 7.17 ppm (t, J=8.0 Hz, 1 H); 13C NMR (100 MHz, CDCl3): δ=168.4, 166.6, 152.3, 146.3, 138.0, 132.4, 130.8, 130.6, 129.7, 129.3, 128.5, 128.0, 126.4, 121.2, 120.4, 117.8, 78.9, 66.5, 51.1, 29.7, 27.9, 25.8, 25.7, 18.4, 17.9, −4.1, −4.5, −5.0 ppm; IR (neat, NaCl): max=2972, 2861, 1732, 1648, 1561, 1152, 1056, 963 cm−1; HRMS (ESI) m/z calcd for C32H48NaO6Si2 [M+Na]+: 607.2887, found: 607.2861.
Methyl (Z)‐4‐((3S,4S,5Z,7Z,9E)‐4,14‐dihydroxy‐1‐oxo‐3,4‐dihydro‐1H‐benzo[c][1]oxacyclododecin‐3‐yl)but‐2‐enoate (5 b). To a THF solution (0.220 mL) of bis‐TBS ester 5 a (13 mg, 0.022 mmol) at RT was added HF/pyridine solution (5 m, 0.135 mL). The reaction was stirred for 24 h, and then quenched with phosphate (pH 7) buffer. The aqueous layer was extracted with EtOAc (3×), the organic extracts washed with brine and dried over MgSO4. Purification of the crude material by flash column chromatography (hexanes/EtOAc, 80:20–50:50) gave 7 mg of a colorless oil (5 b, 89 %): TLC (hexanes/EtOAc, 60:40): R f=0.10; 1H NMR (400 MHz, CDCl3): δ=1.28 (br, 1 H), 2.88 (br, 1 H), 3.10–3.04 (m, 1 H), 3.26 (br, 1 H), 3.68 (s, 3 H), 4.42 (br, 1 H), 5.59 (br, 1 H), 5.81 (dd, J=12, 4.9 Hz, 1 H), 5.87 (d, J=11 Hz, 1 H), 6.07 (br d, J=12 Hz, 1 H), 6.22 (br, 1 H), 6.38 (br, 1 H), 6.77–6.71 (br m, 3 H), 6.88 (d, J=8.2 Hz, 1 H), 7.30 (t, J=8.0 Hz, 1 H), 10.30 ppm (br, 1 H); HRMS (ESI), m/z calcd for C20H21O6 [M+H]+: 357.1338, found: 357.1323; LC–MS analysis revealed 94.6 % purity prior to biological testing.
(3S,4S,5Z,7Z,9E)‐4,14‐Bis(tert‐butyldimethylsilyloxy)‐3‐((Z)‐4‐hydroxybut‐2‐enyl)‐3,4‐dihydro‐1H‐benzo[c][1]oxacyclododecin‐1‐one (6 a). Ester 5 a (9.5 mg, 0.016 mmol) was dissolved in anhydrous CH2Cl2 (0.160 mL) and cooled to −78 °C. To this cooled solution was added DIBAl‐H (0.037 mL, 0.037 mmol, 1 m solution in CH2Cl2) dropwise (Note: very slow addition of DIBAl‐H is required to prevent side reactions.) After 1.5 h the cooling bath was removed, and the reaction was quenched with EtOAc and then Rochelle′s salt solution was added. The mixture was warmed to RT and stirred for 2 h, or until both aqueous and organic layers were clear. The aqueous layer was extracted with EtOAc (3×), and the organic extracts were combined, washed with brine, and dried over MgSO4. Purification via flash column chromatography (hexanes/EtOAc, 95:5–85:15) yielded 8 mg of pure allylic alcohol 6 a as a colorless oil (90 %): TLC (hexanes/EtOAc, 80:20): R f=0.30; =−94.5 (c=1.32, CHCl3); 1H NMR (400 MHz, CDCl3): δ=0.01 (s, 3 H), 0.10 (s, 3 H), 0.17 (s, 3 H), 0.19 (s, 3 H), 0.86 (s, 9 H), 0.99 (s, 9 H), 1.63 (br, 1 H), 2.56–2.63 (m, 1 H), 2.74–2.79 (m, 1 H), 4.10–4.16 (m, 1 H), 4.28–4.32 (m, 1 H), 4.63 (dd, J=10, 2.8 Hz, 1 H), 5.08 (br d, J=10 Hz, 1 H), 5.71–5.81 (m, 3 H), 5.87 (dd, J=11, 4.0 Hz, 1 H), 5.97 (dd, J=12, 4.0 Hz, 1 H), 6.34 (t, J=11 Hz, 1 H), 6.63 (d, J=16 Hz, 1 H), 6.76 (d, J=8.2 Hz, 1 H), 6.80 (d, J=7.6 Hz, 1 H), 7.05 (dd, J=16, 11 Hz, 1 H), 7.17 ppm (t, J=8.0 Hz, 1 H); 13C NMR (100 MHz, CDCl3): δ=168.4, 152.0, 138.8, 132.2, 131.3, 130.7, 130.6, 129.7, 129.3, 128.7, 128.5, 127.9, 126.7, 121.6, 118.5, 79.6, 66.4, 58.5, 25.9, 25.8, 25.7 18.4, 17.9, −4.0, −4.4, −4.5, −4.9 ppm; IR (neat, NaCl): max=3430 (br), 2959, 1729, 1256, 745 cm−1; HRMS (ESI) m/z calcd for C31H48NaO5Si2 [M+Na]+: 579.2938, found: 579.2909.
(3S,4S,5Z,7Z,9E)‐4,14‐Dihydroxy‐3‐((Z)‐4‐hydroxybut‐2‐enyl)‐3,4‐dihydro‐1H‐benzo[c][1]oxacyclododecin‐1‐one (6 b). To a THF solution (0.230 mL) of bis‐TBS Z‐allylic alcohol 6 a (13 mg, 0.023 mmol) at RT was added HF/pyridine solution (5 m, 0.143 mL). The reaction was stirred for 24 h, and then quenched with phosphate (pH 7) buffer. The aqueous layer was extracted with EtOAc (3×), the organic extracts washed with brine and dried over MgSO4. Purification of the crude material by flash column chromatography (hexanes/EtOAc, 50:50–30:70) gave 6 mg of a white solid (6 b, 80 %): TLC (hexanes/EtOAc, 40:60): R f=0.15; 1H NMR (400 MHz, [D6]DMSO): δ=2.39–2.31 (m, 1 H), 2.73 (m, 1 H), 4.10–3.98 (m, 2 H), 4.36 (td, J=10, 4.0 Hz, 1 H), 4.58 (t, J=5.2 Hz, 1 H), 5.04 (td, J=12, 2.0 Hz, 1 H), 5.31 (d, J=4.6 Hz, 1 H), 5.61–5.49 (m, 2 H), 5.79 (t, J=11 Hz, 1 H), 5.96 (ddd, J=17, 11, 4.2 Hz, 2 H), 6.35 (t, J=11 Hz, 1 H), 6.68 (t, J=8.4 Hz, 1 H), 6.70 (s, 1 H), 6.75 (d, J=8.0 Hz, 1 H), 6.92 (dd, J=16, 12 Hz, 1 H), 7.16 (t, J=7.9 Hz, 1 H), 9.34 ppm (s, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=168.2, 153.8, 137.4, 132.5, 132.1, 130.7, 129.8, 129.6, 129.5, 128.5, 128.4, 125.8, 121.8, 118.6, 114.1, 79.0, 64.5, 57.0, 24.9 ppm; HRMS (ESI) m/z calcd for C19H21O5 [M+H]+: 329.1389, found: 329.1393; LC–MS analysis revealed 82.1 % purity prior to biological testing.
1‐((Z)‐4‐((3S,4S,5Z,7Z,9E)‐4,14‐Bis(tert‐butyldimethylsilyloxy)‐1‐oxo‐3,4‐dihydro‐1H‐benzo[c][1]oxacyclododecin‐3‐yl)but‐2‐enyl)‐1H‐pyrrole‐2,5‐dione (7). Allylic alcohol 6 a (27 mg, 0.048 mmol), maleimide (5 mg, 0.053 mmol) and PPh3 (23 mg, 0.086 mmol) were dissolved in anhydrous THF (0.480 mL) and cooled to 0 °C. A solution of diethyl azodicarboxylate (DEAD; 0.039 mL, 0.086 mmol, 40 % w/w solution in toluene) was added dropwise, watching the initial yellow color to dissipate before an additional drop was added. The reaction was stirred for 15 min and then the volatiles were removed by rotary evaporation. Purification of the crude material by flash column chromatography (hexanes/CH2Cl2, 80:20–30:70, then hexanes/EtOAc, 80:20) yielded 19 mg of the desired allylic maleimide derivative 7 and 2 mg of the allylic alcohol 6a (62 %, 67 % based on recovered starting material): TLC (hexanes/EtOAc, 80:20): R f=0.35; =−110 (c=0.31, CHCl3); 1H NMR (400 MHz, CDCl3): δ=0.02 (s, 3 H), 0.11 (s, 3 H), 0.18 (s, 3 H), 0.19 (s, 3 H), 0.89 (s, 9 H), 0.97 (s, 9 H), 2.68–2.76 (m, 1 H), 2.91–2.97 (m, 1 H), 4.13 (dd, J=15, 7.1 Hz, 1 H), 4.24 (dd, J=15, 7.1 Hz, 1 H), 4.63 (dd, J=10, 3.0 Hz, 1 H), 5.14 (td, J=10, 3.0 Hz, 1 H), 5.50–5.56 (m, 1 H), 5.76–5.81 (m, 1 H), 5.85 (t, J=12 Hz, 1 H), 5.87 (dd, J=11, 4.1 Hz, 1 H), 6.00 (dd, J=12, 4.0 Hz, 1 H), 6.34 (t, J=11 Hz, 1 H), 6.59 (s, 2 H), 6.63 (d, J=16 Hz, 1 H), 6.74 (d, J=8.0 Hz, 1 H), 6.79 (d, J=7.7 Hz, 1 H), 7.06 (dd, J=16, 11 Hz, 1 H), 7.16 ppm (t, J=8.0 Hz, 1 H); 13C NMR (100 MHz, CDCl3): δ=170.2, 168.4, 152.4, 137.8, 134.0, 132.2, 130.8, 130.70, 130.68, 129.7, 129.2, 128.3, 128.0, 126.6, 125.2, 121.2, 118.1, 79.6, 66.6, 34.5, 29.7, 25.82, 25.77, 18.4, 17.9, −4.1, −4.2, −4.5, −4.9 ppm; IR (neat, NaCl): max=1729, 1155 cm−1; HRMS (ESI) m/z calcd for C35H50NO6Si2: 636.3177 [M+H]+, found: 636.3169.
1‐((Z)‐4‐((3S,4S,5Z,7Z,9E)‐4,14‐Bis(tert‐butyldimethylsilyloxy)‐1‐oxo‐3,4‐dihydro‐1H‐benzo[c][1]oxacyclododecin‐3‐yl)but‐2‐enyl)‐5‐hydroxy‐1H‐pyrrol‐2(5H)‐one (8 a). Allyl maleimide 7 (15 mg, 0.0236 mmol) and CeCl3⋅7 H2O (6 mg, 0.0236 mmol) were suspended in MeOH (0.100 mL) and cooled to 0 °C. NaBH4 (0.9 mg, 0.0236 mmol) was added in one portion and the reaction stirred for 30 min. The reaction was diluted with MeOH (5 mL) and the volatiles removed by rotary evaporation. This process was repeated two more times, then the crude material was purified by flash column chromatography (hexanes/EtOAc, 90:10–50:50) to yield 13 mg of a dark‐yellow oil (8 a), determined by 1H NMR and LC–MS to be a 55:45 ratio of diastereomers (86 %). These diastereomers were not separated, and the mixture was used in the subsequent step: TLC (hexanes/EtOAc, 70:30): R f=0.20; IR (neat, NaCl): max=3337 (br), 1732, 1690, 1596, 1455, 1253, 1168, 701 cm−1; HRMS (ESI) m/z calcd for C35H52NO6Si2: 638.3333 [M+H]+, found: 638.3342.
1‐((Z)‐4‐((3S,4S,5Z,7Z,9E)‐4,14‐Dihydroxy‐1‐oxo‐3,4‐dihydro‐1H‐benzo[c][1]oxacyclododecin‐3‐yl)but‐2‐enyl)‐5‐hydroxy‐1H‐pyrrol‐2(5H)‐one (8 b). To a THF solution (0.141 mL) of bis‐TBS hydroxypyrrole 8 a (9 mg, 0.0141 mmol) at RT was added HF/pyridine solution (5 m, 0.086 mL). The reaction was stirred for 24 h, and then quenched with phosphate (pH 7) buffer. The aqueous layer was extracted with EtOAc (3×), the organic extracts washed with brine and dried over MgSO4. Purification of the crude material by flash column chromatography (hexanes/EtOAc, 70:30–0:100) gave 4 mg of a tan solid (69 %): TLC (hexanes/EtOAc, 40:60): R f=0.04; 1H NMR (400 MHz, [D6]DMSO): δ=2.58–2.50 (m, 1 H), 2.86–2.78 (m, 1 H), 3.81 (dd, J=15, 7.8 Hz, 1 H), 4.12–4.07 (m, 1 H), 4.38 (td, J=10, 3.8 Hz, 1 H), 5.08 (td, J=9.3, 5.0 Hz, 1 H), 5.36–5.32 (m, 2 H), 5.51–5.45 (m, 1 H), 5.67–5.61 (m, 1 H), 5.81 (t, J=11 Hz, 1 H), 5.97 (ddd, J=15, 11, 4.0 Hz, 2 H), 6.09 (dd, J=7.8, 6.0 Hz, 1 H), 6.19 (dd, J=9.1, 6.5 Hz, 1 H), 6.36 (t, J=11 Hz, 1 H), 6.68 (t, J=7.0 Hz, 1 H), 6.70 (s, 1 H), 6.76 (dd, J=8.0, 5.0 Hz, 1 H), 6.98–6.90 (m, 1 H), 7.01 (d, J=6.0 Hz, 1 H), 7.16 (t, J=7.9 Hz, 1 H), 9.93 ppm (d, J=12 Hz, 1 H); 13C NMR (100 MHz, [D6]DMSO): δ=168.4, 168.2, 153.9, 147.6, 137.4, 132.1, 130.7, 129.9, 129.6, 129.5, 128.6, 128.5, 128.4, 127.1, 121.8, 118.7, 114.2, 82.0, 81.9, 64.6, 35.4, 24.7, 20.8 ppm; HRMS (ESI) m/z calcd for C23H24NO6 [M+H]+: 410.1604, found: 410.1591; LC–MS analysis revealed 85.5 % purity prior to biological testing.
