

McLaughlin et al. uncover a motoneuronal Toll-6–directed pathway that functions via dSARM and FoxO to attenuate microtubule stability through repression of Pavarotti/MKLP1 transcription. Genetic and pharmacological strategies reveal a novel requirement for dynamic synaptic microtubules in structural plasticity, which are established by Toll-6–FoxO signaling.
Abstract
FoxO proteins are evolutionarily conserved regulators of neuronal structure and function, yet the neuron-specific pathways within which they act are poorly understood. To elucidate neuronal FoxO function in Drosophila melanogaster, we first screened for FoxO’s upstream regulators and downstream effectors. On the upstream side, we present genetic and molecular pathway analyses indicating that the Toll-6 receptor, the Toll/interleukin-1 receptor domain adaptor dSARM, and FoxO function in a linear pathway. On the downstream side, we find that Toll-6–FoxO signaling represses the mitotic kinesin Pavarotti/MKLP1 (Pav-KLP), which itself attenuates microtubule (MT) dynamics. We next probed in vivo functions for this novel pathway and found that it is essential for axon transport and structural plasticity in motoneurons. We demonstrate that elevated expression of Pav-KLP underlies transport and plasticity phenotypes in pathway mutants, indicating that Toll-6–FoxO signaling promotes MT dynamics by limiting Pav-KLP expression. In addition to uncovering a novel molecular pathway, our work reveals an unexpected function for dynamic MTs in enabling rapid activity-dependent structural plasticity.
Introduction
The cytoskeleton establishes and maintains key features of the neuronal phenotype. Neuronal microtubules (MTs) direct processes ranging from polarization and migration to axon guidance and dendrite branching (Dent et al., 2011; Kalil and Dent, 2014; Liu and Dwyer, 2014; Kapitein and Hoogenraad, 2015). Dynamic remodeling of the MT cytoskeleton is fundamental to its ability to control neuronal morphology and function. MT dynamics are regulated by posttranslational modifications of tubulin, behavior of MT motors, and interactions with MT-associated proteins (Janke and Kneussel, 2010; Kapitein and Hoogenraad, 2015). Of particular interest, mitotic kinesins have recently been shown to play key roles in anchoring, cross-linking, and sliding of neuronal MTs (Lin et al., 2012; del Castillo et al., 2015). The signaling mechanisms that modulate expression and function of these crucial MT regulators across development are not well defined.
FoxO proteins are evolutionarily conserved transcription factors active within receptor-mediated signaling cascades. Neuronal functions for FoxO transcription factors are of increasing interest because several FoxOs display prominent neuronal expression patterns, and foxO mutant neurons display anatomical and functional defects. FoxO proteins regulate neuronal polarity and neurite branching in mammalian development (de la Torre-Ubieta et al., 2010; Christensen et al., 2011) and maintain neuronal morphology and promote regeneration in adult Caenorhabditis elegans (Tank et al., 2011; Byrne et al., 2014). In Drosophila melanogaster, the sole FoxO orthologue directs neuronal MT organization (Nechipurenko and Broihier, 2012). Although FoxO is linked to insulin/insulin-like growth factor signaling in adult neurons, the neurodevelopmental pathways within which it acts are largely unknown.
Neurodevelopmental roles for Toll-like receptor (TLR) family members are just coming into focus. TLRs are best known as regulators of innate immunity and dorsoventral patterning (Ferrandon et al., 2004; Barak et al., 2014; Lindsay and Wasserman, 2014). Intriguingly, many TLRs are expressed in developing neurons (Okun et al., 2011). In Drosophila, Toll-6 and Toll-7 are expressed in motoneurons and have neuroprotective function (McIlroy et al., 2013). In the canonical pathway, TLRs inhibit Cactus/IκB to permit Dorsal/nuclear factor κB (NF-κB)–dependent transcription (Valanne et al., 2011; Troutman et al., 2012; Lindsay and Wasserman, 2014). Interestingly, neither Dorsal nor Cactus is present in motoneurons (Heckscher et al., 2007), implying that neuronal Toll receptors engage noncanonical pathways. Supporting the existence of novel signaling mechanisms, Toll-6 and Toll-7 act independently of canonical signaling mechanisms to instruct axon and dendrite targeting in the Drosophila olfactory system (Ward et al., 2015).
TLRs are composed of extracellular leucine-rich repeats and intracellular Toll/interleukin-1 receptor (TIR) domains. Cytoplasmic TIR domains of TLRs interact with TIR domain–containing adaptors to drive downstream signaling. Signaling specificity arises from distinct recruitment and activation of five mammalian TIR domain–containing adaptors (O’Neill and Bowie, 2007). However, only two of these adaptors, Myd88 and SARM, have been identified in Drosophila. Drosophila Myd88 has been studied for its role in innate immunity, but its functions are not well characterized in the central nervous system (CNS; Horng and Medzhitov, 2001; Tauszig-Delamasure et al., 2002). dSARM/SARM-1 is expressed in the fly and mouse nervous systems, where it is required for degeneration after axonal injury (Osterloh et al., 2012; Gerdts et al., 2013). Recent studies have elucidated neuronal functions of SARM family members during development. In hippocampal neurons, SARM1 interacts with Syndecan-2 to control dendrite morphology (Chen et al., 2011). TIR-1, the C. elegans orthologue of SARM, controls neuronal asymmetry and regulates odorant receptor expression (Chuang and Bargmann, 2005; Chang et al., 2011). Neurodevelopmental functions for Drosophila dSARM have not been described.
In this study, we first set out to define the FoxO-dependent neuronal pathway in Drosophila. Unexpectedly, we found that FoxO is regulated not by insulin/insulin-like growth factor signaling, but rather by a TLR signaling pathway. We present evidence that Toll-6 promotes FoxO activity via the neuronal TIR adaptor dSARM to regulate dynamic synaptic MTs. To provide mechanistic insight into pathway function, we screened a collection of cytoskeletal regulators for altered expression in pathway mutants. We found that Toll-6–FoxO signaling represses MKLP1/Pavarotti (Pav-KLP), a mitotic kinesin that bundles MTs during cytokinesis and has emerging functions in postmitotic neurons. MKLP1/Pav-KLP plays a conserved role in restraining axon length and attenuating MT dynamics (Lin et al., 2012; del Castillo et al., 2015). The elevated MT dynamics observed in Pav-KLP loss of function (LOF) mutants results, at least in part, from increased Kinesin-1–dependent sliding of MTs relative to each other, implying that Pav-KLP normally slows dynamics by braking MT–MT sliding (del Castillo et al., 2015).
Having uncovered this novel pathway, we next defined its in vivo functions. We found that Toll-6, dSARM, and FoxO control the distribution of stable MTs in the presynaptic compartment at the neuromuscular junction (NMJ). We show that these MT defects impair the behavior of presynaptic terminals, as loss of Toll-6, dSARM, or FoxO eliminates activity-dependent structural plasticity. Moreover, presynaptic plasticity is restored in mutant backgrounds by either reducing levels of Pav-KLP or pharmacologically destabilizing MTs, indicating that impaired MT dynamics are responsible for these phenotypes. Finally, we found that this pathway also limits Pav-KLP expression to promote proper axon transport. Thus, Toll-6–FoxO signaling promotes MT dynamics in both axons and presynaptic terminals via Pav-KLP to enable key neuronal functions.
Results
Toll-6 and dSARM promote nuclear localization of FoxO in the CNS
Transcriptional activity of FoxO proteins is regulated at the level of nuclear localization. The shuttling of FoxO proteins between the nucleus and the cytoplasm is controlled by the activity of several signaling pathways (Calnan and Brunet, 2008). However, the receptor-mediated signaling pathways controlling FoxO activity in developing neurons have not been defined. To identify receptors upstream of Drosophila FoxO, we tested whether mutations in receptors expressed on neurons alter FoxO’s subcellular distribution, using a FoxO antibody we previously generated and validated (Nechipurenko and Broihier, 2012). We quantified FoxO-positive nuclei in a defined region of interest (ROI) encompassing three abdominal segments of third-instar larval ventral nerve cords (VNCs). Although in other contexts FoxO is activated within insulin, TGF-β, and Wnt receptor pathways (Lee et al., 2003; Essers et al., 2005; Naka et al., 2010), we did not detect appreciable changes to nuclear FoxO levels in neurons in these mutant backgrounds (unpublished data).
Unexpectedly, we found that Toll-6 promotes nuclear FoxO localization because loss of Toll-6 results in a 37% decrease in FoxO-positive nuclei relative to wild type (Fig. 1, A–C). Among Toll-6 VNCs, some display a moderate loss of FoxO-positive nuclei (Fig. 1 B), whereas others have a more severe reduction (Fig. 1 C). We observed a concomitant increase in cells with enriched cytoplasmic FoxO in all Toll-6 mutant VNCs (Fig. 1, B and C, insets). Mutations in the related Toll receptor, Toll-7, did not display alterations in the number of neurons with nuclear FoxO (Fig. 1 E). These results suggest that Toll-6, but not Toll-7, promotes FoxO nuclear localization.
Figure 1.
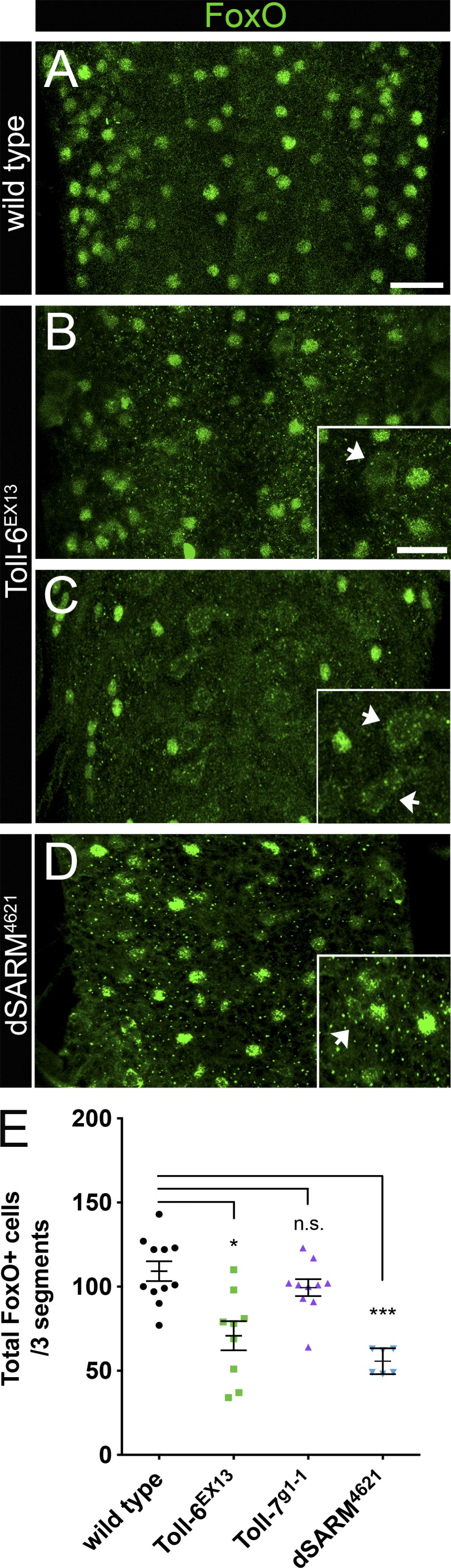
Toll-6 and dSARM promote FoxO nuclear localization in the CNS. (A–D) Representative z projections of three segments of L3 VNCs of the indicated genotypes labeled with anti-FoxO. Bar, 20 µm. (insets) High magnification z projections of Toll-6EX13 and dSARM4621 L3 VNCs. Arrows indicate cytoplasmic FoxO staining. Bar, 10 µm. (E) Quantification of total number of cells with nuclear FoxO. Wild type (OregonR): 109.2 ± 5.9 (n = 11 VNCs); Toll-6EX13: 70.8 ± 8.7 (n = 9 VNCs); Toll-7g1-1: 99.4 ± 5.0 (n = 10 VNCs); and dSARM4621: 55.7 ± 3.1 (n = 6 VNCs). Anterior is up. Error bars are mean ± SEM. n.s., not significantly different. *, P < 0.05; ***, P < 0.001.
