

Abstract
Acid sphingomyelinase deficiency (ASMD; Niemann-Pick disease type A and B) is a lysosomal storage disorder characterized by abnormal intracellular sphingomyelin (SM) accumulation. Prominent liver involvement results in hepatomegaly, fibrosis/cirrhosis, abnormal liver chemistries, and a proatherogenic lipid profile. Olipudase alfa (recombinant human ASM) is in clinical development as an investigational enzyme replacement therapy for the non-neurological manifestations of ASMD. In a phase 1b study conducted to evaluate the safety and tolerability of within-patient dose escalation with olipudase alfa, measurement of SM levels in liver biopsies was used as a pharmacodynamic biomarker of substrate burden. Five adult patients with non neuronopathic ASMD received escalating doses of olipudase alfa every 2 weeks for 26 weeks. Liver biopsies obtained at baseline and 26 weeks after treatment were evaluated for SM storage by histomorphometric analysis, biochemistry, and electron microscopy. Biopsies were also assessed for inflammation and fibrosis, and for the association of SM levels with liver volume, liver function tests, and lipid profiles. At baseline, SM storage present in Kupffer cells and hepatocytes ranged from 9.8% to 53.8% of the microscopic field. After 26 weeks of treatment, statistically significant reductions in SM (P<0.0001) measured by morphometry were seen in 4 patients with evaluable liver biopsies. The 26-week biopsy of the fifth patient was insufficient for morphometric quantitation. Posttreatment SM levels ranged from 1.2% to 9.5% of the microscopic field, corresponding to an 84% to 92% relative reduction from baseline. Improvements in liver volume, liver function tests, and lipid profiles were also observed. This study illustrates the utility of SM assessment by liver biopsy as a pharmacodynamic biomarker of disease burden in these patients.
Key Words: enzyme replacement therapy, Niemann-Pick disease type B, lysosomal storage disease, liver pathology, hyperlipidemia
Acid sphingomyelinase deficiency (ASMD) is a rare lysosomal storage disorder characterized by the pathologic accumulation of sphingomyelin (SM) in cells throughout the body, primarily within the liver, spleen, lungs, and bone marrow, leading to significant clinical disease. The clinical spectrum of ASMD ranges from an infantile-onset visceral and neurodegenerative disease with death in early childhood (Niemann-Pick disease type A; NPD A), to a variable-onset visceral disease with no neurodegeneration and prolonged survival into adulthood (NPD B).1 In NPD B, SM accumulation manifests most prominently as hepatosplenomegaly, interstitial lung disease, and bone marrow infiltration.
The hepatomegaly that develops as a result of longstanding ASMD is a significant cause of morbidity and mortality leading to cirrhosis, liver failure, and death.2,3 SM accumulation occurs within the lysosomes of both macrophage-derived Kupffer cells (foam cells) and in hepatocytes where it cocrystalizes in lamellar structures with cholesterol.3 The liver is a major organ involved in lipoprotein biogenesis, metabolism, and lipid homeostasis.4,5 Therefore, massive hepatocellular accumulation of SM6 is likely to perturb normal cell function and lead to derangements in lipid homeostasis. Natural history studies of patients with ASMD have shown that the majority of the patients have abnormal liver function tests (LFTs) and abnormal lipoprotein profiles with low high-density lipoprotein-C (HDL-C) levels and elevated low-density lipoprotein-C (LDL-C) levels.7–9 Others studies have reported low HDL-C and elevated LDL-C levels, along with elevations in SM and the proinflammatory marker C-reactive protein, and erythrocyte sedimentation rate, in patients with ASMD,10,11 all of which are known risk factors for atherosclerosis previously identified in the general population.12–18 Cardiac computed tomography examination of children with ASMD and lipid abnormalities have revealed that over half have coronary artery calcification, another early risk factor for coronary artery disease.19 It is therefore not surprising that patients with ASMD have been reported to have unstable angina,11 coronary disease, and acute heart failure with death in young adulthood,20 coronary artery disease in early adulthood requiring bypass surgery,2 as well as mitral and aortic valve disease requiring valve replacement.3
On the basis of these observational findings, one could hypothesize that clearance of SM by enzyme replacement therapy (ERT) should be accompanied by reversal and resolution of hepatomegaly and correction of the imbalance in lipid homeostasis. Normalization of the lipoprotein profile would potentially lower the cardiovascular risk of patients. Renal biopsy pathology has been accepted as a validated surrogate biomarker of efficacy in the evaluation of ERT in another lysosomal storage disorder, Fabry disease, and skeletal muscle pathology has proven useful in Pompe disease as well.21–24
We previously reported detailed liver pathology on 17 ASMD patients enrolled in a phase 1 single ascending dose trial of olipudase alfa (recombinant human acid sphingomyelinase).25,26 The current study expands upon these initial single-dose findings and demonstrates the response to repeat-dose therapy. A phase 1b study was conducted to evaluate the safety and tolerability, pharmacokinetics, and pharmacodynamics of within-patient dose escalation and repeat-dosing of olipudase alfa in adult patients with NPD B.27 Here we describe in detail the changes in liver pathology and associated changes in lipid profiles after 26 weeks of repeat dose treatment with olipudase alfa. The current study shows the robust histopathologic clearance of SM from liver biopsies in patients with ASMD after 26 weeks of treatment with olipudase alfa and its association with improvements in hepatomegaly, LFTs, and lipid profiles.
MATERIALS AND METHODS
Study Design and Human Subjects
Five adults with non neuronopathic ASMD were enrolled in a phase 1b trial of olipudase alfa. The protocol was approved by the Institutional Review Board of the Icahn School of Medicine at Mount Sinai (New York, NY) and the Ethic Committee at St Mary’s Hospital (Manchester, UK). The study was conducted according to Good Clinical Practice and in accordance with the principles of the Declaration of Helsinki. All patients provided written informed consent before any study-related procedures. The main inclusion criteria for enrollment in the study were: adults with non neuronopathic ASMD between the ages of 18 and 65 years inclusive, spleen volume ≥6 multiples of normal (MN; normal spleen volume is 0.2% of body weight), diffusing capacity of carbon monoxide (DLCO) measured at >20% and ≤80% of the predicted normal value, and preserved liver function characterized by alanine aminotransferase (ALT) and aspartate aminotransferase (AST) ≤250 IU/L, total bilirubin ≤1.5 mg/L, and international normalized ratio ≤1.5. Patients with a history of major organ transplant, or who required medications that could decrease olipudase alfa activity (eg, chlorpromazine, imipramine, or desipramine) were excluded. Patients on a stable dose and regimen of lipid-lowering therapy were eligible.
Six patients were screened for eligibility, and 5 patients were enrolled to receive intravenous infusions of olipudase alfa every 2 weeks for 26 weeks using a within-patient dose escalation schedule. One patient was not eligible to enroll in the study due to prohibited medication use and excluded baseline hematology and bilirubin levels. Patients started with a 0.1 mg/kg dose, followed by a 0.3 mg/kg dose. After tolerating 2 consecutive doses of 0.3 mg/kg, patients continued dose escalation to 0.6, 1.0, 2.0 mg/kg, and finally to 3.0 mg/kg for the remaining duration of the 26-week study. Prespecified adverse event and tolerability criteria were used to determine the next step in the dose escalation regimen.