(2Z)‐N‐((Z)‐4‐((3S,4S,5Z,7Z,9E)‐4,14‐Bis(tert‐butyldimethylsilyloxy)‐1‐oxo‐3,4‐dihydro‐1H‐benzo[c][1]oxacyclododecin‐3‐yl)but‐2‐enyl)‐4‐(methoxyimino)but‐2‐enamide (9). Hydroxypyrrolone 8 a (13 mg, 0.020 mmol), NaHCO3 (17 mg, 0.203 mmol), and MeONH3Cl (17 mg, 0.203 mmol) were suspended in MeOH (0.200 mL) in a sealed vial. This suspension was heated at 75 °C for 12 h. Upon cooling, the solvent was removed by rotary evaporation and the crude material purified by flash column chromatography (hexanes/EtOAc, 90:10–50:50) to yield 4.5 mg of the desired oxime amide 9 as a colorless oil (a 3:1 ratio of E/Z oxime ether stereoisomers by 1H NMR) and 5 mg of the starting material (34 %, 54 % based on recovered starting material): TLC (hexanes/EtOAc, 70:30): R f=0.65; 1H NMR (400 MHz, CDCl3): δ=0.01 (s, 3 H), 0.10 (s, 3 H), 0.15 (s, 3 H), 0.17 (s, 3 H), 0.89 (s, 9 H), 0.98 (s, 9 H), 2.68–2.71 (m, 2 H), 3.91 (s, 3 H), 3.85–3.93 (m, 1 H), 3.99–4.09 (m, 1 H), 4.63 (dd, J=10, 3.0 Hz, 1 H), 5.08–5.12 (m, 1 H), 5.46 (dt, J=11, 0.8 Hz, 1 H), 5.64–5.71 (m, 1 H), 5.75 (t, J=11 Hz, 1 H), 5.72–5.79 (m, 1 H), 5.84 (br, 1 H), 5.88 (dd, J=11, 4.2 Hz, 1 H), 5.98 (dd, J=12, 4.1 Hz, 1 H), 6.21 (dd, J=12, 10 Hz, 1 H), 6.34 (t, J=11 Hz, 1 H), 6.63 (d, J=16 Hz, 1 H), 6.77 (d, J=8.5 Hz, 1 H), 6.85 (dd, J=7.7, 3.6 Hz, 1 H), 7.02 (dd, J=16, 11 Hz, 1 H), 7.18–7.23 (m, 1 H), 8.96 (dd, J=10, 0.8 Hz, 1 H). Minor Z‐oxime ether isomer peaks: 3.94 (s, 3 H), 8.42 ppm (dd, J=9.6, 0.9 Hz, 1 H); 13C NMR (100 MHz, CDCl3): δ=168.2, 164.7, 151.9, 147.8, 138.2, 133.2, 132.2, 131.3, 130.7, 130.5, 129.5, 128.7, 128.1, 127.5, 125.5, 122.1, 118.7, 79.5, 66.3, 62.1, 35.9, 29.7, 25.9, 25.7, 18.4, 17.9, −4.0, −4.4, −4.5, −4.9 ppm; IR (neat, NaCl): max=3308 (br), 1733, 1674, 1569, 1539, 1049, 700 cm−1; HRMS (ESI) m/z calcd for C36H55N2O6Si2: 667.3599 [M+H]+, found: 667.3607.
(2Z)‐N‐((Z)‐4‐((3S,4S,5Z,7Z,9E)‐4,14‐Dihydroxy‐1‐oxo‐3,4‐dihydro‐1H‐benzo[c][1]oxacyclododecin‐3‐yl)but‐2‐enyl)‐4‐(methoxyimino)but‐2‐enamide (10). To a THF solution (0.09 mL) of bis‐TBS oxime ether 9 (6 mg, 0.009 mmol) at RT was added 0.055 mL of 5 m HF/pyridine solution. The reaction was stirred for 24 h and then quenched with a pH 7 buffer solution. The aqueous layer was extracted with EtOAc (3×), the organic extracts washed with brine and dried over MgSO4. Purification of the crude material by flash column chromatography (hexanes/EtOAc, 80:20–40:60) gave 3.5 mg of a tan solid (89 %): TLC (hexanes/EtOAc, 50:50): R f=0.10; 1H NMR (400 MHz, CDCl3): δ=1.29 (br, 1 H), 2.51 (br, 1 H), 2.67 (br, 1 H), 3.02 (br, 1 H), 3.87 (br, 1 H), 3.93 (s, 3 H), 4.40 (br, 1 H), 5.47 (br, 1 H), 5.58 (br, 1 H), 5.61–5.65 (m, 1 H), 5.75 (d, J=7.5 Hz, 1 H), 5.79 (br, 1 H), 5.93 (br, 1 H), 6.08 (d, J=13 Hz, 1 H), 6.30 (br, 1 H), 6.40 (t, J=11 Hz, 2 H), 6.76–6.78 (m, 3 H), 6.90 (d, J=7.9 Hz, 1 H), 7.32 (br, 1 H), 8.99 (dd, J=10, 0.6 Hz, 1 H), 10.48 (br, 1 H) ppm; HRMS (ESI) m/z calcd for C24H27N2O6: 439.1869 [M+H]+, found: 439.1863; UPLC–MS analysis established 95.4 % purity prior to biological testing.
(E)‐5‐(Hexa‐1,5‐dienyl)‐2,2‐dimethyl‐4H‐benzo[d][1,3]dioxin‐4‐one (31). To a flask containing Pd2dba3 (50.3 mg, 0.0549 mmol), tricyclohexylphosphonium tetrafluoroborate (79.2 mg, 0.215 mmol) and iPr2NEt (0.30 mL, 1.7 mmol) was added CH2Cl2 (100 mL) and the resulting mixture was stirred at RT for 10 min. 4‐Pentyn‐1‐ol (1 mL, 11 mmol) was added and the reaction mixture was cooled to 0 °C. Bu3SnH (3.45 mL, 13.0 mmol) diluted in CH2Cl2 (30 mL) was added dropwise via syringe over 15 min. The reaction was then allowed to stir at 0 °C for 4 h. The reaction mixture was condensed to ∼30 mL of organic mixture and was used directly to the next step without any workup. The crude product showed only (E)‐5‐(tributylstannyl)pent‐4‐en‐1‐ol (27), identical with 1H NMR and 13C properties as reported by Darwish.43
To crude 27 was added Et3N (7.6 mL, 54 mmol) and DMSO (30 mL). The mixture was cooled to 0 °C, and SO3⋅Py (52.0 mg, 0.324 mmol) was added in portions over 2 min. The mixture was stirred at 0 °C for 15 min, and then diluted with hexanes (100 mL) and phosphate buffer pH 7 (20 mL). The organic layer was separated, and then the aqueous layer was extracted with hexanes (3×30 mL). The combined organic layer was concentrated in vacuo to afford a bright‐yellow crude product. The crude aldehyde 28 was used directly in the next step without further purification.
To a mixture of CH3PPh3Br (8.50 g, 23.8 mmol) in anhydrous THF (30 mL), cooled at 0 °C under N2 atmosphere, was added dropwise a solution of tBuOK (2.42 g, 21.6 mmol) over 3 min. After the addition was completed, the mixture turned bright yellow. The mixture was stirred at 0 °C for 30 min and a solution of crude aldehyde 28 diluted in THF (30 mL) was added dropwise over 10 min. The reaction mixture was stirred at 0 °C and warmed to RT overnight, and then diluted with hexanes (100 mL) and quenched with phosphate buffer pH 7 (50 mL). The organic layer was separated and the aqueous layer was extracted with hexanes (3×100 mL), dried over NaSO4 and concentrated in vacuo to afford crude stannyl diene 29 as a yellow oil, which was used directly in the next reaction.
To a flask containing Pd(PPh3)4 (129.0 mg, 0.1116 mmol), LiCl (475.0 mg, 11.21 mmol), and 2,2‐dimethyl‐4‐oxo‐4H‐benzo[d][1,3]dioxin‐5‐yl trifluoromethanesulfonate 30 48 (731.0 mg, 2.241 mmol) was added anhydrous and degassed THF (10 mL) at RT under a N2 atmosphere. Then the crude stannyl diene 29 (∼2.7 mmol) dissolved in anhydrous and degassed DMF was added dropwise over 15 min. The resulting mixture was stirred and heated at 70 °C for 90 h, and then cooled to RT. After 10 % NH4OH (15 mL) was added, the mixture was extracted with hexanes (5×50 mL), washed with brine (20 mL), dried over Na2SO4, and concentrated in vacuo. Purification by column chromatography (10 % EtOAc in hexanes) provided 538.4 mg (2.084 mmol, 93 %) of dienyl acetonide 31 as an opaque oil: 1H NMR (400 MHz, CDCl3): δ=1.71 (s, 6 H), 2.27 (m, 2 H), 2.39 (m, 2 H), 5.00 (dd, J=1.86, 10.2 Hz, 1 H), 5.08 (dd, J=1.7, 17.1 Hz, 1 H), 5.88 (tdd, J=6.6, 10.4, 17.0 Hz, 1 H), 6.23 (td, J=6.8, 15.6 Hz, 1 H), 6.82 (dd, J=0.9, 8.1 Hz, 1 H), 7.22 (d, J=7.8 Hz, 1 H), 7.42 (t, J=8.0 Hz, 1 H), 7.48 ppm (d, J=15.7 Hz, 1 H); 13C NMR (100 MHz, CDCl3): δ=160.4, 156.8, 142.5, 138.0, 135.0, 134.6, 128.5, 121.3, 115.6, 115.0, 110.6, 105.1, 33.3, 32.5, 25.6 ppm; IR (neat, NaCl): =2990, 1735, 1598, 1467, 1320 cm−1; HRMS (ESI+) m/z calcd for C16H18NaO3 [M+Na]+: 281.1148, found: 281.1149.
(3S,4S)‐1‐(4‐Methoxybenzyloxy)‐4‐(methoxymethoxy)hex‐5‐en‐3‐ol (14). To a flask containing 3‐(methoxymethoxy)prop‐1‐ene (13)42 (1.27 g, 12.4 mmol) dissolved in anhydrous THF (25 mL) was added sec‐butyllithium in cyclohexane (1.40 m, 7.3 mL, 10.3 mmol) at −78 °C dropwise. The resulting orange solution was stirred at −78 °C for an additional 30 min, and (+)‐β‐methoxydiisopinocamphenylborane (3.26 g, 10.3 mmol) dissolved in THF (25 mL) was added dropwise. The reaction mixture was stirred at −78 °C for 1 h, then cooled to −90 °C (using MeOH and liquid N2 to prepare the cooling bath). The boron trifluoride etherate (1.72 mL, 13.7 mmol) was added dropwise. Immediately afterward, 3‐(4‐methoxybenzyloxy)propanal 11 49 (2 g, 10.3 mmol) was added dropwise and the mixture was kept at −90 °C for 3 h. The reaction was warmed to RT over 15 h to give a clear colorless solution, and was then quenched with a mixture of 35 % H2O2 (12.5 mL) and saturated NaHCO3 solution (20.0 mL). After stirring at RT for 30 min, the mixture was extracted with Et2O (2×15 mL). The combined organic extracts were washed with brine, dried over Na2SO4, and concentrated in vacuo. Purification by column chromatography (30 % EtOAc in hexanes) provided 2.44 g (8.23 mmol, 80 %) of alcohol 14 as a colorless oil: =+50.6 (c=1.50, CHCl3), (90 % ee);30 1H NMR (400 MHz, CDCl3): δ=1.79 (m, 2 H), 3.22 (br s, 1 H), 3.39 (s, 1 H), 3.66 (m, 2 H), 3.80 (s, 4 H), 3.82 (m, 1 H), 3.92 (dd, J=7.0, 7.0 Hz, 1 H), 4.46 (s, 2 H), 4.60 (d, J=6.7 Hz, 1 H), 4.73 (d, J=6.7 Hz, 1 H), 5.31 (d, J=16.9 Hz, 1 H), 5.31 (d, J=11.0 Hz, 1 H), 5.72 (ddd, J=7.7, 11.4, 16.3 Hz, 1 H), 6.87 (d, J=8.6 Hz, 2 H), 7.25 ppm (d, J=8.6 Hz, 2 H); 13C NMR (100 MHz, CDCl3): δ=159.2, 134.4, 130.1, 129.4, 120.1, 113.8, 94.0, 81.0, 72.8, 71.8, 67.5, 55.7, 55.3, 32.4 ppm; IR (neat, NaCl): =2955 (br), 2953 (br), 1613, 1515, 1460, 1249, 1170 cm−1; HRMS (ESI+) m/z calcd for C16H24NaO5 [M+Na]+: 319.1516, found: 319.1518.
(4S,5E,9E)‐14‐(tert‐Butyldimethylsilyloxy)‐3‐(2‐(4‐methoxybenzyloxy)ethyl)‐4‐(methoxymethoxy)‐3,4,7,8‐tetrahydro‐1H‐benzo[c][1]oxacyclododecin‐1‐one (18). Alcohol 14 (500 mg, 1.69 mmol) was subjected to azeotropic distillation in vacuo with benzene (2×5 mL), dissolved in anhydrous THF (5.0 mL) and cooled to 0 °C. NaHMDS (1.0 m, 3.20 mL, 3.20 mmol) was added dropwise. After stirring at 0 °C for 30 min, salicylate 31 (413 mg, 1.60 mmol) dissolved in distilled THF (5.0 mL) was added dropwise. The reaction was warmed to RT and stirred for 2 h, and then TBSCl (482 mg, 3.20 mmol) and imidazole (218 mg, 3.20 mmol) were added to the reaction mixture as solids. The reaction was stirred overnight and diluted with hexanes (30 mL). The mixture was filtered through a thin silica gel plug, which was rinsed with 20 % Et2O in hexanes (2×35 mL). The combined organic layer was concentrated in vacuo and subjected to azeotropic distillation with benzene (2×5 mL). The crude ester 16 was used directly in the next step without further purification.
To a flask containing crude ester 16 was added anhydrous and degassed toluene (1 L). Ruthenium Grubbs second‐generation catalyst (50.0 mg, 0.0589 mmol) in degassed toluene (1.0 mL) was added in one portion under a nitrogen atmosphere. The solution was heated at reflux and stirred for 20 h. The reaction mixture was allowed to cool to RT and DMSO (15 mL) was added into the reaction flask prior the removal of toluene in vacuo to obtain a dark‐brown liquid. The crude product was loaded directly on to a silica column for purification (0 to 20 % EtOAc in hexanes) to afford 553.3 mg (0.9528 mmol, 60 %) of macrocyclic diene 18 as a light‐yellow oil: =+76.3 (c=1.53, CHCl3); 1H NMR (400 MHz, CDCl3): δ=0.23 (s, 3 H), 0.24 (s, 3 H), 0.98 (s, 9 H), 2.18 (m, 4 H), 2.36 (m, 2 H), 3.35 (s, 3 H), 3.64 (m, 2 H), 3.81 (s, 3 H), 4.39 (q, J=2.6 Hz, 1 H), 4.43 (d, J=11.4 Hz, 1 H), 4.51 (d, J=11.4 Hz, 1 H), 4.57 (d, J=6.8 Hz, 1 H), 4.67 (d, J=6.8 Hz, 1 H), 5.29 (td, J=3.35, 6.7 Hz, 1 H), 5.32 (dd, J=5.5, 15.9 Hz, 1 H), 5.50 (ddd, J=5.6, 6.3, 15.9 Hz, 1 H), 5.77 (td, J=6.2, 15.9 Hz, 1 H), 6.22 (d, J=15.9 Hz, 1 H), 6.68 (d, J=8. Hz, 1 H), 6.87 (m, 3 H), 7.13 (t, J=7.8 Hz, 1 H), 7.29 ppm (d, J=8.6 Hz, 2 H); 13C NMR (100 MHz, CDCl3): δ=168.2, 159.1, 152.1, 137.6, 132.2, 131.5, 130.9, 130.6, 129.9, 129.6, 129.3, 125.7, 118.7, 117.1, 113.8, 94.9, 76.3, 75.3, 72.4, 66.6, 56.1, 55.3, 30.63, 30.61, 30.0, 25.8, 18.3, −4.0, −4.2 ppm; IR (neat, NaCl): =2910, 2857, 1730, 1613, 1600, 1565, 1505, 1463, 1250, 1160, 1033 cm−1; HRMS (ESI+) m/z calcd for C33H46NaO7Si [M+Na]+: 605.2905, found: 605.2907.