We next investigated whether a TIR adaptor protein may be downstream of Toll-6 in this setting. TLRs signal through cytoplasmic TIR adaptor proteins (O’Neill and Bowie, 2007). In Drosophila, there are two known TIR adaptor proteins, Myd88 and dSARM. dSARM is expressed in the Drosophila CNS, so we evaluated whether dSARM may act downstream of Toll-6. dSARM mutant VNCs display a 49% decrease in FoxO-positive nuclei (Fig. 1, D and E). Similar to Toll-6, dSARM mutants have enriched cytoplasmic FoxO labeling (Fig. 1 D, inset). These data raise the possibility that dSARM transduces Toll-6 signals to promote FoxO nuclear localization.
To investigate molecular links between Toll-6 and FoxO, we tested whether Toll-6 regulates known FoxO kinases. Multiple kinases differentially phosphorylate FoxO and influence its subcellular distribution. Akt antagonizes nuclear localization of FoxO and is itself activated via phosphorylation (Brunet et al., 1999; Puig et al., 2003). Thus, we hypothesized that Toll-6 may promote nuclear FoxO by inhibiting Akt activation. Consistent with our hypothesis, phospho-Akt (pAkt) levels are increased 75% in the segmental nerves of Toll-6 mutants relative to wild type (Fig. S1, A–C). pAkt levels are similarly increased in Toll-6 VNCs compared with wild type on an immunoblot (Fig. S1 D). Arguing for specificity of the Toll-6–FoxO relationship, we did not observe a significant increase in pAkt in Toll-7 mutants on tissue or immunoblots (Fig. S1 C and not depicted). These findings provide evidence that Toll-6 inhibits Akt activation to license FoxO activity in the nucleus.
Does Toll-6 also stimulate activity of kinases that activate FoxO? JNK-dependent phosphorylation of FoxO on sites distinct from Akt resulted in FoxO nuclear translocation (Wang et al., 2005), prompting us to test whether Toll-6 promotes JNK activation. Consistent with our hypothesis, loss of Toll-6 results in a 40% reduction in phospho-JNK (pJNK) at the NMJ, indicating that Toll-6 promotes JNK activation (Fig. S1, E–G). In contrast, loss of Toll-7 does not alter levels of pJNK at the NMJ (Fig. S1 G). Collectively, these data are consistent with the model that Toll-6 can regulate FoxO’s subcellular distribution by promoting JNK activity and antagonizing Akt activity.
Toll-6 acts via dSARM and FoxO to drive NMJ overgrowth
To begin to establish whether Toll-6, dSARM, and FoxO act in a common genetic pathway, we turned to the NMJ. FoxO overexpression in motoneurons increases bouton number at NMJ4 and NMJ6/7 relative to controls (Fig. 2, A, B, and I; Fig. S2 A; Nechipurenko and Broihier, 2012), whereas consequences of Toll-6 and dSARM overexpression have not been described. Thus, we tested whether their overexpression in motoneurons increases bouton number. Indeed, Toll-6 overexpression in motoneurons results in an ~40% increase in boutons at NMJ4 and NMJ6/7 (Fig. 2, C and I; and Fig. S2 A), indicating that, like FoxO, Toll-6 promotes NMJ growth. We next examined whether FoxO is necessary for this Toll-6–dependent phenotype. Toll-6 overexpression does not result in NMJ overgrowth in a foxO LOF background (Fig. 2, D and I; and Fig. S2 A), implying that FoxO is genetically downstream of Toll-6 in this context. Given the structural similarity between Toll-6 and Toll-7, we likewise probed the function of the Toll-7 receptor. We found that Toll-7 overexpression also increases bouton number (Fig. S2, A and B). However, this phenotype is independent of FoxO, as Toll-7 still promotes bouton overgrowth in a foxO null background (Fig. S2, A and B). These results are consistent with our findings that Toll-7 does not regulate FoxO subcellular localization (Fig. 1 E). Therefore, from this point on, we focused on the relationship between Toll-6 and FoxO.
Figure 2.

Toll-6 acts via dSARM and FoxO to drive NMJ overgrowth. (A–H) Representative confocal projections of HRP-labeled NMJ4 of indicated genotypes. Bar, 20 µm. (I) Quantification of bouton numbers at NMJ4 in segments A2–A4. Control: 20.5 ± 0.6 (n = 15); OK6>foxO: 35.4 ± 1.5 (n = 17); OK6>Toll-6: 28.2 ± 1.0 (n = 25); OK6>Toll-6, foxOΔ94: 19.8 ± 0.5 (n = 39); OK6>Toll-6ΔCYTO: 20.3 ± 0.7 (n = 12); OK6>Toll-6TIRdead: 19.3 ± 0.7 (n = 20); OK6>Toll-6, dSARM4621: 21.9 ± 1.0 (n = 33); OK6>dSARM: 30.3 ± 1.4 (n = 15); and OK6>dSARM, foxOΔ94: 19.5 ± 0.5 (n = 34). Error bars are mean ± SEM. (J) Schematic representation of the TIR domain in wild-type Toll-6, Toll-6TIRdead, and Toll-6ΔCYTO. Control is OK6Gal4/+. n.s., not significantly different from control. ***, P < 0.001.
TLRs contain cytoplasmic TIR domains required for canonical signaling (Gay and Gangloff, 2007). A recent study found that the intracellular TIR domain of Toll-6 is dispensable for its function in olfactory projection neurons (Ward et al., 2015). To test whether this domain is required for Toll-6 activity in motoneurons, we overexpressed Toll-6 mutant variants with either a mutated nonfunctional TIR domain (TIR-dead) or a deletion of the entire cytoplasmic domain (ΔCyto; Fig. 2 J; Ward et al., 2015). Expression of the TIR-dead or ΔCyto variants of Toll-6 does not alter bouton number (Fig. 2, E and I; and Fig. S2 A), indicating that the TIR domain is necessary for progrowth functions of Toll-6.
The requirement for Toll-6’s TIR domain supports the involvement of a TIR adaptor protein. Thus, we tested whether dSARM can drive NMJ growth. dSARM overexpression in motoneurons results in increased bouton number at NMJ4 and NMJ6/7 (Fig. 2, F and I; and Fig. S2 A). Moreover, FoxO is required for dSARM-dependent NMJ overgrowth, as dSARM overexpression does not drive overgrowth without FoxO (Fig. 2, G and I; and Fig. S2 A). Finally, we interrogated whether dSARM is required for Toll-6–dependent overgrowth. We did not observe Toll-6–dependent overgrowth in the absence of dSARM, indicating that dSARM is required for Toll-6’s progrowth activity (Fig. 2, H and I; and Fig. S2 A). Together, these findings indicate that the TIR domain of Toll-6 is required for its progrowth activity, and that dSARM and FoxO mediate Toll-6’s progrowth function.
Although FoxO overexpression increases bouton number, bouton number is not altered by loss of foxO (Fig. S2, C and D; Nechipurenko and Broihier, 2012). Similarly, loss of neither Toll-6 nor dSARM alters bouton number (Fig. S2, C and D). However, mutant NMJs appeared to be smaller than controls, so we quantified total NMJ length. Consistent with our initial impression, loss of Toll-6 or foxO results in a 20% reduction in total arbor length of NMJ6/7 (Fig. S2 E). Although dSARM mutant arbors also tended to be smaller, the difference did not reach statistical significance (Fig. S2 E). Collectively, these analyses suggest that Toll-6, dSARM, and FoxO have similar functions in motoneurons.
Toll-6, dSARM, and FoxO regulate presynaptic MT stability
Cytoskeletal architecture is a key determinant of synaptic growth and morphology. We previously reported that foxO nulls display aberrant MT organization and enhanced MT stability at the NMJ (Nechipurenko and Broihier, 2012). Hence, we asked whether loss of Toll-6 or dSARM recapitulates this phenotype. Futsch/MAP1B is a neuron-specific MT-associated protein that binds α-tubulin and marks stable MTs (Hummel et al., 2000; Roos et al., 2000). Futsch-positive MT loops are a normal feature of presynaptic cytoskeletal architecture: ∼12 loops are present at wild-type NMJ6/7, and increased numbers of looped MTs are observed in mutants with enhanced MT stability (Nechipurenko and Broihier, 2012; Nahm et al., 2013). Like foxO mutants, Toll-6 and dSARM mutants display 18–20 MT loops per NMJ6/7, representing a 50% increase (Fig. 3, A–E). Toll-6, foxO double mutants exhibit numbers of Futsch-positive loops similar to those of individual single mutants (Fig. 3 E). Supporting presynaptic functions, motoneuron-selective RNAi-mediated knockdown of Toll-6, dSARM, or foxO results in indistinguishable increases in MT loops (Table S1).
Figure 3.

Toll-6, dSARM, and FoxO regulate presynaptic MT stability. (A–D) Representative confocal projections of Futsch-positive MT loops at NMJ6/7 of indicated genotypes. Bar, 10 µm. (E) Quantification of Futsch-positive MT loops at NMJ6/7 in segment A2. Wild type: 12.6 ± 0.5 (n = 22); foxOΔ94: 18.7 ± 1.0 (n = 19); Toll-6EX13: 17.6 ± 1.1 (n = 34); Toll-6EX13, foxOΔ94: 19.1 ± 0.9 (n = 16); and dSARM4621: 19.5 ± 1.0 (n = 20). (F–I) Representative confocal projections of Ac-Tub and HRP-labeled NMJ4s of indicated genotypes. Bar, 5 µm. (J) Quantification of proportion of boutons at NMJ4 with expanded Ac-Tub labeling. Wild type (OregonR): 0.06 ± 0.03 (n = 17); foxOΔ94: 0.33 ± 0.04 (n = 9); Toll-6EX13: 0.33 ± 0.02 (n = 10); Toll-6EX13, foxOΔ94: 0.26 ± 0.04 (n = 20); and dSARM4621: 0.23 ± 0.03 (n = 19). Error bars are mean ± SEM. n.s., not significantly different. **, P < 0.01; ***, P < 0.001.