ASM activity in leukocytes and dried blood spots and fasting lipid profiles were measured as previously described.27 All liver biopsies were performed percutaneously using a Jamshidi needle and were immediately processed at bedside. Liver biopsies were obtained at baseline and at week 26.
Fresh liver samples were divided into 3 pieces at each clinical site and fixed or frozen as follows, before shipment. The study protocol requested that 1 piece (minimum size 1.5 cm or larger) be placed into 10% neutral buffered formalin for later standard paraffin processing and evaluation for fibrosis; the second piece (minimum size 0.5 cm or larger) was placed into a fresh mixture of 2% glutaraldehyde/2% paraformaldehyde in 0.2 mol/L sodium cacodylate buffer, pH 7.3 for later epoxy resin processing, high-resolution light microscopy, morphometric analysis of SM, and electron microscopy examination (in the case of patient 1, the posttreatment sample was inadequate for analysis, consisting entirely of fibrous tissue). A third piece (minimum size 0.5 cm or larger) was frozen in liquid nitrogen for later biochemical quantification of SM.
Paraffin Processing of Liver Biopsies for Special Stains and Immunohistochemistry
The liver biopsy specimens that were immersion fixed in 10% neutral buffered formalin were dehydrated in ascending grades of reagent alcohol, cleared in xylenes, infiltrated, and embedded into Paraplast X-TRA paraffin (McCormick Scientific, LLC, St Louis, MO) on a Tissue-Tek VIP tissue processor (Sacura Finetek Japan Co., Tokyo, Japan). Sections of 5 µm thickness were cut on a Leica microtome and mounted onto Fisherbrand Superfrost Plus Slides (Fisher Scientific, Pittsburgh, PA). Slides were stained with modified hematoxylin and eosin, Masson trichrome, and Gordon-Sweets reticulin stains according to standard methods.28 Immunohistochemistry for the macrophage-specific marker, CD68, was performed using a mouse anti-human CD68 antibody (Dako; clone PG-M1, cat# M0876) at a concentration of 0.4 μg/mL. Slides were stained with Dako diaminobenzidine chromagen solution and a hematoxylin counterstain (Dako).
Tissue Processing for High-resolution Light Microscopy and Electron Microscopy
One-millimeter cubes of each liver specimen were fixed for a minimum of 24 hours in 2% glutaraldehyde, 2% paraformaldehyde in 0.2 mol/L sodium cacodylate buffer, pH 7.3. The tissues were then washed in 0.2 mol/L sodium cacodylate buffer overnight and placed in a working solution of 2.5% potassium dichromate and 0.5% osmium tetroxide in reverse osmosis/deionized water for 6 hours at room temperature (RT), followed by washing in running water for 1 hour. The tissues were then stored overnight in 30% ethanol at 4°C, followed by dehydration in acetones (70%, 100% acetones for 30 min each). After dehydration, the tissues were infiltrated in 70% resin (15 mL DDSA, 5 mL LX-112, 5 mL Araldite 502, and 0.5 mL DMP-30 [Ladd Research Industries, Williston, VT]) in acetone for 1 hour. Tissue was then infiltrated in 2 changes of 100% resin, 1 at 3 hours and 1 overnight. Embedding was performed in “00” Beem capsule with 100% resin and polymerized at 60°C for a minimum of 48 hours. Sections of 1 µm thickness were cut using a Leica EM UC6 microtome (Leica Microsystems) with an ultradiamond knife (Diatome, Hatfield, PA). The sections were collected onto Superfrost Plus Slides and dried overnight on a 40°C slide warmer.
Modified Toluidene Blue Staining of Epon-araldite Sections for High-resolution Light Microscopy
Epon-araldite sections were stained as described previously.29 Briefly, slides were hydrated in water for 1 minute. The slides were then treated with a filtered solution of 0.5% of tannic acid in Ringer solution buffer, pH 6.8 (Electron Microscopy Sciences, Hatfield, PA) for 15 minutes, followed by rinsing in running water for 5 minutes. Slides were then stained for 2 minutes with a 1% solution of toluidine blue/borax pH 8.4 (heated to 60°C). After staining, sections were washed in running water for 5 minutes, dried completely in a 60°C oven, and coverslipped with TBS SHUR/Mount Xylene-Based Mounting Medium (Triangle Biomedical Systems Inc., Durham, NC). Areas containing SM stained purple, nuclei stained dark blue, and cytoplasm and other elements stained light blue, thus rendering the sections suitable for MetaMorph image analysis. All patient samples were stained for SM using this direct staining method, as well as the indirect staining method described below, with comparable results. Metamorph analysis was performed on these direct-stained samples in all cases except those in which SM levels were very low, rendering the purple/blue color separation less distinct. In such cases (eg, patient 3) MetaMorph was performed on the lysenin-stained sections in which the red/blue color separation was more distinct.
Lysenin Affinity Staining on Epon-araldite Sections
Epon sections were hydrated in tris buffered saline (TBS) (Dako) with 0.1% Tween-20 (TBS-T; Dako) for 5 minutes. For SM staining, a 5 μg/mL solution of lysenin protein (Peptides International, Louisville, KY) was diluted in TBS with 0.5% BSA (TBS-B; Dako), placed onto the slides and incubated at 37°C for 1 hour. After rinsing in TBS-T, a 1:2500 dilution of rabbit antilysenin antiserum (Peptides International) diluted in TBS-B was added to the slides for 30 minutes at RT. After another TBS-T rinse, MACH2 rabbit alkaline phosphatase polymer (Biocare Medical, Concord, CA) was applied for 30 minutes at RT. After rinsing in TBS-T, slides were developed in Vulcan Fast Red chromagen (Biocare Medical) and counterstained in a 1:10 dilution of Richardson stain (heated to 60°C) for 15 seconds. Once thoroughly rinsed in running tap water, the slides were air dried, mounted with TBS SHUR/Mount Xylene-Based Mounting Medium (Triangle Biomedical Systems Inc.), and coverslipped. Using this method, SM appeared red against a light blue background, rendering the sections suitable for MetaMorph image analysis. In cases with very low levels of SM (eg, patient 3) MetaMorph was performed on lysenin-stained sections, in which the red/blue color separation was more distinct than the purple/blue color separation of the direct method described above.
Transmission Electron Microscopy
Ultrathin sections of 70 nm thickness were cut from epon blocks and mounted on 200-mesh copper grids. The grids were stained with Leica EM Stain (Leica Microsystems) containing 0.5% aqueous uranyl acetate and 3% lead citrate solution. Images were acquired on a JEOL JEM-1400 transmission electron microscope (JEOL, Peabody, MA) using a Gatan 785 Erlangshan ESW1000 digital camera (Gatan Inc., Pleasanton, CA).