(3S,4S,5E,9E)‐14‐(tert‐Butyldimethylsilyloxy)‐3‐(2‐hydroxyethyl)‐4‐(methoxymethoxy)‐3,4,7,8‐tetrahydro‐1H‐benzo[c][1]oxacyclododecin‐1‐one (20). To a flask containing macrocyclic diene 18 (177.7 mg, 0.3049 mmol) was added CH2Cl2 (10.0 mL) and H2O (1.0 mL). The mixture was cooled to 0 °C, and 2,3‐dichloro‐5,6‐dicyano‐1,4‐benzoquinone (DDQ; 74.9 mg, 0.330 mmol) was added in one portion. The cooling bath was removed and the reaction was stirred at RT for 1 h. The reaction mixture was diluted with saturated NaHCO3 (20 mL), and then extracted with CH2Cl2 (2×20 mL). The organic layer was washed with saturated NaHCO3 (10 mL) and brine (10 mL). The crude mixture was dried over Na2SO4, and concentrated in vacuo. Purification by column chromatography (30 % EtOAc in hexanes) provided 70.9 mg (0.153 mmol, 50 %) of alcohol 20 as a colorless oil: =+108 (c=2.86, CHCl3); 1H NMR (400 MHz, CDCl3): δ=0.24 (s, 3 H), 0.27 (s, 3 H), 0.98 (s, 9 H), 2.09 (m, 2 H), 2.18 (m, 2 H), 2.39 (m, 2 H), 3.39 (s, 3 H), 3.78 (m, 2 H), 4.39 (br, 1 H), 4.58 (d, J=6.8 Hz, 1 H), 4.69 (d, J=6.8 Hz, 1 H), 5.31 (d, J=4.2 Hz, 1 H), 5.34 (dd, J=5.04, 15.8 Hz, 1 H), 5.54 (td, J=5.9, 15.8 Hz, 1 H), 5.80 (td, J=5.9, 15.9 Hz, 1 H), 6.21 (d, J=15.9 Hz, 1 H), 6.70 (d, J=8.2 Hz, 1 H), 6.90 (d, J=7.7 Hz, 1 H), 7.14 ppm (t, J=8.0 Hz, 1 H); 13C NMR (100 MHz, CDCl3): δ=169.4, 152.1, 137.5, 132.2, 131.4, 130.8, 130.1, 129.9, 125.2, 118.6, 117.2, 94.8, 77.2, 75.5, 58.8, 56.1, 34.0, 30.4, 29.8, 25.8, 18.5, −4.0, −4.1 ppm; IR (neat, NaCl): =3450 (br), 3005, 2930, 1731, 1460, 1380 cm−1; HRMS (ESI+) m/z calcd for C25H38NaO6Si [M+Na]+: 485.2330, found: 485.2335.
(3S,4S,5E,9E)‐14‐(tert‐Butyldimethylsilyloxy)‐4‐(methoxymethoxy)‐3‐((E)‐3‐(phenylsulfonyl)allyl)‐3,4,7,8‐tetrahydro‐1H‐benzo[c][1]oxacyclododecin‐1‐one (22). To a flask containing alcohol 20 (50.0 mg, 0.108 mmol) was added CH2Cl2 (0.5 mL), Et3N (75.0 mL, 0.540 mmol) and DMSO (0.5 mL). The mixture was cooled to 0 °C, and SO3⋅Py (52.0 mg, 0.324 mmol) was added in one portion. The mixture was stirred at 0 °C for 20 min, diluted with Et2O (4 mL) and phosphate buffer pH 7 (2 mL). The organic layer was separated, and then the aqueous layer was extracted with Et2O (3×4 mL). The combined organic layer was washed with saturated NH4Cl (4 mL), and then with saturated CuSO4 (2×4 mL). The organic layer was concentrated in vacuo to afford an opaque oil of the corresponding aldehyde. The crude aldehyde was used directly in the next step without further purification.
To a solution of diethyl [(phenylsulfonyl)methyl]phosphonate (26; 31.4 mg, 0.108 mmol) in anhydrous THF (1.0 mL) was added 60 % NaH (5.0 mg, 0.12 mmol). The mixture was cooled to 0 °C, and was stirred for 5 min. The aldehyde in THF (1.0 mL) was added dropwise over 3 min. The mixture was stirred at 0 °C for 40 min, and then diluted with 10 % HCl (0.25 mL) and phosphate (pH 7) buffer (2.0 mL). The crude mixture was extracted with EtOAc (2.0 mL). The organic layer was separated, and washed with saturated NaHCO3 (1.0 mL). The organic layer was washed with brine (1.0 mL), dried over MgSO4, and concentrated in vacuo. Purification by column chromatography (20 % EtOAc in hexanes) provided 31.0 mg (0.0518 mmol, 48 %) of phenylsulfone 22 as an opaque oil: =+100 (c=1.20, CHCl3); 1H NMR (400 MHz, CDCl3): δ=0.21 (s, 3 H), 0.22 (s, 3 H), 0.94 (s, 9 H), 2.10 (m, 2 H), 2.37 (m, 2 H), 2.72 (m, 1 H), 2.94 (m, 1 H), 3.30 (s, 3 H), 4.35 (q, J=3.0 Hz, 1 H), 4.49 (d, J=6.7 Hz, 1 H), 4.63 (d, J=6.7 Hz, 1 H), 5.08 (m, J=2.9 Hz, 1 H), 5.25 (dd, J=6.1, 15.8 Hz, 1 H), 5.52 (td, J=6.2, 15.4 Hz, 1 H), 5.75 (td, J=6.2, 15.7 Hz, 1 H), 6.12 (d, J=15.9 Hz, 1 H), 6.47 (td, J=1.3, 15.2 Hz, 1 H), 6.68 (d, J=8.2 Hz, 1 H), 6.87 (d, J=7.7 Hz, 1 H), 7.05 (td, J=7.4, 14.9 Hz, 1 H), 7.15 (t, J=8.0 Hz, 1 H), 7.56 (m, 3 H), 7.88 ppm (m, 2 H); 13C NMR (100 MHz, CDCl3): δ=168.1, 152.0, 141.5, 140.4, 137.7, 133.4, 133.2, 132.4, 132.2, 131.0, 130.1, 129.6, 129.3, 127.7, 125.1, 118.8, 117.0, 94.6, 75.9, 75.5, 56.2, 32.3, 30.8, 30.1, 25.7, 18.2, −4.0, −4.5 ppm; IR (neat, NaCl): =2935, 1731, 1570, 1460, 1292, 1270, 1148, 1101 cm−1; HRMS (ESI+) m/z calcd for C32H42NaO7SSi [M+Na]+: 621.2313, found: 621.2317.
(3S,4S,5E,9E)‐4,14‐Dihydroxy‐3‐((E)‐3‐(phenylsulfonyl)allyl)‐3,4,7,8‐tetrahydro‐1H‐benzo[c][1]oxacyclododecin‐1‐one (24). To a flask containing phenylsulfone 22 (19.0 mg, 0.0318 mmol) was added 4 m HCl in MeOH (1.0 mL). The mixture was stirred at RT for 120 h, and then concentrated in vacuo. Purification by column chromatography (50 % EtOAc in hexanes) provided 11.5 mg (0.0262 mmol, 82 %) of the desired analogue 24 as a colorless oil: =−35.0 (c=0.575, CHCl3); 1H NMR (400 MHz, CDCl3): δ=1.98 (br s, 1 H) 2.20 (m, 2 H), 2.41 (m, 2 H), 2.72 (m, 1 H), 2.95 (m, 1 H), 4.38 (br s, 1 H), 5.27 (m, 1 H), 5.45 (dd, J=5.8, 15.9 Hz, 1 H), 5.62 (qd, J=4.49, 15.8 Hz, 2 H), 6.44 (d, J=15.0 Hz, 1 H), 6.52 (d, J=15.8 Hz, 1 H), 6.74 (d, J=7.4 Hz, 1 H), 6.85 (dd, J=0.9, 8.3 Hz, 1 H), 7.05 (ddd, J=7.1, 8.0, 15.1 Hz, 1 H), 7.30 (t, J=7.9 Hz, 1 H), 7.46 (m, 3 H), 7.81 (m, 2 H), 10.41 ppm (s, 1 H); 13C NMR (100 MHz, CDCl3): δ=170.5, 161.5, 142.1, 141.7, 140.0, 135.0, 134.8, 134.3, 133.3, 133.1, 129.7, 129.2, 128.8, 127.5, 120.0, 116.3, 111.4, 75.0, 72.5, 32.4, 31.7, 29.8 ppm; IR (neat, NaCl): =3415 (br), 3050, 2920, 2865, 1737, 1630, 1464, 1406, 1044 cm−1; HRMS (ESI+) m/z calcd for C24H24NaO6S [M+Na]+: 463.1186, found: 463.1193.
(3S,4S)‐7‐(4‐Methoxybenzyloxy)‐3‐(methoxymethoxy)hept‐1‐en‐4‐ol (15). To a flask containing 3‐(methoxymethoxy)prop‐1‐ene (13)4 (4.98 g, 48.8 mmol) dissolved in anhydrous THF (100 mL) was added sec‐butyllithium in cyclohexane (1.40 m, 29.0 mL, 40.7 mmol) at −78 °C dropwise. The resulting orange solution was stirred at −78 °C for an additional 30 min, and (+)‐β‐methoxydiisopinocamphenylborane (12.9 g, 40.7 mmol) dissolved in THF (100 mL) was added dropwise. After the reaction mixture was stirred at −78 °C for 1 h, it was cooled to −90 °C (using MeOH and liquid N2 to prepare the cooling bath). Then boron trifluoride etherate (6.80 mL, 54.1 mmol) was added dropwise. Immediately afterward, aldehyde 12 5 (7.90, 40.7 mmol) was added dropwise and the mixture was kept at −90 °C for 3 h. The reaction was warmed to RT over 18 h to give a clear colorless solution, and quenched with a mixture of 35 % H2O2 (30 mL) and saturated NaHCO3 solution (60 mL). After stirring at RT for 30 min, the mixture was extracted with EtOAc (3×200 mL). The combined organic extracts were washed with brine, dried over Na2SO4, and concentrated in vacuo. Purification by column chromatography (30 % EtOAc in hexanes) provided 11.14 g (35.89 mmol, 88 %) of alcohol 15 as a colorless oil: =+44.1 (c=1.31, CHCl3); 1H NMR (400 MHz, CDCl3): δ=1.48 (m, J=4.9 Hz, 1 H), 1.69 (m, J=3.0 Hz, 2 H), 1.83 (m, J=3.1 Hz, 1 H), 3.05 (br s, 1 H), 3.39 (s, 3 H), 3.48 (t, J=6.1 Hz, 2 H), 3.58 (m, 1 H), 3.80 (s, 3 H), 3.88 (t, J=7.3 Hz, 1 H), 4.45 (s, 2 H), 4.59 (d, J=6.6 Hz, 1 H), 4.74 (d, J=6.6 Hz, 1 H), 5.31 (d, J=16.8 Hz, 1 H), 5.32 (d, J=10.3 Hz, 1 H), 5.69 (m, 1 H), 6.87 (d, J=8.6 Hz, 2 H), 7.25 ppm (d, J=8.5 Hz, 2 H);13C NMR (100 MHz, CDCl3): δ=159.2, 134.6, 130.3, 129.4, 120.1, 113.8, 94.0, 81.3, 73.3, 72.6, 69.9, 55.8, 55.3, 29.5, 25.8 ppm; IR (neat, NaCl): =3450 (br), 2950 (br), 1610, 1580, 1455, 1420, 1302, 1240 cm−1; HRMS (ESI+) m/z calcd for C17H26NaO5 [M+Na]+: 333.1672, found: 333.1677.
(3S,4S,5E,9E)‐14‐(tert‐Butyldimethylsilyloxy)‐3‐(3‐(4‐methoxybenzyloxy)propyl)‐4‐(methoxymethoxy)‐3,4,7,8‐tetrahydro‐1H‐benzo[c][1]oxacyclododecin‐1‐one (19). Alcohol 15 (1.19 g, 3.83 mmol) was subjected to azeotropic distillation in vacuo with benzene (2×10 mL), dissolved in anhydrous THF (15.0 mL) and cooled to 0 °C. NaHMDS (1.0 m, 7.26 mL, 7.26 mmol) was added dropwise. After stirring at 0 °C for 30 min, salicylate 31 (0.93 g, 3.63 mmol) dissolved in distilled THF (15.0 mL) was added dropwise. The reaction was warmed to RT and stirred for 2 h, and then TBSCl (1.09 g, 7.26 mmol) and imidazole (0.50 g, 7.26 mmol) were added to the reaction mixture as solids. The reaction was stirred overnight and filtered through a thin silica gel plug, which was rinsed with 20 % Et2O in hexanes (2×50 mL). The combined organic layer was concentrated in vacuo and subjected to azeotropic distillation with benzene (2×10 mL). The crude ester 17 was used directly in the next step without further purification.
To a flask containing crude ester 17 was added anhydrous and degassed toluene (2 L). Ruthenium Grubbs second‐generation catalyst (98.0 mg, 0.116 mmol) in degassed toluene (3.0 mL) was added in one portion under a nitrogen atmosphere. The solution was heated at reflux and stirred for 4 h. The reaction mixture was allowed to cool to RT and DMSO (20 mL) was added into the reaction flask prior the removal of toluene in vacuo to obtain a dark‐brown liquid. The crude product was loaded directly on to silica column for purification (0 to 10 % EtOAc in hexanes) to afford 1.321 g (2.212 mmol, 61 %) of macrocyclic diene 19 as a light‐yellow oil: =+97.7 (c=1.33, CHCl3); 1H NMR (400 MHz, CDCl3): δ=0.21 (s, 3 H), 0.22 (s, 3 H), 0.95 (s, 9 H), 1.81 (m, 3 H), 2.00 (m, 1 H), 2.29 (m, 4 H), 3.37 (s, 3 H), 3.49 (m, 2 H), 3.79 (s, 3 H), 4.40 (m, 1 H), 4.43 (s, 2 H), 4.57 (d, J=6.8 Hz, 1 H), 4.68 (d, J=6.8 Hz, 1 H), 5.17 (m, 1 H), 5.32 (dd, J=4.8, 15.7 Hz, 1 H), 5.47 (td, J=6.0, 15.7 Hz, 1 H), 5.76 (td, J=5.9, 15.9 Hz, 1 H), 6.24 (d, J=15.9 Hz, 1 H), 6.67 (d, J=8.2 Hz, 1 H), 6.85 (t, J=7.3 Hz, 3 H), 7.11 (t, J=8.0 Hz, 1 H), 7.25 ppm (d, J=8.5 Hz, 2 H); 13C NMR (100 MHz, CDCl3): δ=168.2, 159.1, 152.1, 137.5, 132.3, 131.6, 130.7, 129.5, 129.4, 129.2, 125.8, 118.9, 117.1, 113.7, 94.9, 77.1, 75.9, 72.5, 69.9, 60.4, 56.1, 55.2, 30.6, 30.1, 27.4, 25.9, 25.8, 18.3, −4.1, −4.3 ppm; IR (neat, NaCl): =2915, 2865, 1735, 1620, 1600, 1565, 1525, 1460, 1245, 11 450, 1083 cm−1; HRMS (ESI+) m/z [M+Na]+ calcd for C34H48NaO7Si: 619.3062, found: 619.3065.