To further elaborate the MT defects in pathway mutants, we labeled Toll-6, dSARM, and foxO mutants with acetylated α-tubulin (Ac-Tub). Acetylation of α-tubulin at lysine 40 is a hallmark of stable MTs and a metric of presynaptic MT stability (Janke and Bulinski, 2011). In wild-type boutons, Ac-Tub labels a tightly bundled MT filament extending through the bouton center, whereas the bouton periphery is devoid of Ac-Tub signal (Fig. 3 F). We previously demonstrated that foxO null NMJs have an expanded distribution of Ac-Tub labeling at NMJ6/7 (Nechipurenko and Broihier, 2012). Thus, we asked whether loss of Toll-6 or dSARM alters Ac-Tub distribution. We found that Toll-6, foxO, and dSARM mutant NMJs all exhibit comparably expanded Ac-Tub distributions in presynaptic terminals (Fig. 3, G–I). In mutant NMJs, Ac-Tub labeling takes the form of a thicker bundle, or alternatively, entirely fills the bouton. In Toll-6, dSARM, and foxO single mutants, as well as Toll-6, foxO double mutants, the mean percentage of boutons at NMJ4 with expanded Ac-Tub distributions is ∼30% relative to 6% of wild-type boutons (Fig. 3 J). These studies extend the phenotypic similarities between Toll-6, dSARM, and FoxO and suggest they act in a common pathway to regulate MT organization at the NMJ.
The genetic interactions and phenotypic similarities between Toll-6, dSARM, and foxO suggest that Toll-6 signals via dSARM and FoxO in motoneurons. However, these findings do not rule out the possibility that Toll-6 also engages a canonical TLR pathway. To explore this possibility, we asked whether loss of established components of TLR pathways leads to presynaptic MT defects. In canonical TLR signaling, Toll receptors signal via adaptors to regulate activity of Dorsal/NF-κB and Cactus/I-κB (Valanne et al., 2011; Troutman et al., 2012; Lindsay and Wasserman, 2014). Using LOF alleles and motoneuron-specific RNAi-mediated knockdown, we quantified bouton number and presynaptic MT loops when Dorsal, Cactus, Myd88, Pellino, Weckle, and Relish are lost. We did not detect significant changes to bouton number or MT loops in any of these backgrounds (Table S1). Importantly, we did not observe changes in bouton number or MT loops with neuronal expression of four dorsal RNAi lines or three cactus RNAi lines (Table S1). Finally, we did not observe changes in bouton number or MT loops in Myd88 null mutants (Table S1). These findings suggest that Toll-6 signaling does not engage canonical downstream components to regulate MT dynamics in motoneurons.
Toll-6 and FoxO limit Pav-KLP expression in motoneurons
Transcriptional pathways are predicted to control cytoskeletal dynamics by modulating expression of cytoskeletal regulatory genes. Therefore, we sought to identify transcriptional targets of Toll-6 and FoxO underlying their ability to direct MT dynamics. We assumed a targeted gene expression approach using quantitative RT-PCR (qRT-PCR) in third instar larval VNCs to identify putative targets. We tested whether expression of 50 known cytoskeletal regulatory mRNAs was altered in Toll-6 LOF VNCs relative to wild-type (Fig. 4 A). Given that we were isolating RNA from total VNCs, we reasoned that we were more likely to identify differentially regulated genes in Toll-6 mutants, because Toll-6 is more broadly expressed than FoxO (Nechipurenko and Broihier, 2012; McIlroy et al., 2013).
Figure 4.

Toll-6 and FoxO limit Pav-KLP expression in motoneurons. (A) qRT-PCR analyses of relative mRNA levels in Toll-6EX13 L3 VNCs represented by a heat map of fold change compared with wild type. Green, increase in mRNA levels compared with wild type; black, no change in mRNA levels compared with wild type; purple, decrease in mRNA compared with wild type. Fold change was calculated based on two to six biological replicates per mRNA (two to three technical replicates per biological replicate). (B and C) Representative confocal projections of HRP-labeled NMJ4 in segments A2–A4 of indicated genotypes. Bar, 20 µm. (D) Quantification of bouton numbers at NMJ4 in segments A2–A4. Control (D42>dcr-2): 19.3 ± 0.9 (n = 15); D42>dcr-2; Pav RNAi#1: 28.5 ± 0.9 (n = 30); D42>dcr-2; Pav RNAi#2: 28.6 ± 1.1 (n = 21); and D42>dcr-2; Pav RNAi#3: 27.9 ± 1.3 (n = 25). (E–G) Representative confocal projections of NMJ4 in segments A2–A4 labeled with HRP and Pav. Bar, 5 µm. (H) Quantification of Pav intensity normalized to wild type. Wild type: 1.0 ± 0.06 (n = 31); D42>dcr-2; Pav RNAi#1: 0.5 ± 0.1 (n = 11); Toll-6EX13: 2.0 ± 0.2 (n = 13); and foxOΔ94: 2.0 ± 0.2 (n = 8). Wild type is OregonR. Error bars are mean ± SEM. n.s., not significantly different. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
The mitotic kinesin Pavarotti (Pav-KLP) emerged as the most differentially expressed gene in Toll-6 null VNCs relative to wild-type controls. In six biological replicates, pav mRNA was, on average, 4.5 ± 0.2-fold up-regulated in Toll-6 nulls (Fig. 4 A). Several other kinesin family members including Klp3A, Klp61F, and Klp67A were up-regulated between 1.5- and 3.0-fold in Toll-6 mutants (Fig. 4 A). The Spectraplakin short stop (shot) was the most down-regulated gene in Toll-6 VNCs (Fig. 4 A); however, its NMJ phenotypes do not easily align with those of our pathway mutants (Valakh et al., 2013), so it was excluded from further analyses. To assess whether any of the four up-regulated kinesins are functionally downstream of Toll-6, we analyzed their LOF phenotypes at the NMJ. Genes inhibited by Toll-6 are predicted to display LOF phenotypes similar to Toll-6 overexpression phenotypes. Thus, we tested whether RNAi-mediated knockdown of the kinesins in motoneurons increases bouton number because Toll-6 overexpression drives NMJ overgrowth. We did not observe significant changes in bouton number with RNAi-mediated knockdown of Klp3A, Klp61F, or Klp67A (Table S2). However, motoneuronal expression of three pav RNAi lines increased bouton number similarly to Toll-6 overexpression, consistent with repression of Pav-KLP by Toll-6 (Fig. 2, A–C and I; Fig. 4, B–D; and Table S2). We note that the cytokinesis defects in homozygous pav mutant embryos (Adams et al., 1998) preclude an analysis of NMJ development in pav homozygous mutant larvae. We next tested whether pav mRNA is also de-repressed in foxO LOF VNCs. In six biological replicates, we found that Pav-KLP RNA expression is increased 1.6 ± 0.3-fold in foxO LOF VNCs. These data raise the possibility that Pav-KLP is a functional target of Toll-6 and FoxO.
Pav-KLP/MKLP1 is a mitotic kinesin that bundles MTs and stabilizes the mitotic spindle during cell division (Sommi et al., 2010). In neurons, it brakes Kinesin-1–mediated MT-MT sliding in Drosophila axons (del Castillo et al., 2015) and regulates MT polarity in mammalian dendrites (Lin et al., 2012). Yet Pav-KLP’s role at the NMJ has not been explored. Thus, we probed its function at the NMJ, beginning by examining whether it is expressed at the NMJ. Indeed, Pav-KLP protein localizes to presynaptic boutons of wild-type larvae (Fig. 4 E). Confirming antibody specificity, Pav-KLP immunofluorescence is reduced 55% relative to wild-type NMJs when Pav is knocked down in neurons using two separate RNAi lines (Fig. 4 H and not depicted). Based on our qRT-PCR data, we hypothesized that Pav-KLP protein expression would be increased in Toll-6 and foxO LOF NMJs compared with wild type. In line with our hypothesis, we observed a twofold increase in Pav-KLP immunofluorescence at the NMJs of Toll-6 and foxO mutants relative to wild type, indicating that pathway mutants have increased Pav-KLP protein expression (Fig. 4, F–H). Finally, we quantified baseline bouton number and presynaptic MT loops with Pav-KLP overexpression in motoneurons. Analogous to Toll-6 and foxO LOF mutants, Pav-KLP overexpression does not alter bouton number but increases MT loops by ∼50% (Fig. S3, A–D). Collectively, these data support the hypothesis that Toll-6–FoxO signaling represses Pav-KLP expression to promote MT dynamics at the NMJ.
Toll-6, dSARM, and FoxO are required for rapid presynaptic structural plasticity
Next, we sought to define functions for this proposed signaling pathway at the NMJ. We were particularly interested in presynaptic plasticity because in other systems, dynamic MTs are linked to postsynaptic plasticity (Dent et al., 2011). Spaced high K+ depolarization paradigms induce activity-dependent synaptic growth in the form of nascent or ghost boutons over the course of minutes (Ataman et al., 2008; Piccioli and Littleton, 2014). These boutons are incompletely differentiated and are labeled by the presynaptic neuronal membrane marker HRP but lack the postsynaptic PSD-95 homologue Discs-large (Dlg). Nascent boutons are round and have a very thin axonal connection to the parent bouton. Beyond the role of actin remodeling in this form of presynaptic structural plasticity (Piccioli and Littleton, 2014), little is known regarding the underlying cytoskeletal mechanisms.
We began by examining whether Toll-6, dSARM, and foxO are required for rapid activity-dependent structural growth. Using a published protocol (Piccioli and Littleton, 2014), we established the frequency of new bouton formation at wild-type NMJs. We found 7.6 ± 0.6 new boutons at NMJ 6/7 after three 2-min depolarizations in 90 mM K+, in line with published articles (Piccioli and Littleton, 2014). We then investigated whether Toll-6, dSARM, and foxO are required for rapid new bouton addition. The number of new boutons in mutants is equivalent to that of nonstimulated controls, indicating that they are essential for activity-dependent growth (Fig. 5, A–D and F). Moreover, motoneuron expression of RNAi targeting Toll-6, FoxO, or dSARM also eliminates rapid activity-dependent bouton addition (Fig. 5 G), indicating presynaptic pathway activity. Implying linearity of Toll-6–FoxO signaling, the Toll-6, foxO double mutant phenocopies the individual single mutants (Fig. 5 F).
Figure 5.