MetaMorph Digital Analysis
MetaMorph analysis was performed as described elsewhere.25,30 Briefly, 1 representative field from each slide was photographed with a Nikon DXM1200 digital camera and acquired with the Nikon Act 1 photo image capture software for the DXM1200 digital camera, version 1.12 (Nikon Inc., Instrument Group, Melville, NY). Each digital image was photographed with the ×60 objective and formatted at a fixed pixel density (8×10 inches at 150 dpi) using Adobe PhotoShop software (version 5.5). Each digital image was then opened using the MetaMorph Imaging Processing and Analysis software (version 7.7, Universal Imaging Corporation) for histomorphometric analysis.
MetaMorph was used to quantify the amount of SM staining as a percentage of total tissue area in each digital image. The area of total tissue (designated as IMAGE A and represented by purple and blue staining together from the modified toluidine blue staining, or red and blue from lysenin staining) and the area occupied by SM (designated as IMAGE B and represented by purple staining only from modified toluidine blue staining, or red only from lysenin staining) were calculated in terms of pixels. The colocalization function of MetaMorph then calculated the percentage of pixels in common between IMAGE A and IMAGE B and automatically exported the calculation into an Excel spreadsheet.
Up to a maximum of 10 blocks are produced and analyzed from each patient liver biopsy (sample size permitting). One section from each block was cut, stained, mounted on a slide, and analyzed as described above. This “breadloaf” approach ensured the evaluation of SM in up to 10 slides representative of the distribution across the entire length of sample. The average and SD were calculated from each set of sections. The SM identified and measured by this method represents the disease process; therefore, the normal value for this parameter in a non-ASMD liver sample is zero.
Biochemical Analysis of SM Levels
SM levels were measured by liquid chromatography-tandem mass spectrometry as described elsewhere.25
Biochemical Analysis of ASM Activity in Patient Fibroblasts
ASM activity in patient fibroblasts was measured using Amplex Red as described elsewhere.25
Statistical Methods
Mean changes in SM levels from baseline to 26 weeks were measured by computer morphometry and assessed for statistical significance (P<0.05) using the unpaired Student t test (GraphPad PRISM, version 5.0, GraphPad Software).
RESULTS
Liver SM Content
A statistically significant reduction in total histopathologic SM was observed in all evaluable 26-week liver biopsies as measured by computer morphometry and was paralleled by reductions in biochemical measurements as well as liver and spleen volumes (Fig. 1 and Table 1). The glutaraldehyde-fixed, posttreatment biopsy segment from patient 1 designated for high-resolution light microscopy and morphometric quantitation of SM was insufficient for evaluation due to its small size and the absence of hepatic parenchyma (this piece consisted of fibrous tissue only). By week 26, liver volumes in MN, which are normalized to the patient’s weight, had decreased in all patients, corresponding to a mean reduction of 21.9%. Similarly, spleen volumes decreased in all patients with a mean reduction of 29.4% in MN. At baseline, SM storage was present in both Kupffer cells and hepatocytes and ranged from 9.8% to 58.8% of the microscopic field. After 26 weeks of treatment, all 4 patients with evaluable posttreatment liver biopsies showed statistically significant (P<0.0001) reductions in SM. SM storage (as measured by computer morphometry) in posttreatment biopsies ranged from 1.2% to 9.5% of the microscopic field, which corresponded to 84% to 92% relative reductions from baseline.
FIGURE 1.
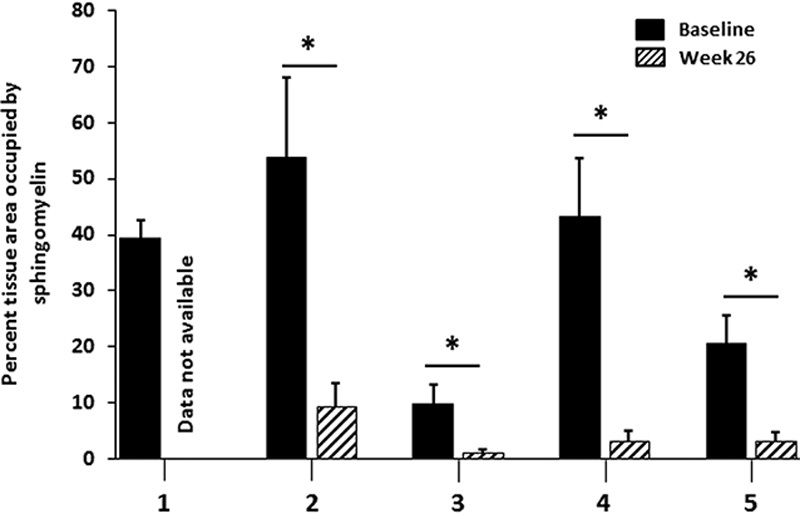
MetaMorph quantification of SM in liver biopsies before and after olipudase alfa treatment. This staining and quantification method identifies and measures only abnormal SM accumulation, which is part of the disease process. The normal value in a non-ASMD liver is 0. The “n” for number of blocks analyzed by MetaMorph at baseline and week 26, respectively, for each patient are as follows: Patient 1, n=2, n=0; patient 2, n=4, n=8; patient 3, n=8, n=9; patient 4, n=5, n=6; patient 5, n=7, n=7. *P<0.0001.
TABLE 1.
Summary of Patient Characteristics
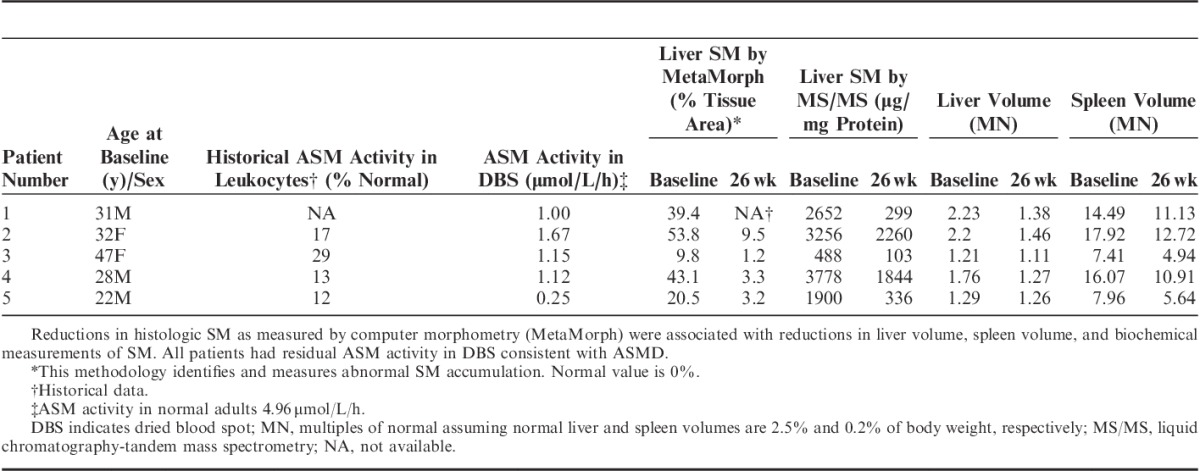
Two complementary staining methods were used to assess changes in SM content in epoxy resin–embedded tissue sections: modified toluidine blue and lysenin affinity staining (Fig. 2). The modified toluidine blue stain identified SM as dark purple cell deposits at the light microscopy level (Figs. 2A, B). Compared with baseline (Fig. 2A), individual Kupffer cells (K) within the sinusoids and clusters of macrophages around the portal tracts appeared completely cleared of SM, and levels of SM in hepatocytes (H) were significantly reduced after 26 weeks of olipudase alfa treatment (Fig. 2B). Comparable identification of SM and its clearance by olipudase alfa was observed in sections stained with lysenin affinity staining (Figs. 2C, D). Remaining lipofuscin debris, visible as blue globules within Kupffer cells, persisted after 26 weeks and is a reflection of the general phagocytic function of these cells in response to longstanding disease and long tissue residence time.31,32
FIGURE 2.