(3S,4S,5E,9E)‐14‐(tert‐Butyldimethylsilyloxy)‐3‐(3‐hydroxypropyl)‐4‐(methoxymethoxy)‐3,4,7,8‐tetrahydro‐1H‐benzo[c][1]oxacyclododecin‐1‐one (21). To a flask containing macrocyclic diene 19 (404.6 mg, 0.6779 mmol) was added CH2Cl2 (15.0 mL) and H2O (1.5 mL). The mixture was cooled to 0 °C, and DDQ (185.0 mg, 0.8149 mmol) was added in one portion. The cooling bath was removed and the reaction was stirred at RT for 1 h and 10 min. The reaction mixture was diluted with EtOAc (100 mL), and then washed with 10 % NaHCO3 solution (2×15 mL). The organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuo. Purification by column chromatography (30 % EtOAc in hexanes) provided 204.5 mg (0.4290 mmol, 63 %) of alcohol 21 as a colorless oil: =+112 (c=1.51, CHCl3); 1H NMR (400 MHz, CDCl3): δ=0.22 (s, 3 H), 0.22 (s, 3 H), 0.96 (s, 9 H), 1.71 (m, 3 H), 1.87 (m, 1 H), 2.00 (m, 1 H), 2.22 (m, 2 H), 2.34 (m, 2 H), 3.39 (s, 3 H), 3.69 (m, 2 H), 4.42 (t, 1 H), 4.58 (d, J=6.8 Hz, 1 H), 4.69 (d, J=6.8 Hz, 1 H), 5.16 (td, J=3.47, 9.9 Hz, 1 H), 5.33 (dd, J=5.0, 15.7 Hz, 1 H), 5.50 (td, J=6.2, 15.1 Hz, 1 H), 5.77 (td, J=6.2, 15.9 Hz, 1 H), 6.23 (d, J=15.9 Hz, 1 H), 6.67 (d, J=8.1 Hz, 1 H), 6.85 (d, J=7.7 Hz, 1 H), 7.12 ppm (t, J=8.0 Hz, 1 H); 13C NMR (100 MHz, CDCl3): δ=168.4, 152.1, 137.6, 132.3, 131.8, 130.8, 129.7, 129.3, 125.6, 118.9, 117.1, 94.9, 77.2, 76.0, 62.7, 56.1, 30.6, 30.1, 28.7, 26.7, 25.7, 18.3, −4.1, −4.3 ppm; IR (neat, NaCl): =3455 (br), 3015, 2935, 1733, 1466, 1380, 1115 cm−1; HRMS (ESI+) m/z calcd for C26H40NaO6Si [M+Na]+: 499.2486, found: 499.2489.
(3S,4S,5E,9E)‐14‐(tert‐Butyldimethylsilyloxy)‐4‐(methoxymethoxy)‐3‐((E)‐4‐(phenylsulfonyl)but‐3‐enyl)‐3,4,7,8‐tetrahydro‐1H‐benzo[c][1]oxacyclododecin‐1‐one (23). To a flask containing alcohol 21 (70.3 mg, 0.147 mmol) was added CH2Cl2 (1.0 mL), Et3N (0.15 mL, 1.07 mmol) and DMSO (1.0 mL). The mixture was cooled to 0 °C, and SO3⋅Py (73.0 mg, 0.459 mmol) was added in three portions over 3 min. The mixture was stirred at 0 °C for 30 min, and then diluted with Et2O (2.5 mL) and phosphate buffer pH 7 (2.5 mL). The organic layer was separated, and then the aqueous layer was extracted with Et2O (5×1 mL). The combined organic layer was washed with saturated NH4Cl (5 mL), and then washed with H2O (2×5 mL) and saturated CuSO4 (2×5 mL). The organic layer was dried over MgSO4 and concentrated in vacuo to afford an opaque oil of the corresponding aldehyde. The crude aldehyde was used directly in the next step without further purification.
To a solution of diethyl [(phenylsulfonyl)methyl]phosphonate (26; 43.7 mg, 0.179 mmol) in anhydrous THF (2.0 mL) was added 60 % NaH (6.8 mg, 0.17 mmol). The mixture was cooled to 0 °C, and stirred for 10 min. The aldehyde in THF (1.0 mL) was added dropwise over 3 min. The mixture was stirred at 0 °C for 40 min, and then diluted with 10 % HCl (0.5 mL) and phosphate buffer pH 7 (4.0 mL). The crude mixture was extracted with Et2O (3×15 mL). The combined organic layer was washed with 10 % NaHCO3 (20.0 mL) and brine (10.0 mL), dried over MgSO4, and concentrated in vacuo. Purification by column chromatography (30 % EtOAc in hexanes) provided 60.1 mg (0.0959 mmol, 65 %) of phenylsulfone 23 as an opaque oil: =+90.3 (c=2.43, CHCl3); 1H NMR (400 MHz, CDCl3): δ=0.18 (s, 3 H), 0.19 (s, 3 H), 0.89 (s, 9 H), 1.98 (m, 2 H), 2.19 (m, 2 H), 2.38 (m, 4 H), 3.34 (s, 3 H), 4.37 (m, 1 H), 4.55 (d, J=6.8 Hz, 1 H), 4.67 (d, J=6.8 Hz, 1 H), 5.09 (td, J=3.4, 10.0 Hz, 1 H), 5.27 (dd, J=5.4, 15.6 Hz, 1 H), 5.48 (td, J=6.3, 15.2 Hz, 1 H), 5.75 (td, J=6.1, 15.9 Hz, 1 H), 6.19 (d, J=15.9 Hz, 1 H), 6.36 (td, J=1.5, 15.0 Hz, 1 H), 6.67 (d, J=7.7 Hz, 1 H), 6.85 (d, J=7.7 Hz, 1 H), 7.02 (td, J=6.4, 15.1 Hz, 1 H), 7.13 (t, J=8.0 Hz, 1 H), 7.56 (m, 3 H), 7.88 ppm (m, 2 H); 13C NMR (100 MHz, CDCl3): δ=168.2, 152.0, 145.8, 140.6, 137.6, 133.3, 132.5, 132.4, 130.9, 130.8, 129.8, 129.3, 128.8, 127.6, 125.3, 119.0, 117.2, 94.8, 76.2, 76.0, 56.2, 30.5, 30.1, 28.2, 27.7, 25.7, 18.3, −4.1, −4.3 ppm; IR (neat, NaCl): =2933, 1735, 1550, 1480, 1282, 1255, 1138, 1111 cm−1; HRMS (ESI+) m/z calcd for C33H44NaO7SSi [M+Na]+: 635.2469, found: 635.2471.
(3S,4S,5E,9E)‐4,14‐Dihydroxy‐3‐((E)‐4‐(phenylsulfonyl)but‐3‐enyl)‐3,4,7,8‐tetrahydro‐1H‐benzo[c][1]oxacyclododecin‐1‐one (25). To a flask containing phenylsulfone 23 (48.7 mg, 0.0777 mmol) was added 4 m HCl in MeOH (5.0 mL). The mixture was stirred at RT for 99 h, and then concentrated in vacuo. Purification by column chromatography (50 % EtOAc in hexanes) provided 31.0 mg (0.0662 mmol, 85 %) of the desired analogue 25 as a colorless oil: =−13.8 (c 0.975, CHCl3); 1H NMR (400 MHz, CDCl3): δ=1.78 (br s, 1 H), 1.91 (m, 1 H), 2.20 (m, 3 H), 2.40 (m, 4 H), 4.33 (t, J=4.8 Hz, 1 H), 5.18 (m, 1 H), 5.47 (dd, J=6.10, 15.6 Hz, 1 H), 5.61 (m, 2 H), 6.41 (d, J=15.2 Hz, 1 H), 6.61 (d, J=15.7 Hz, 1 H), 6.78 (d, J=7.5 Hz, 1 H), 6.86 (d, J=8.3 Hz, 1 H), 7.02 (td, J=6.7, 14.7 Hz, 1 H), 7.31 (t, J=7.9 Hz, 1 H), 7.57 (m, 3 H), 7.88 (d, J=7.9 Hz, 2 H), 10.47 ppm (s, 1 H); 13C NMR (100 MHz, CDCl3): δ=169.7, 160.4, 144.5, 140.7, 139.4, 134.2, 133.8, 133.3, 132.3, 130.2, 128.7, 128.3, 128.0, 126.6, 119.0, 115.2, 110.6, 75.1, 71.6, 31.5, 28.7, 26.9, 26.2 ppm; IR (neat, NaCl): =3415 (br), 3055, 2910, 2855, 1735, 1632, 1484, 1426, 1134 cm−1; HRMS (ESI+) m/z calcd for C25H26NaO6S [M+Na]+: 477.1342, found: 477.1348.
(3S,4S,5Z,7Z,9E)‐3‐(2‐Hydroxyethyl)‐14‐methoxy‐4‐(methoxymethoxy)‐3,4‐dihydro‐1H‐benzo[c][1]oxacyclododecin‐1‐one (33). To a flask containing macrocyclic triene 32 1 (47.4 mg, 0.0792 mmol) was added anhydrous THF (1.0 mL), and then TBAF (1 m in THF, 87.0 mL, 0.0870 mmol) was added dropwise over 5 min at RT. The mixture was stirred at RT for 1.5 h, and then concentrated in vacuo. Purification by column chromatography (40 % EtOAc in hexanes) provided 26.6 mg (0.0694 mmol, 88 %) of alcohol 33 as a colorless oil: =−207 (c=1.33, CHCl3); 1H NMR (400 MHz, CDCl3): δ=2.18 (m, 2 H), 3.37 (s, 3 H), 3.84 (s, 3 H), 3.90 (s, 2 H), 4.53 (d, J=6.4 Hz, 1 H), 4.58 (d, J=6.4 Hz, 1 H), 4.66 (dd, J=3.4, 10.8 Hz, 1 H), 5.55 (ddd, J=3.2, 3.2, 10.7 Hz, 1 H), 5.71 (dd, J=11.3, 11.3 Hz, 1 H), 5.90 (dd, J=3.9, 11.0 Hz, 1 H), 6.21 (dd, J=3.8, 11.9 Hz, 1 H), 6.36 (dd, J=11.3, 11.3 Hz, 1 H), 6.68 (d, J=15.9 Hz, 1 H), 6.82 (t, J=8.3 Hz, 2 H), 7.00 (dd, J=11.5, 15.9 Hz, 1 H), 7.27 ppm (d, J=5.0 Hz, 1 H); 13C NMR (100 MHz, CDCl3): δ=168.2, 155.7, 138.5, 133.3, 132.6, 130.7, 129.9, 129.5, 127.8, 126.9, 123.3, 120.8, 109.4, 93.3, 77.3, 68.4, 61.2, 55.8, 55.5, 30.7 ppm; IR (neat, NaCl): =3432 (br), 3010, 2943, 1729, 1599, 1571, 1468, 1438, 1154, 1089, 1058 cm−1; HRMS (ESI+) m/z calcd for C20H24NaO6 [M+Na]+: 383.1465, found: 383.1462.
(3S,4S,5Z,7Z,9E)‐14‐Methoxy‐4‐(methoxymethoxy)‐3‐((E)‐3‐(phenylsulfonyl)allyl)‐3,4‐dihydro‐1H‐benzo[c][1]oxacyclododecin‐1‐one (34). To a flask containing alcohol 33 (25.0 mg, 0.0694 mmol) were added anhydrous CH2Cl2 (1.2 mL) and molecular sieves 4 Å (70 mg). The mixture was cooled to 0 °C, and pyridinium dichromate (PDC; 104.0 mg, 0.2764 mmol) was added in one portion. The cooling bath was removed and the reaction was stirred at RT for 1.5 h The reaction mixture was diluted with hexanes (3 mL), then filtered through a silica gel plug and rinsed with 40 % EtOAc in hexanes (20 mL). The combined organic layer was concentrated in vacuo to afford an opaque oil of the corresponding aldehyde. The crude aldehyde was used directly in the next step without further purification.
To a solution of diethyl [(phenylsulfonyl)methyl]phosphonate (26)43 (22.0 mg, 0.0759 mmol) in anhydrous THF (1.0 mL) was added 60 % NaH (3.4 mg, 0.086 mmol). The mixture was cooled to 0 °C, and was stirred for 5 min. The aldehyde in THF (1.0 mL) was added dropwise over 3 min. The mixture was stirred at 0 °C for 30 min, and then diluted with saturated NH4Cl (3.0 mL) and phosphate buffer pH 7 (4.0 mL). The crude mixture was extracted with EtOAc (3×4 mL). The combined organic layer was washed with brine (5 mL), dried over Na2SO4, and concentrated in vacuo. Purification by column chromatography (30 % EtOAc in hexanes) provided 17.8 mg (0.0358 mmol, 47 %) of phenylsulfone 34 as an opaque oil: =−179 (c=0.890, CHCl3); 1H NMR (400 MHz, CDCl3): δ=2.83 (m, J=3.0 Hz, 2 H), 3.33 (s, 3 H), 3.95 (s, 3 H), 4.49 (d, J=6.52 Hz, 1 H), 4.54 (d, J=6.5 Hz, 1 H), 4.66 (dd, J=3.36, 10.8 Hz, 1 H), 5.44 (ddd, J=3.1, 3.1, 10.8 Hz, 1 H), 5.59 (dd, J=11.3, 11.3 Hz, 1 H), 5.88 (dd, J=3.7, 11.4 Hz, 1 H), 6.2 (dd, J=3.8, 11.8 Hz, 1 H), 6.37 (dd, J=11.3, 11.3 Hz, 1 H), 6.52 (d, J=15.2 Hz, 1 H), 6.69 (d, J=16.0 Hz, 1 H), 6.83 (dd, J=8.0, 13.8 Hz, 2 H), 6.99 (dd, J=11.7, 15.7 Hz, 1 H), 7.19 (ddd, J=5.5, 7.9, 15.2 Hz, 1 H), 7.29 (t, J=8.0 Hz, 1 H), 7.57 (m, 3 H), 7.90 ppm (m, 2 H); 13C NMR (100 MHz, CDCl3): δ=168.3, 156.4, 143.0, 140.6, 138.1, 133.29, 133.25, 132.8, 132.3, 131.1, 130.0, 129.9, 129.3, 127.6, 127.3, 126.0, 123.0, 120.4, 109.5, 93.3, 75.5, 68.3, 56.0, 55.6, 30.0 ppm; IR (neat, NaCl): =3012, 2945, 2842, 1732, 1635, 1572, 1469, 1318, 1305, 1105, 1087, 1059, 953 cm−1; HRMS (ESI+) m/z calcd for C27H28NaO7S [M+Na]+: 519.1448, found: 519.1454.