Toll-6, dSARM, and FoxO are required for rapid presynaptic structural plasticity. (A–E) Representative confocal projections of NMJ6/7 in A2-3 labeled with HRP and Dlg after high K+ stimulation. Arrows denote nascent boutons. Bar, 10 µm. (F) Quantification of nascent boutons after high K+ and control stimulation paradigms. Wild type2mM: 3.3 ± 0.2 (n = 27); wild type90mM: 7.6 ± 0.6 (n = 31); foxOΔ942mM: 3.6 ± 0.3 (n = 14); foxOΔ9490mM: 4.5 ± 0.4 (n = 26); Toll-6EX132mM: 3.4 ± 0.4 (n = 16); Toll-6EX1390mM: 4.2 ± 0.4 (n = 31); Toll-6EX13, foxOΔ942mM: 3.2 ± 0.5 (n = 10); Toll-6EX13, foxOΔ9490mM: 3.2 ± 0.5 (n = 11); dSARM46212mM: 2.9 ± 0.6 (n = 7); and dSARM462190mM: 2.9 ± 0.5 (n = 16). Wild type is OregonR. (G) Quantification of nascent boutons after high K+ and control stimulation paradigms. Control2mM: 2.4 ± 0.3 (n = 17); control90mM: 7.1 ± 0.8 (n = 18); D42>dcr-2; foxO RNAi2mM: 2.9 ± 0.5 (n = 17); D42>dcr-2; foxO RNAi90mM: 4.2 ± 1.1 (n = 17); D42>dcr-2; Toll-6 RNAi#12mM: 3.2 ± 0.4 (n = 22); D42>dcr-2; Toll-6 RNAi#190mM: 3.3 ± 0.5 (n = 18); D42>dcr-2; Toll-6 RNAi#22mM: 3.0 ± 0.5 (n = 15); D42>dcr-2; Toll-6 RNAi#290mM: 2.7 ± 0.4 (n = 20); D42>dcr-2; dSARM RNAi#12mM: 3.7 ± 0.6 (n = 25); D42>dcr-2; dSARM RNAi#190mM: 4.6 ± 0.7 (n = 29); D42>dcr-2; dSARM RNAi#22mM: 3.7 ± 0.7 (n = 17); and D42>dcr-2; dSARM RNAi#290mM: 5.4 ± 1.6 (n = 14). Control is D42>dcr-2. (H) Quantification of nascent boutons after high K+ and control stimulation paradigms. Control2mM: 2.9 ± 0.3 (n = 27); control90mM: 6.9 ± 0.8 (n = 29); OK6>Pav#12mM: 2.5 ± 0.5 (n = 11); OK6>Pav#190mM: 3.2 ± 0.6 (n = 20); OK6>Pav#22mM: 3.5 ± 0.4 (n = 17); and OK6>Pav#290mM: 3.3 ± 0.6 (n = 12). Control is OK6Gal4/+. Error bars are mean ± SEM. n.s., not significantly different. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Statistical comparisons above 90 mM bars are to 90 mM measurements of wild type or control.
Is excessive MT stability responsible for impaired activity-dependent structural plasticity in pathway mutants? Using the MT-stabilizing drug taxol, we investigated whether acute pharmacological MT stabilization impairs structural plasticity. We used nanomolar taxol concentrations that do not interfere with axonal transport in our hands (see below and Fig. S5). We found that 20-min pretreatment with either 1.2 or 6 nM taxol eliminates activity-dependent bouton addition (Fig. S4), highlighting the importance of dynamic MTs for new bouton addition. Moreover, these results support our model that increased MT stability in Toll-6, dSARM, and foxO mutants interferes with rapid presynaptic structural plasticity.
Finally, we asked whether Pav-KLP overexpression also blocks this form of structural plasticity, because our data indicate that Pav-KLP protein expression is enhanced in Toll-6 and foxO null NMJs. Indeed, motoneuron overexpression of Pav-KLP eliminates rapid structural plasticity (Fig. 5, E and H). These data support the hypothesis that Pav-KLP is normally repressed by Toll-6–mediated signaling.
pav dominantly suppresses plasticity defects observed with loss of Toll-6, foxO, or dSARM function
If Pav-KLP overexpression eliminates structural plasticity in Toll-6 pathway mutants, the phenotype is predicted to be sensitive to pav dosage. Accordingly, using a strong allele of pav (pavB200; Salzberg et al., 1994), we investigated whether pav dominantly suppresses acute structural plasticity in pathway mutant backgrounds. pav heterozygosity dominantly suppresses the structural plasticity defects observed with RNAi-mediated knockdown of Toll-6, foxO, or dSARM in motoneurons (Fig. 6, A–E). We observed complete rescue of nascent bouton formation in Toll-6 and foxO backgrounds by halving the genetic dose of pav, and partial rescue in a dSARM background (Fig. 6 E). These data imply that elevated expression of Pav-KLP in mutant backgrounds contributes to the structural plasticity defects.
Figure 6.

pav dominantly suppresses structural plasticity defects observed with loss of Toll-6, foxO, or dSARM function. (A–D) Representative confocal projections of NMJ6/7 in A2 and A3 labeled with HRP and Dlg after high K+ stimulation. Arrows denote nascent boutons. Bar, 10 µm. (E) Quantification of nascent boutons. Control90mM: 10.7 ± 1.0 (n = 24); D42>dcr-2; foxO RNAi90mM: 4.2 ± 1.1 (n = 17); D42>dcr-2; foxO RNAi; PavB200/+90mM: 8.6 ± 0.9 (n = 20); D42>dcr-2; Toll-6 RNAi#190mM: 3.3 ± 0.5 (n = 18); D42>dcr-2; Toll-6 RNAi#1; PavB200/+90mM: 11.8 ± 2.0 (n = 21); D42>dcr-2; dSARM RNAi#190mM: 4.6 ± 0.7 (n = 29); and D42>dcr-2; dSARM RNAi#1; PavB200/+90mM: 6.9 ± 0.5 (n = 23). Control is D42>dcr-2; PavB200/+. Error bars are mean ± SEM. n.s., not significantly different from control90mm. *, P < 0.05; ***, P < 0.001. Statistical comparisons above individual bars are made to the control.
Excessive MT stability impairs rapid structural plasticity in Toll-6 pathway mutants
Pav-KLP restrains MT-MT sliding in neurons (del Castillo et al., 2015). Thus, we propose that Pav-KLP overexpression in Toll-6 pathway mutants leads to reduced MT dynamics and eliminates rapid activity-induced bouton formation. If so, we hypothesized that presynaptic plasticity would be restored via direct pharmacological MT destabilization. Specifically, we tested whether acute pharmacological destabilization of MTs using vinblastine (VBL; Jordan et al., 1992) rescues structural plasticity defects in Toll-6, dSARM, and foxO mutants. We found that 30-min VBL pretreatment immediately before high K+ stimulation does not alter bouton growth of wild-type NMJs (Fig. 7 A). We similarly incubated Toll-6, foxO, and dSARM NMJs with VBL. Strikingly, acute VBL pretreatment completely rescues structural plasticity defects in Toll-6, foxO, and dSARM mutants (Fig. 7 A). Again, implying presynaptic function of this pathway, VBL pretreatment rescues plasticity defects in Toll-6, foxO, and dSARM knockdown NMJs (Fig. 7 B). This provides direct evidence that enhanced MT stability in mutant NMJs eliminates structural plasticity. This result prompted us to ask whether acute VBL pretreatment could rescue plasticity defects at Pav-KLP overexpression NMJs. Indeed, VBL pretreatment completely rescues nascent bouton formation defects when Pav-KLP is overexpressed (Fig. 7 C). These results imply that Toll-6 signaling represses Pav-KLP to maintain a population of dynamic MTs and enable rapid structural plasticity.
Figure 7.

Excessive MT stability impairs rapid structural plasticity in Toll-6 pathway mutants. (A) Quantification of nascent boutons at NMJ6/7 treated with VBL or 0.2% DMSO before high K+ stimulation. Wild typeDMSO: 7.7 ± 0.7 (n = 6); wild typeVBL: 7.3 ± 0.5 (n = 27); foxOΔ94DMSO: 3.6 ± 0.3 (n = 13); foxOΔ94VBL: 6.8 ± 0.8 (n = 18); Toll-6EX13DMSO: 2.9 ± 0.7 (n = 15); Toll-6EX13VBL: 9.4 ± 1.4 (n = 18); Toll-6EX13, foxOΔ94DMSO: 2.8 ± 0.3 (n = 16); Toll-6EX13, foxOΔ94VBL: 7.0 ± 0.8 (n = 15); dSARM4621DMSO: 3.3 ± 0.3 (n = 6); and dSARM4621VBL: 6.3 ± 0.7 (n = 11). Wild type is OregonR. (B) Quantification of nascent boutons at NMJ6/7 treated with VBL or 0.2% DMSO before high K+ stimulation. ControlDMSO: 7.5 ± 1.2 (n = 21); controlVBL: 8.1 ± 0.8 (n = 16); D42>dcr-2; foxO RNAiDMSO: 3.8 ± 0.9 (n = 18); D42>dcr-2; foxO RNAiVBL: 9.0 ± 1.0 (n = 13); D42>dcr-2; Toll-6 RNAi#1DMSO: 4.3 ± 1.0 (n = 12); D42>dcr-2; Toll-6 RNAi#1VBL: 7.8 ± 1.0 (n = 19); D42>dcr-2; dSARM RNAi#1DMSO: 2.7 ± 0.5 (n = 9); and D42>dcr-2; dSARM RNAi #1VBL: 9.0 ± 1.0 (n = 13). Control is D42>dcr-2. (C) Quantification of nascent boutons at NMJ6/7 treated with VBL or 0.2% DMSO before high K+ stimulation. ControlDMSO: 7.9 ± 1.3 (n = 11); controlVBL: 7.3 ± 1.1 (n = 16); OK6>Pav#1DMSO: 2.8 ± 0.9 (n = 10); OK6>Pav#1VBL: 15.4 ± 2.8 (n = 11); OK6>Pav#2DMSO: 3.8 ± 1.0 (n = 19); and OK6>Pav#2VBL: 9.5 ± 1.4 (n = 11). Control is OK6Gal4/+. Error bars are mean ± SEM. n.s., not significantly different. *, P < 0.05, **, P < 0.01; ***, P < 0.001.
Elevated MT stability impairs axonal transport in Toll-6 pathway mutants
Finally, we investigated whether the MT deficits in this proposed pathway are confined to the presynaptic compartment, or whether the pathway also regulates MT behavior in axons. Molecular motors transport their cargo along neuronal MTs, and MT defects frequently lead to impaired transport (Gornstein and Schwarz, 2014). Using an antibody to the active zone component Bruchpilot (Brp), we assessed protein transport in wild-type and mutant nerves. We quantified the number of Brp-positive aggregates per 30 µM of axon and found that Toll-6, foxO, and dSARM mutants all display a 2–2.5-fold increase in accumulation of axonal Brp compared with wild-type (Fig. 8, A–C, E, and K). Moreover, Toll-6, foxO double mutants have transport defects indistinguishable from single mutants (Fig. 8, D and K), again implying a linear pathway. Supporting this analysis, quantitatively similar trafficking impairments are observed with motoneuronal expression of RNAi constructs targeting Toll-6, foxO, and dSARM (Fig. 8 L).
Figure 8.

Elevated MT stability impairs axonal transport in Toll-6 pathway mutants. (A–J) Representative confocal projections of L3 nerves of indicated genotypes labeled with anti-Brp. Bar, 20 µm. (K) Quantification of Brp accumulations per 30 µm of nerve. Wild type (OregonR): 11.1 ± 2.3 (n = 20); foxOΔ94: 28.7 ± 1.8 (n = 50); Toll-6EX13: 23.5 ± 1.5 (n = 52); Toll-6EX13, foxOΔ94: 25.6 ± 3.0 (n = 34); and dSARM4621: 21.7 ± 1.4 (n = 35). (L) Quantification of Brp accumulations per 30 µm of nerve. Control (D42>dcr-2): 14.1 ± 1.1 (n = 52); D42>dcr-2; foxO RNAi: 26.0 ± 2.2 (n = 25); D42>dcr-2; Toll-6 RNAi#1: 21.5 ± 2.2 (n = 17); D42>dcr-2; Toll-6 RNAi#2: 28.0 ± 3.4 (n = 20); D42>dcr-2; dSARM RNAi#1: 26.4 ± 2.0 (n = 27); and D42>dcr-2; dSARM RNAi#2: 23.0 ± 2.9 (n = 22). (M) Quantification of Brp accumulations per 30 µm of nerve. Control (D42>dcr-2; PavB200/+): 11.7 ± 1.2 (n = 24); OK6>Pav#1: 23.0 ± 1.5 (n = 21); OK6>Pav#2: 24.6 ± 2.2 (n = 33). Control is OK6Gal4/+. (N) Quantification of Brp accumulations per 30 µm of nerve in control: 13.5 ± 0.9 (n = 39); D42>dcr-2; foxO RNAi: 26.0 ± 2.2 (n = 25); D42>dcr-2; foxO RNAi; PavB200/+: 13.3 ± 1.1 (n = 24); D42>dcr-2; Toll-6 RNAi#1: 21.5 ± 2.2 (n = 17); D42>dcr-2; Toll-6 RNAi#1; PavB200/+: 13.0 ± 1.4 (n = 18); D42>dcr-2; dSARM RNAi#1: 26.4 ± 2.0 (n = 27); and D42>dcr-2; dSARM RNAi#1; PavB200/+: 13.0 ± 1.4 (n = 22). Error bars are mean ± SEM. n.s., not significantly different. **, P < 0.01; ***, P < 0.001.
These trafficking impairments might result directly from increased neuronal MT stability in the mutants, or alternatively, could reflect an independent function of Toll-6–dependent signaling in transport. To assess whether increased MT stability can lead to accumulations of axonal Brp, we investigated whether acute treatment with taxol results in axonal aggregates of Brp. Although application of taxol at concentrations less than 40 nM does not alter the number of axonal Brp aggregates, acute treatment with either 40 or 60 nM taxol recapitulates the transport phenotypes in Toll-6, dSARM, and foxO mutants (Fig. S5, A–E). Thus, excessive MT stability can impair axonal transport of Brp.
pav dominantly suppresses axon transport defects observed with loss of Toll-6, foxO, or dSARM function
Does Pav-KLP also regulate MT dynamics impinging on axonal transport? To address this possibility, we asked whether Pav-KLP overexpression leads to Brp trafficking defects similar to Toll-6, dSARM, and foxO LOF mutants. Indeed, motoneuronal Pav-KLP overexpression produces Brp accumulations comparable to Toll-6, dSARM, and foxO mutants (Fig. 8, F and M), indicating that Pav-KLP expression can impair Brp trafficking. Because Toll-6 LOF mutants display elevated levels of Pav-KLP mRNA (Fig. 4 A), we hypothesized that increased Pav-KLP expression is responsible for trafficking defects in pathway mutants. To interrogate this hypothesis, we asked whether pav dominantly suppresses the trafficking defects in Toll-6, foxO, or dSARM knockdown backgrounds. In line with our hypothesis, heterozygosity for pavB200 completely restores Brp trafficking in knockdown nerves (Fig. 8, G–J and N). These data argue that excessive MT stability underlies trafficking defects in Toll-6 pathway mutants. Collectively, our findings indicate that a key function of Toll-6–FoxO signaling is to repress Pav-KLP transcription to enable MT dynamics required for structural plasticity and axon transport.
Discussion
Here we demonstrate that Toll-6 signaling establishes a dynamic MT network required for axon transport and rapid activity-dependent synaptic plasticity in motoneurons (Fig. 9). We further show that Toll-6 signals via a novel pathway including the TIR adaptor dSARM and the transcription factor FoxO. Finally, we identify the mitotic kinesin MKLP1/Pav as a key effector of this pathway, as elevated Pav expression underlies the deficits in pathway mutants. These findings not only characterize a novel pathway from cell surface receptor to transcriptional effector, but also point to a new molecular mechanism for modulating neuronal MT dynamics, function, and plasticity.
Figure 9.

A Toll-6–dSARM–FoxO pathway transcriptionally represses Pavarotti/MKLP1 to regulate core features of the neuronal phenotype.
A neuronal TLR–dSARM–FoxO pathway
TLRs are expressed in astrocytes, microglia, oligodendrocytes, and neurons, and known functions for TLRs include neurogenesis, neurite outgrowth, and branching (Okun et al., 2011; Barak et al., 2014). Although TLR-dependent pathways in glial subtypes are beginning to be defined, TLR-dependent pathways in neurons are largely unresolved. However, recent studies support the existence of diverse, noncanonical TLR pathways in neurons (Ballard et al., 2014; Ward et al., 2015). Toll-8 (Tollo) regulates developmental NMJ growth via a Dorsal/NF-κB and Cactus/IκB independent pathway (Ballard et al., 2014). Like the Toll-6–dependent pathway investigated here, Toll-8 signals through JNK (Ballard et al., 2014); however, these pathways differ functionally because Toll-8 is necessary for developmental NMJ growth whereas Toll-6 is not. Here we demonstrate that Toll-6 and Toll-7 pathways are also distinct. Importantly, only Toll-6 regulates FoxO. In particular, loss of FoxO suppresses the excessive bouton formation observed with Toll-6, but not Toll-7, and nuclear localization of FoxO depends only on Toll-6. Unlike Toll-8, which is proposed to interact with the Spz3 ligand (Ballard et al., 2014), the ligands for Toll-6 and Toll-7 at the NMJ have not been identified. Collectively, these studies imply that Toll-6, Toll-7, and Toll-8 have separable activities in motoneurons. The idea that TLRs have distinct neuronal functions is further supported by a recent study of Toll-6 and Toll-7 function in the olfactory system (Ward et al., 2015). Those authors found that Toll-6 and Toll-7 have unique functions in the olfactory system that are independent of their cytoplasmic domains (Ward et al., 2015).
The cytoplasmic domain of Toll-6 contains a TIR domain, which mediates interactions with TIR domain–containing adaptor proteins. In contrast to Toll-6 function in the olfactory system, we found that Toll-6 activity in motoneurons requires its cytoplasmic domain. Thus, we tested whether either Drosophila TIR domain–containing adaptor protein is active in Toll-6 signaling. Although loss of Myd88 does not result in phenotypes consistent with a function in Toll-6 signaling, loss of dSARM results in phenotypes indistinguishable from those of Toll-6. These results argue that dSARM, not Myd88, mediates Toll-6 signaling in motoneurons. This conclusion is in line with recent studies demonstrating that dSARM/SARM1 mediates TLR-dependent signaling. In particular, dSARM mediates Tollo signaling in the respiratory epithelium (Akhouayri et al., 2011), and dSARM/SARM1 is downstream of TLRs in pro-apoptotic pathways in neurons (Mukherjee et al., 2015) as well as epithelial cells (Meyer et al., 2014). Although our data point to Toll-6 as an upstream regulator of dSARM, further experimentation is necessary to establish the directness of this regulation.
Surprisingly, Toll-6 signals through FoxO in motoneurons. In canonical TLR signaling, Toll receptors signal via Dorsal/NF-κB and Cactus/IκB. However, neither Dorsal nor Cactus is expressed in motoneurons (Cantera et al., 1999; Heckscher et al., 2007), and loss of either dorsal or cactus does not yield NMJ phenotypes (Ballard et al., 2014). It will be critical to establish whether TLRs are regulators of FoxO activity in other neuronal populations. Hinting that the TLR-FoxO link may be evolutionarily conserved, FoxO1/3 has recently been shown to regulate innate immunity downstream of TLR3 in the respiratory epithelium (Seiler et al., 2013).
Pav/MKLP1 mediates pathway function in axon transport and presynaptic plasticity
We first identified the mitotic kinesin MKLP1/Pav as a likely downstream transcriptional target of Toll-6 signaling in our qRT-PCR screen. Confirming Pav repression by Toll-6–FoxO in motoneurons, we demonstrated that Pav-KLP protein levels at the NMJ are increased roughly twofold in both Toll-6 and foxO LOF mutants. RNAi-mediated knockdown of pav in motoneurons results in NMJ overgrowth similar to that observed with overexpression of either Toll-6 or FoxO, suggesting that Pav-KLP is a functionally relevant effector. And most tellingly, pav dominantly suppresses axonal and synaptic phenotypes observed in Toll-6, dSARM, and foxO knockdown mutants, indicating that elevated Pav-KLP expression underlies the mutant phenotypes. We identified three putative FoxO binding sites (5′-TTGTTTAT-3′) upstream of the pav transcriptional start site (−2,340, −3,528, and −4,320 bp) and two putative FoxO binding sites downstream of pav (3,879 and 3,995 bp), raising the possibility that FoxO directly represses pav transcription. It is alternately possible that FoxO indirectly regulates pav transcription, for example, by regulating the expression of another transcription factor. Regardless of the directness of the FoxO–pav interaction, our data demonstrate that Pav-KLP is a key effector of FoxO-dependent functions in developing motoneurons.
The FoxO orthologue daf-16 has recently been shown to promote expression of the kinesin unc-104/KIF1A in adult motoneurons in C. elegans (Li et al., 2016). Although it is not known whether this regulation is direct, Daf-16–mediated expression of Unc-104 is important for maintaining synapse integrity over the lifespan. We tested for differential expression of Drosophila unc-104 in our qRT-PCR screen but did not detect significant differences in unc-104 expression levels in total VNCs in Toll-6 or foxO mutants (Fig. 4 A), suggesting that Drosophila FoxO does not regulate unc-104 during neuronal development. Regardless, it is noteworthy that FoxO proteins play conserved roles in regulating expression of MT motors in motoneurons. It will be important to test whether FoxO proteins also regulate MT motors in other neuronal populations, and whether FoxO proteins may regulate distinct motors during development and in the adult.
This work significantly expands neuronal functions for Pav/MKLP1. Pav-KLP is a Kinesin-6 family member and restrains axon length in both murine and Drosophila neurons (Lin et al., 2012; del Castillo et al., 2015), at least in part by braking Kinesin-1–dependent MT–MT sliding (del Castillo et al., 2015). Kinesin-1–dependent sliding of MTs against each other is critical for neurite growth in young neurons and for axon regeneration after injury (Lu et al., 2013, 2015). Pav-KLP expression in neurons after neurite extension has been proposed to block the morphologic instability that would arise from uncontrolled MT-MT sliding (del Castillo et al., 2015). Our data argue that Pav-KLP levels are precisely developmentally controlled in axons and synaptic terminals after initial outgrowth to enable sufficient MT dynamics for ongoing neuronal functions such as transport and plasticity.
We found that Pav-KLP is regulated by Toll-6–FoxO signaling. The finding that a protein critical for rapid structural remodeling of synapses is controlled at the level of transcription argues that this regulation precedes elevated synaptic activity. In this study, we used a model for rapid plasticity in which NMJs rapidly bud new boutons after spaced high K+ stimulation (Ataman et al., 2008; Piccioli and Littleton, 2014). This form of plasticity persists when the axon is severed from the nucleus (Piccioli and Littleton, 2014). This result implies that the requirement for FoxO-dependent regulation of Pav-KLP precedes elevated activity and establishes competence of motoneurons for rapid bouton addition. We propose that FoxO normally limits Pav-KLP levels in motoneurons, setting the stage for dynamic MT rearrangements required during remodeling of synaptic terminals in response to elevated activity. In the future, it will be essential to determine whether FoxO controls dynamic MTs via Pav-KLP in other neuronal populations during development and in the adult.
Materials and methods
Fly stocks
OregonR is wild type. The following stocks were used: FoxOΔ94 (a gift from L. Partridge, University College, London, England, UK), UAS-FoxO (a gift from R. Tjian, University of California, Berkeley, Berkeley, CA), Toll-6EX13, UAS-Toll-6, Toll-7g1-1, and UAS-Toll-7 (gifts from Y. Ip, University of Massachusetts Medical School, Worcester, MA), UAS-Toll-6ΔCyto, Myd88C03881http://www.ncbi.nlm.nih.gov/nucest/1467132, and UAS-Toll-6TIRdead (gifts from L. Luo, Stanford University, Stanford, CA), OK6Gal4 (Aberle et al., 2002), D42Gal4 (Sanyal, 2009), ElavGal4 (a gift from A. DiAntonio, Washington University, St. Louis, MO), UAS-dSARM and dSARM4621 (gifts from M. Freeman, University of Massachusetts Medical School), UAS-Pav#1 (X) and UAS-Pav#2 (II; gifts from V. Gelfand, Northwestern University, Chicago, IL), and PavB200 (4384; Bloomington Drosophila Stock Center [BDSC]). The following RNAi lines were obtained from BDSC or the Vienna Drosophila Resource Center (VDRC): dSARM RNAi #1 (VDRC 102044), dSARM RNAi #2 (VDRC 105369), dSARM RNAi #3 (BDSC 31175), dSARM RNAi #4 (VDRC 104812), dSARM RNAi #5 (VDRC 105521), FoxO RNAi (VDRC 107786), Myd88 RNAi (BDSC 36107), Toll-6 RNAi #1 (VDRC 928), Toll-6 RNAi #2 (BDSC 56048), Weckle RNAi (BDSC 36580), Pellino RNAi (BDSC 36787), dl RNAi #1 (BDSC 36650), dl RNAi #2 (BDSC 32934), dl RNAi #3 (BDSC 34938), dl RNAi #4 (BDSC 27650), cact RNAi #1 (BDSC 31713), cact RNAi #2 (BDSC 34775), cact RNAi #3 (BDSC 34784), Pav RNAi #1 (VDRC 110330), Pav RNAi #2 (BDSC 35649), Pav RNAi #3 (BDSC 42573), Klp3A RNAi #1 (BDSC 43230), Klp3A RNAi #2 (BDSC 40944), Klp61F RNAi (BDSC 35804), Klp67A RNAi #1 (BDSC 36268), and Klp67A RNAi #2 (BDSC 35606).
Immunohistochemistry and immunofluorescence
Wandering L3 larvae were obtained by crossing a 2:1 ratio of adult virgin females to adult males in either vials or bottles. The adult flies were transferred to a new vial or bottle every 3–5 d to control for overcrowding. Wandering L3 larvae were dissected and processed for immunofluorescence as previously described (Nechipurenko and Broihier, 2012). In brief, wandering L3 larvae were dissected in PBS and fixed in either 4% PFA for 20 min or Bouin’s fixative for 5–10 min. After fixation, larval fillets were washed in PTX (PBS and 0.01% Triton X-100) and blocked in PBT (PBS, 0.01% Triton X-100, and 1% BSA) on a nutator and incubated with primary antibody for either 2 h at RT or overnight at 4°C without agitation. The following concentrations and primary antibodies were used: affinity-purified guinea pig anti-FoxO serum at 1:20, mouse anti-Futsch (22C10; Developmental Studies Hybridoma Bank) at 1:10, mouse anti-Bruchpilot (NC82; Developmental Studies Hybridoma Bank) at 1:100, mouse anti-Dlg (4F3; Developmental Studies Hybridoma Bank) at 1:1,000, mouse anti–Ac-Tub (Sigma-Aldrich) at 1:250, rabbit anti–phospho-Drosophila-Akt (Ser505; Cell Signaling Technology) at 1:200, rabbit anti-pJNK (81E11; Cell Signaling Technology) at 1:500, rabbit anti-Pav (a gift from A. Mogilner and I. Brust-Mascher, University of California Davis, Davis, CA) at 1:100, and Dylite or Alexa Fluor 594–conjugated anti-HRP (Jackson ImmunoResearch Laboratories, Inc.) at 1:300. The following species-specific secondary antibodies were used: Alexa Fluor 488 and Alexa Fluor 568 (Invitrogen) at 1:300.
Pharmacology
The protocol for taxol treatment was previously described (Trotta et al., 2004; Newquist et al., 2013). In brief, relaxed wandering L3 fillets (described in the next section) were incubated in HL-3 containing either 0.2% DMSO or 1.2, 6, 12, 30, 40, or 60 nM taxol concentration (Paclitaxel; Sigma-Aldrich) for 1 h at RT. After taxol treatment, larvae were rapidly washed in HL-3, dissected, and fixed for 10 min in Bouin’s fixative (for use with anti-Brp or anti-Dlg antibodies).
High K+ depolarization paradigm
High K+ depolarization paradigm was adapted from Piccioli and Littleton (2014). To summarize, wandering L3 larvae were dissected in HL-3 saline solution. Larvae were dissected and pinned loosely at the head and tail, and then fully stretched into fillets after stimulation. Relaxed larval fillets were subjected to three 2-min incubations in 90 mM K+ Jan’s saline solution (45 mM NaCl, 90 mM KCl, 4 mM MgCl2, 36 mM sucrose, 5 mM Hepes, and 2 mM CaCl2, pH 7.3) spaced by two 10-min HL-3 washes. After a final 2-min HL-3 wash, larval fillets were fully stretched, pinned, and fixed for 10 min in Bouin's fixative. Nascent boutons were identified via confocal microscopy by the presence of a presynaptic bouton lacking postsynaptic Dlg immunoreactivity at NMJ6/7 in abdominal segments 2 and 3. Data were pooled from these segments, as the number of nascent boutons formed in segments 2 and 3 of wild-type larvae did not differ. 2 mM K+ Jan’s saline solution (45 mM NaCl, 2 mM KCl, 4 mM MgCl2, 36 mM sucrose, 5 mM Hepes, and 2 mM CaCl2, pH 7.3) was used to acquire a baseline nascent bouton number.
For taxol pretreatment experiments, relaxed L3 fillets were incubated in 1.2, 6, 12, 30, 40, or 60 nM taxol in HL-3 or 0.2% DMSO in HL-3 for a 20-min prestimulation; additionally, the HL-3 wash solution was modified to contain taxol or 0.2% DMSO. Fresh taxol solutions were made for each experiment.
For VBL pretreatment experiments, relaxed L3 fillets were incubated in either 1 µM VBL (Sigma-Aldrich) in HL-3 or 0.2% DMSO in HL-3 for a 30-min prestimulation. Additionally, HL-3 wash solution was modified to contain either VBL or 0.2% DMSO. Fresh VBL solutions were made for each experiment.
Image acquisition and quantification
Fluorescent images were acquired on an inverted confocal microscope (TCS SP8; Leica Biosystems) at RT using Application Suite X software (Leica Biosystems). Futsch-labeled NMJ images were deconvolved using Huygens Professional Software (Scientific Volume Imaging). The following oil-immersion objectives were used on the microscope: HC PL APO CS2 40× (1.3 NA), HC PL APO CS2 63× (1.4 NA), and HCX PL APO 100× (1.4 NA). A white-light laser was used for all imaging. Futsch loops and bouton number were manually quantified using the Axioplan 2 equipped with a Colibri 2 illumination system using either PL Apochromat 63× (1.4 NA) or 100× PL Neofluar (1.3 NA) oil-immersion objectives. NMJs imaged on the Axioplan 2 were acquired at RT using AxioVision microscopy software, a FLUAR 40× (1.3 NA) oil-immersion objective, and an AxioCam MRc5 camera (ZEISS). All samples were mounted in 60% glycerol.
For quantification of pJNK, pAKT, and Pav immunofluorescence, control and mutant genotypes were uniformly processed and imaged using identical acquisition settings. Complete z-stacks were acquired with optimized confocal settings to ensure that oversaturation did not occur. Intensity was quantified in complete z-stack projections of nerves or NMJ4 using ImageJ64 (National Institutes of Health) by selecting and measuring fluorescence intensity in a ROI (as defined by anti-HRP immunoreactivity). Background fluorescence was averaged and subtracted from each projection only in the anti-Pav experiments. Intensity measurements of mutant NMJs or nerves are normalized to wild-type or control measurements. No modifications to any images were made before quantification.
Evaluation of anti–Ac-Tub staining was performed at NMJ4 after identically processing control and mutant genotypes. Complete z stacks were acquired with optimized confocal settings to ensure that oversaturation did not occur. The total number of boutons was counted based on anti-HRP immunoreactivity, and then the number of boutons with expanded Ac-Tub was quantified.
Analysis of Bruchpilot accumulation in nerves from segments A2 to A5 of control and mutant genotypes was performed after z stacks were acquired. The number of Bruchpilot-positive puncta per 30 µm of axon was quantified by projecting images and using the cell counter plugin in ImageJ64. Quantifications were either performed blinded or scored by another person.
Nascent bouton quantification was performed from z projections from control and mutant genotypes. Nascent boutons were identified as presynaptic boutons (labeled with anti-HRP) lacking postsynaptic specializations (labeled with anti-Dlg). Nascent boutons were quantified at NMJ6/7 in abdominal segments 2 and 3 in L3 larvae. Quantifications were performed blinded.