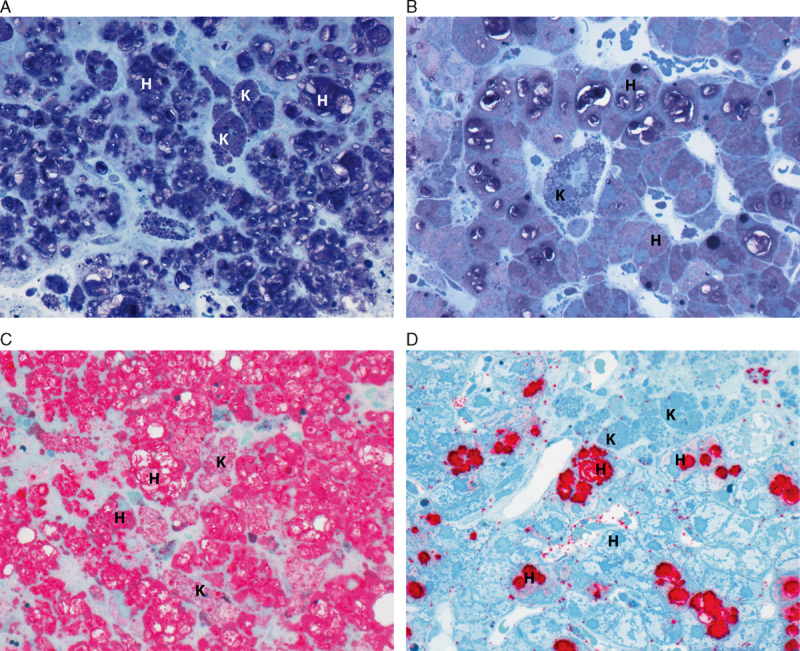
SM was cleared in Kupffer cells (K) and markedly reduced in hepatocytes (H). A modified toluidene blue stain highlights pretreatment (A) and posttreatment (B) SM in dark purple. Comparable identification of SM is also achieved with lysenin affinity staining, which highlights pretreatment (C) and posttreatment (D) SM in red (patient 2, high-resolution light microscopy, epon semithin sections, 600x).
The reduction in morphometric SM was confirmed by biochemical quantification in separate segments of frozen liver biopsy collected for each patient timepoint. At baseline, the SM levels ranged from 488 to 3778 μg/mg tissue and at week 26 from 103 to 2260 μg/mg tissue, corresponding to relative reductions of −30.6% to −88.7%. The MetaMorph and biochemical measurements of SM were generally well correlated (r=0.84).
When the liver distribution of SM accumulation was compared across the patients with relatively low (eg, patient 3), moderate (patient 5), and high (patients 1, 2, and 4) baseline levels of SM as measured by computer morphometry, an evolving pattern of the disease began to emerge visibly by light microscopy observation (Fig. 3, top panels). Patients with low levels of SM (eg, patient 3) exhibited accumulation within Kupffer cells scattered evenly across the biopsy as single cells (stage 1). As histologic SM increased in patients with moderate levels (eg, patient 5), accumulation in hepatocytes became more prominent, along with that in Kupffer cells, with denser cellular accumulation occurring around central veins (zones 2 and 3), and less dense accumulation in zone 1 cells (stage 2). The biopsies of patients with high levels of SM (patients 1, 2, and 4) exhibited equally dense accumulation of SM in all cells across all acinar zones (stage 3) with cells fully engorged with SM. After 26 weeks of ERT there was variable reduction of SM from each acinar zone. For example, in patient biopsies with moderate SM at baseline (patient 5, Fig. 3, bottom left), there was complete clearance from zone 1 hepatocytes, significant clearance from zone 2, but only partial clearance from zone 3 (pericentral) hepatocytes in posttreatment biopsies (Fig. 3, bottom right). Given the small sample size reported here, this observed pattern of clearance merits further confirmation in larger patient studies.
FIGURE 3.
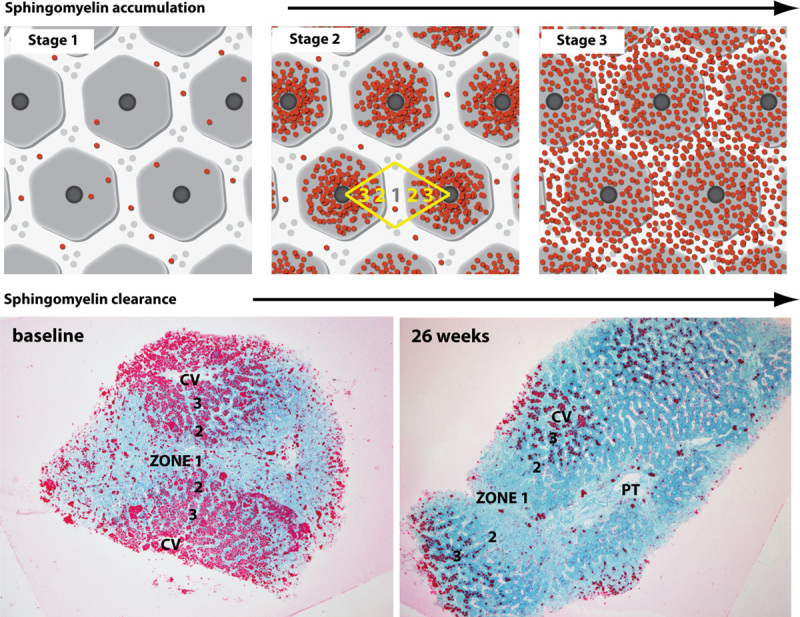
Patterns of hepatic SM accumulation and clearance. The top 3 panels illustrate in cartoon format the 3 primary stages of SM accumulation observed in the patient biopsies. Acinar zones are marked in yellow. The bottom 2 panels show the pattern of SM clearance in a patient biopsy with moderate SM levels (stage 2) at baseline. Pretreatment and posttreatment biopsy images are shown for patient 5. At baseline, substrate accumulation was heaviest around central veins (CV), particularly acinar zones 3 and 2. After 26 weeks of olipudase alfa, there was noticeable clearance of SM in zones 1 and 2, with reduction in zone 3 (Epon semithin sections, lysenin affinity stain). PT indicates portal triad, 20x.