(3S,4S,5Z,7Z,9E)‐4‐Hydroxy‐14‐methoxy‐3‐((E)‐3‐(phenylsulfonyl)allyl)‐3,4‐dihydro‐1H‐benzo[c][1]oxacyclododecin‐1‐one (35). To a flask containing phenyl sulfone 34 (17.0 mg, 0.0359 mmol) was added iPrOH (0.37 mL), and CBr4 (48.0 mg, 0.144 mmol) in one portion. The mixture was heated at 75 °C for 2 h, and then concentrated in vacuo. Purification by column chromatography (40 % EtOAc in hexanes) provided 12.6 mg (0.0278 mmol, 78 %) of alcohol 35 as a bright‐yellow oil: =−183 (c=0.630, CHCl3); 1H NMR (400 MHz, CDCl3): δ=2.78 (m, J=3.50 Hz, 1 H), 2.93 (dd, J=8.1, 15.5 Hz, 1 H), 3.95 (s, 3 H), 4.77 (dd, J=3.2, 10.5 Hz, 1 H), 5.40 (td, J=2.6, 11.5 Hz, 1 H), 5.76 (t, J=11.2 Hz, 1 H), 5.87 (dd, J=3.8, 11.0 Hz, 1 H), 6.10 (dd, J=3.8, 11.9 Hz, 1 H), 6.34 (t, J=11.2 Hz, 1 H), 6.52 (d, J=15.2 Hz, 1 H), 6.69 (d, J=15.9 Hz, 1 H), 6.83 (dd, J=8.0, 15.0 Hz, 2 H), 6.98 (dd, J=11.6, 15.9 Hz, 1 H), 7.18 (ddd, J=5.4, 8.1, 15.1 Hz, 1 H), 7.29 (t, J=8.0 Hz, 1 H), 7.57 (m, 3 H), 7.89 ppm (m, 2 H); 13C NMR (100 MHz, CDCl3): δ=168.5, 156.3, 143.1, 140.6, 138.0, 133.3, 132.8, 132.3, 131.6, 131.1, 130.1, 130.0, 129.3, 128.7, 127.6, 127.2, 122.9, 120.4, 109.6, 76.3, 65.5, 56.0, 29.3 ppm; IR (neat, NaCl): =3470, 3014, 2927, 1731, 1634, 1572, 1469, 1305, 1271, 1107, 1060, 961 cm−1; HRMS (ESI+) m/z calcd for C25H24NaO6S [M+Na]+: 475.1186, found: 475.1181.
(3S,4S,5Z,7Z,9E)‐4,14‐Dihydroxy‐3‐((E)‐3‐(phenylsulfonyl)allyl)‐3,4‐dihydro‐1H‐benzo[c][1]oxacyclododecin‐1‐one (36). To a flask containing alcohol 35 (10.0 mg, 0.0221 mmol) was added anhydrous CH2Cl2 (2.2 mL). The mixture was cooled to −78 °C, and BCl3 1 m in CH2Cl2 (0.22 mL, 0.22 mmol) was added dropwise over 1 min. The mixture was stirred at −78 °C for 10 min, and warmed to RT and stirred for 15 min. The reaction mixture was quenched with H2O (2 mL) and extracted with EtOAc (3×5 mL). The combined organic layer was washed with brine and dried over MgSO4. The crude mixture was concentrated in vacuo. Purification by column chromatography (50 % EtOAc in hexanes) provided 7.2 mg (0.016 mmol, 75 %) of the desired analogue 36 as a bright‐yellow oil: =−130 (c=0.36, CHCl3); 1H NMR (400 MHz, CD3OD): δ=2.70 (m, 1 H), 2.82 (m, 1 H), 4.53 (dd, J=3.3, 10.5 Hz, 1 H), 5.18 (td, J=2.8, 10.9 Hz, 1 H), 5.69 (dd, J=11.2, 11.2 Hz, 1 H), 5.81 (dd, J=3.9, 11.1 Hz, 1 H), 5.98 (dd, J=3.8, 11.9 Hz, 1 H), 6.25 (dd, J=11.2, 11.2 Hz, 1 H), 6.56 (d, J=16.0 Hz, 1 H), 6.65 (m, 3 H), 6.90 (dd, J=11.4, 15.9 Hz, 1 H), 7.08 (ddd, J=7.2, 7.2, 15.2 Hz, 2 H), 7.51 (m, 3 H), 7.82 ppm (m, 2 H); 13C NMR (100 MHz, CD3OD): δ=170.8, 155.6, 145.2, 142.1, 139.4, 134.5, 133.9, 133.8, 131.62, 131.57, 131.1, 130.9, 130.53, 130.48, 130.4, 128.7, 123.1, 120.2, 115.1, 78.6, 66.3, 30.3 ppm; IR (neat, NaCl): =3408 (br), 3052, 3014, 2927, 2854, 1727, 1635, 1464, 1219, 1144, 1085, 1057 cm−1; HRMS (ESI+) m/z calcd for C24H22NaO6S [M+Na]+: 461.1029, found: 461.1037.
(5S,6S)‐6‐(3‐(4‐Methoxybenzyloxy)propyl)‐8,8,9,9‐tetramethyl‐5‐vinyl‐2,4,7‐trioxa‐8‐siladecane (37). To a stirred solution of alcohol 15 (3.00 g, 9.65 mmol) dissolved in anhydrous DMF (5.0 mL) were added TBSCl (10.5 g, 69.7 mmol) and imidazole (4.74 g, 69.6 mmol) as solids. The resulting mixture was heated at 75 °C for 4 h. The reaction mixture was cooled to RT and quenched with phosphate buffer pH 7 (100 mL). The mixture was extracted with CH2Cl2 (3×100 mL). The combined organic layer was dried over Na2SO4 and concentrated in vacuo. Purification by column chromatography (30 % EtOAc in hexanes) provided 2.90 g (6.84 mmol, 71 % yield) of alkene 37 as a colorless oil: =+3.86 (c=1.01, CHCl3); 1H NMR (400 MHz, CDCl3): δ=0.06 (s, 3 H), 0.08 (s, 3 H), 0.89 (s, 9 H), 1.39 (m, 1 H), 1.66 (m, 3 H), 3.35 (s, 3 H), 3.42 (t, J=6.5 Hz, 2 H), 3.72 (ddd, J=3.3, 4.9, 8.0 Hz, 1 H), 3.80 (s, 3 H), 3.99 (dd, J=5.4, 6.5 Hz, 1 H), 4.42 (s, 2 H), 4.58 (d, J=6.6 Hz, 1 H), 4.67 (d, J=6.6 Hz, 1 H), 5.25 (d, J=10.5 Hz, 1 H), 5.27 (d, J=16.7 Hz, 1 H), 5.78 (m, 1 H), 6.87 (d, J=8.6 Hz, 2 H), 7.25 ppm (d, J=8.2 Hz, 2 H); 13C NMR (100 MHz, CDCl3): δ=159.1, 134.8, 130.8, 129.2, 117.9, 113.7, 94.6, 79.9, 74.0, 72.4, 70.3, 55.5, 55.3, 29.1, 25.9, 25.6, 17.7, −4.3, −4.7 ppm; IR (neat, NaCl): =2930, 2880, 2853, 1620, 1510, 1471, 1250, 1100 cm−1; HRMS (ESI+) m/z calcd for C23H40NaO5Si [M+Na]+: 447.2537, found: 447.2539.
(4S,5S,Z )‐1‐(4‐Methoxybenzyloxy)‐5‐(methoxymethoxy)nona‐6,8‐dien‐4‐ol (40). To a stirred solution of alkene 37 (2.90 g, 6.84 mmol) dissolved in THF (54 mL) and H2O (27 mL) was added NMO (0.96 g, 8.2 mmol) and OsO4 in toluene (1.0 m, 0.74 mL, 0.74 mmol). The resulting solution was stirred at RT for 24 h, then quenched with a saturated Na2S2O3 solution (20 mL) and extracted with EtOAc (3×50 mL). The combined organic extracts were dried over Na2SO4 and concentrated in vacuo. The crude product was dried under high vacuum for 4 h before use in the next step. To a flask containing the crude diol was added anhydrous MeOH (60 mL) and the reaction was cooled to 0 °C. To this mixture was added K2CO3 (1.54 g, 11.1 mmol) and Pb(OAc)4 (3.96 g, 8.93 mmol) and the resulting solution was stirred at 0 °C for 15 min before being quenched with saturated NaHCO3 (40 mL). The reaction mixture was extracted with EtOAc (3×50 mL), and the combined organic extracts were washed with brine (20 mL), dried over Na2SO4, and concentrated in vacuo. The crude aldehyde 38 was used directly in the next step without further purification.
To a flask containing allyldiphenylphosphine (2.62 mL, 8.21 mmol) dissolved in anhydrous THF (30 mL) was added tert‐butyllithium (1.70 m, 4.83 mL, 8.21 mmol) dropwise at −78 °C, and this mixture was stirred at 0 °C for 30 min. Ti(OiPr)4 (2.63 mL, 8.90 mmol) was added dropwise at −78 °C, and the resulting solution was stirred for 5 min. The crude aldehyde 38, dissolved in THF (3.0 mL), was added over 4 min via syringe at −78 °C, and this mixture was stirred at −78 °C for 10 min and then at 0 °C for 1 h. Methyl iodide (1.28 mL, 20.6 mmol) was added at 0 °C and the reaction mixture was stirred at RT overnight. The reaction mixture was diluted with hexanes (130 mL), filtered through a silica gel plug (13 g), washed with 20 % EtOAc in hexanes (100 mL), and concentrated in vacuo. The resulting cis‐diene 39 was used directly in the next step without further purification.
The crude cis‐diene 39 was dissolved in anhydrous THF (18 mL) and treated with TBAF in THF (1.0 m, 10.3 mL, 10.3 mmol). The reaction mixture was stirred at RT for 25 h before being diluted with H2O (20 mL) and extracted with EtOAc (3×20 mL). The combined organic extracts were washed with brine (20 mL), dried over Na2SO4, and concentrated in vacuo. Purification by column chromatography (30 % EtOAc in hexanes) provided 0.43 g (1.3 mmol, 19 %) of alcohol 40 as a colorless oil: =+46.9 (c=1.30, CHCl3); 1H NMR (400 MHz, CDCl3): δ=1.43 (m, 1 H), 1.63 (m, 1 H), 1.72 (m, 1 H), 1.82 (m, 1 H), 2.86 (br s, 1 H), 3.38 (s, 3 H), 3.46 (m, 2 H), 3.57 (m, 1 H), 3.80 (s, 3 H), 4.36 (dd, J=7.10, 9.70 Hz, 1 H), 4.42 (s, 2 H), 4.54 (d, J=6.64 Hz, 1 H), 4.69 (d, J=6.7 Hz, 1 H), 5.27 (m, 3 H), 6.29 (dd, J=11.1, 11.8 Hz, 1 H), 6.67 (ddd, J=10.3, 11.7, 16.7 Hz, 1 H), 6.86 (d, J=8.7 Hz, 2 H), 7.24 ppm (d, J=8.6 Hz, 2 H); 13C NMR (100 MHz, CDCl3): δ=159.1, 134.9, 131.7, 130.6, 129.2, 127.5, 120.4, 113.7, 93.6, 74.8, 73.5, 72.4, 69.9, 55.6, 55.2, 29.6, 26.0 ppm; IR (neat, NaCl): =3430 (br), 2940 (br), 1630, 1509, 1473, 1250, 1120 cm−1; HRMS (ESI+) m/z calcd for C19H28NaO5 [M+Na]+: 359.1829, found: 359.1831.
(4S,5S,Z )‐1‐(4‐Methoxybenzyloxy)‐5‐(methoxymethoxy)nona‐6,8‐dien‐4‐yl 2‐(tert‐Butyldimethylsilyloxy)‐6‐((1E,3E)‐penta‐1,3‐dienyl)benzoate (42). Alcohol 40 (350 mg, 1.04 mmol) was subjected to azeotropic distillation in vacuo with benzene (2×4 mL), dissolved in anhydrous THF (4.0 mL) and cooled to 0 °C. NaHMDS (1.0 m, 1.98 mL, 1.98 mmol) was added dropwise. After stirring at 0 °C for 30 min, salicylate 416 (242 mg, 0.991 mmol) dissolved in distilled THF (4.0 mL) was added dropwise. The reaction was warmed to RT and stirred for 2 h, and then TBSCl (298 mg, 1.98 mmol) and imidazole (135 mg, 1.98 mmol) were added to the reaction mixture as solids. The reaction was stirred overnight and then quenched by an ice‐cold solution of 5 % HCl (30 mL). The mixture was extracted with Et2O (2×30 mL) and the combined organic layer was washed with 10 % NaHCO3 (50 mL). The organic layer was washed with brine (30 mL), dried over MgSO4 and concentrated in vacuo. Purification by column chromatography (20 % EtOAc in hexanes) provided 0.578 g (0.907 mmol, 91 % yield) of ester 42 as a colorless oil: =+32.9 (c=1.50, CHCl3); 1H NMR (400 MHz, CDCl3): δ=0.22 (s, 6 H), 0.96 (s, 9 H), 1.61 (m, 1 H), 1.78 (m, 5 H), 1.92 (m, 1 H), 3.27 (s, 3 H), 3.46 (m, 2 H), 3.79 (s, 3 H), 4.42 (s, 2 H), 4.50 (d, J=6.7 Hz, 1 H), 4.63 (d, J=6.8 Hz, 1 H), 4.72 (dd, J=5.5, 9.7 Hz, 1 H), 5.20 (m, 2 H), 5.28 (d, J=16.7 Hz, 1 H), 5.39 (dd, J=10.4, 10.4 Hz, 1 H), 5.81 (m, 1 H), 6.15 (dd, J=10.5, 15.0 Hz, 1 H), 6.27 (dd, J=11.1, 11.1 Hz, 1 H), 6.46 (d, J=15.5 Hz, 1 H), 6.69 (m, 3 H), 6.86 (d, J=8.6 Hz, 2 H), 7.13 (m, 2 H), 7.24 ppm (d, J=8.5 Hz, 2 H); 13C NMR (100 MHz, CDCl3): δ=171.1, 167.8, 159.1, 152.5, 136.4, 134.7, 131.9, 131.7, 131.6, 131.2, 130.7, 129.6, 129.1, 126.9, 126.7, 120.4, 117.6, 117.4, 113.7, 93.4, 76.4, 72.4, 70.5, 69.7, 55.5, 55.2, 27.5, 25.8, 25.6, 25.5, 18.3, −4.1, −4.2 ppm; IR (neat, NaCl): =2950, 2873, 1720, 1565, 1505, 1440, 1280, 1100 cm−1; HRMS (ESI+) m/z calcd for C37H52NaO7Si [M+Na]+: 659.3375, found: 659.3381.
(3S,4S,5Z,7Z,9E)‐14‐(tert‐Butyldimethylsilyloxy)‐3‐(3‐(4‐methoxybenzyloxy)propyl)‐4‐(methoxymethoxy)‐3,4‐dihydro‐1H‐benzo[c][1]oxacyclododecin‐1‐one (43). To a flask containing bis‐diene 42 (100 mg, 0.157 mmol) was added anhydrous and degassed toluene (80 mL). Ruthenium Grubbs second‐generation catalyst (6.7 mg, 0.0079 mmol) in degassed toluene was added in one portion under a nitrogen atmosphere. The solution was heated at reflux and stirred for 60 min. The reaction mixture was allowed to cool to RT and DMSO (5 mL) was added into the reaction flask prior to the removal of toluene in vacuo to obtain a dark‐brown liquid. The crude product was loaded directly on to a silica column for combi‐flash purification (0 to 10 % EtOAc in hexanes) to afford 20.6 mg (0.0346 mmol, 22 %) of the macrocyclic triene 43 as a light‐yellow oil: =−81.9 (c=1.03, CHCl3); 1H NMR (400 MHz, CDCl3): δ=0.19 (s, 3 H), 0.20 (s, 3 H), 0.96 (s, 9 H), 1.73 (m, 2 H), 1.86 (m, 1 H), 1.97 (m, 1 H), 2.07 (m, 1 H), 3.35 (s, 3 H), 3.49 (m, 2 H), 3.80 (s, 3 H), 4.52 (d, J=6.6 Hz, 1 H), 4.55 (d, J=6.4 Hz, 1 H), 4.60 (dd, J=2.9, 10.7 Hz, 1 H), 5.32 (ddd, J=2.9, 2.88, 10.3 Hz, 1 H), 5.68 (dd, J=11.4, 11.4 Hz, 2 H), 5.87 (dd, J=4.1, 11.0 Hz, 1 H), 6.18 (dd, J=4.1, 12.0 Hz, 1 H), 6.34 (t, J=11.2 Hz, 1 H), 6.64 (d, J=16.0 Hz, 1 H), 6.74 (d, J=8.20 Hz, 1 H), 6.78 (d, J=7.7 Hz, 1 H), 6.87 (d, J=8.5 Hz, 2 H), 7.01 (dd, J=11.5, 16.01 Hz, 1 H), 7.16 (dd, J=7.9, 7.9 Hz, 1 H), 7.26 ppm (d, J=8.6 Hz, 2 H); 13C NMR (100 MHz, CDCl3): δ=168.4, 159.0, 152.3, 137.9, 132.2, 131.9, 131.0, 130.8, 129.9, 129.2, 129.1, 127.2, 127.1, 126.6, 121.2, 117.9, 113.7, 93.1, 78.2, 72.5, 69.8, 69.0, 55.4, 55.3, 26.9, 25.8, 25.5, 18.4, −4.0, −4.2 ppm; IR (neat, NaCl): =2930, 2857, 1730, 1612, 1570, 1513, 1463, 1362, 1292, 1250, 1172, 1155, 1102, 1032, 1001, 927, 863, 839, 807, 783, 753, 714, 670 cm−1; HRMS (ESI+) m/z calcd for C34H46NaO7Si [M+Na]+: 617.2905, found: 617.2907.
