

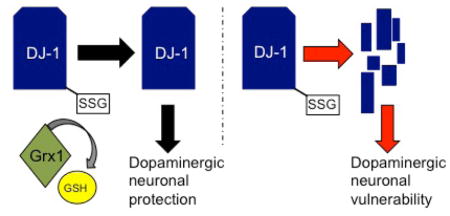
Abstract
Parkinson’s disease (PD) is the second most common neurodegenerative disease worldwide, caused by the degeneration of the dopaminergic neurons in the substantia nigra. Mutations in PARK7 (DJ-1) result in early onset autosomal recessive PD, and oxidative modification of DJ-1 has been reported to regulate the protective activity of DJ-1 in vitro. Glutathionylation is a prevalent redox modification of proteins resulting from the disulfide adduction of the glutathione moiety to a reactive cysteine-SH; and glutathionylation of specific proteins has been implicated in regulation of cell viability. Glutaredoxin 1 (Grx1) is the principal deglutathionylating enzyme within cells, and it has been reported to mediate protection of dopaminergic neurons in C. elegans, however many of the functional downstream targets of Grx1 in vivo remain unknown. Previously, DJ-1 protein content was shown to decrease concomitantly with diminution of Grx1 protein content in cell culture of model neurons (SH-SY5Y and Neuro-2A lines). In the current study we aimed to investigate the regulation of DJ-1 by Grx1 in vivo and characterize its glutathionylation in vitro. Here, with Grx−/− mice we provide evidence that Grx1 regulates protein levels of DJ-1 in vivo. Furthermore, with model neuronal cells (SH-SY5Y) we observed decreased DJ-1 protein content in response to treatment with known glutathionylating agents; and with isolated DJ-1 we identified two distinct sites of glutathionylation. Finally, we found that overexpression of DJ-1 in the dopaminergic neurons partly compensates for the loss of the Grx1 homolog in a C. elegans in vivo model of PD. Therefore; our results reveal a novel redox modification of DJ-1 and suggest a novel regulatory mechanism for DJ-1 content in vivo.
Keywords: Redox, Parkinson’s disease, glutaredoxin, DJ-1, glutathionylation, C. elegans
Graphical Abstract
Introduction
Parkinson’s disease (PD) is a debilitating neurodegenerative disease caused by the loss of dopaminergic neurons in the substantia nigra region of the midbrain [1, 2]. Although the etiology of PD is unclear, PD can be categorized into two groups: familial and sporadic. These classifications are based on whether PD is attributed to a genetic mutation or if the cause of the disease is undefined. In both cases oxidative stress and dysregulation of redox homeostasis have been implicated.
LRRK2 (Leucine-Rich Repeat Kinase 2) and DJ-1 (PARK7) are two proteins in which genetic mutations are linked to development of PD [3, 4]. Mutations in LRRK2 are shown to cause autosomal dominant PD in humans, and over-expression of mutant LRRK2 results in dysfunction of dopamine signaling or degeneration of dopaminergic neurons in animal models of PD [5, 6, 7, 8, 9]. Mutations in DJ-1 result in autosomal recessive, early onset PD in humans, through destabilization and loss of DJ-1 protein content [10]. In addition to mutations driving familial PD, post mortem brain samples from patients with sporadic PD have been reported to have decreased levels of DJ-1 protein within several areas of the brain including the nigral region [ 11]. Although studies have implicated DJ-1 as an anti-oxidant factor [12, 13], the molecular mechanism(s) by which DJ-1 protects cells from oxidative stress-induced death is currently unresolved [14]. Genetic manipulation has revealed a key residue on DJ-1, which is critical for the protection against oxidative stressors. Substitution of cysteine 106 with alanine (C106A) results in a “non-functional” DJ-1; over-expression of WT, but not C106A DJ-1, protects Park7−/− mouse embryonic fibroblasts from H2O2-induced cell death [15]. In addition, it has been found that cysteine 106 can be oxidatively modified to the sulfinic acid. This modification has been reported to play a critical role in protecting M17 neuroblastoma cells from MPP+ toxicity [ 16]. These data suggest that sufficient levels of DJ-1 protein and specific redox modification of DJ-1 play important roles in mediating neuronal protection.
Protein S-glutathionylation (Protein-SSG) is a prevalent oxidative modification involving the formation of a disulfide bond between glutathione (GSH) and a reactive protein-cysteine-SH [ 2]. Protein glutathionylation can result in changes in subcellular localization, protein function and/or protein levels [17, 18, 19]. Glutaredoxin 1 (Grx1) is the principal deglutathionylating enzyme in the cell, restoring the protein-SH and its function [2]. Grx1 has been shown to mediate neuronal protection both in vitro and in vivo [19, 20, 21, 22]. We recently reported evidence that PD patients have diminished levels of Grx1 specifically within the remaining dopaminergic neurons of the midbrain compared to controls [20]. Further we found that Caenorhabditis elegans (C. elegans) lacking the homolog of Grx1 displayed exacerbated losses of dopamine-dependent behavior and dopaminergic neurons in models of both familial and sporadic PD [20]. Thus, it appears that Grx1 mediates protection of dopaminergic neurons in vitro and in vivo, however the downstream molecular mechanism(s) that accomplish this protection remain to be elucidated. In this regard, knockdown of Grx1 in model dopaminergic neurons was previously reported to increase cell death [21, 22], and to decrease DJ-1 protein content [22]. Over-expression of WT, but not C53S or C106S DJ-1, abolished the cell toxicity associated with diminished Grx1 function, suggesting that the content of DJ-1, its cysteine redox status, and its effect on neuronal cell viability are regulated via Grx1 [22]. Since this interpretation may also apply to our observations that loss of the Grx1 homolog in C. elegans exacerbates PD-like phenotypes [ 20], we set out in the current study to determine whether Grx1 regulates DJ -1 in vivo.
Here, we report evidence supporting a post-translational mechanism for regulation of DJ-1 protein content involving reversible glutathionylation and regulation by Grx1 in vivo. Accordingly, over-expression of DJ-1 partially compensates for neuronal toxicity due to the loss of Grx1 in models of PD.
Methods
Western blot analysis of mouse brain tissue
Mouse brain tissues from two independent cohorts obtained from Case Western Reserve University and The University of Vermont (a kind gift from Dr. Y. Janssen-Heininger), were dissected into three sections, midbrain, cortex and cerebellum. Midbrain portions of the brains were snap frozen in liquid nitrogen. Prior to use, midbrains were homogenized by physical disruption using a pestle in a 1.5 ml Eppendorf tube with Cell Lysis Buffer (Cell Signaling) containing protease inhibitors (Roche). Tubes were incubated on ice for 15 min followed by sonication three times for 5 sec each. Samples were clarified by centrifugation at 4°C for 30 min at 16,000 x g. Aliquots of the supernatant (70 μg of protein content) were run on a 15% SDS-PAGE gel, transferred to PVDF membrane using a semi-dry transfer apparatus (Bio-Rad) at 20 V for 1 h, blocked with 5% milk in Tris-buffered saline with 0.05% Tween-20 (TBST) for 1 h, and probed overnight with anti-actin (Sigma, diluted 1:5000), DJ-1 (Abcam,1:5000) and Grx1 (Genscript, 1:1000) antibodies. Blots were washed three times for 10 min each in TBST and probed with secondary antibodies (Bio-Rad, 1:5000) for 1 h. Blots were developed using enhanced chemiluminescence (Thermo Fisher). Rabbit polyclonal anti-Grx1 was generated by Genscript against recombinant human Grx1 produced in E. coli [ 20]. The western blots were scanned and band densities were quantified using ImageJ software (NIH).
Western blot analysis of human brain tissue
Postmortem brain tissue samples from histopathologically diagnosed PD and non-PD cases (Supplemental Table A), were used for this study according to approved IRB protocols. Frozen tissues of brainstem or pons were obtained from the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland (Baltimore, MD), and from the Case Western Reserve University/University Hospitals of Cleveland Brain Bank (Cleveland, OH). Areas of the frozen brainstem samples containing the substantia nigra were carefully dissected and were homogenized with Cell Lysis Buffer (Cell Signaling) plus 1 mM phenylmethanesulfonyl fluoride (Sigma) and Protease Inhibitor Cocktail (Sigma). Equal amounts of total protein extract were run on a 15% SDS–PAGE gel and transferred to PVDF (Millipore). Membranes were blocked in 5% milk in TBST for 1 h. Primary antibodies were incubated overnight in 5% milk in TBST, and washed three times in TBST prior to addition of the secondary antibody. Secondary antibodies were incubated for 1 h in 5% milk in TBST, washed three times in TBST and developed using ECL (Pierce). Antibodies used included anti-actin (Sigma), anti-DJ-1 (Abcam) and anti-Grx1 [20].
Western blot analysis of human blood cells
Human blood samples used for this study were collected according to approved IRB protocols (see patient information Supplemental Table B). PBMCs from 1.5 ml of cryogenically frozen blood were isolated by lysis of the red blood cells in 10 ml red blood cell lysis buffer (0.155 M ammonium chloride, 0.01 M potassium bicarbonate, and 0.00013 M EDTA) for 15 min on a rocker. The suspension was spun at 1,500g on a table-top centrifuge, the supernatant was removed, and 10 ml of red cell lysis buffer was added. This process was repeated two more times with incubation on the rocker shortened to 5 min each. The resulting PBMCs were lysed and were analyzed via western blotting in the same manner as described for the western blot analysis of mouse brain tissue except 35 μg of lysate was loaded per sample.
