

Abstract
A versatile and efficient modular synthetic platform was developed for assembling multifunctional conjugates and targeted forms of platinum–(benz)acridines, a class of highly cytotoxic DNA-targeted hybrid agents. The synthetic strategy involved amide coupling beween succinyl ester-modified platinum compounds (P1, P2) and a set of 11 biologically relevant primary and secondary amines (N1–N11). To demonstrate the feasibility and versatility of the approach, a structurally and functionally diverse range of amines was introduced. These include biologically active molecules, such as rucaparib (a PARP inhibitor), E/Z-endoxifen (an estrogen receptor antagonist), and a quinazoline-based thyrosine kinase inhibitor. Micro-scale reactions in Eppendorf tubes or on 96-well plates were used to screen for optimal coupling conditions in DMF solution with carbodiimide-, uronium-, and phosphonium-based compounds, as well as other common coupling reagents. Reactions with the phosphonium-based coupling reagent PyBOP produced the highest yields and gave the cleanest conversions. Furthermore, it was demonstrated that the chemistry can also be performed in aqueous media and is amenable to parallel synthesis based on multiple consecutive reactions in DMF in a “one-tube” format. In-line LC-MS was used to assess the stability of the conjugates in physiologically relevant buffers. Hydrolysis of the conjugates occurs at the ester moiety and is facilitated by the aquated metal moiety under low-chloride ion conditions. The rate of ester cleavage greatly depends on the nature of the amine component. Potential applications of the linker technology are discussed.
Introduction
Platinum-based cytotoxics have a wide range of applications as curative and palliative oncology drugs.1 Despite their overall clinical success, these agents suffer from unfavorable ADME (absorption, distribution, metabolism, excretion) properties and high systemic toxicities.2 In addition to overcoming the dose-limiting toxicities of these drugs, improving the response of notoriously chemoresistant forms of cancer to platinum-based therapeutics remain high-priority goals.3 To address the latter drawback, several classes of non-classical platinum-containing pharmacophores, such as platinum–acridine hybrid agents, have been developed.4-6 The most active members in this class of DNA adduct-forming/intercalating agents (e.g., compound 1, Fig. 1) show an up to three orders of magnitude higher potency than cisplatin in non-small-cell lung cancer (NSCLC) cells where they induce apoptosis at low-nanomolar concentrations.7, 8 Unfortunately, platinum–acridines also produce major dose-limiting toxicity and weight loss, as demonstrated in mice.7 Current work, therefore, is focused on improving the selectivity and tolerability of these agents.
Fig. 1.

Structures of the most cytotoxic platinum–acridine agent (1), functionalized derivatives (2a–c), and ester-modified precursors (2d,e).
Several strategies are being pursued to improve the pharmacological properties (systemic circulation and clearance, target-selective action) of chemotherapeutic agents. These include delivery of a cytotoxic payload in form of prodrugs, receptor-targeted conjugates, and nanoformulations, as well as co-delivery of agents that sensitize the affected tissue to the cytotoxic agent.9 Polymer-based delivery platforms and (liposomal) nanoencapsulation are currently under (pre)clinical investigation with the goal of improving the pharmacological properties of cisplatin and its second-generation derivatives.10-13 The majority of targeted nanodelivery vehicles contain the kinetically more inert platinum(IV) forms of the clinical platinum drugs.14 Release of active drug from these carriers is achieved by bio-reductive loss of axial ligation in the Pt(IV) precursors, while activation/release of Pt(II)-based formulations is achieved by simple, or acidic pH-promoted, ligand substitution.10, 14 In addition to serving as attachment points for passive or receptor-mediated delivery, the axial positions in Pt(IV) prodrugs have also been exploited to generate “dual-threat” conjugates containing bioactive ligands, such as chemosensitizers, or a second cytotoxic component.14
To allow incorporation of our Pt(II)-based platinum–acridines as cytotoxic “warheads” into multifunctional scaffolds, we have designed analogues containing conjugatable functional groups on the nonleaving intercalator moiety (e.g., OH and COOH in R2 and R3 in compounds 2a–c, Fig. 1).8 We have previously demonstrated that ester derivatives of compound 2c (e.g, 2d, Fig. 1) are activated by platinum-promoted ester hydrolysis.15 The mechanism takes advantage of the concentration differential that exists between serum and intracellular chloride (Scheme 1), and the rate of cleavage depends on the length of the linker and the nature of R3. Derivatives such as valproic ester 2e (Fig. 1),15 which are chemically robust but selectively cleaved by carboxylesterase-2 (CES-2), a prodrug-activating enzyme expressed at high levels in the liver, gastrointestinal tract, and certain solid tumors,16-18 can also be generated.
Scheme 1. Platinum-promoted ester cleavage in a low-chloride environment (X = chlorido or aqua ligand).

The goal of the current study was to develop versatile modular chemistry that combines our ester technology with amide coupling chemistry under conditions compatible with the reactive Pt(II) moiety in platinum–acridines. To demonstrate the utility of this strategy, a diverse set of multifunctional model pharmacophores was generated and the linker stability tested under physiologically relevant conditions.
Results and discussion
Development and optimization of coupling chemistry
Initial attempts to directly couple carboxylic acid-modified derivative 2a (Fig. 1) with biologically relevant amines were unsuccessful. No amide bond formation was observed in the presence of coupling reagents routinely used in peptide synthesis (unpublished data). We attributed this lack of reactivity to the steric hindrance produced by the platinum and acridine moieties adjacent to residue R2 (see Fig. 1). To reduce the steric bulk of R2, which also improves the biological activity of the DNA-targeted agent,8, 15 we chose R1 on the amidine carbon as the point of linker attachment. Previously, we identified compound 2d as a butyric ester-masked lipophilic prodrug of the highly cytotoxic hydroxyl-modified platinum–acridine 2c (Fig. 1).15 To enable amide coupling chemistry at R1, we introduced a carboxylic acid functionality in 2c by extending the terminal hydroxyl group with a succinyl group. Two derivatives, acridine-based P1 and benz[c]acridine-based19 P2, were generated (Fig. 2). Both analogues were used as the carboxylic acid-containing components in coupling reactions with 11 different primary and secondary amines, N1–N11 (Fig. 2). P1 and P2 were generated in a simple three-step “one-pot” synthesis in DMF. The reactions involved substitution of one chlorido ligand in silver ion-activated [Pt(pn)Cl2] (pn = propane-1,3-diamine) with succinyl-modified nitrile (4-(3-cyanopropoxy)-4-oxobutanoic acid, 3) and subsequent addition of N1-(acridin-9-yl)-N2-methylethane-1,2-diamine) (A1) and N1-(benzo[c]acridin-7-yl)-N2-methylethane-1,2-diamine (B1), respectively (see the Experimental Section and the ESI for details).