(3S,4S,5Z,7Z,9E)‐14‐(tert‐Butyldimethylsilyloxy)‐3‐(3‐hydroxypropyl)‐4‐(methoxymethoxy)‐3,4‐dihydro‐1H‐benzo[c][1]oxacyclododecin‐1‐one (44). To a flask containing macrocyclic triene 43 (15.9 mg, 0.0274 mmol) was added CH2Cl2 (1.0 mL) and H2O (0.1 mL). The mixture was cooled to 0 °C, and DDQ (7.5 mg, 0.033 mmol) was added in one portion. The cooling bath was removed and the reaction was stirred at RT for 1 h. The reaction mixture was diluted with EtOAc (10 mL), and then washed with 10 % NaHCO3 solution (2×5 mL). The organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuo. Purification by column chromatography (30 % EtOAc in hexanes) provided 12.7 mg (0.0267 mmol, 98 %) of alcohol 44 as a colorless oil: =−88.1 (c=0.630, CHCl3); 1H NMR (400 MHz, CDCl3): δ=0.20 (s, 3 H), 0.21 (s, 3 H), 0.97 (s, 9 H), 1.66 (m, 2 H), 1.91 (m, 2 H), 3.36 (s, 3 H), 3.72 (m, 2 H), 4.53 (d, J=6.44 Hz, 1 H), 4.56 (d, J=6.5 Hz, 1 H), 4.61 (dd, J=2.9, 10.7 Hz, 1 H), 5.34 (td, J=2.8, 10.3 Hz, 1 H), 5.68 (dd, J=11.3, 11.3 Hz, 1 H), 5.87 (dd, J=3.6, 11.1 Hz, 1 H), 6.19 (dd, J=4.1, 12.0 Hz, 1 H), 6.35 (dd, J=11.2, 11.2 Hz, 1 H), 6.64 (d, J=16.0 Hz, 1 H), 6.75 (d, J=8.2 Hz, 1 H), 6.79 (d, J=7.6 Hz, 1 H), 7.01 (ddd, J=0.5, 11.3, 16.0 Hz, 1 H), 7.16 ppm (t, J=8.0 Hz, 1 H); 13C NMR (100 MHz, CDCl3): δ=168.3, 152.3, 137.9, 132.2, 132.1, 131.1, 129.9, 129.2, 127.2, 127.0, 121.3, 117.9, 93.2, 78.1, 69.0, 62.7, 60.4, 55.5, 29.9, 25.8, 25.0, 18.4, −4.0, −4.2 ppm; IR (neat, NaCl): =3438.92 (br), 2931, 2859, 1731, 1570, 1255, 1153, 1102, 1030 cm−1; HRMS (ESI+) m/z calcd for C26H38NaO6Si [M+Na]+: 497.2330, found: 497.2337.
(3S,4S,5Z,7Z,9E)‐14‐(tert‐Butyldimethylsilyloxy)‐4‐(methoxymethoxy)‐3‐((E)‐4‐(phenylsulfonyl)but‐3‐enyl)‐3,4‐dihydro‐1H‐benzo[c][1]oxacyclododecin‐1‐one (45). To a flask containing alcohol 44 (10.0 mg, 0.0210 mmol) was added anhydrous CH2Cl2 (0.4 mL) and molecular sieves 4 Å (21 mg). The mixture was cooled to 0 °C, and PDC (32.0 mg, 0.0842 mmol) was added in one portion. The cooling bath was removed and the reaction was stirred at RT for 1.5 h. The reaction mixture was diluted with hexanes (1 mL), and then filtered through a silica gel plug and rinsed with 40 % EtOAc in hexanes (7 mL). The combined organic layer was concentrated in vacuo to afford an opaque oil of the corresponding aldehyde (6.0 mg). The crude aldehyde was used directly in the next step without further purification.
To a solution of diethyl [(phenylsulfonyl)methyl]phosphonate 26 (6.7 mg, 0.023 mmol) in anhydrous THF (0.3 mL) was added 60 % NaH (1.0 mg, 0.026 mmol). The mixture was cooled to 0 °C, and was stirred for 5 min. The aldehyde in THF (1.0 mL) was added dropwise over 3 min. The mixture was stirred at 0 °C for 30 min, and then diluted with saturated NH4Cl (4.0 mL) and phosphate buffer pH 7 (4.0 mL). The crude mixture was extracted with EtOAc (3×4 mL). The combined organic layer was washed with brine (5.0 mL), dried over Na2SO4, and concentrated in vacuo. Purification by column chromatography (30 % EtOAc in hexanes) provided 6.0 mg (0.0098 mmol, 47 %) of phenylsulfone 45 as an opaque oil: =−66 (c=0.30, CHCl3); 1H NMR (400 MHz, CDCl3): δ=0.17 (s, 3 H), 0.19 (s, 3 H), 0.93 (s, 9 H), 1.97 (m, 1 H), 2.11 (m, 1 H), 2.37 (m, 1 H), 2.63 (m, 1 H), 3.30 (s, 3 H), 4.47 (d, J=6.5 Hz, 1 H), 4.51 (d, J=6.5 Hz, 1 H), 4.59 (dd, J=2.9, 10.7 Hz, 1 H), 5.29 (td, J=2.9, 10.4 Hz, 1 H), 5.60 (dd, J=11.3, 11.3 Hz, 1 H), 5.87 (dd, J=3.9, 11 Hz, 1 H), 6.20 (dd, J=4.0, 11.84 Hz, 1 H), 6.33 (d, J=11.3 Hz, 1 H), 6.39 (d, J=15.1 Hz, 1 H), 6.64 (d, J=16.0 Hz, 1 H), 6.74 (d, J=8.2 Hz, 1 H), 6.79 (d, J=7.6 Hz, 1 H), 6.95 (dd, J=11.5, 16.0 Hz, 1 H), 7.05 (td, J=6.4, 15.1 Hz, 1 H), 7.17 (t, J=8.0 Hz, 1 H), 7.56 (m, 3 H), 7.88 ppm (d, J=7.2 Hz, 2 H); 13C NMR (100 MHz, CDCl3): δ=168.2, 152.2, 145.7, 140.6, 137.9, 133.3, 132.6, 132.2, 131.1, 130.9, 129.9, 129.4, 129.2, 127.6, 127.1, 126.4, 126.1, 121.3, 117.9, 93.1, 77.2, 68.7,55.5, 28.5, 26.4, 25.8, 18.4, −4.0, −4.1 ppm; IR (neat, NaCl): =2930, 1731, 1570, 1463, 1292, 1252, 1148, 1101, 1027 cm−1; HRMS (ESI+) m/z calcd for C33H42NaO7SSi [M+Na]+: 633.2313, found: 633.2303.
(3S,4S,5Z,7Z,9E)‐4,14‐Dihydroxy‐3‐((E)‐4‐(phenylsulfonyl)but‐3‐enyl)‐3,4‐dihydro‐1H‐benzo[c][1]oxacyclododecin‐1‐one (46). To a flask containing phenylsulfone 45 (6.0 mg, 0.0098 mmol) was added 4 m HCl in MeOH (0.7 mL). The mixture was stirred at RT for 211 h, and then concentrated in vacuo. Purification by column chromatography (60 % EtOAc in hexanes) provided 3.0 mg (68 %) of the desired analogue 46 as a colorless oil: =−120 (c=0.15, CHCl3); 1H NMR (400 MHz, CD3OD): δ=1.87 (m, 1 H), 2.03 (m, 1 H), 2.38 (m, 1 H), 2.48 (m, 1 H), 4.48 (dd, J=3.6, 10.5 Hz, 1 H), 5.07 (td, J=2.7, 11.8 Hz, 1 H), 5.72 (dd, J=11.3, 11.3 Hz, 1 H), 5.81 (dd, J=4.0, 11.0 Hz, 1 H), 5.96 (dd, J=4.0, 12.0 Hz, 1 H), 6.23 (dd, J=11.4, 11.4 Hz, 1 H), 6.53 (d, J=15.9 Hz, 1 H), 6.61 (t, J=7.1 Hz, 3 H), 6.88 (dd, J=11.5, 15.8 Hz, 1 H), 6.95 (ddd, J=6.1, 8.5, 14.8 Hz, 1 H), 7.05 (t, J=7.9 Hz, 1 H), 7.51 (m, J=5.0 Hz, 3 H), 7.77 ppm (m, J=2.2 Hz, 2 H); 13C NMR (100 MHz, CD3OD): δ=170.8, 155.3, 147.7, 142.1, 139.4, 134.5, 134.1, 132.9, 131.5, 131.2, 130.9, 130.83, 130.75, 130.5, 128.9, 128.5, 123.4, 120.3, 115.0, 78.6, 66.4, 28.6, 26.3 ppm; IR (neat, NaCl): =3440 (br), 3060, 2950, 2854, 1730, 1640, 1457, 1115, 1070 cm−1; HRMS (ESI+) m/z calcd for C25H24NaO6S [M+Na]+: 475.1186, found: 475.1186.
2‐((3S,4S,5E,9E)‐14‐((tert‐Butyldimethylsilyl)oxy)‐4‐(methoxymethoxy)‐1‐oxo‐3,4,7,8‐tetrahydro‐1H‐benzo[c][1]oxacyclododecin‐3‐yl)acetaldehyde (47). To a solution of alcohol 20 (188 mg, 0.406 mmol) in CH2Cl2 (4.1 mL) was added Dess–Martin periodinane (259 mg, 0.610 mmol) as a solid. The resultant mixture was stirred at RT for 2.5 h before it was filtered. The filtrate was concentrated by rotary evaporation. Purification by column chromatography (20 % EtOAc in hexanes) provided 165 mg (0.0358 mmol, 88 %) of aldehyde 47 as a colorless oil: 1H NMR (400 MHz, CDCl3): δ=0.230 (s, 3 H), 0.234 (s, 3 H), 0.96 (s, 9 H), 2.05–2.16 (m, 2 H), 2.34–2.43 (m, 2 H), 2.83 (ddd, J=17.3, 4.8, 2.1 Hz, 1 H), 3.07 (ddd, J=17.3, 4.8, 2.1 Hz, 1 H), 3.32 (s, 3 H), 4.49–4.51 (m, 2 H), 4.66 (d, J=6.7 Hz, 1 H), 5.30 (dd, J=15.8, 6.3 Hz, 1 H), 5.48–5.52 (m, 1 H), 5.57 (dt, J=15.9, 6.2 Hz, 1 H), 5.76 (dt, J=15.9, 6.2 Hz, 1 H), 6.14 (d, J=15.9 Hz, 1 H), 6.69 (d, J=8.2 Hz, 1 H), 6.88 (d, J=7.6 Hz, 1 H), 7.16 (t, J=8.0, 1 H), 9.82 ppm (s, 1 H).
(3S,4S,5E,9E)‐14‐((tert‐Butyldimethylsilyl)oxy)‐3‐((E)‐3‐iodoallyl)‐4‐(methoxymethoxy)‐3,4,7,8‐tetrahydro‐1H‐benzo[c][1]oxacyclododecin‐1‐one (49). To a suspension of CrCl2 (350 mg, 2.84 mmol) in THF (1.7 mL) was added a solution of aldehyde 47 (109 mg, 0.237 mmol) and iodoform (373 mg, 0.947 mmol) in dioxane (10.2 mL). The resultant mixture was stirred at RT for 22 h and then quenched with brine and extracted with EtOAc. The combined organic layer was washed with brine, dried with MgSO4, and concentrated by rotary evaporation. Purification by column chromatography (10 % EtOAc in hexanes) provided 120 mg (0.205 mmol, 86 %) of iodide 49 as a colorless oil (9:1 ratio of E/Z vinyl iodide isomers by 1H NMR): =+96.7 (c=1.19, CHCl3); 1H NMR (400 MHz, CDCl3): δ=0.237 (s, 3 H), 0.244 (s, 3 H), 0.98 (s, 9 H), 2.12–2.21 (m, 2 H), 2.32–2.42 (m, 2 H), 2.52–2.59 (m, 1 H), 2.70–2.78 (m, 1 H), 3.41 (s, 3 H), 4.40–4.42 (m, 1 H), 4.58 (d, J=6.7, 1 H), 4.70 (d, J=6.8 Hz, 1 H), 5.04 (ddd, 8.5, 5.4, 3.1 Hz, 1 H), 5.30 (dd, J=15.8, 5.6 Hz, 1 H), 5.52 (dt, J=15.7, 6.1 Hz, 1 H), 5.77 (dt, J=15.7, 6.1 Hz, 1 H), 6.17 (d, J=15.9 Hz, 1 H), 6.25 (d, J=14.4 Hz, 1 H), 6.60 (dt, J=14.4, 7.2 Hz, 1 H), 6.68 (d, J=8.0 Hz, 1 H), 6.86 (d, J=7.7 Hz, 1 H), 7.14 ppm (t, J=8.0 Hz, 1 H); 13C NMR (100 MHz, CDCl3): δ=168.0, 152.0, 140.8, 137.6, 132.2, 131.9, 130.9, 129.8, 129.6, 125.3, 118.7, 117.0, 94.9, 78.5, 76.1, 75.4, 56.2, 36.5, 30.7, 30.0, 25.7, 18.2, −4.0, −4.4 ppm; IR (neat, NaCl): =3064, 2928, 2856, 1730, 1574, 1466, 1290, 1255, 1107, 1036, 841 cm−1; HRMS (ESI+) m/z calcd for C26H37INaO5Si [M+Na]+: 607.1347, found: 607.1357.