Electron microscopy was utilized to further confirm the presence of SM at the ultrastructural level. Electron microscopy images from all baseline liver biopsies demonstrate the typical loose “fingerprint” whorls of SM present in abundance within Kupffer cells, occasionally admixed with electron-dense lipofuscin debris (Fig. 4A). After 26 weeks of olipudase alfa, only lipofuscin (also observed at the light level in Fig. 2) and other cell debris remained behind in those cells with complete clearance (Fig. 4B). Lipofuscin deposits also persisted in the endothelial cells lining the sinusoids (data not shown). Similar changes in SM were also observed within hepatocytes (Figs. 5A, B).
FIGURE 4.
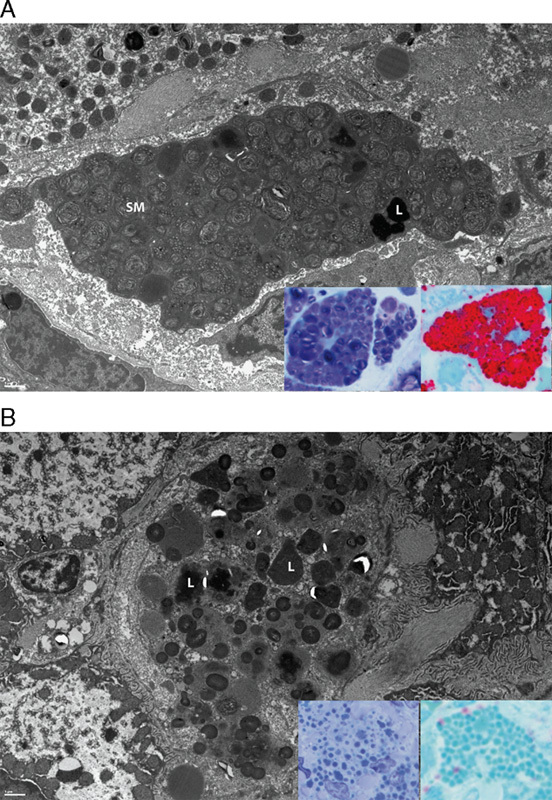
Electron microscopy and high-resolution light microscopy examination of SM accumulation and clearance from Kupffer cells. A, The characteristic “fingerprint” whorls of SM present in Kupffer cells at baseline in patient 3. Electron dense lipofuscin (L) is also present. Insets in A: Kupffer cell SM is dark purple in modified toluidene blue sections (left) and red in lysenin affinity sections (right). B, The clearance of SM, with lipofuscin remaining (L). Insets in B: This residual lipofuscin appears light blue in modified toluidene blue sections (left) and blue-green in lysenin affinity sections (right). Electron microscopy scale bars=1 μm.
FIGURE 5.
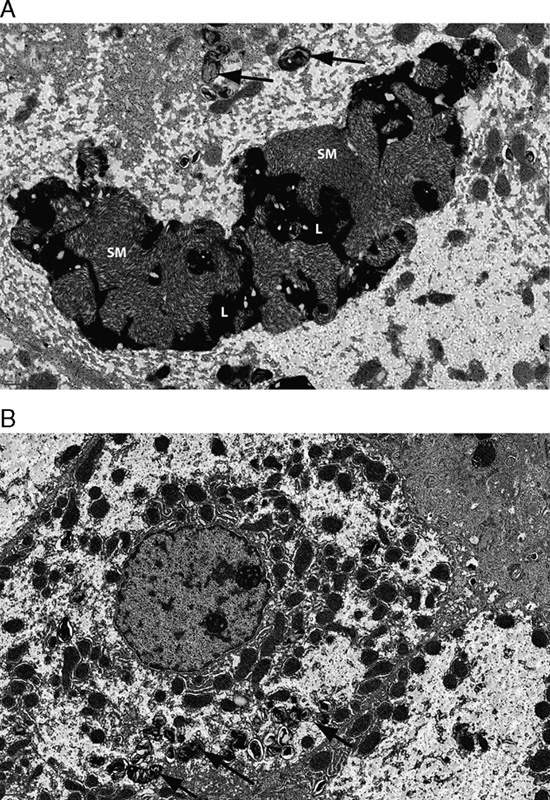
Electron microscopy examination of SM accumulation and clearance from hepatocytes. A, The presence of large SM masses mixed with lipofuscin (L) within hepatocytes at baseline in patient 5. Smaller whorls of SM are also present (black arrows). B, The reduction of SM after 26 weeks of olipudase alfa treatment. Black arrows indicate residual SM. Electron microscopy scale bars=1 μm.
Secondary Liver Pathology
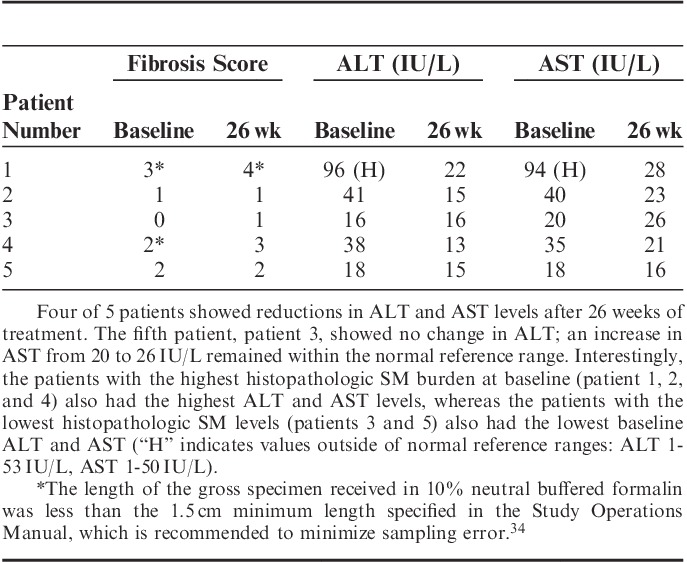
The formalin-fixed, paraffin-embedded segment of each liver biopsy was assessed for fibrosis according to the Laennec scoring system,33 which grades the extent of fibrosis on a scale from 0 to 4 (0, none; 1, minimal; 2, mild; 3, moderate; 4, cirrhosis). This scoring system was used to assess all patient biopsies at screening for cirrhosis, as patients with cirrhosis were excluded from entry into the trial, per protocol. Posttreatment biopsy samples were similarly assessed. Two of 5 patients exhibited no change in fibrosis scores from baseline to 26 weeks, whereas 3 patients had a 1-point increase (Table 2). In 2 of the 3 latter cases, biopsy samples at 1 or both time points did not meet the minimum size requirement (≥1.5 cm) specified by the Study Operations Manual to ensure adequate tissue sampling for scoring fibrosis due to its inherent heterogenous distribution.33–35 The patient with the highest fibrosis score at baseline (patient 1) had the highest AST/ALT levels, which were above the normal ranges; both returned to within the normal range after 26 weeks of treatment. Reductions in AST/ALT levels to within the normal range were also observed in 3 of the other 4 patients (patients 2, 4, 5) after 26 weeks of treatment. Patient 3 showed no change in ALT; an increase in AST from 20 to 26 IU/L remained within the normal reference range. The normalization of liver enzymes after treatment suggests that the 1-point differences in fibrosis scores were likely the result of tissue sampling variation rather than worsening disease. In addition, the 26-week biopsy of patient 1 contained a single focus of dense lymphocytic infiltrate measuring approximately 0.3 mm in diameter and colocalized within an area of portal fibrosis. This inflammatory change was not observed in the patient’s baseline biopsy.