Quantification of FoxO-positive VNC cells was performed after z projections of the larval VNC were obtained. An identical ROI was used for each VNC, which typically encompassed three segments, and the number of FoxO-positive nuclei was quantified in abdominal segments using the cell counter function of ImageJ64. Larvae of each genotype were stained together, and images were acquired with identical settings.
Quantification of total NMJ length was performed after images were obtained of NMJ6/7 in segment A2 using either an Axioplan 2 (Leica) or TCS SP8 confocal microscope. NMJs were labeled with anti-HRP, and total length was traced in ImageJ64. Traces were analyzed, and NMJ length was computed using NeuronStudio (Computational Neurobiology and Imaging Center, Mount Sinai School of Medicine, New York, NY).
Drosophila protein extracts and immunoblots
CNS lysates were prepared from ∼15 wandering L3 larvae by extracting and immediately homogenizing brains and VNCs in 2× Laemmli sample buffer (Bio-Rad Laboratories). Lysates were subsequently heated for 5 min at 95°C. Approximately 15 CNS equivalents were loaded per well onto 12% SDS-PAGE gels (Bio-Rad Laboratories). The following antibodies and concentrations were used: rabbit anti–phospho-Drosophila Akt (Ser505; Cell Signaling Technology) at 1:1,000 and goat anti-GAPDH (Imgenex) at 1:10,000. Species-specific HRP-conjugated secondary antibodies were used at 1:10,000. Immunoblots were probed with anti-GAPDH as a loading control.
qRT-PCR
Approximately 55–60 L3 VNCs were dissected, placed on ice, and washed in cold PBS. Tissue was homogenized in TRIzol (Thermo Fisher Scientific), and RNA was isolated using chloroform extraction. cDNA was prepared from ∼1 µg RNA using SuperScript III First-Strand Synthesis SuperMix (Thermo Fisher Scientific), and real-time PCR was performed for 40 cycles with the StepOne Plus Real Time PCR system (Thermo Fisher Scientific). All TaqMan probes and reagents were purchased from the Life Technologies database. CT values of experimental genes were normalized to CT values for the housekeeping gene RpII140. Relative gene expression was determined using the ΔΔCT method. Statistical measures were performed on ΔCT values. We performed two to six biological replicates per mRNA and two to three technical replicates per biological replicate.
Statistical analysis
All statistical analyses were performed and graphs were generated using Prism 6 (GraphPad Software). In all graphs, data are presented as mean ± SEM, unless otherwise stated. All pairwise sample comparisons were performed using a Mann-Whitney test. A Kruskal-Wallis test was used to compare each sample with other samples in a group of three or more, and Dunn’s multiple comparisons test was subsequently performed. In all figures, p-values for statistical tests are as follows: *, P < 0.05; **, P < 0.01; and ***, P < 0.001.
Online supplemental material
Fig. S1 demonstrates that pAkt levels are increased in Toll-6EX13 nerves and VNCs and pJNK levels are decreased in Toll-6EX13 NMJs. Fig. S2 contains bouton counts at NMJ6/7 and NMJ4, as well as NMJ6/7 length measurements. Fig. S3 shows that motoneuronal overexpression of Pav results in bouton and MT loop phenotypes similar to pathway mutant phenotypes. Fig. S4 demonstrates that pretreatment of wild-type NMJs with taxol impairs activity-dependent structural plasticity. Fig. S5 illustrates that taxol treatment can impair transport of Bruchpilot in wild-type nerves. Table S1 contains bouton numbers and MT loop quantifications for canonical TLR pathway members, as well as Toll-6, FoxO, and dSARM RNAi lines. Table S2 contains quantification of bouton numbers in RNAi lines of mRNAs up-regulated in Toll-6EX13 VNCs. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201601014/DC1. Additional data are available in the JCB DataViewer at http://dx.doi.org/10.1083/jcb.201601014.dv.
Supplementary Material
Acknowledgments
We thank the Bloomington Drosophila Stock Center, the Vienna Drosophila RNAi Center, the Developmental Studies Hybridoma Bank at the University of Iowa, and Aaron DiAntonio, Marc Freeman, Vladimir Gelfand, Tony Ip, Christian Klämbt, Liqun Luo, Linda Partridge, and Robert Tjian for fly strains and/or antibodies. We also thank Alexander Ward for assistance with Toll-6 stocks. We thank Kelsey Herrmann, Jahci Perry-Richardson, Kendall Hoover, Karlie Fedder, Regan Brady, Dallas Eckman, Priya Tumuluru, and James Ferguson for technical assistance. We thank other members of the Broihier laboratory for helpful discussions.
This work was supported by National Institutes of Health grant T32AG00271 to C.N. McLaughlin and National Institutes of Health grants R21NS090369 and R01NS095895 to H.T. Broihier.
The authors declare no competing financial interests.
Footnotes
Abbreviations used:
- Ac-Tub
- acetylated α-tubulin
- CNS
- central nervous system
- LOF
- loss of function
- MT
- microtubule
- NMJ
- neuromuscular junction
- qRT-PCR
- quantitative RT-PCR
- ROI
- region of interest
- TIR
- Toll/interleukin-1 receptor
- TLR
- Toll-like receptor
- VBL
- vinblastine
- VNC
- ventral nerve cord
References
- Aberle H., Haghighi A.P., Fetter R.D., McCabe B.D., Magalhães T.R., and Goodman C.S.. 2002. wishful thinking encodes a BMP type II receptor that regulates synaptic growth in Drosophila. Neuron. 33:545–558. 10.1016/S0896-6273(02)00589-5 [DOI] [PubMed] [Google Scholar]
- Adams R.R., Tavares A.A., Salzberg A., Bellen H.J., and Glover D.M.. 1998. pavarotti encodes a kinesin-like protein required to organize the central spindle and contractile ring for cytokinesis. Genes Dev. 12:1483–1494. 10.1101/gad.12.10.1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhouayri I., Turc C., Royet J., and Charroux B.. 2011. Toll-8/Tollo negatively regulates antimicrobial response in the Drosophila respiratory epithelium. PLoS Pathog. 7:e1002319 10.1371/journal.ppat.1002319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ataman B., Ashley J., Gorczyca M., Ramachandran P., Fouquet W., Sigrist S.J., and Budnik V.. 2008. Rapid activity-dependent modifications in synaptic structure and function require bidirectional Wnt signaling. Neuron. 57:705–718. 10.1016/j.neuron.2008.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard S.L., Miller D.L., and Ganetzky B.. 2014. Retrograde neurotrophin signaling through Tollo regulates synaptic growth in Drosophila. J. Cell Biol. 204:1157–1172. 10.1083/jcb.201308115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak B., Feldman N., and Okun E.. 2014. Toll-like receptors as developmental tools that regulate neurogenesis during development: An update. Front. Neurosci. 8:272 10.3389/fnins.2014.00272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A., Bonni A., Zigmond M.J., Lin M.Z., Juo P., Hu L.S., Anderson M.J., Arden K.C., Blenis J., and Greenberg M.E.. 1999. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 96:857–868. 10.1016/S0092-8674(00)80595-4 [DOI] [PubMed] [Google Scholar]
- Byrne A.B., Walradt T., Gardner K.E., Hubbert A., Reinke V., and Hammarlund M.. 2014. Insulin/IGF1 signaling inhibits age-dependent axon regeneration. Neuron. 81:561–573. 10.1016/j.neuron.2013.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calnan D.R., and Brunet A.. 2008. The FoxO code. Oncogene. 27:2276–2288. 10.1038/onc.2008.21 [DOI] [PubMed] [Google Scholar]
- Cantera R., Kozlova T., Barillas-Mury C., and Kafatos F.C.. 1999. Muscle structure and innervation are affected by loss of Dorsal in the fruit fly, Drosophila melanogaster. Mol. Cell. Neurosci. 13:131–141. 10.1006/mcne.1999.0739 [DOI] [PubMed] [Google Scholar]
- Chang C., Hsieh Y.-W., Lesch B.J., Bargmann C.I., and Chuang C.-F.. 2011. Microtubule-based localization of a synaptic calcium-signaling complex is required for left-right neuronal asymmetry in C. elegans. Development. 138:3509–3518. 10.1242/dev.069740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.-Y., Lin C.-W., Chang C.-Y., Jiang S.-T., and Hsueh Y.-P.. 2011. Sarm1, a negative regulator of innate immunity, interacts with syndecan-2 and regulates neuronal morphology. J. Cell Biol. 193:769–784. 10.1083/jcb.201008050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen R., de la Torre-Ubieta L., Bonni A., and Colón-Ramos D.A.. 2011. A conserved PTEN/FOXO pathway regulates neuronal morphology during C. elegans development. Development. 138:5257–5267. 10.1242/dev.069062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang C.-F., and Bargmann C.I.. 2005. A Toll-interleukin 1 repeat protein at the synapse specifies asymmetric odorant receptor expression via ASK1 MAPKKK signaling. Genes Dev. 19:270–281. 10.1101/gad.1276505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre-Ubieta L., Gaudillière B., Yang Y., Ikeuchi Y., Yamada T., DiBacco S., Stegmüller J., Schüller U., Salih D.A., Rowitch D., et al. 2010. A FOXO-Pak1 transcriptional pathway controls neuronal polarity. Genes Dev. 24:799–813. 10.1101/gad.1880510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Castillo U., Lu W., Winding M., Lakonishok M., and Gelfand V.I.. 2015. Pavarotti/MKLP1 regulates microtubule sliding and neurite outgrowth in Drosophila neurons. Curr. Biol. 25:200–205. 10.1016/j.cub.2014.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent E.W., Merriam E.B., and Hu X.. 2011. The dynamic cytoskeleton: Backbone of dendritic spine plasticity. Curr. Opin. Neurobiol. 21:175–181. 10.1016/j.conb.2010.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essers M.A.G., de Vries-Smits L.M.M., Barker N., Polderman P.E., Burgering B.M.T., and Korswagen H.C.. 2005. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science. 308:1181–1184. 10.1126/science.1109083 [DOI] [PubMed] [Google Scholar]
- Ferrandon D., Imler J.-L., and Hoffmann J.A.. 2004. Sensing infection in Drosophila: Toll and beyond. Semin. Immunol. 16:43–53. 10.1016/j.smim.2003.10.008 [DOI] [PubMed] [Google Scholar]
- Gay N.J., and Gangloff M.. 2007. Structure and function of Toll receptors and their ligands. Annu. Rev. Biochem. 76:141–165. 10.1146/annurev.biochem.76.060305.151318 [DOI] [PubMed] [Google Scholar]