(3S,4S,5E,9E)‐14‐((tert‐Butyldimethylsilyl)oxy)‐4‐hydroxy‐3‐((E)‐3‐iodoallyl)‐3,4,7,8‐tetrahydro‐1H‐benzo[c][1]oxacyclododecin‐1‐one (51). To a flask containing iodide 49 (117 mg, 0.200 mmol) was added iPrOH (4 mL), and CBr4 (266 mg, 0.801 mmol) in one portion. The mixture was heated at 75 °C for 2 h, cooled to RT and then concentrated by rotary evaporation. Purification by column chromatography (15 % EtOAc in hexanes) provided 93.1 mg (0.172 mmol, 86 %) of alcohol 51 as a colorless oil: =+37.6 (c=1.22, CHCl3); 1H NMR (400 MHz, CDCl3): δ=0.24 (3 H), 0.25 (s, 3 H), 0.98 (s, 9 H), 1.73 (br, 1 H), 2.23–2.38 (m, 4 H), 2.43–2.50 (m, 1 H), 2.69–2.77 (m, 1 H), 4.36 (s, 1 H), 5.13 (ddd, J=9.9, 4.9, 2.0 Hz, 1 H), 5.47 (s, 2 H), 5.75 (dt, J=15.8, 5.8 Hz, 1 H), 6.21 (d, J=16.1 Hz, 1 H), 6.28 (d, J=14.4 Hz, 1 H), 6.57 (ddd, J=15.0, 8.5, 6.8 Hz, 1 H), 6.70 (d, J=8.3 Hz, 1 H), 6.87 (d, J=7.7 Hz, 1 H), 7.16 ppm (t, J=8.0 Hz, 1 H); 13C NMR (100 MHz, CDCl3): δ=167.6, 152.2, 140.4, 137.3, 132.9, 132.7, 130.2, 129.9, 129.2, 125.2, 118.8, 117.0, 79.1, 75.5, 70.8, 36.9, 30.7, 29.8, 25.7, 18.2, −4.0, −4.4 ppm; IR (neat, NaCl): =3326 (br), 3435, 2955, 2929, 2856, 1726, 1645, 1573, 1466, 1253, 1106, 840, 783 cm−1; HRMS (ESI+) m/z calcd for C24H33INaO4Si [M+Na]+: 563.1085, found: 563.1085.
(3S,4S,5E,9E)‐4,14‐Bis((tert‐butyldimethylsilyl)oxy)‐3‐((E)‐3‐iodoallyl)‐3,4,7,8‐tetrahydro‐1H‐benzo[c][1]oxacyclododecin‐1‐one (53). A solution of alcohol 51 (76.9 mg, 0.142 mmol) and 2,6‐lutidine (33 μL, 0.28 mmol) in CH2Cl2 (1.4 mL) was cool to −78 °C and TBSOTf (65 μL, 0.28 mmol) was added. The solution was stirred at −78 °C for 3 h and then left at 0 °C overnight. EtOAc was added and the organic mixture was washed with saturated NaHCO3 solution. The aqueous layer was re‐extracted with EtOAc. The combined organic layer was washed with brine, dried with MgSO4, and concentrated by rotary evaporation. Purification by column chromatography (5 % EtOAc in hexanes) provided 72.3 mg (0.110 mmol, 78 %) of iodide 53 as a colorless oil: =+79.8 (c=1.02, CHCl3); 1H NMR (400 MHz, CDCl3): δ=0.01 (s, 3 H), 0.06 (s, 3 H), 0.24 (s, 6 H), 0.88 (s, 9 H), 0.98 (s, 9 H), 1.91–2.21 (m, 2 H), 2.25–2.41 (m, 2 H), 2.43–2.59 (m, 1 H), 2.61–2.74 (m, 1 H), 4.46 (dd, J=6.6, 3.7 Hz, 1 H), 4.84 (ddd, J=7.4, 6.2, 3.7 Hz, 1 H), 5.32 (dd, J=15.7, 6.7 Hz, 1 H), 5.39–5.48 (m, 1 H), 5.69–5.76 (m, 1 H), 6.13 (d, J=15.8 Hz, 1 H), 6.18 (d, J=14.5 Hz, 1 H), 6.54 (dt, J=14.6, 7.3 Hz, 1 H), 6.71 (d, J=8.2 Hz, 1 H), 6.88 (d, J=7.5 Hz, 1 H), 7.17 ppm (t, J=7.9 Hz, 1 H); 13C NMR (100 MHz, CDCl3): δ=168.3, 151.8, 141.6, 137.9, 133.3, 132.1, 131.3, 130.4, 129.8, 125.8, 118.9, 117.3, 78.4, 77.8, 72.6, 36.0, 30.9, 30.4, 25.9, 25.8, 18.2, 18.1, −3.8, −4.1, −4.3, −4.9 ppm.
(2Z,4E)‐N‐((E)‐3‐((3S,4S,5E,9E)‐4,14‐Dihydroxy‐1‐oxo‐3,4,7,8‐tetrahydro‐1H‐benzo[c][1]oxacyclododecin‐3‐yl)prop‐1‐en‐1‐yl)‐4‐(methoxyimino)but‐2‐enamide (55). To a flask containing iodide 53 (71.4 mg, 0.109 mmol), amide 57 (69.8 mg, 0.545 mmol), CuI (41.5 mg, 0.218 mmol) and Cs2CO3 (178 mg, 0.545 mmol) was charged dimethylacetamide (DMA; 11 mL). The resultant mixture was degassed under high vacuum for 5 min before N,N′‐dimethylethylenediamine (DMEDA; 59 μL, 0.55 mmol) was added. The reaction was stirred at RT for 24 h and then H2O was added. The crude mixture was extracted with EtOAc. The combined organic layer was washed with brine, dried over MgSO4, and concentrated by rotary evaporation. The crude bis‐TBS enamide was directly used in the next step without further purification.
The crude bis‐TBS enamide was dissolved in THF (1 mL) and 5 m HF⋅Py solution (HF⋅Py (70 % HF, Aldrich): pyridine/THF=1:2:4.7, 1.3 mL, 6.5 mmol) was added. The reaction was stirred for 48 h and quenched with phosphate (pH 7) buffer. The aqueous layer was extracted with EtOAc. The organic extracts were washed with brine, dried over MgSO4, and concentrated by rotary evaporation. Purification by column chromatography (60 % EtOAc in hexanes) provided 18.6 mg (0.0436 mmol, 40 %) of analogue 55 as a white solid: =−45.4 (c=0.372, CHCl3); 1H NMR (400 MHz, [D6]acetone): δ=2.20–2.42 (m, 4 H), 2.50–2.60 (m, 1 H), 2.67–2.74 (m, 1 H), 3.87 (s, 3 H), 4.20 (d, J=6.4 Hz, 1 H), 4.38 (s, 1 H), 5.17 (ddd, J=7.7, 5.9, 3.2 Hz, 1 H), 5.37 (dt, J=14.4, 7.2 Hz, 1 H), 5.51–5.64 (m, 2 H), 5.67–5.76 (m, 1 H), 6.12 (d, J=11.5 Hz, 1 H), 6.47 (t, J=10.8 Hz, 1 H), 6.60 (d, J=15.8 Hz, 1 H), 6.79 (d, J=8.2 Hz, 1 H), 6.81 (d, J=7.4 Hz, 1 H), 6.90 (dd, J=14.4, 10.3 Hz, 1 H), 7.27 (t, J=8.0 Hz, 1 H), 9.08 (d, J=10.2 Hz, 1 H), 9.33 (d, J=10.4 Hz, 1 H), 9.90 ppm (s, 1 H); 13C NMR (100 MHz, [D6]acetone): δ=170.4, 162.6, 159.4, 148.5, 141.1, 134.9, 134.2, 133.3, 132.5, 132.1, 131.5, 126.5, 126.1, 119.5, 117.0, 115.9, 109.8, 78.5, 72.5, 62.5, 32.5, 31.4, 30.7 ppm; IR (neat, NaCl): =3306 (br), 3419, 2928, 2852, 1652, 1526, 1465, 1259, 1118, 1043, 966, 819 cm−1; HRMS (ESI+) m/z calcd for C23H26N2NaO6 [M+Na]+: 449.1683, found: 449.1686.
3‐((3S,4S,5E,9E)‐14‐((tert‐Butyldimethylsilyl)oxy)‐4‐(methoxymethoxy)‐1‐oxo‐3,4,7,8‐tetrahydro‐1H‐benzo[c][1]oxacyclododecin‐3‐yl)propanal (48). To a solution of alcohol 21 (28.4 mg, 0.0596 mmol) in CH2Cl2 (1.2 mL) was added Dess–Martin periodinane (37.9 mg, 0.0894 mmol) as a solid. The resultant mixture was stirred at RT for 3 h before it was filtered. The filtrate was concentrated by rotary evaporation. Purification by column chromatography (20 % EtOAc in hexanes) provided 20.3 mg (0.0428 mmol, 72 %) of aldehyde 48 as a colorless oil: =+150 (c=1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ=0.23 (s, 3 H), 0.24 (s, 3 H), 0.98 (s, 9 H), 2.13–2.23 (m, 4 H), 2.30–2.41 (m, 2 H), 2.63–2.68 (m, 2 H), 3.39 (s, 3 H), 4.41–4.43 (m, 1 H), 4.58 (d, J=6.8 Hz, 1 H), 4.69 (d, J=6.8 Hz, 1 H), 5.16 (ddd, J=7.8, 5.8, 3.2 Hz, 1 H), 5.32 (dd, J=15.6, 5.3 Hz, 1 H), 5.48–5.55 (m, 1 H), 5.76 (dt, J=15.9, 6.2 Hz, 1 H), 6.23 (d, J=15.9 Hz, 1 H), 6.70 (d, J=8.2 Hz, 1 H), 6.86 (d, J=7.7 Hz, 1 H), 7.14 (t, J=7.9 Hz, 1 H), 9.80 ppm (s, 1 H).
(3S,4S,5E,9E)‐14‐((tert‐Butyldimethylsilyl)oxy)‐3‐((E)‐4‐iodobut‐3‐en‐1‐yl)‐4‐(methoxymethoxy)‐3,4,7,8‐tetrahydro‐1H‐benzo[c][1]oxacyclododecin‐1‐one (50). To a suspension of CrCl2 (114 mg, 0.924 mmol) in THF (0.55 mL) was added a solution of aldehyde 48 (36.7 mg, 0.0770 mmol) and iodoform (121 mg, 0.308 mmol) in dioxane (3.3 mL). The resultant mixture was stirred at RT for 23 h and then quenched with brine and extracted with EtOAc. The combined organic layer was washed with brine, dried with MgSO4, and concentrated by rotary evaporation. Purification by column chromatography (10 % EtOAc in hexanes) provided 38.9 mg (0.0650 mmol, 84 %) of iodide 50 as a colorless oil (9:1 ratio of E/Z vinyl iodide isomers by 1H NMR): =+110 (c=1.0, CHCl3); 1H NMR (400 MHz, CDCl3): δ=0.22 (s, 3 H), 0.23 (s, 3 H), 0.97 (s, 9 H), 1.89–2.00 (m, 2 H), 2.13–2.42 (m, 6 H), 3.39 (s, 3 H), 4.38–4.40 (m, 1 H), 4.57 (d, J=6.8 Hz, 1 H), 4.69 (d, J=6.8 Hz, 1 H), 5.14 (td, J=6.9, 3.1 Hz, 1 H), 5.32 (dd, J=15.7, 5.1 Hz, 1 H), 5.46–5.53 (m, 1 H), 5.77 (dt, J=15.8, 6.1 Hz, 1 H), 6.08 (d, J=14.4 Hz, 1 H), 6.24 (d, J=15.7 Hz, 1 H), 6.55 (dt, J=14.1, 6.9 Hz, 1 H), 6.68 (d, J=8.1 Hz, 1 H), 6.85 (d, J=7.6 Hz, 1 H), 7.13 ppm (t, J=8.0 Hz, 1 H); 13C NMR (100 MHz, CDCl3): δ=168.1, 152.1, 145.3, 137.5, 132.4, 132.2, 130.7, 129.7, 128.9, 125.5, 118.9, 117.1, 94.9, 76.2, 76.0, 75.2, 56.2, 32.2, 30.5, 30.1, 29.0, 25.7, 18.3, −4.1, −4.3 ppm; IR (neat, NaCl): =3065, 2929, 2857, 1728, 1573, 1466, 1032, 841 cm−1; HRMS (ESI+) m/z calcd for C27H39INaO5Si [M+Na]+: 621.1504, found: 621.1509.
(3S,4S,5E,9E)‐14‐((tert‐Butyldimethylsilyl)oxy)‐4‐hydroxy‐3‐((E)‐4‐iodobut‐3‐en‐1‐yl)‐3,4,7,8‐tetrahydro‐1H‐benzo[c][1]oxacyclododecin‐1‐one (52). To a flask containing iodide 50 (38.9 mg, 0.0650 mmol) was added iPrOH (1.3 mL), and CBr4 (86.2 mg, 0.260 mmol) in one portion. The mixture was heated at 75 °C for 2 h, cooled to RT and then concentrated by rotary evaporation. Purification by column chromatography (15 % EtOAc in hexanes) provided 34.5 mg (0.0622 mmol, 96 %) of alcohol 52 as a colorless oil: =+31 (c=0.50, CHCl3); 1H NMR (400 MHz, CDCl3): δ=0.227 (s, 3 H), 0.233 (s, 3 H), 0.97 (s, 9 H), 1.65 (d, J=9.7 Hz, 1 H), 1.86–1.98 (m, 2 H), 2.13–2.41 (m, 6 H), 4.31 (d, J=8.8 Hz, 1 H), 5.19 (td, J=7.1, 2.0 Hz, 1 H), 5.43–5.55 (m, 2 H), 5.72–5.79 (m, 1 H), 6.08 (d, J=14.4 Hz, 1 H), 6.26 (d, J=15.6 Hz, 1 H), 6.54 (dt, J=14.1, 7.0 Hz, 1 H), 6.72 (d, J=8.2 Hz, 1 H), 6.86 (d, J=7.7 Hz, 1 H), 7.16 ppm (t, J=8.0 Hz, 1 H); 13C NMR (100 MHz, CDCl3): δ=167.8, 152.3, 145.1, 137.3, 133.1, 132.6, 130.1, 129.8, 129.4, 125.4, 119.0, 117.3, 76.3, 75.4, 72.1, 32.2, 30.6, 29.9, 29.4, 25.8, 18.4, −4.1, −4.2 ppm; IR (neat, NaCl): =3326 (br), 3065, 2954, 2928, 2856, 1730, 1572, 1465, 1106, 840 cm−1; HRMS (ESI+) m/z calcd for C25H35INaO4Si [M+Na]+: 577.1242, found: 577.1246.
(3S,4S,5E,9E)‐4,14‐Bis((tert‐butyldimethylsilyl)oxy)‐3‐((E)‐4‐iodobut‐3‐en‐1‐yl)‐3,4,7,8‐tetrahydro‐1H‐benzo[c][1]oxacyclododecin‐1‐one (54). A solution of alcohol 52 (34.5 mg, 0.0622 mmol) and 2,6‐lutidine (14 μL, 0.12 mmol) in CH2Cl2 (0.6 mL) was cool to −78 °C and TBSOTf (29 μL, 0.12 mmol) was added. The solution was stirred at −78 °C for 3 h and then left at 0 °C overnight. EtOAc was added and the organic mixture was washed with saturated NaHCO3 solution. The aqueous layer was re‐extracted with EtOAc. The combined organic layer was washed with brine, dried with MgSO4, and concentrated by rotary evaporation. Purification by column chromatography (5 % EtOAc in hexanes) provided 35.1 mg (0.0525 mmol, 84 %) of iodide 54 as a colorless oil: =+75 (c=0.50, CHCl3); 1H NMR (400 MHz, CDCl3): δ=0.02 (s, 3 H), 0.06 (s, 3 H), 0.22 (s, 3 H), 0.23 (s, 3 H), 0.89 (s, 9 H), 0.97 (s, 9 H), 1.80–1.95 (m, 2 H), 2.05–2.37 (m, 6 H), 4.43 (dd, J=5.5, 4.1, 1 H), 4.95 (ddd, J=7.8, 5.5, 3.6 Hz, 1 H), 5.36 (dd, J=15.6, 5.6 Hz, 1 H), 5.43 (dt, J=15.7, 6.1 Hz, 1 H), 5.73 (dt, J=15.9, 6.5 Hz, 1 H), 6.04 (d, J=14.4 Hz, 1 H), 6.22 (d, J=16.1 Hz, 1 H), 6.54 (dt, J=14.1, 6.9 Hz, 1 H), 6.71 (d, J=8.2 Hz, 1 H), 6.86 (d, J=7.7 Hz, 1 H), 7.16 ppm (t, J=8.0 Hz, 1 H); 13C NMR (100 MHz, CDCl3): δ=168.2, 151.9, 145.6, 137.8, 132.28, 132.26, 131.1, 131.0, 129.6, 126.0, 119.1, 117.3, 77.9, 75.0, 73.3, 32.5, 30.6, 30.4, 28.5, 25.9, 25.8, 18.3, 18.2, −4.0, −4.1, −4.2, −4.9 ppm; IR (neat, NaCl): =3066, 2955, 2929, 2857, 1728, 1465, 1107, 840, 779 cm−1; HRMS (ESI+) m/z calcd for C31H49INaO4Si2 [M+Na]+: 691.2106, found: 691.2120.