TABLE 2.
Liver Fibrosis Scores and Liver Function Assessments
Lipid Profiles
Changes in the lipid profiles for all 5 patients before and after treatment with olipudase alfa are shown in Table 3. Patients 1 and 2 were on stable doses of lipid-lowering therapies (statins) before and during the course of the trial. Therefore, changes observed in these patients during the course of the trial occurred with the addition of ERT. The levels of the proatherogenic lipid markers including total cholesterol, LDL-C, very low-density lipoprotein-C (VLDL-C), triglycerides, and apoB100 (the major apolipoprotein present on LDL and VLDL particles) declined in all patients. Meanwhile, the antiatherogenic lipid markers, HDL-C and apoA-I (the major apolipoprotein present on HDL particles), increased in all patients. The relative increases in HDL-C ranged from 13% to 84%, and apoA-I ranged from 1.5% to 33% over baseline levels. These improvements paralleled the improvements in liver SM content, hepatomegaly, and ALT/AST levels.
TABLE 3.
Lipid Profiles Improved in All Patients After Olipudase Alfa Treatment

Interestingly, patient 3 exhibited the mildest liver pathology (lowest SM levels by MetaMorph analysis and fibrosis score=0 on baseline biopsy) of the 5 patients at both baseline and 26 weeks and had the highest HDL-C and apoA-I levels, which were within the normal range at both timepoints. Further, and more remarkable, are the improvements in HDL-C levels in patients 1, 2, and 4 (patients with low HDL-C and higher liver SM content at baseline) who demonstrated increases of 84%, 58%, and 32%, respectively. Patient 5, who had a low HDL-C and moderate liver SM content at baseline, had a 60% improvement in HDL-C, which raised this patient’s HDL-C levels to within the normal range by 26 weeks.
DISCUSSION
The current study shows the ability of olipudase alfa ERT to reverse SM storage in the livers of patients with ASMD. The liver is the main organ responsible for lipoprotein metabolism. Patients with ASMD have proatherogenic lipid profiles with HDL-C levels that are among the lowest reported in the general population.36,37 Only recently, however, has cardiovascular disease been recognized as an important comorbidity associated with this rare disease.8,20 In this study, the robust histopathologic clearance of SM from liver biopsies was accompanied by reductions in hepatomegaly and improvements in lipid profiles. Although the body of evidence is still emerging, the improved lipid profiles after administration of olipudase alfa might be expected to have an impact on the cardiovascular disease in this patient population.
The differential zonal accumulation and clearance of SM observed in liver biopsies parallels the oxygen concentration gradient within the hepatic acinus. The partial pressure of oxygen in the blood declines from 60 to 65 mm Hg as it enters the portal areas (zone 1, high O2) to 30 to 35 mm Hg as it exits the central veins (zone 3, low O2).38–40 This oxygen gradient is a modulator of the metabolic zonation in which hepatocytes in different acinar regions are known to have different metabolic functions.38–40 The metabolic zonation may affect the functional efficiency with which the hepatocytes take up and utilize circulating enzyme to breakdown and clear intracellular SM.
The liver’s critical role in lipoprotein metabolism is likely disrupted by the markedly abnormal SM accumulation present in baseline biopsies of patients with ASMD. SM is the second most abundant phospholipid in plasma lipoproteins, forming the surface together with phosphatidylcholine, cholesterol, and apolipoproteins.41 Differences in the surface lipid composition of plasma lipoproteins, such as elevated SM, have been shown to influence the biogenesis and metabolism of these macromolecules.42–45 In ASMD, it is likely that as different lipoprotein species are being variously assembled and secreted, or taken up, metabolized, reassembled, and resecreted as modified particles by an SM-laden liver, the excess hepatic SM is being incorporated into those particle surfaces.
This concept is supported by findings from studies on HDL biogenesis. HDL biogenesis takes place in the liver where nascent apoA-I-containing discoid lipid particles are assembled, secreted into the extracellular space, and then converted into mature spherical HDL particles by the enzyme lecithin:cholesterol acyltransferase.5 Increased levels of SM have been detected in nascent HDL particles from patients with ASMD and found to inhibit the activity of lecithin:cholesterol acyltransferase,43,44 thus contributing to the low HDL-C and low HDL particle (as reflected by low apolipoprotein A-I levels) levels observed in this patient population.10 This inhibitory property of SM has also been confirmed in a number of in vivo and in vitro studies.14,42,46 Twenty-six weeks of olipudase alfa treatment appeared to reverse the inhibitory effect of excess SM, resulting in increases in HDL-C and apoA-I (reflective of HDL particles) measurements in all patients. These antiatherogenic species mediate reverse cholesterol transport, a process whereby cholesterol is removed from peripheral tissues and transported back to the liver for excretion in bile salts.47 Improvements in HDL-C and apoA-I would therefore be potentially athero-protective.47
The hepatic metabolism of VLDL depends on apoE-mediated uptake by either the LDL receptor or LDL-receptor–related protein. Synthetic particle uptake studies in HepG2 cell culture demonstrated that increasing SM content in apoE-containing particles reduced this apoE-mediated binding and uptake.45 This suggests that SM enrichment of VLDL particles in vivo contributes to decreased uptake and metabolism by the liver and leads to increases in circulating plasma levels of these proatherogenic species, as observed in patients with ASMD.19 The reduction in VLDL-C levels observed in all 5 patients in this study after 26 weeks of olipudase alfa supports these models.
Sphingomyelin has also been shown to inhibit the activity of lipoprotein lipase, which plays a role in the hydrolysis of triglycerides in chylomicrons and VLDL particles,48 two lipoprotein species that also undergo significant uptake and remodeling by the liver.4 This finding is consistent with the elevated triglyceride levels observed in the patients at baseline and the reduction observed after 26 weeks of olipudase alfa treatment.
The dysregulation of lipoprotein metabolism by elevated SM is further illustrated by studies in apoE knockout mice, a model of dyslipidemia and atherosclerosis. When SM synthesis was inhibited by administration of myriocin, an inhibitor of serine palmitoyltransferase, dose-dependent reductions in VLDL-C and LDL-C and elevations in HDL-C were observed, along with reductions in atherosclerotic lesions.49 We observed similar changes in the lipoprotein profiles of our patients when SM storage was reduced by administration of olipudase alfa.
In this clinical study of 5 patients with ASMD, the changes in LFTs (ALT/AST) and hepatomegaly, along with the reduction of histopathologic and biochemical SM burden in all patients, suggested an improvement in overall liver health after olipudase alfa treatment. The normal levels of liver enzymes after treatment suggests that the 1-point differences in fibrosis scores for 3 of the patients were likely the result of tissue sampling variation of fibrosis, which is an inherently heterogenous process in the liver that is difficult to accurately capture in small samples33–35 rather than worsening disease. Furthermore, subsequent improvements in lipoprotein profiles (decreases in proatherogenic species and increases in antiatherogenic species) suggest that normal metabolic function is being restored to the liver by the clearance of SM. The improvement in both proatherogenic and antiatherogenic lipid markers in parallel with reductions in SM burden is consistent with epidemiological studies, which have demonstrated that absolute and relative plasma SM levels are independent risk factors for coronary heart disease.13,50 Although the findings in this study are based on data from a small number of patients, further research on the dyslipidemia and cardiovascular risk in this discrete, monogenic metabolic disorder may shed light on the known association between elevated SM levels and atherosclerosis in the general population and reveal targetable pathways for future lipid-lowering therapeutics.