Fig. 2.

(A) Structures of platinum (P) and amine (N) components used for methodology development and library assembly. (B) Schematic representation of the coupling strategy for amide–ester linkers. The benz[c]acridine derivative in (A) contains an additional fused ring highlighted with dashed bonds.
Using carboxyl-functionalized building block P1 and the simple primary amine 2-azidoethan-1-amine (N1, Fig. 2), initially 14 solution-phase micro-scale reactions were assembled to screen a wide range of coupling reagents under varying conditions. Conversion to the desired amide in the mixtures was monitored by in-line LC-MS (see the ESI), and the yield of each reaction was determined from HPLC traces (Table 1, entries 1–14). Surprisingly, the carbodiimide-based reagents, DCC‡ and EDC,20-22 completely failed to mediate amide bond formation. Likewise, no product was observed when coupling reactions with these reagents were performed in the presence of base (diisopropylethylamine, DIPEA, “Hünig's base”) or the carboxylic acid-activating reagent N-hydroxysuccinimide (NHS). By contrast, the desired amide was produced with conversion yields of greater than 90% when uronium-based HBTU and phosphonium-based PyBOP were used. This outcome is consistent with the rapid formation of the activated benzotriazolyl esters by these coupling reagents under mild reaction conditions.21 On the other hand, COMU, another uronium-based coupling reagent, which usually performs better than HBTU in solution-phase and solid-phase peptide synthesis,22 was not as efficient under the same conditions in our system. Finally, good conversion was also achieved in the presence of excess CDI (10 equiv.), which required removal of unreacted coupling reagent prior to adding the amine component. While cost-efficient, this additional step can be considered a major disadvantage when assembling libraries of conjugates for high-throughput screening.
Table 1. Conditions for Coupling Reactions with Model Primary (Top) and Secondary (Bottom) Amines.
| Entrya | Coupling reagent | Base | Molar ratio of reactants (Pt:amine:coupling reagent:base) | Pt concentration (mM) | Conversionb (%) ± S.D. |
|---|---|---|---|---|---|
|
| |||||
|
| |||||
| 1 | DCC | -- | 1:1.2:1.2:0 | 5 | no reaction |
| 2 | DCC | DIPEA | 1:1.2:1.2:1 | 5 | no reaction |
| 3 | DCC/NHS | DIPEA | 1:1.2:1.2(DCC):1.2(NHS):1 | 5 | no reaction |
| 4 | EDC | -- | 1:1.2:1.2:0 | 5 | no reaction |
| 5 | EDC | DIPEA | 1:1.2:1.2:1 | 5 | no reaction |
| 6 | EDC/NHS | DIPEA | 1:1.2:1.2(EDC):1.2(NHS):1 | 5 | no reaction |
| 7 | CDI | -- | 1:1.2:1.2:0 | 5 | 76 ± 2 |
| 8c | CDI | -- | 1:10:1.2:0 | 5 | 98 ± 2 |
| 9 | HBTU | DIPEA | 1:1.1:1.1:1 | 5 | 94 ± 3 |
| 10 | PyBOP | DIPEA | 1:1.1:1.1:1 | 5 | 99 ± 1 |
| 11 | COMU | DIPEA | 1:1.1:1.1:1 | 5 | 83 ± 1 |
| 12 | HBTU | DIPEA | 1:1.1:1.1:1 | 0.5 | 22 ± 3 |
| 13 | PyBOP | DIPEA | 1:1.1:1.1:1 | 0.5 | 17 ± 2 |
| 14 | COMU | DIPEA | 1:1.1:1.1:1 | 0.5 | 18 ± 1 |
|
| |||||
|
| |||||
|
| |||||
| 15c | CDI | -- | 1:10:1.2:0 | 5 | 67 ± 3 |
| 16 | HBTU | DIPEA | 1:1.1:1.1:1 | 5 | 92 ± 1 |
| 17 | PyBOP | DIPEA | 1:1.1:1.1:1 | 5 | 90 ± 2 |
| 18 | COMU | DIPEA | 1:1.1:1.1:1 | 5 | 73 ± 1 |
| 19 | HBTU | DIPEA | 1:1.1:1.5:1 | 5 | 88 ± 1 |
| 20 | PyBOP | DIPEA | 1:1.1:1.5:1 | 5 | 99 ± 1 |
Each reaction was performed in triplicate and reaction mixtures were characterized by LC-MS without purification.
The conversion yields were determined from HPLC traces recorded in an acridine-specific wavelength range.
CDI was pre-incubated with Pt complexes for 5 h before the intermediate was precipitated with diethyl ether to remove unreacted CDI before mixing with amine.
To test if the above coupling chemistry can be applied to secondary amino groups in bulkier amines, 6 additional reactions were assembled with (E/Z)-4-hydroxy-N-desmethyltamoxifen (endoxifen, N6, Fig. 2). Similar to the primary amine N1, N6 showed the best conversion in the presence of PyBOP and HBTU (> 90%) (Table 1, entries 15–20). Since HBTU and other uronium-based coupling reagents also react with amines, resulting in guanidinium side products,20, 23 PyBOP was selected as the coupling reagent of choice for assembling the final, extended library of conjugates. Overall, the highest conversion yields were observed for reaction mixtures containing 1.0 equiv. of carboxylic acid, 1.1 equiv. of amine, 1.0 equiv. of base, 1.5 equiv. of PyBOP, and when incubations were run for 16 h at room temperature at a concentration of P1 of 5 mM or higher.
Assembly of an extended library in a 96-well plate format
With the optimized coupling conditions we set forth to validate the modular fragment-based conjugate assembly for the 2 functionalized platinum precursors and 10 amine components (P1, P2, N1–N10, Fig. 2). Micro-scale coupling reactions (200 μL) were performed in parallel and analyzed by fully automated LC-MS. To demonstrate the scope and utility of the amide/ester-based conjugation chemistry, a set of structurally and functionally diverse amino group containing, biologically relevant molecules were studied. Coupling of azido–amine N1 introduces a handle into the conjugate allowing additional chemical ligation (click chemistry).8 Lipophilic hexadecylamine (N2) and the phospholipid 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE, N3) were chosen to turn the cationic, highly water soluble platinum–(benz)acridines into amphiphilic molecules. Such conjugates have the potential to self-assemble into micellar structures24 or liposomal vesicles,25 which are known to passively accumulate in tumor tissue.26 The arginine derivative N4 was introduced as a model of amino acids and oligopeptides that might potentially facilitate uptake of platinum–(benz)acridines into cancer cells.27 Multifunctional conjugates containing other clinically approved drugs or their key pharmacophores targeted at specific forms of cancer were also generated. The amine components introduced include (i) rucaparib28 (Clovis Oncology) (N5), a member of the class of poly-ADP-ribose polymerase-1 (PARP-1) inhibitors, which have been shown to sensitize cancer cells to DNA-targeted chemotherapies,29 (ii) endoxifen (N6), the active form of the estrogen receptor (ER) antagonist and breast cancer treatment tamoxifen,30 and (iii) the epidermal growth factor receptor (EGFR) kinase-targeted quinazoline-based inhibitor N7,31 a derivative of the lung cancer therapy gefitinib (Astra Zeneca).32 Finally, amide coupling of P1 and P2 with mitochondria-targeted triphenylphosphonium (TPP) salts33 (N8, N9) and with a poly(amidoamine) dendrimer (PAMAM-G3-NH2)34 (N10) was also studied.
Using the previously optimized conditions for PyBOP-mediated amide coupling, 20 reactions were assembled in Eppendorf tubes and analyzed by LC-MS (see the ESI). The majority of reactions went virtually to completion (14 out of 20) without producing undesired side products. The outcome of coupling reactions deemed successful (yields > 95%) and the structures of the resulting conjugates (confirmed by high-resolution mass spectrometry, HR-MS) are summarized in the ESI. A comparison of the results obtained for the TPP salts, N8 and N9, shows that PyBOP coupling is limited by the steric hindrance of the amine component. While yields for N8 did not exceed 70%, introduction of an extended 11-aminoundecanoic acid linker in N9 to separate the bulky TPP moiety from the reactive primary amino group led to quantitative conversion under the same conditions. No reaction of P1/P2 with the dendrimer–amine (N10) and DSPE (N3) was observed, which is most likely due to steric crowding and lack of solubility in DMF, respectively.
To demonstrate that the coupling reactions can be performed on a useful preparative scale, we resynthesized a representative conjugate, P1-N7, and isolated the pure product with a yield of 83%. To confirm formation of the desired conjugate by establishing scalar connectivity within the linker, P1-N7 was fully characterized by 2-D NMR spectroscopy (see the ESI).
Towards aqueous coupling chemistry
While the coupling chemistry in DMF solvent provides a versatile synthetic platform, methodology would be desirable by which amide bonds with P1 and P2 can be formed in aqueous buffers or biological media. One potential application would be the attachment of platinum–(benz)acridines to cancer-targeted peptides or proteins that are insoluble in DMF. As a model reaction we chose amide coupling between compound P2 and 8-aminooctanoic acid (N11, Fig. 2). In peptide synthesis, this scenario typically requires protecting group chemistry to avoid cross-reactions between unprotected carboxyl group and coupling reagent. Because the conditions used for the deprotection steps would result in the decomposition of P2, a two-step synthesis was designed (Scheme 2). The reaction involves conversion of the carboxylic acid group in P2 with 1.5 equiv. of TSTU, a uronium-based coupling reagent,35 to produce an NHS-activated ester, which is stable against hydrolysis, but reactive with amines in aqueous solution. The resulting intermediate, P2-NHS, was isolated by ether precipitation and reacted with N11 in phosphate-buffered saline (PBS). The aqueous reaction step was conducted at a high chloride ion concentration to avoid aquation of the Pt(II) moiety and prevent coordination of the primary amino group in N11 with the metal. P2-NHS and N11 cleanly converted (yields > 70%) to the desired conjugate, P2-N11, after the mixture was incubated for 16 h at room temperature (see LC-MS analysis, ESI). It is noteworthy to mention that no hydrolysis of the succinyl ester was observed during the incubation in PBS. Thus, P2-NHS appears to be a chemically robust reagent that should allow efficient modification of primary amines, such as lysine residues in peptides or proteins,36 with platinum–(benz)acridines.
Scheme 2. Aqueous amide coupling using NHS-activated ester.

Multistep “one-tube” assembly of conjugates for combinatorial library design
We have previously used a combinatorial approach for generating and screening modular libraries of functionalized platinum–acridines.8 We are now devising an efficient four-step reaction scheme that allows assembly of ester–amide linked multifunctional conjugates in DMF solution starting from simple diam(m)inedichlorido platinum precursor. The synthesis involves two simple precipitation/centrifugation steps, but requires no additional purification of intermediates. To demonstrate the feasibility of the proposed “one-tube” assembly, conjugate P1-N5 was generated. The reaction sequence along with a schematic diagram is shown in Fig. 3. Briefly, [PtCl2(pn)] was reacted with one equiv. of AgNO3 (Step 1), AgCl was removed by centrifugation, and the activated complex was treated with excess 4-(3-cyanopropoxy)-4-oxobutanoic acid (3). To the resulting platinum–nitrile intermediate was then added acridine–amine A1 to generate P1, which was recovered quantitatively and separated from unreacted A1 by ether precipitation prior to the amide coupling step. LC-MS analysis performed after steps 1–3 showed that P1 had been generated with a conversion yield of 85% (Fig. 4A). Finally, a coupling reaction using PyBOP was assembled that was able to convert 72% of P1 into the desired rucaparib (N5) conjugate, P1-N5 (Fig. 4B). The only minor side product detected in the LC-MS traces recorded after steps 3 and 4 (Fig. 4C) resulted from direct reaction of A1 with residual [PtCl(NO3)pn]. With further optimization of the procedure it should be possible to generate even higher yields and purities that will allow for direct screening of the resulting library mixtures in cell-based assays. For applications requiring removal of unreacted precursor molecules, pure batches of the conjugates can be generated by passing the final reaction mixtures through gel filtration columns (Sephadex LH-20, methanol:acetonitrile, 1:1).
Figure 3.
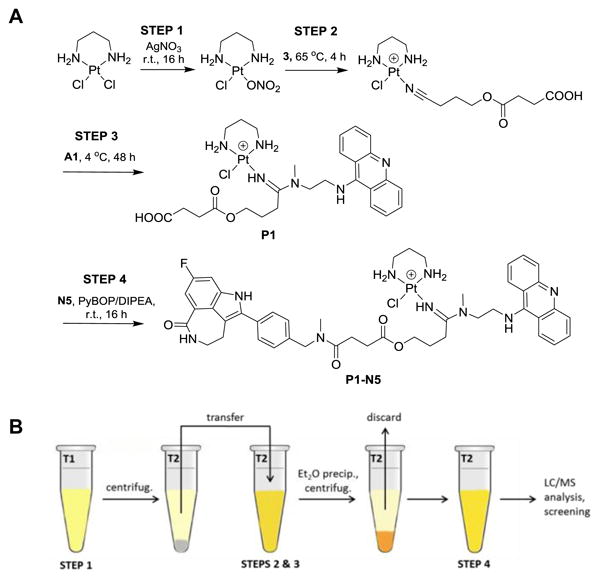
Synthetic scheme (A) and schematic representation (B) of four-step “one-tube” assembly of conjugate P1-N5.
Figure 4.
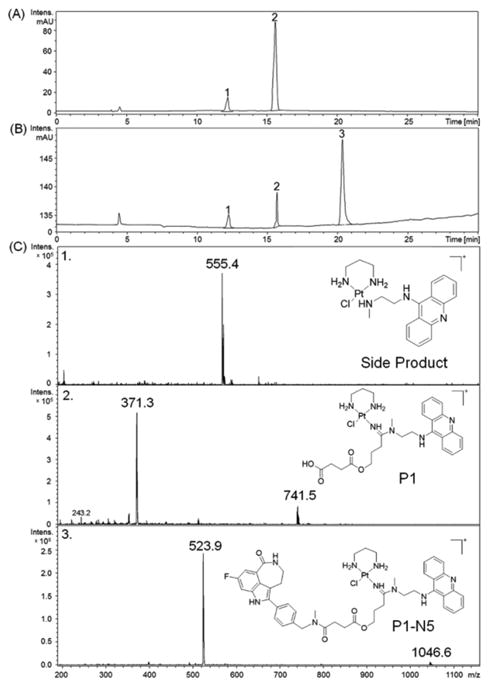
“One-tube” coupling of P1 with N5 monitored by LC-MS. HPLC traces recorded after steps 3 (A) and 4 (B) and mass spectra recorded in positive-ion mode with molecular ions [M]+ and [M]2+ labeled (C) are shown.
Conjugate stability and hydrolytic linker cleavage
Depending on the mechanism of action of a particular conjugate, the timing of linker cleavage is a critical parameter that assures maximum target selectivity and/or synergistic activity of its components. Suitable linkers are stable in circulation but undergo cleavage in target tissue as the result of a chemical or biological stimulus.36 Pendant ester groups in platinum–acridines have been demonstrated to be chemically significantly more reactive than amide and carbamate groups.8, 15, 37 To test the hydrolytic stability of the conjugates, 7 representative derivatives were incubated in physiologically relevant buffers, and linker cleavage was monitored by LC-MS (see the ESI for a detailed product analysis). All conjugates show selective cleavage at the ester bond resulting in hydroxyl-modified platinum–acridine 2c (Fig. 1) or the analogous benz[c]acridine hybrid, which undergo partial exchange of the chlorido ligand by water or formate ion under the conditions of this assay (see Fig. 5 for P1-N1 as an example). Decomposition of conjugates unrelated to linker cleavage was observed after incubation periods of longer than 48 h, but only to a minor extent and only for conjugates otherwise stable to hydrolysis (see the ESI).
Figure 5.
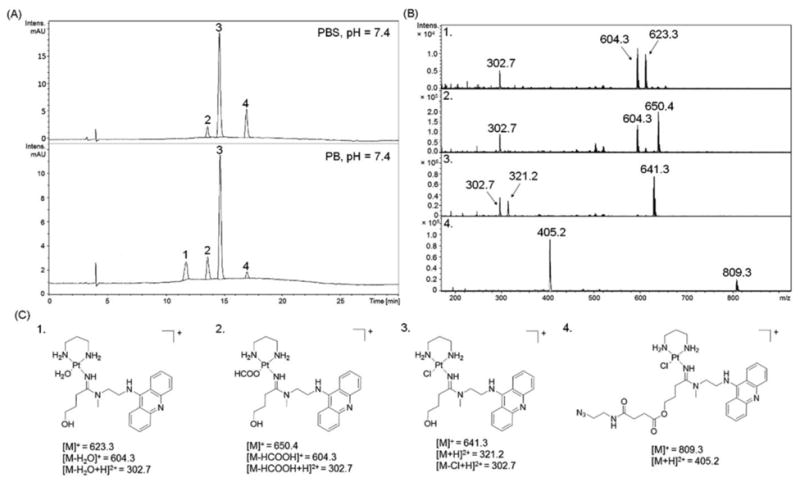
Product analysis by LC-MS for ester cleavage in conjugate P1-N1 (12 h, 37 °C, pH 7.4). (A) HPLC traces, mass spectra recorded in positive-ion mode with molecular ions [M]+ and [M]2+ labeled (C), and the corresponding product structures (C) are shown.