- Gerdts J., Summers D.W., Sasaki Y., DiAntonio A., and Milbrandt J.. 2013. Sarm1-mediated axon degeneration requires both SAM and TIR interactions. J. Neurosci. 33:13569–13580. 10.1523/JNEUROSCI.1197-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gornstein E., and Schwarz T.L.. 2014. The paradox of paclitaxel neurotoxicity: Mechanisms and unanswered questions. Neuropharmacology. 76:175–183. 10.1016/j.neuropharm.2013.08.016 [DOI] [PubMed] [Google Scholar]
- Heckscher E.S., Fetter R.D., Marek K.W., Albin S.D., and Davis G.W.. 2007. NF-kappaB, IkappaB, and IRAK control glutamate receptor density at the Drosophila NMJ. Neuron. 55:859–873. 10.1016/j.neuron.2007.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horng T., and Medzhitov R.. 2001. Drosophila MyD88 is an adapter in the Toll signaling pathway. Proc. Natl. Acad. Sci. USA. 98:12654–12658. 10.1073/pnas.231471798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummel T., Krukkert K., Roos J., Davis G., and Klämbt C.. 2000. Drosophila Futsch/22C10 is a MAP1B-like protein required for dendritic and axonal development. Neuron. 26:357–370. 10.1016/S0896-6273(00)81169-1 [DOI] [PubMed] [Google Scholar]
- Janke C., and Bulinski J.C.. 2011. Post-translational regulation of the microtubule cytoskeleton: Mechanisms and functions. Nat. Rev. Mol. Cell Biol. 12:773–786. 10.1038/nrm3227 [DOI] [PubMed] [Google Scholar]
- Janke C., and Kneussel M.. 2010. Tubulin post-translational modifications: encoding functions on the neuronal microtubule cytoskeleton. Trends Neurosci. 33:362–372. 10.1016/j.tins.2010.05.001 [DOI] [PubMed] [Google Scholar]
- Jordan M.A., Thrower D., and Wilson L.. 1992. Effects of vinblastine, podophyllotoxin and nocodazole on mitotic spindles. Implications for the role of microtubule dynamics in mitosis. J. Cell Sci. 102:401–416. [DOI] [PubMed] [Google Scholar]
- Kalil K., and Dent E.W.. 2014. Branch management: mechanisms of axon branching in the developing vertebrate CNS. Nat. Rev. Neurosci. 15:7–18. 10.1038/nrn3650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitein L.C., and Hoogenraad C.C.. 2015. Building the neuronal microtubule cytoskeleton. Neuron. 87:492–506. 10.1016/j.neuron.2015.05.046 [DOI] [PubMed] [Google Scholar]
- Lee S.S., Kennedy S., Tolonen A.C., and Ruvkun G.. 2003. DAF-16 target genes that control C. elegans life-span and metabolism. Science. 300:644–647. 10.1126/science.1083614 [DOI] [PubMed] [Google Scholar]
- Li L.-B., Lei H., Arey R.N., Li P., Liu J., Murphy C.T., Xu X.Z.S., and Shen K.. 2016. The neuronal kinesin Unc-104/KIF1A is a key regulator of synaptic aging and insulin signaling-regulated memory. Curr. Biol. 26:605–615. 10.1016/j.cub.2015.12.068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S., Liu M., Mozgova O.I., Yu W., and Baas P.W.. 2012. Mitotic motors coregulate microtubule patterns in axons and dendrites. J. Neurosci. 32:14033–14049. 10.1523/JNEUROSCI.3070-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay S.A., and Wasserman S.A.. 2014. Conventional and non-conventional Drosophila Toll signaling. Dev. Comp. Immunol. 42:16–24. 10.1016/j.dci.2013.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G., and Dwyer T.. 2014. Microtubule dynamics in axon guidance. Neurosci. Bull. 30:569–583. 10.1007/s12264-014-1444-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W., Fox P., Lakonishok M., Davidson M.W., and Gelfand V.I.. 2013. Initial neurite outgrowth in Drosophila neurons is driven by kinesin-powered microtubule sliding. Curr. Biol. 23:1018–1023. 10.1016/j.cub.2013.04.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W., Lakonishok M., and Gelfand V.I.. 2015. Kinesin-1-powered microtubule sliding initiates axonal regeneration in Drosophila cultured neurons. Mol. Biol. Cell. 26:1296–1307. 10.1091/mbc.E14-10-1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlroy G., Foldi I., Aurikko J., Wentzell J.S., Lim M.A., Fenton J.C., Gay N.J., and Hidalgo A.. 2013. Toll-6 and Toll-7 function as neurotrophin receptors in the Drosophila melanogaster CNS. Nat. Neurosci. 16:1248–1256. 10.1038/nn.3474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer S.N., Amoyel M., Bergantiños C., de la Cova C., Schertel C., Basler K., and Johnston L.A.. 2014. An ancient defense system eliminates unfit cells from developing tissues during cell competition. Science. 346:1258236 10.1126/science.1258236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee P., Winkler C.W., Taylor K.G., Woods T.A., Nair V., Khan B.A., and Peterson K.E.. 2015. SARM1, not MyD88, mediates TLR7/TLR9-induced apoptosis in neurons. J. Immunol. 195:4913–4921. 10.4049/jimmunol.1500953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahm M., Lee M.-J., Parkinson W., Lee M., Kim H., Kim Y.-J., Kim S., Cho Y.S., Min B.-M., Bae Y.C., et al. 2013. Spartin regulates synaptic growth and neuronal survival by inhibiting BMP-mediated microtubule stabilization. Neuron. 77:680–695. 10.1016/j.neuron.2012.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naka K., Hoshii T., Muraguchi T., Tadokoro Y., Ooshio T., Kondo Y., Nakao S., Motoyama N., and Hirao A.. 2010. TGF-β-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature. 463:676–680. 10.1038/nature08734 [DOI] [PubMed] [Google Scholar]
- Nechipurenko I.V., and Broihier H.T.. 2012. FoxO limits microtubule stability and is itself negatively regulated by microtubule disruption. J. Cell Biol. 196:345–362. 10.1083/jcb.201105154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newquist G., Drennan J.M., Lamanuzzi M., Walker K., Clemens J.C., and Kidd T.. 2013. Blocking apoptotic signaling rescues axon guidance in Netrin mutants. Cell Reports. 3:595–606. 10.1016/j.celrep.2013.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill L.A.J., and Bowie A.G.. 2007. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 7:353–364. 10.1038/nri2079 [DOI] [PubMed] [Google Scholar]
- Okun E., Griffioen K.J., and Mattson M.P.. 2011. Toll-like receptor signaling in neural plasticity and disease. Trends Neurosci. 34:269–281. 10.1016/j.tins.2011.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterloh J.M., Yang J., Rooney T.M., Fox A.N., Adalbert R., Powell E.H., Sheehan A.E., Avery M.A., Hackett R., Logan M.A., et al. 2012. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science. 337:481–484. 10.1126/science.1223899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccioli Z.D., and Littleton J.T.. 2014. Retrograde BMP signaling modulates rapid activity-dependent synaptic growth via presynaptic LIM kinase regulation of cofilin. J. Neurosci. 34:4371–4381. 10.1523/JNEUROSCI.4943-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig O., Marr M.T., Ruhf M.L., and Tjian R.. 2003. Control of cell number by Drosophila FOXO: Downstream and feedback regulation of the insulin receptor pathway. Genes Dev. 17:2006–2020. 10.1101/gad.1098703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos J., Hummel T., Ng N., Klämbt C., and Davis G.W.. 2000. Drosophila Futsch regulates synaptic microtubule organization and is necessary for synaptic growth. Neuron. 26:371–382. 10.1016/S0896-6273(00)81170-8 [DOI] [PubMed] [Google Scholar]
- Salzberg A., D’Evelyn D., Schulze K.L., Lee J.K., Strumpf D., Tsai L., and Bellen H.J.. 1994. Mutations affecting the pattern of the PNS in Drosophila reveal novel aspects of neuronal development. Neuron. 13:269–287. 10.1016/0896-6273(94)90346-8 [DOI] [PubMed] [Google Scholar]
- Sanyal S. 2009. Genomic mapping and expression patterns of C380, OK6 and D42 enhancer trap lines in the larval nervous system of Drosophila. Gene Expr. Patterns. 9:371–380. 10.1016/j.gep.2009.01.002 [DOI] [PubMed] [Google Scholar]
- Seiler F., Hellberg J., Lepper P.M., Kamyschnikow A., Herr C., Bischoff M., Langer F., Schäfers H.-J., Lammert F., Menger M.D., et al. 2013. FOXO transcription factors regulate innate immune mechanisms in respiratory epithelial cells. J. Immunol. 190:1603–1613. 10.4049/jimmunol.1200596 [DOI] [PubMed] [Google Scholar]
- Sommi P., Ananthakrishnan R., Cheerambathur D.K., Kwon M., Morales-Mulia S., Brust-Mascher I., and Mogilner A.. 2010. A mitotic kinesin-6, Pav-KLP, mediates interdependent cortical reorganization and spindle dynamics in Drosophila embryos. J. Cell Sci. 123:1862–1872. 10.1242/jcs.064048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tank E.M.H., Rodgers K.E., and Kenyon C.. 2011. Spontaneous age-related neurite branching in Caenorhabditis elegans. J. Neurosci. 31:9279–9288. 10.1523/JNEUROSCI.6606-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauszig-Delamasure S., Bilak H., Capovilla M., Hoffmann J.A., and Imler J.-L.. 2002. Drosophila MyD88 is required for the response to fungal and Gram-positive bacterial infections. Nat. Immunol. 3:91–97. 10.1038/ni747 [DOI] [PubMed] [Google Scholar]
- Trotta N., Orso G., Rossetto M.G., Daga A., and Broadie K.. 2004. The hereditary spastic paraplegia gene, spastin, regulates microtubule stability to modulate synaptic structure and function. Curr. Biol. 14:1135–1147. 10.1016/j.cub.2004.06.058 [DOI] [PubMed] [Google Scholar]
- Troutman T.D., Bazan J.F., and Pasare C.. 2012. Toll-like receptors, signaling adapters and regulation of the pro-inflammatory response by PI3K. Cell Cycle. 11:3559–3567. 10.4161/cc.21572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valakh V., Walker L.J., Skeath J.B., and DiAntonio A.. 2013. Loss of the spectraplakin short stop activates the DLK injury response pathway in Drosophila. J. Neurosci. 33:17863–17873. 10.1523/JNEUROSCI.2196-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valanne S., Wang J.-H., and Rämet M.. 2011. The Drosophila Toll signaling pathway. J. Immunol. 186:649–656. 10.4049/jimmunol.1002302 [DOI] [PubMed] [Google Scholar]
- Wang M.C., Bohmann D., and Jasper H.. 2005. JNK extends life span and limits growth by antagonizing cellular and organism-wide responses to insulin signaling. Cell. 121:115–125. 10.1016/j.cell.2005.02.030 [DOI] [PubMed] [Google Scholar]
- Ward A., Hong W., Favaloro V., and Luo L.. 2015. Toll receptors instruct axon and dendrite targeting and participate in synaptic partner matching in a Drosophila olfactory circuit. Neuron. 85:1013–1028. 10.1016/j.neuron.2015.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.