ACKNOWLEDGMENTS
The authors thank all the patients who have participated in this study as well as the members of Sanofi Genzyme’s Clinical Research and Clinical and Exploratory Pharmacology Departments for contributing their expertise in rare clinical trial development and conduction, and the Department of Pathology for their expertise in lysosomal disease pathology.
Footnotes
Clintrials.gov trial registration # NCT01722526.
Conflicts of Interest and Source of Funding: Genzyme Corporation was the sponsor and provided support for the design and conduct of the study. The study was supported in part by Grant Number #UL1TR000067 from the National Center for Advancing Translational Sciences (NCATS), a component of the National Institutes of Health (NIH), and contents are solely the responsibility of the authors and do not necessarily represent the official views of NCATS or NIH. M.P.W. has received research support from Genzyme for the conduct of this study; researchers at Mount Sinai have developed and patented olipudase alfa, which they licensed to Genzyme Corporation. S.A.J. has received honoraria for consulting and lectures, travel assistance, and medical writing assistance from Sanofi Genzyme; CMFT entered into a clinical trial agreement to complete the work described in this manuscript. B.L.T., G.F.C., and A.C.P. are employees of Sanofi Genzyme. For the remaining author none was declared.
REFERENCES
- 1.Schuchman EH, Desnick RJ.Valle D, Beaudet A, Vogelstein B, Kinzler K, Antonarakis S, Ballabio A, Gibson K, Mitchell G. Niemann-Pick disease types A and B: acid sphingomyelinase deficiencies. OMMBID-The Online Metabolic and Molecular Bases of Inherited Disease. New York: McGraw Hill; 2013. http://ommbid.mhmedical.com/content.aspx?bookid=474&Sectionid=45374145. Accessed May 2015. [Google Scholar]
- 2.McGovern MM, Lippa N, Bagiella E, et al. Morbidity and mortality in type B Niemann-Pick disease. Genet Med. 2013;15:618–623. [DOI] [PubMed] [Google Scholar]
- 3.Lidove O, Sedel F, Charlotte F, et al. Cirrhosis and liver failure: expanding phenotype of acid sphingomyelinase-deficient Niemann-Pick disease in adulthood. JIMD Rep. 2015;15:117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hegele RA. Plasma lipoproteins: genetic influences and clinical implications. Nat Rev Genet. 2009;10:109–121. [DOI] [PubMed] [Google Scholar]
- 5.Rye K-A, Barter PJ. Regulation of high-density lipoprotein metabolism. Circ Res. 2014;114:143–156. [DOI] [PubMed] [Google Scholar]
- 6.Vanier MT. Biochemical studies in Niemann-Pick disease: major sphingolipids of liver and spleen. Biochim Biophys Acta. 1983;750:178–184. [DOI] [PubMed] [Google Scholar]
- 7.Wasserstein MP, Desnick RJ, Schuchmann EH, et al. The natural history of type B Niemann-Pick disease: results from a 10-year longitudinal study. Pediatrics. 2004;114:e672–e677. [DOI] [PubMed] [Google Scholar]
- 8.McGovern MM, Wasserstein MP, Giugliani R, et al. A prospective, cross-sectional survey study of the natural history of Niemann-Pick Disease Type B. Pediatrics. 2008;122:e341–e349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hollak CEM, de Sonnaville ESV, Cassiman D, et al. Acid sphingomyelinase (Asm) deficiency patients in The Netherlands and Belgium: disease sprectrum and natural course in attenuated patients. Mol Genet Metab. 2012;107:526–533. [DOI] [PubMed] [Google Scholar]
- 10.Viana MB, Giugliani R, Leite VHR, et al. Very low levels of high density lipoprotein cholesterol in four sibs of a family with non-neuropathic Niemann-Pick disease and sea-blue histiocytosis. J Med Genet. 1990;27:499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nicholson AG, Florio R, Hansell DM, et al. Pulmonary involvement by Niemann-Pick disease. A report of six cases. Histopathology. 2006;48:596–603. [DOI] [PubMed] [Google Scholar]
- 12.Jiang X-C, Paultre F, Pearson TA, et al. Plasma sphingomyelin level as a risk factor for coronary artery disease. Arterioscler Thromb Vasc Biol. 2000;20:2614–2618. [DOI] [PubMed] [Google Scholar]
- 13.Nelson JC, Jiang X-C, Tabas I, et al. Plasma sphingomyelin and subclinical atherosclerosis: findings from the multi-ethnic study of atherosclerosis. Am J Epidemiol. 2006;163:903–912. [DOI] [PubMed] [Google Scholar]
- 14.Worgall TS. Sphingolipids: major regulators of lipid metabolism. Curr Opin Clin Nutr Metab Care. 2007;10:149–155. [DOI] [PubMed] [Google Scholar]
- 15.Van Wijk DF, Boekholdt SM, Wareham NJ, et al. C-reactive protein, fatal and nonfatal coronary artery disease, stroke, and peripheral artery disease in the prospective EPIC-Norfolk cohort study. Arterioscler Thromb Vasc Biol. 2013;33:2888–2894. [DOI] [PubMed] [Google Scholar]
- 16.Bian F, Yang X, Zhou F, et al. C-reactive protein promotes atherosclerosis by increasing LDL transcytosis across endothelial cells. Br J Pharmacol. 2014;171:2671–2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Erikssen G, Liestøl K, Bjørnholt JV, et al. Erythrocyte sedimentation rate: a possible marker of atherosclerosis and a strong predictor of coronary heart disease mortality. Eur Heart J. 2000;21:1614–1620. [DOI] [PubMed] [Google Scholar]
- 18.Natali A, L’Abbate A, Ferrannini E. Erythrocyte sedimentation rate, coronary atherosclerosis, and cardiac mortality. Eur Heart J. 2003;24:639–648. [DOI] [PubMed] [Google Scholar]
- 19.McGovern MM, Pohl-Worgall T, Deckelbaum RJ, et al. Lipid abnormalities in children with types A and B Niemann-Pick Disease. J Pediatr. 2004;145:77–81. [DOI] [PubMed] [Google Scholar]
- 20.Ishii TT, Toyono M, Tamure M, et al. Acid sphingomyelinase deficiency: cardiac dysfunction and characteristic findings of the coronary arteries. J Inherit Metab Dis. 2006;29:232–234. [DOI] [PubMed] [Google Scholar]
- 21.Thurberg BL, Rennke H, Colvin RB, et al. Globotriaosylceramide accumulation in the fabry kidney is cleared from multiple cell types after enzyme replacement therapy. Kidney Int. 2002;62:1933–1946. [DOI] [PubMed] [Google Scholar]
- 22.Thurberg BL, H.Byers HR, Granter SR, et al. Monitoring the three year efficacy of enzyme replacement therapy in fabry disease by repeated skin biopsies. J Invest Dermatol. 2004;122:900–908. [DOI] [PubMed] [Google Scholar]
- 23.Thurberg BL, Lynch Maloney C, Vaccaro C, et al. Characterization of pre- and post-treatment pathology after enzyme replacement for pompe disease. Lab Invest. 2006;86:1208–1220. [DOI] [PubMed] [Google Scholar]
- 24.Thurberg BL, Fallon JT, Mitchell R, et al. Cardiac microvascular pathology in Fabry disease: evaluation of endomyocardial biopsies before and after enzyme replacement therapy. Circulation. 2009;118:2561–2567. [DOI] [PubMed] [Google Scholar]
- 25.Thurberg BL, Wasserstein MP, Schiano T, et al. Liver and skin histopathology in adults with acid sphingomyelinase deficiency (Niemann-Pick Disease Type B). Am J Surg Pathol. 2012;36:1234–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McGovern MM, Wasserstein MP, Kirmse B, et al. Novel first-dose adverse drug reactions during a phase I trial of olipudase alfa (recombinant human acid sphingomyelinase) in adults with Niemann-Pick disease type B (acid sphingomyelinase deficiency). Genet Med. 2016;18:34–40. [DOI] [PubMed] [Google Scholar]
- 27.Wasserstein MP, Jones SA, Soran H, et al. Successful within-patient dose escalation oflipudase alfa in acid sphingomyelinase deficiency. Mol Genet Metab. 2015;116:88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sheehan DC, Hrapchak BB. Theory and Practice of Histotechnology, 2nd Ed Columbus OH: Battelle Press; 1980. [Google Scholar]
- 29.Taksir TV, Johnson J, Maloney CL, et al. Optimization of a histopathological biomarker for sphingomyelin accumulation in Niemann-Pick B Disease. J Histochem Biochem. 2012;60:620–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lynch CM, Johnson J, Vaccaro C, et al. High resolution light microscopy (HRLM) and digital analysis of Pompe disease pathology. J Histochem Cytochem. 2005;53:63–73. [DOI] [PubMed] [Google Scholar]
- 31.Bouwens L, Baekeland M, Zanger RD, et al. Quantitation, tissue distribution and proliferation kinetics of Kupffer cells in normal rat liver. Hepatology. 1986;6:718–722. [DOI] [PubMed] [Google Scholar]
- 32.Takahashi K. Development and differentiation of macrophages and related cells: historical review and current concepts. J Clin Exp Hematol. 2001;41:1–33. [Google Scholar]
- 33.Wanless JR, Crawford JM.Odze RD, et al. Chap 37: Cirrhosis. Surgical Pathology of the GI Tract, Liver, Biliary Tract, and Pancreas. Philadelphia: Saunders; 2004:863–882. [Google Scholar]
- 34.Antonio LB, Suriawinata A, Thung SN.Odze RD, et al. Chap 32: Liver tissue processing techniques. Surgical Pathology of the GI Tract, Liver, Biliary Tract, and Pancreas. Philadelphia: Saunders; 2004:739. [Google Scholar]
- 35.Scheuer PJ, Lefkowitch JH. Chap 1: General considerations, and Chap 10: Cirrhosis. Liver Biopsy Interpretation, 7th Ed Philadelphia: Elsevier Saunders; 2006. [Google Scholar]
- 36.Rader DJ, deGoma EM. Approach to the patient with extremely low HDL-cholesterol. J Clin Endocrinol Metab. 2012;97:3399–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Calabresi L, Gomaraschi M, Simonelli S, et al. HDL and atherosclerosis: insights from inherited HDL disorders. Biochim Biophys Acta. 2015;1851:13–18. [DOI] [PubMed] [Google Scholar]
- 38.Gumucio JJ, Miller DL. Functional implications of liver cell heterogeneity. Gastroenterology. 1981;80:393–403. [PubMed] [Google Scholar]
- 39.Jungermann K, Kietzmann T. Oxygen: modulator of metabolic zonation and disease of the liver. Hepatology. 2000;31:255–260. [DOI] [PubMed] [Google Scholar]
- 40.Sato A, Kadokura K, Uchida H, et al. An in vitro hepatic zonation model with a continuous oxygen gradient in a microdevice. Biochem Biophys Res Commun. 2014;453:767–771. [DOI] [PubMed] [Google Scholar]
- 41.Nilsson A, Duan R-D. Absorption and lipoprotein transport of sphingomyelin. J Lipid Res. 2006;47:154–171. [DOI] [PubMed] [Google Scholar]
- 42.Bolin DJ, Jonas A. Sphingomyelin inhibits the lecithin-cholesterol acyltransferase reaction with reconstituted high density lipoproteins by decreasing enzyme binding. J Biol Chem. 1996;32:19152–19158. [DOI] [PubMed] [Google Scholar]
- 43.Lee CY, Lesimple A, Larsen A, et al. ESI-MS quantitation of increased SM in Niemann-Pick disease type B HDL. J Lipid Res. 2005;46:1213–1228. [DOI] [PubMed] [Google Scholar]
- 44.Lee CY, Lesimple A, Denis M, et al. Increased SM content impairs HDL biogenesis and maturation in human Niemann-Pick disease type B. J Lipid Res. 2006;47:622–632. [DOI] [PubMed] [Google Scholar]
- 45.Morita S-Y, Okuhira K, Tsuchimoto N, et al. Effects of sphingomyelin on apolipoprotein E and lipoprotein lipase-mediated cell uptake of lipid particles. Biochim Biophys Acta. 2003;1631:169–176. [DOI] [PubMed] [Google Scholar]
- 46.Subbaiah PV, Jiang X-C, Belikova NA, et al. Regulation of plasma cholesterol esterification by sphingomyelin: Effect of physiological variations of plasma sphingomyelin on lecithin-cholesterol acyltransferase activity. Biochim BioPhys Acta. 2012;1821:908–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barter PJ, Rye KA. The rationale for using apoA-I as a clinical marker of cardiovascular risk. J Int Med. 2006;259:447–454. [DOI] [PubMed] [Google Scholar]
- 48.Arimoto I, Saito H, Kawashima Y, et al. Effects of sphingomyelin and cholesterol on lipoprotein lipase-mediated lipolysis in lipid emulsions. J Lipid Res. 1998;39:143–151. [PubMed] [Google Scholar]
- 49.Park T-S, Panek RL, Rekhter MD, et al. Modulation of lipoprotein metabolism by inhibition of sphingomyelin synthesis in apoE knockout mice. Atherosclerosis. 2006;189:264–272. [DOI] [PubMed] [Google Scholar]
- 50.Schlitt A, Blankenberg S, Yan D, et al. Further evaluation of plasma sphingomyelin levels as a risk factor for coronary artery disease. Nutr Metab (Lond). 2006;3:5. [DOI] [PMC free article] [PubMed] [Google Scholar]