The time course of ester cleavage was monitored by determining the composition of reaction mixtures after 3, 6, 12, 24, and 48 h of continuous incubation (Fig. 6). Surprisingly, while all tested conjugates contain the same succinic ester linkage, -(CH2)3OC(O)(CH2)2C(O)NH-, major differences are observed in their hydrolysis rates. While cleavage in some cases is rapid and virtually complete within 12 h (P1-N1, P2-N1), other conjugates are remarkably stable (e.g., P1-N6) with only a minor amount of cleaved ester observed after 48 h. In general, the sterically least hindered alkyl amines were most reactive, whereas amines with charged (P1-N4) or bulkier groups (P1-N6, P2-N6, P1-N7) were more stable in buffered solution. A significant decrease in rate of ester hydrolysis of up to ∼4-fold is observed in phosphate buffer supplemented with chloride (PBS) relative to chloride-free buffer. This observation confirms the previously established mechanism (Scheme 1) in which aquated platinum facilitates Lewis-acid promoted ester cleavage.15 Although the rate of cleavage primarily depends on the nature of the coupled amine, replacing acridine in P1-N1 and P1-N6 with benz[c]acridine in P2-N1 and P2-N6 also alters the rate of ester hydrolysis. From the LC-MS profiles (see the ESI) it appears that the extent of aquation of platinum in the conjugates correlates well with the rate of linker hydrolysis, further corroborating the proposed metal-assisted cleavage mechanism.
Figure 6.
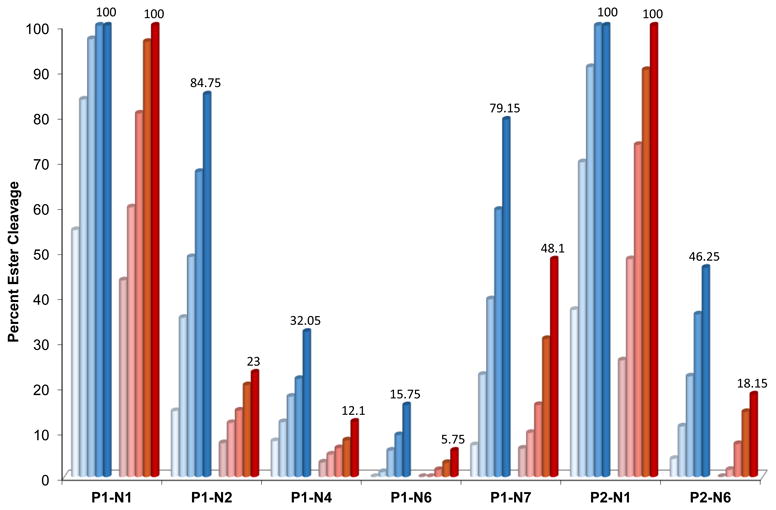
Progress of ester hydrolysis in selected conjugates monitored by LC-MS in phosphate buffer (blue columns) and phosphate-buffered saline (PBS, red columns). The percentages of cleaved ester are plotted for 3, 6, 12, 24, and 48 h of continuous incubation. Data represent averages of three incubations. Numbers on the last column in each data set indicate final % cleavage after 48 h.
It should be noted that incubations of several of the chemically more stable new conjugates with CES-2 did not lead to enhanced linker cleavage (unreported data). Whether the conjugates are susceptible to cleavage by other ubiquitous serum esterases remains to be determined. Likewise, potential proteolytic cleavage of the amide linkage in biological media remains to be investigated.
Conclusions
In conclusion, we have expanded the toolbox of linker and conjugation chemistries for platinum–acridines, a class of highly cytotoxic anticancer agents. Amide coupling appears to be a versatile and efficient platform for assembling multifunctional conjugates containing a Pt(II) moiety. The method is amenable to high-throughput library-based synthesis and is compatible with applications that require conjugation in aqueous media. Combining the coupling chemistry with cleavable linkers of variable reactivity has the potential to lead to the discovery of new multifunctional anticancer drugs with novel, synergistic modes of action. The methodology will also enable conjugation of otherwise too toxic platinum–acridines to targeted carriers, such as monoclonal antibodies. Whether the particular amide–ester type linker used in this study will be sufficiently stable for use in long-circulating, immuno-targeted antibody–drug conjugates (ADCs)36 remains to be established. The linker chemistry developed in this study, in combination with previously introduced carbamate-coupled and azide–alkyne clickable analogues,8,37 should also have broad applicability in other clinical and experimental platinum and non-platinum metallodrugs.
Experimental section
Materials and procedures
The platinum complex [PtCl2(pn)]8 and acridine precursors A18 and B119 were synthesized as described in the literature. 4-(3-Cyanopropoxy)-4-oxobutanoic acid (3)38 and compounds N1,8 N4,39 N6,37 N7,31 and N840 were synthesized according to published procedures. Compound N9 was generated by HBTU/DIPEA coupling of N8 with 11-aminoundecanoic acid. Amines N2, N3, N5 (Cayman Chem. Comp., Ann Arbor, MI), N10 (Dendritech Inc., Midland, MI), and N11 were commercially available from common vendors or the provider of specialty chemicals indicated and used as supplied. All other reagents (incl. coupling reagents) were obtained from common vendors and used without further purification.
1H NMR spectra of newly synthesized target compounds and intermediates were recorded on Bruker Advance DRX-500 and 300 MHz instruments. 13C {1H} NMR spectra were recorded on a Bruker DRX-500 instrument operating at 125.8 MHz. 2-D COSY and 1H-13C gradient-selected Heteronuclear Multiple Bond Coherence (gs-HMBC) data sets were acquired on a Bruker DRX-500 instrument equipped with a TBI probe. HMBC spectra were collected at 25 °C with 2048 pts in t2 (sw = 6510 Hz), 256 pts in t1 (sw = 27670 Hz), 128 scans per t1 increment, and a recycle delay (d1) of 1.5 s. Chemical shifts (δ) are reported in parts per million (ppm) relative to internal standard tetramethylsilane (TMS). 1H NMR data is reported in the conventional format including chemical shifts (δ, ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad), coupling constants (J, Hz), and integrated signal intensities. 13C NMR data are reported as chemical shift listings (δ, ppm). The NMR spectra were processed and analyzed using the MNova software package (Mestrelab Research, version 9.0.0).
LC-MS analysis was performed on an Agilent 1100LC/MSD ion trap mass spectrometer equipped with an atmospheric pressure electrospray (ES) source. HPLC-grade solvents were used for all HPLC separations in mass spectrometry experiments. Eluent nebulization was achieved with a N2 pressure of 50 psi and solvent evaporation was assisted by a flow of N2 drying gas (350 °C). Mass spectra in positive-ion mode were recorded with a capillary voltage of 2800 V and a mass-to-charge (m/z) scan range of 150 to 2200. HR-MS was performed on a Thermo Scientific LTQ Orbitrap XL equipped with an electrospray source, and the data was processed with Xcalibur 2.1 (Thermo Scientific). To determine the purity of target compounds, samples were diluted in methanol containing 0.1 % formic acid and separated using a 4.6 mm × 150 mm reverse-phase Agilent ZORBAX SB-C18 (5 mm) analytical column at 25 °C, using the following solvent system: solvent A, optima water, and solvent B, methanol/0.1 % formic acid, with a flow rate of 0.5 mL/min and a gradient of 95 % A to 5 % A over 30 minutes. HPLC traces were recorded with a monitoring wavelength range of 363-463 nm. Peak integration was done using the LC/MSD Trap Control 4.0 data analysis software (Agilent).
Synthetic procedures and product characterization
Synthesis of [PtClC27H38N6O4](NO3) (P1)
P1 was synthesized using a two-step, one-pot approach. A mixture of 0.678 g (2.00 mmol) of [PtCl2(pn)] and 0.338 g (2.00 mmol) of AgNO3 in 10 mL of anhydrous DMF was stirred at room temperature in the dark for 16 h. Precipitated AgCl was filtered off through a syringe filter. 2.96 g (16 mmol) of nitrile 3 was added to the filtrate, and the suspension was stirred at 65 °C for 4 h in the dark. The reaction mixture was then cooled to 4 °C in an ice bath. Acridine–amine A1 (0.51 g, 2.02 mmol) was added to the solution, and the suspension was stirred at 4 °C for another 48 h. To quench the reaction, the mixture was added dropwise into 200 mL of anhydrous diethyl ether, and the resulting yellow slurry was vigorously stirred for 30 min. The precipitate was recovered by membrane filtration and recrystallized from hot ethanol, affording P1 as yellow a solid. Yield: 704 mg (43 %). 1H NMR (300 MHz, MeOH-d4) δ 8.56 (d, J = 8.7 Hz, 2H), 7.95 (t, J = 7.5 Hz, 2H), 7.81 (d, J = 8.4 Hz, 2H), 7.57 (t, J = 7.6 Hz, 2H), 5.08 (br s, 2H), 4.80 (br s, 2H), 4.38 (t, J = 6.7 Hz, 2H), 4.32–4.22 (m, 3H), 3.94 (t, J = 6.6 Hz, 3H), 3.13–2.96 (m, 5H), 2.82–2.46 (m, 8H), 2.15 (s, 2H), 1.81 (s, 2H). 13C NMR (75 MHz, MeOH-d4) δ 174.33, 172.68, 168.67, 161.75, 161.75, 154.16, 142.51, 131.29, 126.77, 121.35, 119.47, 117.88, 65.29, 63.64, 44.43, 43.58, 33.34, 32.57, 31.97, 28.99, 14.91. HR-MS (positive-ion mode): m/z for C27H38ClN6O4Pt ([M]+), Calculated 740.2291; found, 740.2297 (tolerance: 0.81 ppm). Analytical purity > 98% by LC-MS (see the ESI).
Synthesis of [PtClC31H40N6O4](NO3) (P2)
P2 was synthesized by the same procedure as P1 from 0.678 g (2.00 mmol) of [PtCl2(pn)] and benzo[c]acridine–amine B1 (0.61 g, 2.02 mmol). Yield: 782 mg (46 %). 1H NMR (300 MHz, MeOH-d4) δ 9.06 (d, J = 7.9 Hz, 1H), 8.55 (d, J = 8.6 Hz, 1H), 8.27 (t, J = 9.3 Hz, 2H), 8.14–7.76 (m, 5H), 7.69 (t, J = 7.7 Hz, 1H), 5.08 (br s, 2H), 4.80 (br s, 2H), 4.35 (t, J = 6.5 Hz, 2H), 4.20 (d, J = 5.9 Hz, 2H), 3.93–3.89 (m, 2H), 3.19 – 2.90 (m, 5H), 2.88–2.45 (m, 8H), 2.08 (s, 2H), 1.79 (s, 2H). 13C NMR (75 MHz, MeOH-d4) δ 175.59, 173.30, 168.88, 154.31, 150.58, 143.04, 135.22, 132.17, 131.75, 130.20, 128.42, 127.56, 126.32, 125.37, 123.09, 122.94, 122.07, 116.70, 113.97, 64.04, 44.13, 43.48, 32.77, 31.75, 28. 93, 27.50, 15.44. HR-MS (positive-ion mode): m/z for C27H38ClN6O4Pt ([M]+), Calculated 790.2447; found, 790.2463 (tolerance: 2.02 ppm). Analytical purity > 98% by LC-MS (see the ESI).
Synthesis of [PtCl(pn)C43H47ClFN9O4](NO3)2 (P1-N7)
100 mg of P1 (0.122 mmol), 71 mg of N7 (0.183 mmol), 127 mg of PyBOP (0.244 mmol) and 31 mg of DIPEA were mixed in 3 mL of anhydrous DMF. The reaction mixture was incubated at room temperature for 16 h and then poured into 200 mL of vigorously stirred anhydrous ethyl ether. The precipitate was recovered by membrane filtration and recrystallized from the mixture of ethanol and ethyl acetate (3:1) to give 121 mg of conjugate P1-N7 (yield: 83%) as a yellow solid which was dried in a vacuum. 1H NMR (500 MHz, DMF) δ 13.91 (br s, 1H), 10.05 (br s, 1H), 8.91–8.87 (m, 1H), 8.74–8.68 (m, 2H), 8.17–8.12 (m, 1H), 8.03–7.96 (m, 5H), 7.92-7.84 (m, 1H), 7.68-7.51 (m, 4H), 6.72–6.27 (m, 2H), 5.49 (br s, 2H), 5.05 (br s, 2H), 4.58–4.50 (m, 2H), 4.37–3.99 (m, 6H), 3.86–3.74 (m, 2H), 3.50–3.47(m, 2H), 3.28–3.13 (m, 8H), 2.83–2.53 (m, 8H), 2.30–2.17 (m, 2H), 1.90–1.80 (m, 2H), 1.56–1.43 (m, 3H). 13C NMR (126 MHz, DMF) δ 172.94, 172.91, 172.74, 171.31, 168.33, 158.56, 158.31, 157.43, 157.22, 156.23, 154.28, 154.08, 146.39, 140.75, 139.82, 135.30, 135.16, 126.27, 125.99, 124.58, 123.82, 119.75, 119.60, 118.95, 116.93, 116.76, 109.17, 109.03, 99.03, 98.59, 97.20, 96.77, 65.45, 63.53, 56.69, 47.53, 45.99, 43.45, 42.82, 41.57, 41.08, 33.05, 28.15, 27.87, 14.92, 13.91. HR-ESMS (positive-ion mode): m/z for C46H57Cl2FN11O4Pt ([M]+), Calculated 1111.3604; found, 1111.3615 (tolerance: 0.99 ppm). Analytical purity > 98% by LC-MS (see the ESI).
Micro-scale reactions and library assembly
Stock solutions of the carboxylic acid-modified platinum–(benz)acridines (P1, P2) (25 mM), coupling reagent PyBOP (25 mM), amines to be linked via amide bond formation (N1–N10) (25 mM), and DIPEA (25 mM) were prepared in anhydrous DMF. Micro-scale coupling reactions were carried out in 1.5-mL Eppendorf tubes or 96-well plates by mixing all the reaction components using the following volumes: P1 or P2 (40 μL), PyBOP (60 μL), DIPEA (40 μL), and amines N1–N10 (44 μL). Finally, 16 μL of dry DMF were added to produce a total incubation volume of 200 μL and final platinum concentration of 5 mM. Mixtures were incubated at room temperature for 16 hours in a vortex shaker. To characterize conjugates and to determine conversion yields, 5-μL samples are removed from the solutions and diluted with 995 μL of methanol containing 0.1% formic acid prior to LC-MS analysis. Chromatographic separations and product quantification were performed as described above.
Chemical hydrolysis assay
Selected conjugates were generated using the microscale reactions as described before. To purify the conjugates, the reaction mixtures were precipitated in ethyl ether and separated by size exclusion chromatography (Sephadex LH-20, 10 × 1 cm column) using methanol:acetonitrile (1:1, v/v) as eluent. Ester hydrolysis was carried out by incubating 0.1 mM of each test compound in 10 mM phosphate buffer (pH 7.4) or 1× PBS containing 150 mM NaCl (pH 7.4) at 37 °C. Samples were withdrawn from the reaction mixture at various time points and analyzed by in-line LC-MS using the method and settings described above.
Supplementary Material
Acknowledgments
The authors thank Mu Yang for providing a sample of the quinazoline derivative N7. This work was supported by the National Institutes of Health (grant CA101880). HR-MS spectra were acquired on a Thermo Scientific LTQ Orbitrap instrument purchased with funds provided by the US National Science Foundation (NSF grant 947028).
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
For a complete list of acronyms and systematic names of coupling reagents used in this study and their chemical structures, see the ESI.
Notes and references
- 1.Kelland L. Nat Rev Cancer. 2007;7:573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 2.Farrell NP. Curr Top Med Chem. 2011;11:2623–2631. doi: 10.2174/156802611798040714. [DOI] [PubMed] [Google Scholar]
- 3.Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, Kroemer G. Oncogene. 2012;31:1869–1883. doi: 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- 4.Komeda S. Metallomics. 2011;3:650–655. doi: 10.1039/c1mt00012h. [DOI] [PubMed] [Google Scholar]
- 5.Komeda S, Casini A. Curr Top Med Chem. 2012;12:219–235. doi: 10.2174/156802612799078964. [DOI] [PubMed] [Google Scholar]
- 6.Suryadi J, Bierbach U. Chem Eur J. 2012;18:12926–12934. doi: 10.1002/chem.201202050. [DOI] [PubMed] [Google Scholar]
- 7.Ma Z, Choudhury JR, Wright MW, Day CS, Saluta G, Kucera GL, Bierbach U. J Med Chem. 2008;51:7574–7580. doi: 10.1021/jm800900g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ding S, Qiao X, Kucera GL, Bierbach U. J Med Chem. 2012;55:10198–10203. doi: 10.1021/jm301278c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shapira A, Livney YD, Broxterman HJ, Assaraf YG. Drug Resist Updat. 2011;14:150–163. doi: 10.1016/j.drup.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 10.Ding S, Bierbach U. Future Med Chem. 2015;7:911–927. doi: 10.4155/fmc.15.37. [DOI] [PubMed] [Google Scholar]
- 11.Wang X, Guo Z. Chem Soc Rev. 2013;42:202–224. doi: 10.1039/c2cs35259a. [DOI] [PubMed] [Google Scholar]
- 12.Butler JS, Sadler PJ. Curr Opin Chem Biol. 2013;17:175–188. doi: 10.1016/j.cbpa.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 13.Stathopoulos GP. Anticancer Drugs. 2010;21:732–736. doi: 10.1097/CAD.0b013e32833d9adf. [DOI] [PubMed] [Google Scholar]
- 14.Johnstone TC, Suntharalingam K, Lippard SJ. Chem Rev. 2016;116:3436–3486. doi: 10.1021/acs.chemrev.5b00597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ding S, Pickard AJ, Kucera GL, Bierbach U. Chem Eur J. 2014;20:16164–16173. doi: 10.1002/chem.201404675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pratt SE, Durland-Busbice S, Shepard RL, Heinz-Taheny K, Iversen PW, Dantzig AH. Clin Cancer Res. 2013;19:1159–1168. doi: 10.1158/1078-0432.CCR-12-1184. [DOI] [PubMed] [Google Scholar]
- 17.Wang J, Williams ET, Bourgea J, Wong YN, Patten CJ. Drug Metab Dispos. 2011;39:1329–1333. doi: 10.1124/dmd.111.039628. [DOI] [PubMed] [Google Scholar]
- 18.Imai T, Taketani M, Shii M, Hosokawa M, Chiba K. Drug Metab Dispos. 2006;34:1734–1741. doi: 10.1124/dmd.106.009381. [DOI] [PubMed] [Google Scholar]
- 19.Pickard AJ, Liu F, Bartenstein TF, Haines LG, Levine KE, Kucera GL, Bierbach U. Chem Eur J. 2014;20:16174–16187. doi: 10.1002/chem.201404845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.El-Faham A, Albericio F. Chem Rev. 2011;111:6557–6602. doi: 10.1021/cr100048w. [DOI] [PubMed] [Google Scholar]
- 21.Valeur E, Bradley M. Chem Soc Rev. 2009;38:606–631. doi: 10.1039/b701677h. [DOI] [PubMed] [Google Scholar]
- 22.El-Faham A, Albericio F. J Pept Sci. 2010;16:6–9. doi: 10.1002/psc.1204. [DOI] [PubMed] [Google Scholar]
- 23.Najera C. Synlett. 2002:1388–1403. [Google Scholar]
- 24.Lu JQ, Liu C, Wang PC, Ghazwani M, Xu JN, Huang YX, Ma XC, Zhang PJ, Li S. Biomaterials. 2015;62:176–187. doi: 10.1016/j.biomaterials.2015.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aryal S, Hu CMJ, Zhang LF. Chem Commun. 2012;48:2630–2632. doi: 10.1039/c2cc18176b. [DOI] [PubMed] [Google Scholar]
- 26.Lammers T, Kiessling F, Hennink WE, Storm G. J Control Release. 2012;161:175–187. doi: 10.1016/j.jconrel.2011.09.063. [DOI] [PubMed] [Google Scholar]
- 27.Vargas JR, Stanzl EG, Teng NNH, Wender PA. Mol Pharm. 2014;11:2553–2565. doi: 10.1021/mp500161z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ekblad T, Camaioni E, Schuler H, Macchiarulo A. FEBS J. 2013;280:3563–3575. doi: 10.1111/febs.12298. [DOI] [PubMed] [Google Scholar]
- 29.Znojek P, Willmore E, Curtin NJ. Br J Cancer. 2014;111:1319–1326. doi: 10.1038/bjc.2014.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goetz MP, Rae JM, Suman VJ, Safgren SL, Ames MM, Visscher DW, Reynolds C, Couch FJ, Lingle WL, Flockhart DA, Desta Z, Perez EA, Ingle JN. J Clin Oncol. 2005;23:9312–9318. doi: 10.1200/JCO.2005.03.3266. [DOI] [PubMed] [Google Scholar]
- 31.Yang M, Pickard AJ, Qiao X, Gueble MJ, Day CS, Kucera GL, Bierbach U. Inorg Chem. 2015;54:3316–3324. doi: 10.1021/ic502998a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoshida T, Zhang G, Haura EB. Biochem Pharmacol. 2010;80:613–623. doi: 10.1016/j.bcp.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 33.Milane L, Trivedi M, Singh A, Talekar M, Amiji M. J Control Release. 2015;207:40–58. doi: 10.1016/j.jconrel.2015.03.036. [DOI] [PubMed] [Google Scholar]
- 34.Yellepeddi VK, Vangara KK, Palakurthi S. J Nanopart Res. 2013;15 [Google Scholar]
- 35.Bannwarth W, Knorr R. Tetrahedron Lett. 1991;32:1157–1160. [Google Scholar]
- 36.Alley SC, Okeley NM, Senter PD. Curr Opin Chem Biol. 2010;14:529–537. doi: 10.1016/j.cbpa.2010.06.170. [DOI] [PubMed] [Google Scholar]
- 37.Ding S, Qiao X, Kucera GL, Bierbach U. Chem Commun. 2013;49:2415–2417. doi: 10.1039/c3cc38957j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kita Y, Maeda H, Takahashi F, Fukui S. Chem Comm. 1993:410–412. [Google Scholar]
- 39.Castillo JA, Infante MR, Manresa A, Vinardell MP, Mitjans M, Clapes P. ChemMedChem. 2006;1:1091–1098. doi: 10.1002/cmdc.200600148. [DOI] [PubMed] [Google Scholar]
- 40.Millard M, Gallagher JD, Olenyuk BZ, Neamati N. J Med Chem. 2013;56:9170–9179. doi: 10.1021/jm4012438. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.