Immmunoprecipitation of DJ-1 from mouse brain lysate
Whole snap frozen C57BL/6 or FEB mouse brains were placed in 4 ml cell lysis buffer (Cell Signaling) plus 50 mM iodoacetamide (to block free cysteine-SH). Brains were mechanically homogenized via mortar and pestle, and then subjected to lysis via sonication on ice for 2 min, alternating 10 s on 10 s off. Lysate was then spun down at 16,000g at 4°C in a tabletop centrifuge for 1 hour. The resulting supernatant was divided into aliquots, with addition of either 50 μl Protein G beads (Pierce) and 5 μg anti-goat IgG (Santa Cruz); or 50 μl Protein G beads and 5 μg anti-DJ-1 (Goat, Abcam). Samples were rotated at 4°C for 48 h, washed three times in lysis buffer plus 0.25 M NaCl. Laemmeli sample buffer (without reducing agents) was added and beads were boiled for 10 min. Samples were then run on a 15% non-reducing SDS-PAGE gel, transferred to a PVDF membrane, and probed for glutathionylation using an anti-glutathione antibody (anti-GSH, Millipore). Membranes were stripped by incubation in 0.1 M Tris-HCl pH 6.8, 2% SDS, and 0.11 M β-mercapto ethanol at 50°C for 30 min followed by three washes in TBST for 15 min each. Blots were then probed with rabbit anti-DJ-1 (Cell Signaling) in the same manner as described above.
LC-MS analysis of proteins and peptides
Mass spectrometric (MS) analyses were performed using a LTQ Velos dual-pressure linear ion trap mass spectrometer equipped with an Accela 600 quaternary pump for in-line protein and peptide high performance liquid chromatography (HPLC) separation and an electrospray ionization source (Thermo Scientific, Waltham, MA). The electrospray ionization source was operated in the positive ion mode with the following parameters: normalized collision energy 35 kV, capillary temperature 300 °C, source voltage 5 kV, capillary voltage 43 V, and tube lens 105 V. Mass spectra were collected over a 200–2000 m/z range. To record mass spectra of intact proteins, 0.5 μg of DJ-1 or its glutathionylated form was loaded onto an XBridge BEH300 C4 column (3.5 μm, 2.1 × 50 mm) (Waters, Milford, MA) and eluted with a linear gradient (5–100%) of acetonitrile in water containing 0.1% formic acid. The gradient was developed over 20 min at a flow rate of 0.25 ml/min.. Masses of the proteins were calculated by deconvolution of intact protein ions using ProMass for Xcalibur software (Thermo Scientific). The same instrumentation including column and solvents was employed to separate peptide products of tryptic digestion of DJ-1. However, to ensure adequate peptide separation the gradient of acetonitrile was developed over 40 min. The protein tryptic digestion was performed in 50 mM Tris-HCl, pH 7.5, at 37 °C overnight. Products of the reaction were directly analyzed by LC-MS. Peptides were identified based on a correlation of their tandem-mass spectra (MS/MS) with theoretically predicted ion fragmentation patterns.
The efficiency of protein glutathionylation was assessed by relative quantification of unmodified DJ-1 in the reaction sample. For this purpose, prior to the tryptic digestion, the glutathionylated DJ-1 sample was supplemented with an equal amount (12.5 μg) of 15N-labeled DJ-1. LC-MS analysis of the tryptic peptide mixtures revealed co-eluting native and 15N-labeled peptides. The relative ion intensities for pairs of native and 15N-labeled peptides at the same charge state were used to calculate the relative abundance of unmodified DJ-1 and thus the fraction of glutathionylated protein. Glutathionylation of cysteine residues were estimated using unmodified 15N DJ-1 as an internal standard. Quantification of modified tryptic peptides were estimated by subtraction of the peak areas of the non-modified peptides in the treated 14N-samples from those in the untreated 15N-samples.
C. elegans strains and maintenance
A list of C. elegans strains with their designation, corresponding genotype, and strain nomenclature is shown in Supplemental Table C. C. elegans strains were maintained on nematode growth medium (NGM) plates with E. coli OP50 as a bacterial food, as previously described [6].
Single-worm PCR
Individual worms were placed in PCR tubes containing 10 μl total volume consisting of 1x OneTaq buffer (New England Biolabs) and 0.5 mg/ml proteinase K (Sigma). Worms were frozen on dry ice for 15 min, then lysed via incubation at 65°C for 90 min followed by 95°C for 30 min. PCR amplifications were performed using OneTaq polymerase following manufacturer’s instructions (New England Biolabs). Identification of WT and mutant djr-1.2 DNA was accomplished using the following primers (Integrated DNA Technologies), Forward: 5′-GCAAACTCGAAAGCAGTTGC-3′, Reverse WT: 5′-CGTATTGTTCCGGACGTAGC-3′ Reverse knockout: 5′-TTTGACCGTGTTCTTTGTCG-3′. Identification of LRRK2 and glrx-10 DNA was accomplished using the primers described previously [20].
Generation of C. elegans expressing Pdat-1::mCherry or Pdat-1::DJR1.2::mCherry in dopaminergic neurons
Plasmids containing Pdat-1::mCherry and Pdat-1::DJR-1.2::mCherry were constructed in the backbone of the pFxneRFP vector (a generous gift of Dr. S. Mitani, Tokyo Women’s Hospital, Japan). The Pdat-1 insert was excised from the Pdat-1::pFxneEGFP plasmid [6] using the KpnI and HindIII sites. The DJR-1.2 open reading frame was PCR amplified from the DJR-1.2_pDONR201 plasmid (GE) with primers containing the BamHI and Xho1 sites, Forward 5′-GATATAGGATCCATGGCTGCCCAAAAGAG-3′ and Reverse 5′-GTACTACTCGAGTACTTTGCCAAACACAG-3′. The mCherry insert was obtained by PCR amplification from the pGH8 plasmid (Addgene) with primers containing the NotI and BglII sites, Forward 5′-ATAATAGCGGCCGCTTAATGGTCTCAAAG-3′ and Reverse 5′-GCGCGCGAGATCTACTACTTATACAATTCATCC-3′. Restriction digestion was performed with appropriate selective enzymes according to the manufacturer’s instructions (New England Biolabs). PCR products were purified with the QIAquick PCR purification kit (Qiagen). All DNA fragments were run on 1% agarose gels, followed by extraction with the QIAquick gel extraction kit (Qiagen). DNA inserts were ligated into pFxneRFP vector at room temperature for 2.5 h using T4 ligase (New England Biolabs) to generate the Pdat-1:: mCherry and Pdat-1::DJR-1.2::mCherry plasmids. The plasmids were transformed into E. coli DH5α cells (Life Technologies), propagated, purified on Qiagen miniprep columns, and verified by DNA sequencing. The DNA constructs were microinjected into C. elegans N2 strain by Knudra Transgenic (Murray, Utah). Stable worm lines with red fluorescent dopaminergic neurons expressing mCherry (Pdat-1::mCherry) or DJR-1.2 fused with mCherry (Pdat-1::DJR-1.2::mCherry) were selected, and their genotypes were verified by PCR.
Basal slowing assay
The basal slowing assay was performed as previously described [6]. Briefly, NGM assay plates were seeded with a ring of E. coli OP50. Age-synchronized worms were transferred to a non-seeded NGM plate and washed twice in S basal buffer (100 mM NaCl, 10 μg/ml cholesterol, 50 mM potassium phosphate, pH 6.0). Worms were then transferred to either seeded or non-seeded assay plates, allowed to recover for 5 min, and their body bends were recorded in 20 s intervals.
Dopaminergic neuron counting
Approximately 30 age-synchronized adult worms were placed into 200 μl of 10 mM tetramisole (Sigma) in raised wall four chamber slides (Lab-Tek). GFP neurons were visualized with a Nikon microscope at 20x zoom. Dopaminergic CEP and ADE head neurons were assessed for degeneration in a blinded fashion, according to published protocols [ 20]. Neurons with detectable GFP signal were counted as positive, neurons lacking cell body GFP signal were counted as negative.
Treatment and analysis of SH-SY5Y cells
SH-SY5Y cells (ATCC) were grown in OPTI -MEM (Invitrogen) + 10% fetal bovine serum (Atlanta Biological) at 37 °C with 5% CO2. For H2O2 treatment, cells were treated with increasing concentrations of H2O2 for 2 h as indicated in the figure legend. For diamide treatment experiments, cells were pretreated with either 10 μM MG-132 (Sigma) diluted in DMSO, 1 μM lactacystin (Santa Cruz) diluted in DMSO, 1:200 dilution of cell culture broad protease inhibitors (Sigma) diluted in DMSO, or DMSO (0.5%) for 7 h. Cells were then treated with 0.1 mM diamide or PBS for 2 h. All cells were lysed in Cell Lysis Buffer (Cell Signaling) containing protease inhibitors (Roche) on ice for 15 min with occasional mixing (Vortex). Lysates were spun down at 4°C for 30 min at 16,000 x g in a table-top centrifuge. The supernatant (35 μg of protein) was loaded onto a 15% SDS-PAGE gel and western blotting was completed in the same manner as the mouse midbrain tissue described above.
Validation of glutathionyl mixed disulfide specificity of the anti-GSH Antibody
Glutathionylation of proteins can be reversed by addition of disulfide reducing agents. In order to validate the interpretation of the reactivity of the anti-GSH antibody, anti-DJ-1 beads were used to immunoprecipitate DJ-1 from mouse brains, and the resultant effluent was processed on a non-reducing SDS-PAGE gel and transferred to a PVDF membrane. After transfer the blot was blocked in 5% milk for 1h and cut in half. Half of the blot was incubated with buffer and the other half was incubated with buffer plus 100 mM dithiothreitol (DTT) for 30 min. The blots were then washed and probed with the anti-GSH antibody overnight. The blot incubated in buffer alone revealed the prominent band at 45 kDa while the blot incubated in buffer plus DTT failed to show immunoreactivity at 45 kDa (Supplemental Figure S4). Blots were then stripped and re-probed for DJ-1 confirming the presence of DJ-1 on both halves of the membrane.
Generation of recombinant DJ-1
Recombinant DJ-1 was purified following the protocol described in [23]. Briefly 1.5 L of BL21(DE3) E. coli containing pET15b-DJ-1 was induced with 1 mM IPTG for 4 h. Cells were spun down and lysed via lysozyme treatment followed by sonication. Centrifugally cleared lysate containing 6x-His tagged DJ-1 was bound to Ni2+ His-select resin (Sigma), washed, eluted with 200 mM imidazole, cleaved with thrombin, and dialyzed overnight. Uncleaved DJ-1 was removed using a second pass over the Ni2+ column, thrombin was removed using benzamidine-sepharose resin, and co-purifying nucleic acids were removed from DJ-1 using a Q-sepharose column. DJ-1 (free of the 6x-His tag) was then concentrated and snap frozen in liquid nitrogen.
Generation of 15N labeled DJ-1
Recombinant uniformly 15N-labeled DJ-1 was generated as previously described [23]. Briefly, N-terminally 6x-His tagged DJ-1 was expressed from pET15b in BL21(DE3) E coli. An inoculum of the cells was grown overnight in LB media containing 100 μg/ml ampicillin at 37°C. 50 ml of this culture was spun down and washed twice in 10 ml of sterile 15NH4Cl M9 defined media [23] and transferred to 1 L of 15NH4Cl M9 media. These cells were grown at 37°C and DJ-1 expression was induced by the addition of 0.5 mM IPTG when the A600 = 0.5. Protein purification was performed as described above. 15N labeling efficiency was determined using mass spectrometry as described [23] and the incorporation efficiency was >99%.
RNA isolation and qPCR Methods
Midbrain tissue from C57BL/6 and BALBc Grx1−/− mice and age- and sex-matched controls (when available) was homogenized in 1mL TRIzol (Life Technologies) using a Teflon and glass mortar and pestle. RNA was isolated according to manufacturer’s instructions. Resulting RNA was converted to cDNA using SuperScriptII (Invitrogen) according to manufacturer’s instructions. Park7 (DJ-1) expression (probe Mm00498538_m1) was analyzed using TaqMan Fast Universal PCR mix (Life Technologies) on the StepOnePlus machine (Life Technologies) using StepOne v2 software. Park7 expression was normalized to Gapdh (probe Mm99999915_g1) expression as an internal control.
RESULTS
Grx1 controls DJ-1 protein expression in vivo.
As reported previously, knockdown of Grx1 via shRNA results in decreased DJ-1 protein content in the SH-SY5Y [21] and Neuro2A cell lines[ 22], however it is unknown if Grx1 plays a role in mediating DJ-1 protein content in vivo. Midbrain lysates from Grx1−/− and WT mice in the C57BL/6 background from two independent cohorts were subjected to western blot analysis, in order to investigate the potential control of DJ-1 by Grx1. Western blot analysis indicated that Grx1−/− mice have diminished levels of DJ-1 protein compared to WT controls (Figure 1 A, B). Quantitative PCR was used to measure the mRNA levels of DJ-1 in the Grx1−/− and WT mice in the C57BL/6 background. No statistical difference was found in the mRNA levels between the Grx1−/− and WT mice (Supplemental Figure S1A). We also analyzed DJ-1 content in brain samples (kindly provided by Reiko Matsui, Boston University) from transgenic mice in which hGRx1 was overexpressed so that total Grx1 content (human plus mouse) was about 2-fold higher relative to WT mice [ 24]. In this case the DJ-1 content was not different (transgenic vs. WT), suggesting that the normal content of Grx1 is sufficient to maintain DJ-1 content, at least in the absence of increased oxidative stress (Supplemental Figure S1B). Notably, we also analyzed midbrain lysates from Grx1−/− and WT mice in the BALB/c background and these mice did not show a difference in the DJ-1 protein or mRNA content (Supplemental Figure S1 C, D); thus indicating that differential genetic expression and/or posttranslational regulation in the BALB/c strain disconnects the regulatory effect of Grx1 on DJ-1.
Figure 1. DJ-1 protein content is regulated by Grx1 in vivo.

(A) Western blot analyses of midbrain homogenates from WT and Grx1−/− mice probed with anti-Grx1, anti-DJ-1, and anti-actin antibodies, respectively. (B) Quantification of DJ-1 protein content (normalized to actin) in the lysates of midbrain samples from WT and Grx1−/− mice; n = 6 independent WT and Grx1−/− mice/*p<0.05. Statistical analysis was completed using Student’s T-test. Error bars represent SEM. (C) Positive correlation between the protein contents of DJ-1 and Grx1 (normalized to actin) in human PBMCs; n = 14 independent biological samples/p<0.01, R2=0.54. Statistical analysis was completed using a one-way linear regression test. (D) Positive correlation between the contents of the DJ-1 and Grx1 proteins (normalized to actin) in lysates of human post mortem midbrain tissue; n = 14 independent biological samples/p<0.01, R2=0.63. Statistical analysis was completed using a one-way linear regression test.
In addition, to investigate whether DJ-1 and Grx1 protein levels correlate in human tissue, lysates from human peripheral blood mononuclear cells (PBMCs) and post-mortem midbrain tissue were subjected to western blot analysis. Protein levels of Grx1 and DJ-1 (normalized to actin) showed a significant positive correlation for protein isolated from both PBMCs (Figures 1 C) and midbrain tissue (Figures 1 D; blots are shown in Supplemental Figure 2S A, B). Taken together, these data provide in vivo evidence for the first time that Grx1 influences DJ-1 protein content in the midbrains of mice, and that Grx1 and DJ-1 protein contents are co-variant inhuman tissue s.
Treatment of cells with glutathionylation-inducing agents results in loss of DJ-1 protein
As noted, knockdown of Grx1 in model dopaminergic neuronal (Neuro2A) cells via shRNA resulted in decreased DJ-1 protein, but not mRNA content [22]; however the mechanism by which this loss of DJ-1 protein occurred remains unclear. We hypothesized that loss of Grx1 results in accumulation of glutathionylated DJ-1 (DJ-1-SSG), which is then rapidly degraded. In order to test whether agents that promote DJ-1-SSG formation result in loss of DJ-1 content, SH-SY5Y cells were treated acutely with H2O2, which is known to result in protein-SSG formation [25, 26]. DJ-1 protein content was selectively diminished in a dose dependent manner (relative to actin, which is known to be glutathionylated without affecting its content) (Figure 2 A, B). Since H2O2 can modify cysteine residues to sulfinic and sulfonic acids in addition to protein-SSG, we wanted to limit the modification to glutathionylation of DJ-1. Accordingly, cells were treated separately with diamide, an oxidizing agent characterized to promote selective formation of protein-SSG [27, 28, 29]. Western blot analysis indicated that treatment with diamide also resulted in a significant loss of DJ-1 protein content compared to untreated controls. (Figure 2 C, D). Notably, the extent of diminution in DJ-1 protein content (~40%) was similar for both the H2O2- and diamide-treated cells. It was previously reported that loss of DJ-1 content concomitant with knockdown of Grx1 could be limited by treatment with a cocktail of inhibitors of proteolysis, consistent with the concept of enhanced degradation [22]. In order to learn whether loss of DJ-1 after diamide treatment of SH-SY5Y cells displayed the analogous pattern of non-proteasomal proteolysis as observed in Grx1-dificient Neuro2A cells [22], we pretreated the SH-SY5Y cells with the proteasome inhibitor MG-132 or with a broad protease inhibitor (PI) cocktail prior to diamide treatment [22]. Pretreatment with the broad protease inhibitors, prevented the diamide induced loss of DJ-1 (Figure 2 C, D). Notably, pretreatment of SH-SY5Y cells with either MG-132 or lactacystin inhibited the proteasome (as indicated by increased β-catenin content (Figure 2, E, G), but failed to blunt the diamide-induced loss of DJ-1 protein (Figure 2 E, F). These data suggest that treatment of SH-SY5Y cells with glutathionylation-inducing agents results in DJ-1 degradation in a proteasome-independent manner.
Figure 2. DJ-1 protein content is diminished after treatment with glutathionylation-inducing agents.


(A) Representative western blot analysis of lysates from SH-SY5Y cells treated with increasing concentrations of H2O2. The blot was partitioned and probed with anti-DJ-1 or anti-actin, respectively. (B) Quantification of normalized DJ-1 content after H2O2 treatment; n = 8 independent biological replicates. (C) Representative western blot analysis of lysates from SH-SY5Y cells pretreated with DMSO, or a broad protease inhibitor cocktail (PI, see Methods); then treated with PBS or 0.1 mM diamide. The blot was partitioned and probed with anti-DJ-1 or anti-actin, respectively. Lane 1, DMSO + PBS; lane 2, DMSO + diamide; lane 3, PI + PBS; lane 4, PI + diamide. (D) Quantification of normalized DJ-1 content after diamide and PI treatments; n=8 independent biological replicates. (E) Representative western blot analysis of lysates from SH-SY5Y cells pretreated with DMSO, MG-132, or lactacystin; then treated with PBS or 0.1 mM diamide. The blot was partitioned and probed with anti-β-catenin, anti-DJ-1, and anti-actin, respectively. (F) Quantification of DJ-1 protein content (normalized to actin); n = 8 independent biological replicates. Statistical analyses for quantifications shown in (B), (D), and (F) were completed using ANOVA followed by Tukey’s post hoc test. Error bars on quantification for (B), (D), and (F) represent SEM.
DJ-1 is glutathionylated in vivo
DJ-1 contains three cysteine residues located at positions 46, 53, and 106. Cysteines 53 and 106 have been reported previously to be oxidatively modified [23, 30], but there is no evidence for glutathionylation of DJ-1 in a physiological setting. In order to determine if DJ-1 undergoes glutathionylation in vivo, DJ-1 was immunoprecipitated from whole mouse brain homogenates and subjected to non-reducing SDS-PAGE and western blot analysis. Using an antibody that recognizes glutathionylated moieties (anti-GSH), we observed a prominent band at approximately 45 kDa (Figure 3 A, arrow). The blot was stripped and re-probed with an anti-DJ-1 antibody, which resulted in a band at the same molecular weight as recognized by the anti-GSH antibody. (Figure 3 B). The band located at approximately 45 kDa corresponds to the size of the dimeric DJ-1. This suggests that dimeric DJ-1-SSG can form an intermolecular bond between two DJ-1 monomers either in the cell or while running through the electric field in the gel. In order to validate that the approximately 45 kDa band is DJ-1 and not a non-specific band, mouse brain homogenates were immunoprecipitated with anti-DJ-1 beads to isolate DJ-1, and the sample was split into two aliquots. One aliquot was reduced with 100 mM dithiothreitol (DTT) and the other was left non-reduced; then the two samples were analyzed on western blots. The ~45 kDa band appeared in the non-reduced lane, but in the reduced lane a single band was present at approximately 22 kDa, corresponding to the molecular weight of monomeric DJ-1 (Supplemental Figure S3A). In addition, western blot analyses with the anti-DJ-1 antibody were performed with DTT-treated lysates of brain samples from WT and DJ-1−/− mice (a kind gift from Vera Bonilha, Cleveland Clinic Foundation). An immunoreactive band appeared at approximately 22 kDa as expected for the WT samples; but no band was present for the samples from the DJ-1−/− mice, documenting the specificity of the anti-DJ-1 antibody (Supplemental Figure 3S B).
Figure 3. DJ-1 is glutathionylated in vivo and in vitro.

(A) Representative western blot analysis of immunopreciptated DJ-1 from mouse brain homogenate probed with anti-glutathione (GSH) antibody. Replicate analyses of four independent biological samples gave similar results. Arrow denotes a band of immunoreactivity at approximately 45 kDa. (B) Immunopreciptated DJ-1 recognized by anti-DJ-1 antibody. The blot shown in (A) was stripped and re-probed with anti-DJ-1 antibody. Again, this was replicated with the four different samples. Arrow denotes a band of immunoreactivity at approximately 45 kDa. Overlay of the films shown in panels A and B documented coincidence of the 45 kDa band, representing glutathionylated DJ-1. (C) Electrospray ionization mass spectrum of intact recombinant DJ-1. The mass spectrum shows a distribution of charged states (+11 to +28) for intact DJ-1 protein. Deconvolution of the spectrum reveals a protein mass of 20036.3 Da, which is identical to the theoretical mass of recombinant DJ-1. (D) Electrospray ionization mass spectrum of glutathionylated DJ-1. Incubation of DJ-1 protein with 5 mM GSH and 1 mM diamide led to an increase in protein mass of 305-Da (blue ion series, deconvoluted mass = 20341.5 Da) or 610-Da (red ions series, deconvoluted mass = 20646.7 Da), corresponding to covalent modification of DJ-1 with one or two glutathione moieties, respectively.
Glutathionylation of proteins can be reversed by disulfide reducing agents. To confirm the interpretation of the reactivity of the anti-glutathione antibody, DJ- was immunoprecipitated from mouse brains, run on a non-reducing SDS-PAGE gel and transferred to a PVDF membrane. After transfer half of the blot was then incubated with 100 mM dithiothreitol (DTT) for 30 min to reduce glutathionylated cysteines. The blot incubated with DTT lost immunoreactivty with the anti-GSH antibody(Supplemental Figure 4S). Blots were then stripped and re-probed for DJ-1 confirming the presence of DJ-1 on both halves of the membrane. These data show that treatment with DTT results in ablation of reactivity with the anti-glutathione antibody, consistent with the conclusion that DJ-1 is glutathionylated within the midbrains of mice. It was also confirmed separately that actin which is known to be glutathionylated in intact cells[ 31] and has a similar molecular weight (42 kDa) to dimeric DJ-1 was not detected on the blot of the immunoprecipitated DJ-1 (data not shown).
Preferential glutathionylation of DJ-1 at cysteines 53 and 106
To further characterize the glutathionylation of DJ-1, 12.5 μg of pure recombinant DJ-1 was incubated with 5 mM GSH and 1 mM diamide for 30 min and subjected to liquid chromatography-mass spectrometry (LC-MS) analysis. MS spectra of the untreated (Figure 3 C) and treated (Figure 3 D) intact protein revealed two distinct sets of ions in the treated sample as compared to untreated DJ-1. The deconvoluted spectrum of untreated DJ-1 revealed the expected protein mass of 20036.3 Da, equal to the theoretical mass of the polypeptide based on the amino acid sequence. Reaction with GSH and diamide resulted in increases of 305 and 610 Da in the protein mass to 20341.5 and 20646.7 Da, corresponding to the additional masses of a single and two glutathionyl moieties covalently attached to DJ-1 (Figure 3 D).
To identify the sites of glutathione adduction, the glutathionylation reaction was repeated; the DJ-1 reaction mixture was dialyzed to remove GSH/diamide, digested with trypsin, and subjected to tandem MS/MS analysis. The trypsin digests clearly revealed the presence of two covalently modified peptides DVVIC53PDASLEDAK (m/z = 766.5 [M+2H]+2) and GLIAAIC106AGPTALLAHEIGFGSK (m/z = 756.4 [M+3H]+3) in the GSH/diamide treated sample (Figure 4 C–D) as compared to the unmodified DJ-1 (Figure 4 A–B). The identity of each of the peptides and sites of modification were confirmed by their MS/MS fragmentation patterns. A 305-Da shift in the b- and y-ion series observed between the modified and unmodified peptides indicated glutathionylation on the side chains of the Cys-53 (Figure 4 E) and Cys-106 (Figure 4 F) residues.
Figure 4.

Identification of the glutathionylation sites of DJ-1 by tandem mass spectrometry. Selective Ion chromatographs of tryptic digest of DJ-1 treated with 5 mM GSH and 1 mM diamide revealed two Cys containing peptides (C) DVVIC53PDASLEDAK (m/z = 766.5 [M+2H]+2) and (D) GLIAAIC106AGPTALLAHEIGFGSK (m/z = 1258.2 [M+2H]+2), with corresponding masses increased by 305-Da as compared to the untreated sample shown in (A) and (B). (E) and (F): MS/MS fragmentation pattern of these peptides identifies Cys53 and Cys106 residues as the sites of glutathionylation. According to Biemann nomenclature, the series of b and y ions are identified in the mass fragmentograms and in the diagrams of the Cys-containing peptides at the top of panels E and F. For example, the site of glutathionylation at Cys-53 is identified by the loss of 408 mass units for the y9 ion versus the y10 ion, corresponding to the loss of the cysteinylglutathione moiety.
In order to estimate the relative extents of glutathionylation of the different cysteine residues, “light DJ-1” (14N, natural abundance) was treated with GSH and diamide. An equimolar amount of “heavy DJ-1” (15N-labeled) was untreated and used as an internal standard. Both 14N- and 15N-proteins were digested with trypsin, and subjected to LC-MS analysis (Supplemental Figure 5S, A–E). Quantification of the peak areas for the non-modified peptides indicated that major glutathionylation occurs at cysteine residues Cys53 and Cys106. The extent of glutathionylation was estimated by subtracting the peak area of the treated non-modified peptide (14N) from the peak area of the untreated standard peptide (15N), giving a value for the peak area of the glutathionylated peptide. Accordingly, the extent of glutathionylation of Cys53 and Cys106 were calculated to be 70 and 77%, respectively (Table 1). We did find a very minor fraction (~7%) of glutathionylated Cys46 (Table 1), but this was likely due to heterogeneity within the preparation of DJ-1 and/or unfolding of the protein in solution, as Cys46 is known to be buried in the hydrophobic core of the protein [ 32]. Taken together, our data show that isolated recombinant DJ-1 is preferentially glutathionylated at cysteine resides 53 and 106.
Table 1.
| Site | Peptide | Native (Area) | N15-labeled(Area) | Modified (%) |
|---|---|---|---|---|
| C46 | DPVQCSR | 14,214,288 | 15,332,719 | 7.3 |
| C53 | DVVICPDASLEDAK | 69,277,937 | 229,809,312 | 70.0 |
| C106 | GLIAAICAGPTALLAHEIGFGSK | 268,610,760 | 1,179,067,215 | 77.2A |
Quantitation of estimated glutathionylation of cysteines of DJ-1. Glutathionylation of cysteine residues 46, 53, and 106 were estimated using unmodified 15N DJ-1 as an internal standard. Quantification of modified tryptic peptides were estimated by subtraction of the peak areas of the non-modified peptides in the treated 14N-samples from those in the untreated 15N-samples (Supplemental Figure 3).
Example of calculation of extent of glutathionylation of Cys106: 1,179,067,215 (15N non-modified area) - 268,610,760 (14N non-modified area) = 910,456,455 (estimated area of the glutathionylated peptide). 910,456,455 (estimated are of the glutathionylated peptide) / 1,179,067,215 (15N non-modified area) × 100 = 77.2% (Estimated percentage of glutathionylated peptide as compared to total internal standard).
Loss of DJ-1 homolog exacerbates LRRK2-mediated dysfunction of dopamine-dependent movement in C. elegans
Overexpression of DJ-1 has been shown to mediate dopaminergic neuronal protection against LRRK2 toxicity in vitro [ 33]. In order to investigate the potential neuroprotective role of DJ-1 in vivo, we chose to use C. elegans as a model because of the simplicity of the dopaminergic neuronal system. C. elegans worms contain two distinct homologs to human DJ-1, DJR-1.1 and DJR-1.2. Two independent groups have reported that DJR-1.1 is expressed highly in the intestine and not within the head neurons [34, 35]. DJR-1.2, in contrast, has been reported to be expressed in the dopaminergic neurons of the worms [35]. Therefore, we used DJR-1.2-deficient animals in this study. Genetic crosses were made between the djr-1.2−/− strain and transgenic animals expressing GFP in the dopaminergic neurons (used as WT control). In addition, the djr-1.2−/− strain was crossed with lines that co-express GFP and pathogenic human LRRK2 mutants in the dopaminergic neurons (G2019S-LRRK2 or R1441C-LRRK2, hereafter referred to as G2019S and R1441C, respectively) [6]. Crosses were confirmed via PCR genotyping of djr-1.2 and LRRK2(Figure 5A).
Figure 5. Loss of DJR-1.2 induces PD phenotypes in C. elegans, exacerbates mutant LRRK2-mediated impairment of dopamine dependent behavior, but does not consistently exacerbate mutant LRRK2-mediated dopaminergic neurodegeneration.

(A) Representative PCR analysis of LRRK2 (upper panel) and WT djr-1.2 or mutant djr-1.2 DNA (lower panel) in C. elegans lines. Primers of djr-1.2 were designed to generate two distinct PCR products depending on the inclusion (upper band) or exclusion (lower band) of the first exon of djr-1.2 in the C. elegans lines. (B) Basal slowing data demonstrating that loss of DJR-1.2 results in dysfunction of dopamine-dependent movement, and exacerbates R1441C- and G2019S-mediated dysfunction of dopamine-dependent movement; *p<0.05, comparing control to djr-1.2−/−; #p<0.05, comparing R1441C to R1441C; djr-1.2−/−; &p<0.05, comparing G2019S to G2019S; djr-1.2−/−. (C) Dopaminergic degeneration data demonstrating that loss of DJR-1.2 results in late onset degeneration, but does not exacerbate mutant LRRK2-mediated degeneration consistently. ^p<0.05 comparing control to djr-1.2−/−, $p<0.05 comparing R1441C to R1441C; djr-1.2−/−. For (B) and (C) error bars represent SEM. n = 3 independent groups of worms for all days, ~10 worms (B) or ~30 worms (C) per genotype per experiment. Statistical analysis was completed using one-way ANOVA followed by Tukey’s post-hoc test. Statistical comparisons between WT and LRRK2 expressing lines recapitulated previously published experimental data [20], and were not included to simplify the figure.
We examined the functional integrity of dopaminergic signaling in the C. elegans lines using a well-established assay of dopamine-dependent behavior, termed the basal slowing assay [36]. In agreement with our previous findings, transgenic expression of either G2019S or R1441C in the dopaminergic neurons of C. elegans resulted in age-dependent loss of dopamine-dependent movement behavior [6, 20]. Compared to WT, the mutant djr-1.2−/− worms displayed a late onset loss of dopamine-dependent behavior on adult day 2 (Figure 5B). These data are in agreement with the previous finding that dauer-stage C. elegans lacking djr-1.2 have a dopamine signaling defect [35]. Moreover, the hybrid lines, both deficient in DJR-1.2 and expressing G2019S or R1441C, displayed a more robust loss of dopamine-dependent behavior (Figure 5B). We also examined the morphological integrity of the GFP labeled-dopaminergic neurons via microscopy in young and aged worms. Expression of G2019S or R1441C resulted in an age dependent loss of dopaminergic neurons as previously documented [ 6, 20]. The djr-1.2−/− mutant strain showed decreased survival of dopaminergic neurons compared to WT on adult day 8 (Figure 5C). Exacerbation of dopaminergic neuronal loss in the lines lacking DJR-1.2 and expressing LRRK2 mutants was limited and seen only on adult day 2 in the R1441C; djr-1.2−/− line. These data indicate that loss of DJR-1.2 in vivo results in diminution of dopamine-dependent behavior, exacerbation of mutant LRRK2-induced diminution of dopamine-dependent behavior, but only incomplete dopaminergic degeneration and limited exacerbation of LRRK2 -mediated dopaminergic degeneration.
Over-expression of DJ-1 homolog partially compensates for the loss of Grx1 in R1441C-mediated neurotoxicity
We previously found that loss of GLRX-10, the worm homolog of Grx1, exacerbates LRRK2-mediated dopaminergic dysfunction [20]. Our current data analyzing midbrain homogenate from Grx1−/− mice indicates that loss of Grx1 results in a decreased protein content of DJ-1 in vivo(Figure 1 A, B), indicating that regulation of DJ-1 content is functionally downstream of Grx1. Synthesizing these observations, we hypothesized that the exacerbation of LRRK2 toxicity by deficiency of the Grx1 homolog in C. elegans [20] is due, at least in part, to increased glutathionylation and consequent loss of DJ-1. In order to test if DJ-1 overexpression could rescue the Grx1 deficiency, we generated R1441C; glrx-10−/− worms specifically expressing DJR-1.2 fused to red fluorescent protein mCherry (DJR-1.2-mCherry) or mCherry (vector control) in the dopaminergic neurons. The compound worm lines were confirmed via PCR (Figure 6A) and fluorescence microscopy (Figure 6B). These worms were subjected to the basal slowing assay to assess their dopamine-dependent behavior.
Figure 6. Over-expression of DJR-1.2 partially compensates for the loss of GLRX-10.

(A) PCR analysis of LRRK2 (upper panel), WT or mutant glrx-10 DNA (middle panel), and coding DNA sequence (CDS) of djr-1.2 (lower panel) indicating the generation of R1441C; glrx-10−/−; vector (mCherry) and R1441C; glrx-10−/−; DJR-1.2-mCherry expressing C. elegans lines. (B) Fluorescent images of the head region of the newly generated C. elegans depicting the expression of Pdat-1::mCherry and Pdat-1::DJR-1.2:mCherry in the dopaminergic neurons. Expression of GFP tagged dopaminergic neurons co-localize with expression of both Pdat-1::mCherry and Pdat-1::DJR-1.2::mCherry indicating dopaminergic expression of mCherry and DJR-1.2-mCherry, respectively. (C) Basal slowing data demonstrating that over-expression of DJR-1.2 partially recues the LRRK2-R1441C mediated dysfunction of dopamine-dependent movement in the glrx-10−/− background. *p<0.05 compared to R1441C; glrx-10−/−; vector. Error bars represent SEM. n = 3 independent groups of worms for all days, 10 worms per genotype per experiment. Statistical analysis was completed using one way ANOVA followed by Tukey’s post-hoc test.
Consistent with the previous report [20], R1441C; glrx-10−/− worms displayed a robust loss in dopamine-dependent behavior. Expression of the mCherry control in the R1441C; glrx-10−/− line resulted in no change in basal slowing response compared to its cognate strain (Figure 6C). Overexpression of DJR-1.2-mCherry in the dopaminergic neurons of the R1441C; glrx-10−/− worms showed significant improvement in basal slowing response compared to the R1441C; glrx-10−/− worms expressing the vector control on adult Day 0, and continued to show apparent but not statistically significant improvement on adult days 1 and 2. These data indicate that expression of DJR-1.2 can partially rescue the pathogenic LRRK2 induced dysfunction of dopamine-dependent behavior in C. elegans lacking GLRX-10.
Discussion
DJ-1 plays an important role in mediating dopaminergic neuronal protection. De -stabilizing mutations in DJ-1 result in familial PD, and diminished protein content of DJ-1 has been reported in sporadic cases of PD [ 10, 11]. Furthermore, knockout or knockdown of DJ-1 results in increased susceptibility to toxin-induced dopaminergic cell death in vivo [37, 38]. These studies indicate that protein levels of DJ-1 play an important role in protecting against dopaminergic degeneration. Hence, deciphering the mechanisms of regulation of DJ-1 protein content represents an important focus of study in PD biology.
Reversible glutathionylation of proteins is recognized as an important regulatory mechanism for pathways that mediate cell death/viability [ 39]. For example, the FAS receptor is a signal transducing receptor that upon activation via Fas-ligand drives cell death through activation of the death domain. Fas-induced cell death has been shown to be regulated in part by glutathionylation at cysteine 294 [17]. Treatment of lung fibroblasts with Fas-ligand resulted in cell death when WT-Fas, but not C294A-Fas was expressed [ 17], suggesting that glutathionylation of Fas is a pre-requisite for cell death signal transduction. In addition, glutathionylation of the cell survival kinase Akt has been associated with decreased Akt activation and reduced cell viability in the human fetal retinal pigment epithelial cells when challenged with H2O2 [ 40]. Conversely, overexpression of Grx1 resulted in decreased levels of Akt glutathionylation in response to H2O2 and increased activation of Akt signaling [ 40]. These and other studies demonstrate the functional impact of selective protein glutathionylation on cell viability under stress conditions.
Our previous data revealed that Grx1 protein content was decreased in post-mortem samples of brain tissue from[ 20]. Our current data supports the interpretation that Grx1 regulates DJ-1 protein content in vivo and that DJ-1 is susceptible to glutathionylation in vitro and in vivo. Additional data supporting the idea that DJ-1 is regulated by glutathionylation have been reported. Unlike WT DJ-1, the C106A and C53A mutants of DJ-1 are resistant to degradation in Neuro2A cells in which Grx1 was knocked down [22]. Our current finding that cysteine residues 53 and 106 are more readily glutathionylated than cysteine 46 is consistent with data from the crystal structure of DJ-1 indicating that cysteine 46 is not solvent accessible and buried within the protein [32]. We hypothesize that glutathionylation of DJ-1 regulates its protein level in the cell. Consistent with this interpretation, treatment of SH-SY5Y cells with two known inducers of glutathionylation resulted in decreased DJ-1 protein content. This decrease in DJ-1 protein can be abrogated by pre-treatment with non-specific protease inhibitors suggesting that the loss of DJ-1 is caused by proteolytic degradation following glutathionylation. The result is in agreement with previously published data indicating that loss of Grx1 via shRNA knockdown in Neuro2A cells results in a loss of DJ-1 protein content [22]. In order to identify which component(s) of the proteolytic inhibitor cocktail blocked DJ-1 degradation and therefore which pathway(s) is dysregulated, we treated Neuro2A cells with the individual components and then knocked down Grx1 via shRNA. As expected, knockdown of Grx1 resulted in a loss of DJ-1 protein content; however none of the individual components alone blunted the Grx1 knockdown-mediated loss of DJ-1 (Supplemental Figure 7S, A–F). These data suggest a complex mechanism of degradation likely involving multiple enzymes contributing to regulation of DJ-1 degradation.
In order to test whether DJ-1 plays a functional role in regulating dopaminergic viability in concert with Grx1in vivo we used C. elegans as a model system. We previously found that C. elegans lacking GLRX -10, the Grx1 homolog, displayed an exacerbation of the neurodegenerative phenotype in both familial and sporadic models of PD [ 20]. Our observation of decreased levels of DJ-1 in Grx1−/− mice led us to hypothesize that decreased DJ-1 could be at least partly responsible for the more toxic PD-like phenotype in the C. elegans lines lacking GLRX-10. Protein content of DJ-1 within the glrx-10−/− worms was not determined because the antibodies for human DJ-1 failed to specifically recognize C. elegans DJR-1.2 (data not shown). As an alternative approach to test if an elevated content of DJ-1 could compensate for the loss of GLRX-10 in C. elegans, DJR-1.2 was specifically expressed in the dopaminergic neurons of R1441C-LRRK2; glrx10−/− worms. Using the well-established basal slowing assay to determine integrity of dopamine-dependent behavior, we found that expression of DJR-1.2 could partially compensate for the loss of GLRX-10. Indeed, the protective effect of DJ-1 appears to occur early in the life of the worm and then fades in adulthood (Figure 6C). Fluorescence microscopic analysis was conducted on adult days 0 and 2 to assess levels of mCherry in control worms or in worms expressing exogenous mCherry-tagged DJ-1 protein. Quantification of mCherry/GFP intensity showed no difference in the expression of exogenous mCherry in the control lines on adult days 0 and 2 as expected (Supplemental Figure S6, left panel). Notably there was also no change in the mCherry/GFP ratio in the DJ-1:mCherry worms (Supplemental Figure S6, right), indicating that diminution of DJ-1 mediated protection with age is not due to a loss of the exogenous DJ-1 protein. These data suggest several alternative hypotheses for why the effect of the overexpressed DJ-1 fades with time, including (a) the levels of exogenous DJ-1 fail to sustain protection due to an increased burden of ROS over time in these LRRK2-expressing worms: (b) DJ-1 is functionally inactivated in the worms due to increased ROS; and (c) the DJ-1 mediated dopaminergic neuronal protection involves co-factors that may be deactivated by glutathionylation in the absence of the Grx1 homolog. One or more of these hypotheses may be supported by previous findings. For example, it has been reported that pathogenic LRRK2 drives increased ROS expression in vitro [ 41]; DJ-1 has been reported to exist in a (possibly inactive) sulfonic acid state after oxidative challenge [42]; the glutathionylated protein database [ 43] reports that the DJ-1 binding partner thioredoxin-1 can be glutathionylated. Taken together these observations provide a plausible explanation for the transient protective effect of elevation of DJR-1.2 content, contributing some compensation for loss of the Grx1 homolog and some protection against mutant LRRK2-mediated toxicity in vivo.
Although DJ-1 provides some protection against exacerbation of LRRK2 toxicity by glutaredoxin deficiency, the lack of complete rescue indicates that other protein(s) also likely play a role in mediating Grx1 neuronal protection. For example, Akt1 is a well-established survival kinase that has been shown to play neuroprotective roles in a variety of contexts including PD [ 44]. Notably, the G2019S- and R1441C-LRRK2 mutants showed a decreased ability to phosphorylate and activate Akt1 in vitro, suggesting that pathogenic mutations of LRRK2 drive toxicity at least in part through loss of a critical cell survival signaling mechanisms involving Akt [ 45]. Proper activation/regulation of Akt is critical for Akt mediated protection. Considering the context of Grx1 deficiency, it is important to note that Akt1 has been reported in other contexts to be regulated both directly [40] and likely indirectly [46] through reversible protein glutathionylation. Thus, treatment of human retinal pigment epithelial cells with H2O2 resulted in Akt1 glutathionylation, impaired phosphorylation of Akt1, and cell death [40]. Over-expression of Grx1 resulted in decreased levels of glutathionylated Akt1, normalized phosphorylation/activation of Akt1, and increased cell viability when challenged with H2O2 [40]. These data indicate that glutathionylation of Akt itself or of regulators of Akt may interfere with its cell-survival activity. In particular, Akt1 is positively regulated through phosphorylation by the upstream kinase PKC [47], and PKC itself appears to be negatively regulated by glutathionylation [43]. Thus, purified recombinant PKC treated with diamide and 35S-GSH was found to be glutathionylated and inactivated. Furthermore, glutathionylation of intracellular PKC was confirmed by documenting DTT-reversible incorporation of 35S-radioactivity into PKC that was immunoprecipitated from diamide-treated NIH3T3 cells in which GSH had been radiolabeled [46]. These data suggest that glutathionylation and concomitant inactivation of PKC would decrease Akt1 phosphorylation and result in an impaired ability to respond to stress. Taken all together, loss of Grx1within dopaminergic neurons besides affecting cell viability through diminution of DJ-1 may also result in an impaired stress response due to hyper-glutathionylation of PKC and Akt. The increased glutathionylation and consequent inactivation of DJ-1 and the Akt pathway may combine to explain the exacerbated toxicity seen when the Grx1 homolog is genetically ablated in models of familial and sporadic PD.[ 20]. Hence, further studies are warranted to examine the synergistic impact of dysregulation of glutathionylation on neuronal cell survival mechanisms involving DJ-1 and Akt.
Therapeutic advancement in the general PD population has not changed greatly since the introduction of levodopa as the primary therapy for PD in the 1960’s. Currently there are no therapeutic approaches available to slow the progression of dopaminergic degeneration. The ideal intervention would slow or even reverse the degeneration of dopaminergic neurons by targeting a dysregulated pathway common to both familial and sporadic PD. Although familial mutations represent only approximately 10 percent of all PD cases, post-translational modifications mimicking the functional consequences of the genetic mutations may play a role in the development of sporadic PD. In our previous study post-mortem PD brain tissue was found to have diminished levels of Grx1 [20], supporting the possibility that glutathionylation of key proteins involved in PD development/progression may contribute to PD pathogenicity. Our current data are consistent with the idea that glutathionylation of DJ-1 results in its degradation leading to decreased protein levels. This loss of DJ-1 protein mimics the outcome of the pathogenic L166P mutation that results in reduced steady-state levels of DJ-1 [ 48]. A search of the database dbGSH reveals that several other proteins implicated in the etiology of PD are susceptible to glutathionylation [43]. These proteins include VPS35, UCH-L1, and EIF4G1. Although these proteins can be glutathionylated, it is unclear if glutathionylation affects the function of these proteins in an analogous manner to pathogenic mutations, so further study is warranted. Nevertheless, the potential impact of deficient Grx1 on glutathionylation status and function of proteins implicated in PD suggests that pharmacologically increasing Grx1 activity in neurons may decrease dopaminergic toxicity by regulating not only DJ-1 protein content, but also other downstream targets that are altered via glutathionylation.
Statistical analysis
Statistical analyses of the reported data, as noted in the figure legends, were performed using ANOVA followed by Tukey’s post hoc test, Student’s t-Test, or one-way linear regression. All statistical analyses were run using Microsoft Excel or GraphPad Prism.
Supplementary Material
Acknowledgments
We thank Sandra L. Siedlak (Case Western Reserve University) for expert technical assistance and Dr. Vera Bonilha (Cleveland Clinic Foundation) for brain tissue samples from DJ-1−/− mice. We are grateful to Dr. Shasta Sabo (Case Western Reserve University) for her critical feedback on the design of experiments as well as feedback on the manuscript. The collection of PBMC samples was made possible by the Clinical and Translational Science Collaborative of Cleveland, UL1TR000439 from the National Center for Advancing Translational Sciences component of the National Institutes of Health and NIH roadmap for Medical Research. Part of the human brain tissue was obtained from the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland. The C. elegans djr -1.2−/− strain (tm1346) and glrx-10−/− strain (tm4634) were obtained from National Bioresource Project (Japan). The C. elegans WT reference strain N2 Bristol and E. coli strain OP50 were obtained from the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health Office of Research Infrastructure Programs (P40 OD010440).
Funding
This work was supported in part by the National Institutes of Health (grant NS073170 to A.L.W. and S.G.C.; grant NS085503 to A.L.W., J.J.M. and S.G.C.; grant NS083498 to X.Z.; grant R01GM092999 to M.A.W.; grant EY023948 to M.G.; and pre-doctoral fellowship T32 GM008803 to W.M.J.), Parkinson’s Disease Foundation (summer student fellowship PDF-SFW-1348 to W.M.J. and PDF-SFW-1566 to P.L.C.), the Department of Veterans Affairs (merit review grant BX000290 to J.J.M.), the National Science Foundation Advance Institutional Transformation Program (ACES research opportunity grant to S.G.C.), and the Chinese Overseas, Hong Kong and Macao Scholars Collaborated Researching Fund (grant 81228007 to X.Z.). The contents of publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIH and other funding agencies.
Abbreviations
- ATCC
American Type Culture Collection
- C. elegans
Caenorhabditis elegans
- Grx1
Glutaredoxin 1
- GSH
Glutathione
- GSSG
Glutathione disulfide
- Protein-SSG
Glutathionylated protein
- HPLC
High performance liquid chromatography
- PBMCs
Human peripheral blood mononuclear cells
- DJ-1-SSG
Glutathionylated DJ-1
- LRRK2
Leucine Rich Repeat Kinase 2
- LC-MS
Liquid chromatography-mass spectrometry
- m/z
Mass/charge ratio
- MS
Mass spectrometry (mass spectrometric)
- NGM
Nematode growth media
- PD
Parkinson’s disease
- TBST
Tris-buffered saline with 0.05% Tween-20
Footnotes
Conflict of Interest statement: None declared
Supporting Information Available
Supplemental items of data include western blot and qPCR analyses for Grx1−/− and hGrx1-transgenic mice, western blot analyses of human samples, validation of purity and specificity of antibodies, HPLC-mass spectrometric chromatograms, fluorescence data for quantification of expression of exogenous DJ-1 in C. elegans, and cell culture data for individual components of the proteolytic inhibitor cocktail. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Lees AJ, Hardy J, Revesz T. Parkinson’s disease. Lancet. 2009;373:2055–2066. doi: 10.1016/S0140-6736(09)60492-X. [DOI] [PubMed] [Google Scholar]
- 2.Johnson WM, Wilson-Delfosse AL, Chen SG, Mieyal JJ. The roles of redox enzymes Parkinson’s disease: Focus on glutaredoxin. Ther Targets Neurol Dis. 2015:2. [PMC free article] [PubMed] [Google Scholar]
- 3.Li JQ, Tan L, Yu JT. The role of the LRRK2 gene in Parkinsonism. Mol Neurodegener. 2014:47. doi: 10.1186/1750-1326-9-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ariga H, Takahashi-Niki K, Kato I, Maita H, Niki T, Iguchi-Ariga SM. Neuroprotective function of DJ-1 in Parkinson’s disease. Oxid Med Cell Longev. 2013;2013:683920. doi: 10.1155/2013/683920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Angeles DC, Ho P, Chua LL, Wang C, Yap YW, Ng C, Zhou Z, Lim KL, Wszolek ZK, Wang Y, Tan EK. Thiol peroxidases ameliorate LRRK2 mutant-induced mitochondrial and dopaminergic neuronal degeneration in Drosophila. Hum Mol Genet. 2014;23:3157–3165. doi: 10.1093/hmg/ddu026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yao C, El Khoury R, Wang W, Byrd TA, Pehek EA, Thacker C, Zhu X, Smith MA, Wilson-Delfosse A, Chen SG. LRRK2-mediated neurodegeneration and dysfunction of dopaminergic neurons in a Caenorhabditis elegans model of Parkinson’s disease. Neurobiol Dis. 2010;40:73–81. doi: 10.1016/j.nbd.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Y, Liu W, Oo TF, Wang L, Tang Y, Jackson-Lewis V, Zhou C, Geghman K, Bogdanov M, Przedborski, Beal MF, Burke RE, Li C. Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson’s disease. Nat Neurosci. 2009;12:826–828. doi: 10.1038/nn.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramonet D, Daher JP, Lin BM, Stafa K, Kim J, Banerjee R, Westerlund M, Pletnikova O, Glauser, Yang L, Liu Y, Swing DA, Beal MF, Troncoso JC, McCaffery JM, Jenkins NA, Copeland N, Galter D, Thomas B, Lee MK, Dawson TM, Dawson VL, Moore DJ. Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing mutant LRRK2. PLoS One. 2011;6:e18568. doi: 10.1371/journal.pone.0018568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yue M, Hinkle KM, Davies P, Trushina E, Fiesel FC, Christenson TA, Schroeder AS, Zhang, Bowles E, Behrouz B, Lincoln SJ, Beevers JE, Milnerwood AJ, Kurti A, McLean PJ, Fryer J, Springer W, Dickson DW, Farrer MJ, Melrose HL. Progressive dopaminergic alterations mitochondrial abnormalities in LRRK2 G2019S knock-in mice. Neurobiol Dis. 2015;78:172–195. doi: 10.1016/j.nbd.2015.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alvarez-Castelao B, Munoz C, Sanchez I, Goethals M, Vandekerckhove J, Castano JG. Reduced protein stability of human DJ-1/PARK7 L166P, linked to autosomal recessive Parkinson disease, is due direct endoproteolytic cleavage by the proteasome. Biochim Biophys Acta. 2012;1823:524–533. doi: 10.1016/j.bbamcr.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 11.Kumaran R, Vandrovcova J, Luk C, Sharma S, Renton A, Wood NW, Hardy JA, Lees AJ, Bandopadhyay R. Differential DJ-1 gene expression in Parkinson’s disease. Neurobiol Dis. 2009;36:393–400. doi: 10.1016/j.nbd.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 12.Taira T, Saito Y, Niki T, Iguchi-Ariga SM, Takahashi K, Ariga H. DJ-1 has a role antioxidative stress to prevent cell death. EMBO Rep. 2004;5:213–218. doi: 10.1038/sj.embor.7400074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Im JY, Lee KW, Woo JM, Junn E, Mouradian MM. DJ-1 induces thioredoxin 1 expression through the Nrf2 pathway. Hum Mol Genet. 2012;21:3013–3024. doi: 10.1093/hmg/dds131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilson MA. The role of cysteine oxidation in DJ-1 function and dysfunction. Antioxid Redox. 2011;15:111–122. doi: 10.1089/ars.2010.3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Waak J, Weber SS, Gorner K, Schall C, Ichijo H, Stehle T, Kahle PJ. Oxidizable residues mediating protein stability and cytoprotective interaction of DJ-1 with apoptosis signal-regulating kinase 1. J Biol Chem. 2009;284:14245–14257. doi: 10.1074/jbc.M806902200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Canet-Aviles RM, Wilson MA, Miller DW, Ahmad R, McLendon C, Bandyopadhyay S, Baptista MJ, Ringe D, Petsko GA, Cookson MR. The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc Natl Acad Sci U S A. 2004;101:9103–9108. doi: 10.1073/pnas.0402959101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anathy V, Aesif SW, Guala AS, Havermans M, Reynaert NL, Ho YS, Budd RC, Janssen-Heininger YM. Redox amplification of apoptosis by caspase-dependent cleavage of glutaredoxin 1 and S-glutathionylation of Fas. J Cell Biol. 2009;184:241–252. doi: 10.1083/jcb.200807019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qanungo S, Starke DW, Pai HV, Mieyal JJ, Nieminen AL. Glutathione supplementation potentiates hypoxic apoptosis by S-glutathionylation of p65-NFkappaB. J Biol Chem. 2007;282:18427–18436. doi: 10.1074/jbc.M610934200. [DOI] [PubMed] [Google Scholar]
- 19.Rodriguez-Rocha H, Garcia Garcia A, Zavala-Flores L, Li S, Madayiputhiya N, Franco R. Glutaredoxin 1 protects dopaminergic cells by increased protein glutathionylation in experimental Parkinson’s disease. Antioxid Redox Signal. 2012;17:1676–1693. doi: 10.1089/ars.2011.4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson WM, Yao C, Siedlak SL, Wang W, Zhu X, Caldwell GA, Wilson-Delfosse AL, Mieyal JJ, Chen SG. Glutaredoxin deficiency exacerbates neurodegeneration in C. elegans models of Parkinson’s disease. Hum Mol Genet. 2015;24:1322–1335. doi: 10.1093/hmg/ddu542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sabens EA, Distler AM, Mieyal JJ. Levodopa deactivates enzymes that regulate thiol-disulfide homeostasis and promotes neuronal cell death: implications for therapy of Parkinson’s disease. Biochemistry. 2010;49:2715–2724. doi: 10.1021/bi9018658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saeed U, Ray A, Valli RK, Kumar AM, Ravindranath V. DJ-1 loss by glutaredoxin but not glutathione depletion triggers Daxx translocation and cell death. Antioxid Redox Signal. 2010;13:127–144. doi: 10.1089/ars.2009.2832. [DOI] [PubMed] [Google Scholar]
- 23.Prahlad J, Hauser DN, Milkovic NM, Cookson MR, Wilson MA. Use of cysteine-reactive cross-linkers to probe conformational flexibility of human DJ-1 demonstrates that Glu18 mutations are dimers. J Neurochem. 2014;130:839–853. doi: 10.1111/jnc.12763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murdoch CE, Shuler M, Haeussler DJ, Kikuchi R, Bearelly P, Han J, Watanabe Y, Fuster JJ, Walsh K, Ho YS, Bachschmid MM, Cohen RA, Matsui R. Glutaredoxin-1 up-regulation induces soluble vascular endothelial growth factor receptor 1, attenuating post-ischemia limb revascularization. J Biol Chem. 2014;289:8633–8644. doi: 10.1074/jbc.M113.517219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Y, Shi W, Cui N, Wu Z, Jiang C. Oxidative stress inhibits vascular K(ATP) channels by S-glutathionylation. J Biol Chem. 2010;285:38641–38648. doi: 10.1074/jbc.M110.162578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zmijewski JW, Banerjee S, Abraham E. S-glutathionylation of the Rpn2 regulatory subunit inhibits 26 S proteasomal function. J Biol Chem. 2009;284:22213–22221. doi: 10.1074/jbc.M109.028902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takata T, Tsuchiya Y, Watanabe Y. 90-kDa ribosomal S6 kinase 1 is inhibited by S-glutathionylation of its active-site cysteine residue during oxidative stress. FEBS Lett. 2013;587:1681–1686. doi: 10.1016/j.febslet.2013.04.017. [DOI] [PubMed] [Google Scholar]
- 28.Butturini E, Darra E, Chiavegato G, Cellini B, Cozzolino F, Monti M, Pucci P, Dell’Orco D, Mariotto S. S-Glutathionylation at Cys328 and Cys542 impairs STAT3 phosphorylation. ACS Chem Biol. 2014;9:1885–1893. doi: 10.1021/cb500407d. [DOI] [PubMed] [Google Scholar]
- 29.Barrett WC, DeGnore JP, Konig S, Fales HM, Keng YF, Zhang ZY, Yim MB, Chock PB. Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry. 1999;38:6699–6705. doi: 10.1021/bi990240v. [DOI] [PubMed] [Google Scholar]
- 30.Girotto S, Sturlese M, Bellanda M, Tessari I, Cappellini R, Bisaglia M, Bubacco L, Mammi S. Dopamine-derived quinones affect the structure of the redox sensor DJ-1 through modifications at Cys-106 and Cys-53. J Biol Chem. 2012;287:18738–18749. doi: 10.1074/jbc.M111.311589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang J, Tekle E, Oubrahim H, Mieyal JJ, Stadtman ER, Chock PB. Stable and controllable RNA interference: Investigating the physiological function of glutathionylated actin. Proc Natl Acad Sci U S A. 2003;100:5103–5106. doi: 10.1073/pnas.0931345100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilson MA, Collins JL, Hod Y, Ringe D, Petsko GA. The 1.1-A resolution crystal structure of DJ-1, the protein mutated in autosomal recessive early onset Parkinson’s disease. Proc Natl Acad Sci U S A. 2003;100:9256–9261. doi: 10.1073/pnas.1133288100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heo HY, Park JM, Kim CH, Han BS, Kim KS, Seol W. LRRK2 enhances oxidative stress-induced neurotoxicity via its kinase activity. Exp Cell Res. 2010;316:649–656. doi: 10.1016/j.yexcr.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 34.Lee JY, Song J, Kwon K, Jang S, Kim C, Baek K, Kim J, Park C. Human DJ-1 and its homologs are novel glyoxalases. Hum Mol Genet. 2012;21:3215–3225. doi: 10.1093/hmg/dds155. [DOI] [PubMed] [Google Scholar]
- 35.Chen P, DeWitt MR, Bornhorst J, Soares FA, Mukhopadhyay S, Bowman AB, Aschner M. Age- and manganese-dependent modulation of dopaminergic phenotypes in a C. elegans DJ-1 genetic model of Parkinson’s disease. Metallomics. 2015;7:289–298. doi: 10.1039/c4mt00292j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sawin ER, Ranganathan R, Horvitz HR. C. elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron. 2000;26:619–631. doi: 10.1016/s0896-6273(00)81199-x. [DOI] [PubMed] [Google Scholar]
- 37.Kim RH, Smith PD, Aleyasin H, Hayley S, Mount MP, Pownall S, Wakeham A, You-Ten AJ, Kalia SK, Horne P, Westaway D, Lozano AM, Anisman H, Park DS, Mak TW. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc Natl Acad Sci U S A. 2005;102:5215–5220. doi: 10.1073/pnas.0501282102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiong R, Wang Z, Zhao Z, Li H, Chen W, Zhang B, Wang L, Wu L, Li W, Ding J, Chen S. MicroRNA-494 reduces DJ-1 expression and exacerbates neurodegeneration. Neurobiol Aging. 2014;35:705–714. doi: 10.1016/j.neurobiolaging.2013.09.027. [DOI] [PubMed] [Google Scholar]
- 39.Sabens Liedhegner EA, Gao XH, Mieyal JJ. Mechanisms of altered redox regulation in neurodegenerative diseases--focus on S--glutathionylation. Antioxid Redox Signal. 2012;16:543–566. doi: 10.1089/ars.2011.4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu X, Jann J, Xavier C, Wu H. Glutaredoxin 1 (Grx1) Protects Human Retinal Pigment Epithelial Cells From Oxidative Damage by Preventing AKT Glutathionylation. Invest Ophthalmol Vis Sci. 2015;56:2821–2832. doi: 10.1167/iovs.14-15876. [DOI] [PubMed] [Google Scholar]
- 41.Wang X, Yan MH, Fujioka H, Liu J, Wilson-Delfosse A, Chen SG, Perry G, Casadesus G, Zhu X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum Mol Genet. 2012;21:1931–1944. doi: 10.1093/hmg/dds003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kinumi T, Kimata J, Taira T, Ariga H, Niki E. Cysteine-106 of DJ-1 is the most sensitive cysteine residue to hydrogen peroxide-mediated oxidation in vivo in human umbilical vein endothelial cells. Biochem Biophys Res Commun. 2004;317:722–728. doi: 10.1016/j.bbrc.2004.03.110. [DOI] [PubMed] [Google Scholar]
- 43.Chen YJ, Lu CT, Lee TY. dbGSH: a database of S-glutathionylation. Bioinformatics. 2014;30:2386–2388. doi: 10.1093/bioinformatics/btu301. [DOI] [PubMed] [Google Scholar]
- 44.Aleyasin H, Rousseaux MW, Marcogliese PC, Hewitt SJ, Irrcher I, Joselin AP, Parsanejad M, Kim RH, Rizzu P, Callaghan SM, Slack RS, Mak TW, Park DS. DJ-1 protects the nigrostriatal axis from the neurotoxin MPTP by modulation of the AKT pathway. Proc Natl Acad Sci U S A. 2010;107:3186–3191. doi: 10.1073/pnas.0914876107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ohta E, Kawakami F, Kubo M, Obata F. LRRK2 directly phosphorylates Akt1 as a possible physiological substrate: impairment of the kinase activity by Parkinson’s disease-associated mutations. FEBS Lett. 2011;585:2165–2170. doi: 10.1016/j.febslet.2011.05.044. [DOI] [PubMed] [Google Scholar]
- 46.Ward NE, Stewart JR, Ioannides CG, O’Brian CA. Oxidant-induced S-glutathiolation inactivates protein kinase C-alpha (PKC-alpha): a potential mechanism of PKC isozyme regulation. Biochemistry. 2000;39:10319–10329. doi: 10.1021/bi000781g. [DOI] [PubMed] [Google Scholar]
- 47.Antico Arciuch VG, Alippe Y, Carreras MC, Poderoso JJ. Mitochondrial kinases in cell signaling: Facts and perspectives. Adv Drug Deliv Rev. 2009;61:1234–1249. doi: 10.1016/j.addr.2009.04.025. [DOI] [PubMed] [Google Scholar]
- 48.Moore DJ, Zhang L, Dawson TM, Dawson VL. A missense mutation (L166P) in DJ-1, linked to familial Parkinson’s disease, confers reduced protein stability and impairs homo-oligomerization. J Neurochem. 2003;87:1558–1567. doi: 10.1111/j.1471-4159.2003.02265.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.