(2Z,4E)‐N‐((E)‐4‐((3S,4S,5E,9E)‐4,14‐Dihydroxy‐1‐oxo‐3,4,7,8‐tetrahydro‐1H‐benzo[c][1]oxacyclododecin‐3‐yl)but‐1‐en‐1‐yl)‐4‐(methoxyimino)but‐2‐enamide (56). To a flask containing iodide 54 (18.6 mg, 0.0278 mmol), amide 57 (17.8 mg, 0.139 mmol), CuI (10.6 mg, 0.0556 mmol) and Cs2CO3 (45.3 mg, 0.139 mmol) was charged DMA (2.8 mL). The resultant mixture was degassed under high vacuum for 5 min before DMEDA (15 μL, 0.14 mmol) was added. The reaction was stirred at RT for 24 h and then H2O was added. The crude mixture was extracted with EtOAc. The combined organic layer was washed with brine, dried over MgSO4, and concentrated by rotary evaporation. The crude bis‐TBS enamide was directly used in the next step without further purification.
The crude bis‐TBS enamide was dissolved in THF (0.28 mL) and 5 m HF⋅Py solution (HF⋅Py (70 % HF, Aldrich): pyridine/THF=1:2:4.7, 0.17 mL, 0.85 mmol) was added. The reaction was stirred for 24 h and quenched with phosphate (pH 7) buffer. The aqueous layer was extracted with EtOAc. The organic extracts were washed with brine, dried over MgSO4, and concentrated by rotary evaporation. Purification by column chromatography (60 % EtOAc in hexanes) provided 3.7 mg (0.0084 mmol, 30 %) of analogue 56 as a white solid: =−62 (c=0.050, CHCl3); 1H NMR (400 MHz, CDCl3): δ=1.76 (d, J=5.9 Hz, 1 H), 1.86 (dq, J=14.6, 7.9 Hz, 1 H), 2.03–2.11 (m, 1 H), 2.14–2.28 (m, 4 H), 2.36–2.48 (m, 2 H), 3.94 (s, 3 H), 4.36 (q, J=5.1 Hz, 1 H), 5.21 (ddd, J=7.6, 5.4, 3.9 Hz, 1 H), 5.28 (dt, J=14.1, 7.0 Hz, 1 H), 5.49 (dd, J=15.7, 5.7 Hz, 1 H), 5.57–5.66 (m, 2 H), 5.84 (dd, J=11.5, 1.1 Hz, 1 H), 6.50 (dd, J=11.4, 10.4 Hz, 1 H), 6.65 (d, J=15.7 Hz, 1 H), 6.77–6.88 (m, 3 H), 7.05–7.08 (m, 1 H), 7.31 (t, J=7.9 Hz, 1 H), 9.01 (dd, J=10.3, 1.0 Hz, 1 H), 10.49 ppm (s, 1 H); 13C NMR (100 MHz, CDCl3): δ=170.8, 161.6, 161.2, 147.6, 141.6, 135.4, 135.0, 134.0, 129.7, 129.5, 125.8, 124.2, 122.9, 119.9, 116.2, 113.1, 112.1, 76.9, 72.5, 62.4, 32.4, 29.8, 29.0, 26.2 ppm; IR (neat, NaCl): =3306 (br), 3044, 2954, 2928, 1652, 1603, 1450, 1217, 1122, 1045, 965, 838, 821, 778 cm−1; HRMS (ESI+) m/z calcd for C24H28N2NaO6 [M+Na]+: 463.1840, found: 463.1845.
Cytotoxicity assays
The adherent human melanoma SK‐Mel‐5 and SK‐Mel‐28 and breast cancer MCF‐7 cell lines were obtained from the NCI and grown in normal RPMI 1640 culture medium containing 10 % fetal bovine serum. Human promyelocytic leukemia HL‐60 cells were obtained from ATCC and grown in suspension in IMDM (ATCC 30‐2005) containing 20 % fetal bovine serum. Following growth in exponential‐phase maintenance culture, adherent cells were dissociated with 0.25 % trypsin, and then all cells were harvested by centrifugation at 125 g for 5 min, the supernatant was removed, and the cells were resuspended in fresh culture medium. The cell density for all lines was adjusted to 1×105 cells per mL and dispensed in triplicate on 96‐well plates in 50 μL volumes. After incubation overnight at 37 °C under 5 % CO2, 50 μL of culture medium containing various concentrations of the test compounds were added. Paclitaxel and oximidine II were used as positive controls. After 48 h incubation, the relative cell viability in each well was determined by using the AlamarBlue Assay Kit. Fluorescence intensity (λ excitation=530 nm, λ emission=590 nm) was measured on a SpectraMax plate reader (Molecular Devices). The IC50 value of each compound was determined by fitting the relative viability of the cells to the drug concentration using Prism version 6 (GraphPad).
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
These studies were supported by the University of Minnesota through the Vince and McKnight Endowed Chairs. C.M.S. acknowledges a pre‐doctoral fellowship from the US National Institutes of Health (NIH) (NIH T32 GM 008545) while at the University of Kansas. K.K. thanks the Royal Thai Government for fellowship support. The cytotoxicity assays were performed by Defeng Tian in the Institute for Therapeutics Discovery and Development at the University of Minnesota. Special acknowledgements to Rebecca A. Cuellar and Sara Coulup for their help in preparing the spectra, and Dr. Subhashree Francis for LC–MS analyses.
C. M. Schneider, W. Li, K. Khownium, G. H. Lushington, G. I. Georg, ChemMedChem 2016, 11, 1600.
References
- 1. Erickson K. L., Beutler J. A., J. H. Cardellina II , Boyd M. R., J. Org. Chem. 1997, 62, 8188–8192. [DOI] [PubMed] [Google Scholar]
- 2. Erickson K. L., Beutler J. A., J. H. Cardellina II , Boyd M. R., J. Org. Chem. 2001, 66, 1532. [Google Scholar]
- 3. Galinis D. L., McKee T. C., Pannell L. K., J. H. Cardellina II , Boyd M. R., J. Org. Chem. 1997, 62, 8968–8969. [Google Scholar]
- 4. Kunze B., Jansen R., Sasse F., Hofle G., Reichenbach H., J. Antibiot. 1998, 51, 1075–1080. [DOI] [PubMed] [Google Scholar]
- 5. Dekker K. A., Aiello R. J., Hirai H., Inagaki T., Sakakibara T., Suzuki Y., Thompson J. F., Yamauchi Y., Kojima N., J. Antibiot. 1998, 51, 14–20. [DOI] [PubMed] [Google Scholar]
- 6. Hayakawa Y., Tomikawa T., Shin-ya K., Arao N., Nagai K., Suzuki K.-i., J. Antibiot. 2003, 56, 899–904. [DOI] [PubMed] [Google Scholar]
- 7. Hayakawa Y., Tomikawa T., Shin-ya K., Arao N., Nagai K., Suzuki K.-i., Furihata K., J. Antibiot. 2003, 56, 905–908. [DOI] [PubMed] [Google Scholar]
- 8. Kim J. W., Shin-ya K., Furihata K., Hayakawa Y., Seto H., J. Org. Chem. 1999, 64, 153–155. [DOI] [PubMed] [Google Scholar]
- 9. Boyd M. R., Farina C., Belfiore P., Gagliardi S., Kim J. W., Hayakawa Y., Beutler J. A., McKee T. C., Bowman B. J., Bowman E. J., J. Pharmacol. Exp. Ther. 2001, 297, 114–120. [PubMed] [Google Scholar]
- 10. Boyd M. R., Paull K. D., Drug Dev. Res. 1995, 34, 91–109. [Google Scholar]
- 11. Lafourcade C., Sobo K., Kieffer-Jaquinod S., Garin J., Goot F. G. van der, PLoS One 2008, 3, e2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nishi T., Forgac M., Nat. Rev. Mol. Cell Biol. 2002, 3, 94–103. [DOI] [PubMed] [Google Scholar]
- 13. Forgac M., Nat. Rev. Mol. Cell Biol. 2007, 8, 917–929. [DOI] [PubMed] [Google Scholar]
- 14. Wieczorek H., Brown D., Grinstein S., Ehrenfeld J., Harvey W. R., BioEssays 1999, 21, 637–648. [DOI] [PubMed] [Google Scholar]
- 15. Farina C., Gagliardi S., Drug Discovery Today 1999, 4, 163–172. [DOI] [PubMed] [Google Scholar]
- 16. Hinton A., Bond S., Forgac M., Pfluegers Arch. 2009, 457, 589–598. [DOI] [PubMed] [Google Scholar]
- 17. Pérez-Sayáns M., Somoza-Martín J. M., Barros-Angueira F., Gándara Rey J. M., García-García A., Cancer Treat. Rev. 2009, 35, 707–713. [DOI] [PubMed] [Google Scholar]
- 18. Yet L., Chem. Rev. 2003, 103, 4283–4306. [DOI] [PubMed] [Google Scholar]
- 19. Maier M. E., Org. Biomol. Chem. 2015, 13, 5302–5343. [DOI] [PubMed] [Google Scholar]
- 20. Sugimoto Y., Konoki K., Murata M., Matsushita M., Kanazawa H., Oishi T., J. Med. Chem. 2009, 52, 798–806. [DOI] [PubMed] [Google Scholar]
- 21. Balan D., Burns C. J., Fisk N. G., Huegel H., Huang D. C. S., Segal D., White C., Wagler J., Rizzacasa M. A., Org. Biomol. Chem. 2012, 10, 8147–8153. [DOI] [PubMed] [Google Scholar]
- 22. Xie X.-S., Padron D., Liao X., Wang J., Roth M. G., De Brabander J. K., J. Biol. Chem. 2004, 279, 19755–19763. [DOI] [PubMed] [Google Scholar]
- 23. Lebreton S., Jaunbergs J., Roth M. G., Ferguson D. A., De Brabander J. K., Bioorg. Med. Chem. Lett. 2008, 18, 5879–5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jundt L., Steinmetz H., Luger P., Weber M., Kunze B., Reichenbach H., Hoefle G., Eur. J. Org. Chem. 2006, 5036–5044. [Google Scholar]
- 25. Kunze B., Steinmetz H., Hoefle G., Huss M., Wieczorek H., Reichenbach H., J. Antibiot. 2006, 59, 664–668. [DOI] [PubMed] [Google Scholar]
- 26. Haack T., Haack K., Diederich W. E., Blackman B., Roy S., Pusuluri S., Georg G. I., J. Org. Chem. 2005, 70, 7592–7604. [DOI] [PubMed] [Google Scholar]
- 27. Yang K., Blackman B., Diederich W., Flaherty P. T., Mossman C. J., Roy S., Ahn Y. M., Georg G. I., J. Org. Chem. 2003, 68, 10030–10039. [DOI] [PubMed] [Google Scholar]
- 28. Schneider C. M., Khownium K., Li W., Spletstoser J. T., Haack T., Georg G. I., Angew. Chem. Int. Ed. 2011, 50, 7855–7857; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 8001–8003, S7855/1–S7855/67. [Google Scholar]
- 29. Blackman B., Georg G. I., Lushington G. H., Lett. Drug Des. Discovery 2006, 3, 104–107. [Google Scholar]
- 30. Wang X., J. A. Porco, Jr. , J. Am. Chem. Soc. 2003, 125, 6040–6041. [DOI] [PubMed] [Google Scholar]
- 31. Schoenberg A., Bartoletti I., Heck R. F., J. Org. Chem. 1974, 39, 3318–3326. [Google Scholar]
- 32. Walker M. A., Tetrahedron Lett. 1994, 35, 665–668. [Google Scholar]
- 33. Walker M. A., J. Org. Chem. 1995, 60, 5352–5355. [Google Scholar]
- 34. Wu Y., Liao X., Wang R., Xie X.-S., De Brabander J. K., J. Am. Chem. Soc. 2002, 124, 3245–3253. [DOI] [PubMed] [Google Scholar]
- 35. Palmer J. T., Rasnick D., Klaus J. L., Bromme D., J. Med. Chem. 1995, 38, 3193–3196. [DOI] [PubMed] [Google Scholar]
- 36. Lebreton S., Xie X.-S., Ferguson D., De Brabander J. K., Tetrahedron 2004, 60, 9635–9647. [Google Scholar]
- 37. Nicolaou K. C., Kim David W., Baati R., O′Brate A., Giannakakou P., Chem. Eur. J. 2003, 9, 6177–6191. [DOI] [PubMed] [Google Scholar]
- 38. Klebe G., Abraham U., Mietzner T., J. Med. Chem. 1994, 37, 4130–4146. [DOI] [PubMed] [Google Scholar]
- 39. Clark M., R. D. Cramer III , Van Opdenbosch N., J. Comput. Chem. 1989, 10, 982–1012. [Google Scholar]
- 40. Gasteiger J., Marsili M., Tetrahedron 1980, 36, 3219–3222. [Google Scholar]
- 41. Dragon (version 5): software for the calculation of molecular descriptors, R. Todeschini, V. Consonni, A. Mauri, M. Pavan, Talete SRL, Milan (Italy), 2005: www.talete.mi.it/products/dragon_description.htm (accessed May 2016).
- 42. Brown H. C., Jadhav P. K., Bhat K. S., J. Am. Chem. Soc. 1988, 110, 1535–1538. [Google Scholar]
- 43. Darwish A., Lang A., Kim T., Chong J. M., Org. Lett. 2008, 10, 861–864. [DOI] [PubMed] [Google Scholar]
- 44. Wang X., Bowman E. J., Bowman B. J., J. A. Porco, Jr. , Angew. Chem. Int. Ed. 2004, 43, 3601–3605; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 3685–3689. [Google Scholar]
- 45. Bhattacharjee A., De Brabander J. K., Tetrahedron Lett. 2000, 41, 8069–8073. [Google Scholar]
- 46. VEGA ZZ (version 3.05), molecular modeling toolkit: nova.disfarm.unimi.it/cms/index.php?Software_projects:VEGA_ZZ (accessed May 2016).
- 47.PyMOL Molecular Graphics System (version 1.7.0.1): sourceforge.net/projects/pymol/ (accessed May 2016).
- 48. Fürstner A., Konetzki I., Tetrahedron 1996, 52, 15071–15078. [Google Scholar]
- 49. Hayashi Y., Yamaguchi H., Toyoshima M., Okado K., Toyo T., Shoji M., Org. Lett. 2008, 10, 1405–1408 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary