

Abstract
Herpesviruses establish a chronic infection in the host characterized by intervals of lytic replication, quiescent latency, and reactivation from latency. Murine gammaherpesvirus 68 (MHV68) naturally infects small rodents and has genetic and biologic parallels with the human gammaherpesviruses (gHVs), Kaposi’s sarcoma-associated herpesvirus and Epstein–Barr virus. The murine gammaherpesvirus model pathogen system provides a platform to apply cutting-edge approaches to dissect the interplay of gammaherpesvirus and host determinants that enable colonization of the host, and that shape the latent or lytic fate of an infected cell. This knowledge is critical for the development of novel therapeutic interventions against the oncogenic gHVs. The nuclear factor kappa B (NF-κB) signaling pathway is well-known for its role in the promotion of inflammation and many aspects of B cell biology. Here, we review key aspects of the virus lifecycle in the host, with an emphasis on the route that the virus takes to gain access to the B cell latency reservoir. We highlight how the murine gammaherpesvirus requires components of the NF-κB signaling pathway to promote replication, latency establishment, and maintenance of latency. These studies emphasize the complexity of gammaherpesvirus interactions with NF-κB signaling components that direct innate and adaptive immune responses of the host. Importantly, multiple facets of NF-κB signaling have been identified that might be targeted to reduce the burden of gammaherpesvirus-associated diseases.
Keywords: NF-kappaB, gammaherpesvirus, pathogenesis, latency
Gammaherpesvirus Infection and Associated Malignancies
Features of Gammaherpesviruses
The gammaherpesviruses (gHVs) represent an ancient and highly successful family of viruses that coevolved with their hosts over the course of 60–80 million years (Davison, 2002). As a subfamily of the herpesviridae, the gammaherpesvirinae are characterized by an encapsidated double-stranded DNA genome that encodes 70–80 open reading frames (Parker et al., 1990; Russo et al., 1996; Virgin et al., 1997). In addition to protein coding genes, the gHVs encode non-coding RNAs including miRNAs (Pfeffer et al., 2004, 2005). Herpesvirus virions are surrounded by a lipid envelope that contains numerous glycoproteins that mediate entry into the cell. Another characteristic of the herpesvirus virion is the tegument, a structured proteinaceous layer located between the capsid and the lipid envelope. Tegument proteins are delivered into the cytoplasm of the infected cell immediately upon infection and many play crucial roles in early infection.
A hallmark of herpesvirus infection, including that of the gHVs, is the ability to switch between two distinct phases: lytic infection and latency. Lytic infection is characterized by expression of a majority of viral genes in a regulated cascade of gene expression, replication of viral DNA as linear concatemers, and production of infectious virions. Latency is defined by extremely restricted viral gene expression, the maintenance of the viral genome as a circular non-integrated episome tethered to the cellular genome (Yates and Guan, 1991; Ballestas et al., 1999; Lee et al., 1999; Collins et al., 2002; Habison et al., 2012), and the ability to switch from latent infection to productive virus infection, a process known as reactivation. GHVs infect a wide range of cell types, including epithelial cells (Sixbey et al., 1983, 1984), endothelial cells (Boshoff et al., 1995), monocytes (Weck et al., 1999b), and lymphocytes (Alfieri et al., 1991; Sunil-Chandra et al., 1992a) (Table 1). The predominant cellular reservoir of latency is lymphocytes; the human gHVs target the mature B cell compartment (Ambroziak et al., 1995; Babcock et al., 1998; Hassman et al., 2011).
Table 1.
Comparison of select gammaherpesviruses.
| Murine gammaherpesvirus 68 (MHV68) | Herpesvirus saimiri (HVS) | Kaposi’s sarcoma-associated herpesvirus (KSHV) | Epstein–Barr virus (EBV) | |
|---|---|---|---|---|
| Formal name | Murid herpesvirus 4 | Saimiriine herpesvirus 2 | Human herpesvirus 8 | Human herpesvirus 4 |
| Classification | Rhadinovirus | Rhadinovirus | Rhadinovirus | Lymphocryptovirus |
| Natural host | Wood mice | Squirrel monkey | Humans | Humans |
| Transmission | Likely sexual | Likely by contact, fomites, aerosol | Saliva, sexual | Saliva, sexual |
| Associated diseases | Increased incidence lymphoproliferative disease, lymphoproliferative disease and fibrosis in immune deficient mice | Hematologic malignancies in non-native new world primates | Primary effusion lymphomas, Kaposi’s sarcoma, Multicentric Castleman’s disease, AIDS-associated lymphoproliferative disease | Infectious mononucleosis, Burkitt’s lymphoma, gastric carcinoma, nasopharyngeal carcinoma, Hodgkin’s disease, post-transplant and AIDS-associated lymphoproliferative disease |
| Cell targets | Endothelial cells, epithelial cells, fibroblasts, B lymphocytes, dendritic cells macrophages | Epithelial cells, T lymphocytes | Endothelial cells, B lymphocytes, monocytes, epithelial cells | Epithelial cells, B lymphocytes |
| Major latency reservoir | B lymphocytes, Macrophage cells | T lymphocytes | B lymphocytes | B lymphocytes |
| Latency maintenance | Episome tethering to host chromosome via LANA, chromatin silencing | Episome tethering to host chromosome via LANA, chromatin silencing | Episome tethering to host chromosome via LANA, chromatin silencing | Episome tethering to host chromosome via EBNA1, chromatin silencing |
| Triggers of reactivation | Terminal differentiation to plasma cells, inhibition of histone deacetylases, TLR stimulation, induced expression of viral transactivator RTA | Inhibition of histone deacetylases, induced expression of viral transactivator RTA | Inhibition of histone deacetylases, induced expression of viral transactivator RTA | Terminal differentiation to plasma cells, inhibition of histone deacetylases, induced expression of viral transactivators Zebra and RTA |
The Human Gammaherpesviruses Are Associated with Cancer
Within the gHVs there are two known human gHVs; EBV (Epstein–Barr virus, human herpesvirus-4), is the prototypical member of lymphocryptoviridae, and KSHV (Kaposi’s sarcoma-associated herpesvirus, human herpesvirus-8), is the prototypical member of the rhadinoviridae. EBV infects over 95% of adult humans, while KSHV infection varies by geographical location and HIV infection status. EBV infection is associated with the development of Burkitt’s lymphoma, Hodgkin’s lymphoma, and nasopharyngeal carcinoma (Table 1). KSHV infection is the etiological agent of Kaposi’s sarcoma and is associated with primary effusion lymphoma and multicentric Castleman disease (Verma and Robertson, 2003; Cesarman, 2014). Immunocompromised patients, such as individuals co-infected with HIV and either gHV, have a greatly increased incidence of malignancies (Simard and Engels, 2010; Jha et al., 2016). Despite the effectiveness of HAART therapy in controlling HIV, both gHV-induced lymphomas remain the most common cancer-related cause of death in HIV-infected patients (Simard et al., 2011). EBV infection also causes post-transplant lymphoproliferative disorder (PTLD). Immunosuppressive drugs used to prevent transplant rejection impair T cell control of the latent infection, leading to the emergence of EBV-infected cells with a less restricted latency program that can drive proliferation of virally infected cells (Landgren et al., 2009; Rasul et al., 2012; Tse and Kwong, 2015).
Murine Gammaherpesvirus 68 (MHV68) Infection of Mice As A Model System of Gammaherpesvirus Pathogenesis
Value of a Small Animal Pathogen System
Due to the ineffectiveness of current antiviral therapies in combating latent infections and the lack of vaccines, a detailed understanding of virus–host interactions is needed for the design of targeted therapeutics against gHVs. Cell culture-based studies of the human gHVs have defined the role of many viral and host factors during infection. The mechanisms by which EBV and KSHV promote proliferation and survival of the host cell are likely major risk factors for the development of lymphomas. However, studies on the role of virus–host interactions and how they influence pathogenesis in the host have been hampered due to the inability to perform synchronous de novo infection in cell culture and the lack of tractable small animal models due to strict host tropism. Thus, a natural gammaherpesvirus pathogen of murid rodents provides a relevant and powerful model system for assaying factors that affect gHV pathogenesis (Simas and Efstathiou, 1998; Blackman et al., 2000; Speck and Ganem, 2010; Barton et al., 2011).
Murine Gammaherpesvirus 68 Is Endemic to Murid Rodents
Murine gammaherpesvirus 68 (MHV68, formally identified as murid herpesvirus 4) is a natural pathogen of murid rodents used to study virus–host interactions in the context of a whole animal. MHV68 was originally isolated from bank voles in the former Soviet republic of Czechoslovakia (Blaskovic et al., 1980), and has since been identified in yellow-necked wood mice in England (Blasdell et al., 2003), indicating that MHV68 may be endemic to European rodent populations. MHV68 productively infects, and establishes latency in, all tested strains of Mus musculus.
Genomic Similarities to Primate Gammaherpesviruses
Murine gammaherpesvirus 68 shares genomic, biologic, and pathologic properties with primate and human gHVs (Table 1). MHV68 is classified as a Rhadinovirus along with KSHV and herpesvirus saimiri (HVS, saimiriine herpesvirus 2). The genome of MHV68 is ∼128 kb, and encodes for an estimated 80 ORFs that are largely organized in gene blocks similar to the genomes of HVS, KSHV, and EBV (Efstathiou et al., 1990a,b; Virgin et al., 1997; Simas and Efstathiou, 1998). Transposon mutagenesis screening of MHV68 genes identified a number of genes essential for virus growth that are conserved within the gHV family (Moorman et al., 2004; Song et al., 2005).
As found for other gHVs, MHV68 encodes genes that were likely acquired from the host (Virgin et al., 1997) including a viral homolog of the cellular anti-apoptotic bcl-2 protein (vBcl-2, MHV68 M11), a homolog of an IL-8 receptor (vGPCR, MHV68 ORF74), cyclin D (v-cyclin, MHV68 ORF72), and the complement regulatory protein (MHV68 ORF4; Virgin et al., 1997). MHV68 M11 exhibits both anti-apoptotic and anti-autophagic functions (Ku et al., 2008; Sinha et al., 2008; Xiaofei et al., 2009), and functions to promote the survival of infected immature B cells (Coleman et al., 2014). MHV68 expresses the vGPCR (ORF74), a chemokine receptor with homology to human CXCL2, during latency (Virgin et al., 1997; Wakeling et al., 2001). MHV68 vGPCR is not constitutively active, however, it is activated by a wide array of cytokines containing ELR motifs to activate signaling pathways including nuclear factor kappa B (NF-κB) and Akt (Verzijl et al., 2004), and it can transform NIH3T3 cells in culture (Wakeling et al., 2001). The MHV68 v-cyclin promotes replication and reactivation in vivo (Upton and Speck, 2006), contributes to the immortalization of fetal liver B cells (Liang et al., 2011) and drives lymphomagenesis (van Dyk et al., 1999).
While many proteins involved in lytic replication are largely conserved amongst the gHVs, each gHV encodes unique genes. MHV68 contains both unique genes and non-coding RNAs (Bowden et al., 1997; Virgin et al., 1999; Reese et al., 2010; Zhu et al., 2010). The MHV68 genome encodes 14 unique M genes that are located throughout the viral genome, many have immunomodulatory functions that promote chronic infection (Evans et al., 2008; Siegel et al., 2008; Hughes et al., 2011). The left end of MHV68 encodes eight non-coding RNAs termed TMERS, viral tRNA-like molecules embedded with single or multiple 3’ miRNAs, that are expressed during latency and productive infection (Bogerd et al., 2010; Feldman et al., 2014). Recent work has uncovered a role for the viral TMERs in viral dissemination and latency establishment in wild-type (WT) mice, as well as pneumonia in an immunocompromised host (Feldman et al., 2014, 2016; Diebel et al., 2015). Specific targets for the miRNAs and the function of the tRNA-like molecules have not been determined.
Pathogenic Features Common with Primate and Human Gammaherpesviruses
Murine gammaherpesvirus 68 also shares pathogenic similarities with both the primate and human gHVs (Table 1). MHV68 undergoes a subclinical period of acute replication upon infection of the nasal or respiratory mucosal tissue (Rajcani et al., 1985; Sunil-Chandra et al., 1992b; Milho et al., 2012). Productive lytic infection in the lung causes localized tissue damage that undergoes normal repair (Ehtisham et al., 1993). The initial infection is cleared from the lungs by 12 days post infection in BALB/c and C57BL/6 mice (Sunil-Chandra et al., 1992b; Cardin et al., 1996), then trafficked by B cells to the spleen (Usherwood et al., 1996c). MHV68 infection is associated with an expansion of lymphocyte populations that drives an infectious mononucleosis-like response marked by enlarged lymph nodes and splenomegaly (Usherwood et al., 1996a; Tripp et al., 1997; Flano et al., 2002b).
Upon gaining access to the spleen, MHV68 establishes latency in germinal center B cells (Collins et al., 2009). Levels of latency in the spleen contract by 3–4 weeks post-infection, and the predominant reservoir of viral latency shifts to the memory B cells as found for EBV (Miyashita et al., 1995, 1997; Babcock et al., 1998; Flano et al., 2002a; Willer and Speck, 2003). Immune competent animals typically control latent infection, but long-term studies reported a higher incidence of lymphomas between 6 months and 3 years post infection compared to uninfected mice (Sunil-Chandra et al., 1994). The latently infected B lymphoma cell line, S11, was derived from long-term MHV68-infected mice that spontaneously developed lymphoproliferative disease (Usherwood et al., 1996b). Additional immunosuppression via cyclosporine A administration, the loss of T cells or interferon gamma signaling increases the prevalence of lymphoproliferative disease, mirroring the development of gHV-associated malignancies in immunosuppressed humans (Sunil-Chandra et al., 1994; Tarakanova et al., 2005, 2008; Lee et al., 2009). In addition, MHV68 can drive fetal liver B cell immortalization and latency programs are associated with the development of B cell lymphomas in mice (Liang et al., 2011).
Genetic Manipulation of Virus and Host Determinants
The MHV68 pathogenesis system allows for the mutation of host or viral genes, enabling in vivo analysis of determinants of viral pathogenesis. The MHV68 genome is cloned as a bacterial artificial chromosome system (BAC), allowing for straightforward mutagenesis of the viral genome in E. coli (Adler et al., 2000; Tischer et al., 2010). To better track the course of infection in vivo, several reporter viruses have been generated. These include a constitutively expressed YFP, a lytic promoter-driven luciferase marker, or direct reporter fusion with the major latency protein LANA (Hwang et al., 2008; Collins et al., 2009; Nealy et al., 2010). Likewise, the mouse system offers a wide array of germline and inducible knockouts of host genes to identify host determinants of infection and to facilitate a better understanding of the virus–host interactions that occur during multiple stages of infection. Coupling a virus with a reporter gene flanked by loxP sites with transgenic mice that express Cre in specific cell types has been applied to track the cells that MHV68 infects en route to the latency reservoir (Gaspar et al., 2011).
In addition, the accessibility of lytic and latent tissue culture models and the ability of MHV68 to replicate to high titer in tissue culture facilitate mechanistic studies in vitro. MHV68 replicates in murine fibroblasts, endothelial, epithelial, dendritic cells, and macrophages, with a single round of lytic replication lasting 12–18 h (Speck and Ganem, 2010). B cells can be de novo infected in vitro, albeit with low efficiency, and latent B cell lines have been established (Usherwood et al., 1996b; Forrest and Speck, 2008; Liang et al., 2011). Together, these systems allow for examination of multiple aspects of the viral life cycle in vitro.
Course of MHV68 Infection in Mice
Routes of Transmission
Epstein–Barr virus and KSHV are primarily contracted through the oral mucosa, and initial infection is often subclinical. EBV infection can lead to acute infectious mononucleosis (AIM), especially when contracted in adolescence or early adulthood (Hoagland, 1964). AIM patients generally exhibit symptoms many weeks after infection; thus, the initial kinetics of EBV replication in the oral epithelium and latency establishment of B cells are not well-understood (Sixbey et al., 1984; Dunmire et al., 2015). Natural transmission of MHV68 between animals is not clearly defined. MHV68 can readily infect the nasal and respiratory mucosa upon intranasal infection (Tan et al., 2014), but it is unclear if this occurs in the wild. MHV68 and the closely related MHV72 may undergo vertical transfer from mother to pup, as viral DNA can be detected in the breast milk of infected dams and stomachs of the pups (Raslova et al., 2001; Stiglincova et al., 2011). However, bone fide vertical transmission was not observed since latency was not established in the pups (Francois et al., 2013). Co-housing of latently infected animals with naïve same-sex animals is insufficient for horizontal transmission (Francois et al., 2013).
Recent studies support a sexual mode of transmission for MHV68. Epidemiologic studies of wild mice found the highest prevalence of infection in the heaviest, most sexually active males (Telfer et al., 2007). High titers of virus inoculum applied to the vagina of mice led to intranasal infection of naïve mice (Milho et al., 2012). Intermittent virus replication has been reported in the genitalia of intranasally infected mice during latent infection (Francois et al., 2013). Indeed, MHV68 was transmitted from a female reactivating at the genitalia to a naïve male upon sexual contact (Francois et al., 2013).
Virus Colonization of the Host
Infection of mice with MHV68 has given us insight into how gHVs ultimately gain access to the B cell memory reservoir. Viruses that mark infected cells have been coupled with cell-specific knock-out or ablation approaches to reveal a complex series of events that culminate in colonization of the spleen (Gaspar et al., 2011; Frederico et al., 2012, 2014a,b, 2015). The virus likely undergoes sequential infection of multiple cell types during transit from the site of initial infection- nasal or lung tissue in laboratory settings- to the draining lymph nodes and the spleen (Figure 1). MHV68 infection in vivo does not result in high levels of cell-free virus (Sunil-Chandra et al., 1992a), and the virus depends on cell-to-cell spread for efficient propagation (May et al., 2005). Hematogenous dissemination is likely involved; deletion of MHV68 non-coding RNA TMER4 is sufficient to reduce viral burden in peripheral leukocytes and impair infection of the spleen and peritoneal cavity (Feldman et al., 2016). In the absence of B cells, seeding of the secondary lymphoid tissues is significantly decreased, and mice do not develop an infectious mononucleosis-like syndrome (Usherwood et al., 1996c; Chao et al., 2015). Even in the absence of B cells, latency is detected in the spleen at late times post infection, suggesting alternative routes that allow access to distal reservoirs of latency (Weck et al., 1996). MHV68 latency in non-B cells such as macrophages requires the function of v-cyclin to drive reactivation and maintain the viral reservoir (van Dyk et al., 2003).
FIGURE 1.

Course of an MHV68 infection in mice. Acute lytic replication occurs in the lung (intranasal infection) or in the spleen (intraperitoneal infection). Ongoing replication is cleared typically by 12 days. The peak of latency is measured around 16 days post-infection in secondary lymphoid tissues such as the spleen. Maintenance of latency is typically evaluated 42 days post-infection. B cells are the predominant latency reservoir, followed by macrophages, dendritic cells, and endothelial cells. Lymphomas arise at late stages in immunocompromised animals.
Upper Respiratory Tract Infection
Intranasal infection without the use of anesthetic delivers the viral inoculum into the upper airways (Tan et al., 2014). Upon inoculation of the upper respiratory tract infection, MHV68 first infects the neuroepithelium of the rostral nasal cavity (Frederico et al., 2012; Milho et al., 2012). Infection of the neuroepithelium is dependent on viral heparin sulfate-binding glycoproteins (Milho et al., 2012). From here, infection progresses to the draining superficial cervical lymph nodes via dendritic cells, after which the virus transits to the spleen (Milho et al., 2009; Gaspar et al., 2011). Upper respiratory tract infection leads to similar levels of splenic latency in the spleen as that obtained from deeper lung infection; however, there is a kinetic delay in viral colonization of the spleen (Milho et al., 2009). Subcapsular sinus macrophages that filter the afferent lymph nodes become infected by MHV68, yet produce few infectious progeny virions. Interestingly, their depletion increased splenic B cell infection, and led the authors to propose that this cell type may represent a bottleneck to dissemination (Frederico et al., 2015).
Lower Respiratory Tract Infection
Murine gammaherpesvirus 68 pathogenesis studies typically report the inoculation of mice by autonomic deep inhalation occurring after light anesthesia. A large bolus of liquid is inhaled deep into the lung, infecting both the upper respiratory tract as well as the lung epithelium, but neither the trachea nor bronchi (Milho et al., 2009). Intranasal inoculation leads to viral attachment to type 1 alveolar epithelial cells and initial infection of alveolar macrophages (Lawler et al., 2015), which collect particulate from the alveolar lumen (Geiser, 2002). From these macrophages, infection spreads to type 1 alveolar epithelial cells (Sunil-Chandra et al., 1992a; Nash and Sunil-Chandra, 1994; Milho et al., 2009; Lawler et al., 2015). Viral titer increases in the lungs, peaking at 7 dpi, before being cleared by 12 dpi (Ehtisham et al., 1993), with CD8+ T cells playing a central role in this process (Weck et al., 1996; Doherty et al., 2001). Interestingly, despite the significant expansion of virus in the lungs and evidence of inflammatory infiltrate, tissue repair largely restores lung morphology (Sunil-Chandra et al., 1992a; Ehtisham et al., 1993; Dutia et al., 1997). There is significant infection of macrophages in the lungs by this method, and the virus primarily drains to the mediastinal lymph node via a dendritic cell-independent route (Gaspar et al., 2011). The virus then transits to the spleen in a B cell dependent manner (Usherwood et al., 1996c).
Role of Productive Replication in Dissemination and Latency
The influence of acute replication defects in the lung on the establishment of latency after intranasal inoculation is well documented. MHV68 mutants lacking genes necessary for productive replication or DNA replication fail to establish WT levels of latency in the spleens of infected mice after intranasal inoculation (Coleman et al., 2003; Flano et al., 2005; Moser et al., 2006; Tibbetts et al., 2006; Gill et al., 2009, 2010; Milho et al., 2011; Minkah et al., 2015). Splenic latency could be partially or fully restored after altering the route of infection or dramatically increasing virus dose (Coleman et al., 2003; Flano et al., 2005; Gill et al., 2009, 2010; Milho et al., 2011; Minkah et al., 2015). Together these observations imply a critical role for lytic replication in seeding the latent splenic reservoir after intranasal inoculation. For many mutants, it remains to be determined if phenotypes represent an inability to replicate in mucosal tissues at sites of entry or represent an inability to efficiently infect immune cells critical for the dissemination of MHV68 to the B cells in the spleen. Alternatively, some degree of ‘lytic’ gene expression might be required to establish a latent gene expression in a particular cell-type.
Seeding of the Germinal Center
The route of MHV68 to the germinal center B cell likely involves multiple cellular transitions (Gaspar et al., 2011; Frederico et al., 2012, 2014a, 2015). Blood entering the spleen in a central arteriole first passes through the macrophage-rich marginal sinuses. These macrophages are the first splenocytes to become infected by MHV68, providing an entryway into the white pulp and expanding lymphocyte reservoir (Frederico et al., 2014a). Infection of these marginal zone macrophages leads to lytic infection and the production of virions that are better suited to infect B cells (Frederico et al., 2012). These virions infect adjacent marginal zone B cells that carry the virus to the emerging germinal centers (Frederico et al., 2014a). Marginal zone B cells associate with and deposit virions onto the follicular dendritic cells (Frederico et al., 2014a). The dendritic cells then seed the expanding population of germinal center B cells, where latent infection is detected at the highest frequency (Sunil-Chandra et al., 1992b). Although the germinal center B cell response subsides after the 14–18 dpi peak, latency is maintained in germinal center B cells as well as memory B cells at late times after infection (Flano et al., 2002a; Willer and Speck, 2003; Nealy et al., 2010). T follicular helper cells play a critical role by supporting germinal center formation and infected B cell expansion through the secretion of the pleiotropic cytokine IL-21 (Collins and Speck, 2014, 2015).
ORF50-null viruses that lack the key lytic gene transactivator, replication and transcription activator (RTA), are impaired for splenic latency establishment (Moser et al., 2006; Frederico et al., 2014a). An ORF50-null mutant failed to seed the marginal zone B cells as visualized by fluorescence analysis of infected spleen sections (Frederico et al., 2014a). An ORF27-stop virus, lacking the gp48 glycoprotein, was only slightly attenuated for lytic replication in cell culture but was greatly impaired for latency establishment upon intraperitoneal inoculation (May et al., 2005). As with the ORF50-mutant, the ORF27 mutant remained in the marginal zone macrophage cells and did not seed B cells in the white pulp (Frederico et al., 2014a). Taken together, defects in lytic replication and intercellular spread impact latency establishment in the B cell reservoir, even when the virus is administered by a more direct route. Examination of trafficking defects in additional mutants are needed to validate and extend this developing model for MHV68 dissemination.
While these trafficking studies provide insight into the pathogenesis of gHV infection, it is important to note that there are differences in the splenic architecture of mice and humans. In humans, the marginal sinuses and associated macrophages reside more proximal to the follicle in a region called the perifollicular zone, rather than in the more distal marginal zone where the marginal zone B cells reside in the mouse (Steiniger et al., 1997). While the path to the germinal center may differ slightly between species, MHV68 studies suggest that access to the memory B cell involves transition through multiple cell types, providing additional strategies to interfere with chronic gHV infection.
Maintenance of Latency
Latency is established in dendritic cells, macrophages, epithelial, and endothelial cells (Weck et al., 1999b; Flano et al., 2000; Suarez and van Dyk, 2008), and many B cell subsets (Nealy et al., 2010; Coleman et al., 2014; Feldman et al., 2016). The peak of viral latency occurs around 16 days post-infection, but viral genomes are detectable at late times during chronic infection (Figure 1). Although MHV68 initially gains access to the germinal center B cell reservoir, it is predominantly found in immunoglobulin isotype class-switched memory B cells during long-term latency (Flano et al., 2002a; Marques et al., 2003; Willer and Speck, 2003). Access to, and establishment of, latency in the memory B cell reservoir provides the gHV with a long-lived cellular reservoir that can reside in lymphoid tissue or circulate through the body (Giesecke et al., 2014). However, memory B cells likely undergo homeostatic proliferation and terminal differentiation into plasma cells upon receptor engagement, for instance in the context of co-infections.
During infection, MHV68 also infects pro-B, pre-B, and immature B cells in the bone marrow and is detected in immature and transitional B cells during chronic infection (Coleman et al., 2010). Depletion of developing B cells reduces latency in the mature B cells (Coleman et al., 2014). Normal B cell maturation is dependent on limited survival signals and deletion of self-reactive cells. Interestingly, MHV68 vBcl-2 functions to prevent apoptosis following activation of the BCR (Coleman et al., 2014), perhaps allowing MHV68 infected cells to bypass normal B cell selection checkpoints.
Reactivation from Latency
Latent cells infected with gHVs can be reactivated using chemical or biological agents in vitro, but the stimuli for reactivation in the immune competent host is poorly understood (Murata, 2014). MHV68 has been used to further evaluate reactivation in vivo. A critical event in induction of reactivation of the gHVs is the terminal differentiation of the B cell into a plasma cell (Laichalk and Thorley-Lawson, 2005; Bhende et al., 2007; Sun and Thorley-Lawson, 2007; Wilson et al., 2007; Yu et al., 2007; Siegel et al., 2010). This phenomenon appears to be driven by the transcription factors IRF-4 and XBP-1, which are expressed upon B cell maturation into plasma cells (Crawford and Ando, 1986; Laichalk and Thorley-Lawson, 2005; Matthias and Rolink, 2005; Yu et al., 2007). The human gHVs are dependent on XBP-1 for reactivation (Laichalk and Thorley-Lawson, 2005; Bhende et al., 2007; Sun and Thorley-Lawson, 2007; Wilson et al., 2007; Yu et al., 2007), while MHV68 requires IRF-4 (Matar et al., 2014). MHV68 M2 is a latency-associated gene (Virgin et al., 1999) that promotes B cells differentiation into a plasma cell-like phenotype (Liang et al., 2009). Deletion of the plasma cell lineage causes a reduction in reactivation as well as a contraction of the latent reservoir during long-term infection (Siegel et al., 2010). In vivo injection of TLR agonists into a latently infected animal drives reactivation, and results in a higher latent viral load (Gargano et al., 2009). Stimulation of either CD40 or TLR9 can also drive reactivation in cell culture (Moser et al., 2005; Ptaschinski et al., 2010).
A latent gHV is likely influenced by the activation status of the immune compartment and cytokines in the host. Co-infection of MHV68 infected mice with the pathogenic helminths H. polygyrus or S. mansoni induces reactivation (Reese et al., 2014). The mechanism appears to be IL-4 induction by helminth infection that reduces IFNγ levels, a cytokine that represses the expression of the lytic transactivator RTA to promote the maintenance of viral latency (Reese et al., 2014). Recently, impairment of autophagy in macrophage cells was found to suppress reactivation by increasing inflammation and T cell activation to drive IFN-γ production (Park et al., 2016).
B Cell-Mediated Control of Infection
In addition to serving as the primary reservoir of latency, B cells contribute to immune control of gHV infection. Studies in B cell-deficient mice reveal high levels of viral latency and reactivation in the peritoneum (Weck et al., 1999a), but the lack of B cells also impairs the development of Vβ4 T cells, which secrete IFNγ and control viral reactivation (Weck et al., 1996; Flano et al., 1999). Passive transfer of immune sera into B cell deficient μMT mice reduces the establishment of viral latency (Gangappa et al., 2002). Infection of mice lacking CD28, a costimulatory molecule necessary for T cell support of germinal center B cells, reveals only slightly reduced latency establishment and normal antibody response (Kim et al., 2002). However, this antibody response was poorly neutralizing and rapidly decreased over time. Depletion of T cells from CD28-/- mice led to recrudescence of the virus that could be inhibited by passive transfer of immune sera, indicating that humoral immunity contributes to the control of latent infection (Kim et al., 2002). In addition, immunization of mice with a reactivation-deficient virus generates immune sera that can prevent superinfection by a WT virus (Tibbetts et al., 2003). Interestingly, neutralizing antibody can prevent infection of cell types that do not express Fc receptors, but increases infection of cell types expressing Fc receptors (Rosa et al., 2007). To tease apart the contributions of B cell and T cell interactions from the generation of anti-viral antibody, McClellan et al. (2006) infected mice with a germline rearrangement of an antibody variable region such that all B cells to produce antibodies specific to hen egg lysozyme. While virus-specific antibody was not necessary for control of latent infection, depletion of CD8+ T cells increased latency and reactivation in the peritoneum and depletion of either CD4+ or CD8+ caused increased latency and reactivation in the spleen (McClellan et al., 2006).
T Cell-Mediated Control of Infection
Numerous studies have identified roles for both CD4+ and CD8+ T cells in clearance of acute lytic infection in the lungs (Ehtisham et al., 1993; Cardin et al., 1996; Stevenson et al., 1999b; Molloy et al., 2011). During long-term infection, loss of CD4+ T cells compromises immune control, leading to recrudescence in the lungs, progressive pathology, and death (Cardin et al., 1996; Molloy et al., 2011). Cytotoxic CD8+ T cells also play an important role in control of early gHV infection; however, their loss leads to a much different phenotype than that of CD4+ T cell-deficient animals. In Balb/c mice, depletion of CD8+ T cells after lung infection causes persistent lytic infection in the lung as well as the spleen and is accompanied by significant clinical pathology and increased necrosis of the lung (Ehtisham et al., 1993). This phenomenon is strain-dependent, as C57BL/6 mice depleted of CD8+ T cells suffer delayed clearance from the lung accompanied by increased viral latency in the spleen (Stevenson et al., 1999b). In both backgrounds, simultaneous depletion of CD4+ and CD8+ T cells leads to persistent virus replication and death (Ehtisham et al., 1993; Stevenson et al., 1999b).
While T cells play an important role in initial clearance of viral infection, they are also crucial for control of latent infection. It is telling that much of the burden of human disease caused by the gHVs occurs during T cell immunosuppression (Malnati et al., 2003; Thompson and Kurzrock, 2004; Cesarman, 2014). CD8+ T cells specific to MHV68 are observed in the infected mice as soon as 1 week after infection and are critical for immune surveillance and control of gHV latency (Stevenson and Doherty, 1998; Stevenson et al., 1999a; Gredmark-Russ et al., 2008). This response consists of multiple immunodominant and subdominant T cells that shift over time (Stevenson et al., 1999a; Freeman et al., 2010). During latency, CD8+ T cells serve to control viral reactivation at two important sites of latency: the spleen and the peritoneum (Tibbetts et al., 2002). Latency establishment occurs normally in the absence of CD4+ T cells (Stevenson et al., 1999b). Unlike CD8+ T cell KO animals, CD4+ T cell KO mice fail to contract the latent reservoir in the spleen and suffer a progressive increase in reactivation in the lungs, eventually leading to death (Cardin et al., 1996). This defect can be rescued with anti-CD40 antibody treatment in a CD8+ T cell dependent manner, indicating that CD4+ T cells provide important costimulatory signals to CD8+ T cells (Sarawar et al., 2001; Dias et al., 2010).
The NF-κB Signaling Pathway
Despite the importance of latency in the viral life cycle, the mechanisms that drive latency establishment in some cells types but not others, and the cellular signals that maintain latency or initiate reactivation are poorly understood. Better understanding of the host signaling pathways that contribute to the establishment and maintenance of a latent infection is critical for the development of therapeutics against infection and virus-associated malignancies. The NF-κB pathway is a signaling process that involves upstream phosphorylation events that culminate in the proteasomal processing of regulatory proteins that enable translocation of NF-κB transcription factors into the nucleus. NF-κB signaling plays a critical role in many cellular processes, including cell survival and inflammatory signaling, as well as a central role in B cell maturation.
In resting cells, NF-κB complexes are retained in the cytoplasm by inhibitors of NF-κB (IκBs). IκBs are phosphorylated by IκB kinase (IKK) complexes resulting in activation of canonical or non-canonical NF-κB pathways. The NF-κB family consists of five subunits, p65 (RelA) RelB, cRel, p50/p105 (NF-κB1), and p52/p100 (NF-κB2) that can form homo- or hetero-dimers. Depending on the composition of the dimers and other variables such as post-translational modification and interactions with other factors, NF-κB dimers can either induce or repress gene expression through direct binding to DNA sequences in the regulatory region of target genes and also through interactions with other factors (Oeckinghaus and Ghosh, 2009). NF-κB signaling consists of two distinct pathways; the IKKβ-dependent canonical pathway (Figure 2), and the IKKα-dependent non-canonical pathway (Figure 3).
FIGURE 2.

Canonical NF-κB signaling. Canonical NF-κB signaling is activated by the binding of extracellular ligands. Each receptor family differs in the upstream signaling molecules used to mediate signaling, but they share activation of TRAF2 and TRAF6. The TRAFs ubiquitinate TAK1, causing TAK1 to be activated and associate with IKKγ. TAK1 can then phosphorylate IKKβ, which in turn phosphorylates IκBα. Phosphorylated IκBα is degraded by the proteasome, releasing the canonical transcription factors p65, p50, and cREL. These translocate into the nucleus and activate pro-survival and pro-inflammatory genes.
FIGURE 3.
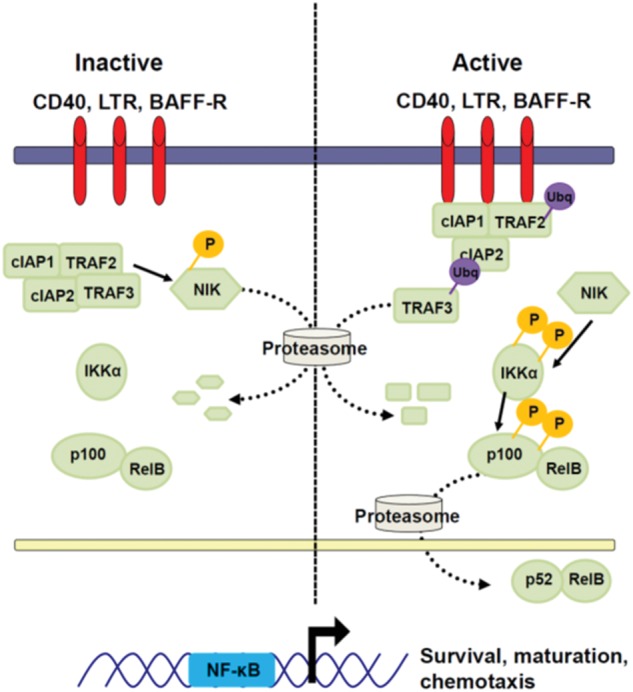
Non-canonical NF-κB signaling. Non-canonical NF-κB signaling is initiated by the binding of cell surface receptors to their ligands. Trimerization of the receptors recruits TRAF2, cIAP1, and cIAP2 to the receptor. This changes the specificity of cIAP1/2 ubiquitin ligation from NIK to TRAF3. TRAF3 is ubiquitinated by this complex and degraded by the proteasome. The absence of TRAF3 prevents degradation of NIK, leading to its accumulation. NIK can phosphorylate IKKα. Activated IKKα phosphorylates p100, inducing its partial degradation by the proteasome. The processed p100 subunit, p52, associates with RelB and translocates into the nucleus where it mediates transcription of pro-survival genes.
Canonical NF-κB Signaling
The canonical pathway is activated by the IKKα/β/γ complex and results in IKKβ-mediated phosphorylation and ubiquitin-mediated proteosomal processing of IκBα proteins, which sequester NF-κB subunits p50/p65 in the cytoplasm. Degradation of IκBα unmasks the nuclear localization signals of the subunits, permitting their nuclear translocation. Canonical subunits have preferential binding partners, p65 with c-Rel or p50, and p50 dimerizing with itself (Vallabhapurapu and Karin, 2009). These dimers have subtly different target sequences, and can associate with other transcription factors to control their target genes (Urban and Baeuerle, 1991; Kunsch et al., 1992; Perkins et al., 1992; Bonizzi et al., 2004). The canonical NF-κB signaling pathway serves a broad range of functions within the body, contributing to immune activation in response to cytokines and pro-inflammatory signaling in response to pathogen detection (Li et al., 2002). These cell surface receptors include: the IL-1R and the TNFR family of receptors which bind cytokines, TLRs that recognize pathogen-associated molecular patterns, and the B and T cell receptors, which provide pro-survival signals. Each receptor transduces its signal with an overlapping set of effector molecules that are summarized in Figure 2.
Non-canonical NF-κB Signaling
Non-canonical/alternative NF-κB signaling mediates organo genesis, cell maturation, and pro-survival signaling. This pathway is activated by specific members of the TNFR family, such as CD40, BAFFR, and TNFRSF3 (LTβR; Matsushima et al., 2001; Sun, 2012) (Figure 3). In contrast to the canonical pathway receptors, activated receptors of the non-canonical NF-κB pathway directly associate with TRAF proteins (Pullen et al., 1999; Hildebrand et al., 2010) through multiple TRAF binding sites on their intracellular tails (Pullen et al., 1998). In resting cells, NF-κB-inducing kinase (NIK), which is the major IKKα-activating kinase in cells, is constitutively active, but is present at very low levels due to its constitutive degradation by cIAP1/2 (Varfolomeev et al., 2007), dependent on TRAF3 and TRAF2 (Liao et al., 2004; Gardam et al., 2008). Relief from constitutive degradation by receptor engagement and TRAF3 degradation allows for activated NIK to accumulate (Dejardin et al., 2002; Gardam et al., 2008). This accumulation drives the phosphorylation of IKKα on two serines in its activation loop (Regnier et al., 1997; Lin et al., 1998; Ling et al., 1998) and licenses IKKα to phosphorylate p100 (Senftleben et al., 2001). Full-length p100 can act similarly to IκBα, sequestering NF-κB subunits in the cytoplasm. Upon phosphorylation, p100 undergoes partial proteolysis to p52 that can associate with the NF-κB transcription factor RelB and translocate to the nucleus to initiate gene transcription (Xiao et al., 2001; Solan et al., 2002; Bonizzi et al., 2004). Activated IKKα also serves to negatively regulate the non-canonical pathway signaling through phosphorylation and destabilization of NIK (Razani et al., 2010).
Crosstalk between Canonical and Non-canonical NF-κB Signaling
Extensive research has defined the signaling pathways for each arm of NF-κB signaling, as well as numerous levels of crosstalk between these two arms. Full-length p100 may sequester canonical subunit dimers in addition to RelB in the cytoplasm, interfering with canonical signaling (Basak et al., 2007). Canonical NF-κB signaling can influence non-canonical pathway signaling through the induction of RelB and p100 transcription (Dejardin et al., 2002; Basak et al., 2008). NF-κB1 subunit p50 may compete with p52 for binding to RelB, (Basak et al., 2008), and p65 can bind p100 and induce its degradation, leading to the formation of p52/p65 heterodimers (Hoffmann et al., 2003). The use of common upstream signaling molecules and the formation of NF-κB heterodimers likely contributes to extensive overlap in target genes between canonical and non-canonical NF-κB pathways (Li et al., 2002).
The Role of NF-κB Signaling in B Cell Development
Nuclear factor-kappa B signaling pathways are utilized by B lymphoid cells, the primary latency reservoir for gHVs, to drive transcription of genes important for B cell maturation, responses to infection, proliferation, protection from apoptosis, and immunoglobulin isotype class switching (Pasparakis et al., 2006). B cells develop in the bone marrow where they undergo VDJ recombination and emerge with a functional heavy and light immunoglobulin chain (Ichii et al., 2014). The B cells emerge into the periphery as transitional 1 (T1) B cells, characterized as CD23lo IgMhi (Hsu et al., 2002), and home to secondary lymphoid organs (Figure 4). Successful entry to the periarteriolar lymphoid sheath of the spleen (Loder et al., 1999) exposes the nascent B cell to peripheral selection by BAFF signaling. BAFF is secreted by numerous cell types in the spleen, including myeloid, dendritic, and stromal cells (Nardelli et al., 2001; Litinskiy et al., 2002; Gorelik et al., 2003). BAFF binds primarily to the BAFF receptor (BAFFR) on B cells, inducing pro-survival Bcl-2 family members (Claudio et al., 2002) and down-regulating apoptotic genes (Craxton et al., 2005) through non-canonical NF-κB, Akt, and ERK signaling in an IKKα-dependent manner (Claudio et al., 2002; Patke et al., 2006; Otipoby et al., 2008; Woodland et al., 2008; De Silva et al., 2016). BAFF, BAFFR, or IKKα- knockout cells mature and exit the bone marrow but undergo apoptosis upon entry to the spleen, reducing the transitional 2 (CD23hi IgMhi) and mature B cells by up to 95% (Lentz et al., 1996; Kaisho et al., 2001; Senftleben et al., 2001; Hsu et al., 2002). Conversely, transgenic expression of BAFF in mice drastically expands the transitional and marginal zone B cell reservoirs (Batten et al., 2000). Upon maturation past the T1 phase into the T2 phase, the nascent B cell relocates to the splenic follicle where it undergoes BCR-dependent selection (Loder et al., 1999) mediated by the NF-κB signaling pathway (Lucas et al., 2001; Wang et al., 2001; Shinohara et al., 2005). Successful activation licenses the T2 B cell to become a marginal zone or naïve mature follicular B cell (Loder et al., 1999).
FIGURE 4.
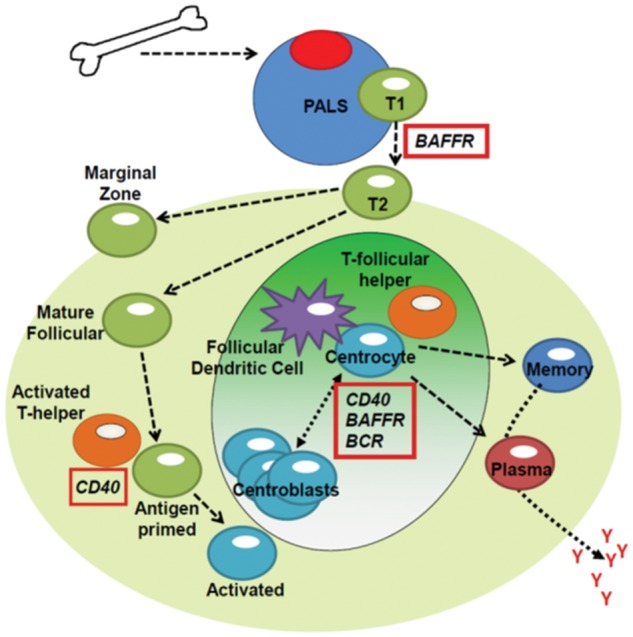
Role of NF-κB signaling in B cell maturation and differentiation. B cells exit the bone marrow and home to secondary lymphoid organs. The B cell exits from the arteriole into the peripheral arteriole lymphoid sheath (PALS) as a T1 B cell. Here, the B cell requires BAFF signaling to survive and mature to a T2 B cell. The T2 B cell can mature to a marginal zone B cell or follicular B cell. The mature follicular B cell can be activated by the detection of cognate antigen and CD40 costimulation. The activated B cell enters the germinal center, where it competes for antigen and costimulatory factors presented on follicular dendritic cells and produced by T follicular helper cells. Strong antigen binding and receipt of costimulatory NF-κB-dependent signaling through CD40 and BAFF allows the B cell to mature into a memory B cell or a plasma cell.
The next step of B cell maturation occurs upon detection of a cognate antigen for the BCR, accompanied by CD40 co-signaling, leading to the formation of the germinal center (Goetz and Baldwin, 2008). CD40 signals through both IKKα and IKKβ (Kosaka et al., 1999). Mice lacking CD40 fail to form germinal centers (Goetz and Baldwin, 2008). Germinal centers can form from the small population of mature B cells in BAFF-/- mice during infection, although these are short-lived (Rahman et al., 2003; Vora et al., 2003). Both of the downstream IKKs are necessary for passage of the B cell through the germinal center of mice (Goetz and Baldwin, 2008). A dominant negative knock-in of IKKα (IKKαSSAA) abrogates germinal center formation (Kaisho et al., 2001; Mills et al., 2007), while conditional deletion of IKKβ drastically impairs global B cell survival (Pasparakis et al., 2002). It has been recently shown that conditional deletion of NIK after passage of the T1 checkpoint impairs the ability of B cells to enter the germinal center, further confirming the necessity of non-canonical signaling in the germinal center reaction (Brightbill et al., 2015).
Disruption of the NF-κB Signaling Pathways Alters MHV68 Pathogenesis
During the course of infection, the gHVs traverse numerous cell types before gaining access to germinal center B cells, and ultimately memory B cells. However, the signaling events that determine the lytic or latent course of infection are not well understood. The NF-κB signaling pathway is activated during B cell maturation and differentiation. Numerous gHV proteins can activate or impair signaling through the expression of viral proteins and miRNAs. While these viral molecules are important for latency and infected cell survival in culture, the role of NF-κB signaling in the gHV lifecycle of the host is not well-defined. The genetic tractability of the MHV68 pathogenesis system has enabled the in vivo role of many NF-κB signaling components to be examined (Table 2).
Table 2.
In vivo studies of the role of NF-κB signaling on MHV68 pathogenesis.
| Gene | Knockout | Immune defects | Route/Dose | Impact on pathogenesis | Changes in host response | Reference |
|---|---|---|---|---|---|---|
| LTα | Germline | No lymph nodes | INa/2 × 105,b | ↓ Clearance from lung | ↑ Lung inflammation | Lee et al., 2000 |
| ↑ Latency | ↓ CD8+ T cell activation | |||||
| CD40 | Germline | No germinal center formation | IN/1 × 103 | ↑ Reactivation | Willer and Speck, 2005 | |
| ↑ Infection of naïve B cell | ||||||
| Recrudescence in lungs | ||||||
| CD40 | Mixed BM chimera | IN/4 × 102 | Equal latency establishment Loss of latency in CD40-/- subset | Exclusion of CD40-/- B cells from germinal center | Kim et al., 2003 | |
| BAFFR | Germline | ↓ Mature B cells Impaired Abc production | IN/1 × 104 | ↓ Latency at 30 dpid | ↓ Germinal centers | Frederico et al., 2014b |
| ↓ MHV68-specific Ab | ||||||
| IPe/1 × 105 | ↑ Viral titer at 3 dpi, | |||||
| ↓ Infection from 8–30 dpi | ||||||
| TLR2/9 | Germline | IN/5 × 104 | ↑ Viral load at 3 dpi | Sun et al., 2015 | ||
| TLR3 | Germline | IN/1 × 103 | None | Gargano et al., 2008 | ||
| TLR7 | Germline | IP/1 × 105 | None | Haas et al., 2014 | ||
| TLR9 | Germline | IP/1 × 105 | ↑ Reactivation | |||
| MyD88 | Germline | ↓ B cell activation | IN/1 × 103 | ↓ Latency in spleen at 16 dpi | ↓ B cell activation | Gargano et al., 2008 |
| No difference in latency at 90 dpi | ↓ Germinal centers | |||||
| ↓ Class switching | ||||||
| Delayed ab production | ||||||
| IP/1 × 103 | ↑ Reactivation in PECs | |||||
| ↓ Reactivation in spleen | ||||||
| Mixed BM f Chimera | IN/1 × 103 | ↓ Latency | ↓ MyD88-/- germinal center entry | |||
| IkBaM | Viral Transgene | IN/1 × 103 | ↓ Latency establishment | Krug et al., 2007 | ||
| ↓ Infected lung B cells | ||||||
| IP/1 × 103 | ↓ Viral titer | |||||
| ↓ Latency | ||||||
| p50 | Germline | ↓ B cell proliferation | IN/1 × 102 | ↑ Viral titers in lung | ↓ Antibody production | Krug et al., 2009 |
| ↓ Ab levels | ↓ Latency at 16 dpi | ↓ Vβ4+ T cell | ||||
| ↓ Germinal centers | ↑ Latency over time | |||||
| Recrudescence in lungs | ||||||
| Mixed BM chimera | None | IN/1 × 102 | Normal lung titers | ↓ p50-/- germinal center B cell | ||
| ↓ Latency in p50-/- B cells | ||||||
| Recrudescence in lungs | ||||||
aIN, intranasal infection under light anesthesia.
bDose described as PFU per animal.
cAb, antibody.
ddpi, days post infection.
eIP, intraperitoneal infection.
fBM, bone marrow.
Role of NF-κB Signaling during Acute Phase of Infection
Infection with a recombinant MHV68 expressing a dominant-negative IκBα that blocks canonical NF-κB subunit activity did not impact viral replication in fibroblasts or lung epithelial cells in culture, and did not alter levels of acute expansion in the lungs of infected mice (Krug et al., 2009, 2010). Likewise viral replication proceeds normally in cells lacking the p50 subunit (Krug et al., 2009). Thus, NF-κB subunits are dispensable for MHV68 replication (Krug et al., 2007, 2009, 2010). In contrast, overexpression of the NF-κB subunit p65 was reported to impair RTA activation and virus replication, and suggested that NF-κB signaling is deleterious to lytic gene expression (Brown et al., 2003a). As detailed in the section describing gHV modulation of NF-κB signaling below, recent studies have determined that the virus subverts upstream signaling events while impairing subunit activation to enhance lytic gene expression (Dong and Feng, 2011; Dong et al., 2012; Karijolich et al., 2015).
The NF-κB p50 subunit is activated during both lytic and latent infection in vitro (Krug et al., 2009). Germline deletion of the NF-κB1 subunit that is processed to p50 in mice leads to higher acute lung replication after intranasal infection (Krug et al., 2009). The p50-/- mice also exhibited reduced B and T cell responses upon MHV68 infection. The increase in lytic replication was rescued in bone-marrow chimeric mice generated from WT and p50-/- donors, indicating the increased viral replication is not a cell-intrinsic effect, but rather due to impaired immune control (Krug et al., 2009). Mice lacking lymphotoxin alpha (LTα), a homolog of TNFα that signals through the canonical NF-κB pathway (Medvedev et al., 1996), lack lymph nodes and germinal centers in the spleen (Lee et al., 2000). Upon infection of LTα-/- mice, viral titers in the lung were increased and clearance was delayed, and mutant mice had a transient increase in virus replication upon colonization of the spleen as compared to WT mice (Lee et al., 2000). The whole body knock-outs emphasize the role of the immune response in control of acute expansion that can impact dissemination to the spleen and long-term immune control of gHV infection.
Interestingly, interferon-γ receptor knockout mice typically succumb to MHV68 infection via viral persistence that drives fibrosis of organs including the lung (Mora et al., 2005). Lung fibrosis and mortality in the IFNγR-/- mice was prevented by overexpression of a dominant-negative IκBα from a recombinant MHV68 (Krug et al., 2010). The inhibition of NF-κB subunit function both reduced viral load and the expression of the inflammatory chemokines MCP-1 and CXCL12 in the infected tissues (Krug et al., 2010). Further investigation is required to determine how NF-κB signaling in the infected tissue contributes to inflammatory cytokines and immune activation during infection.
Role of NF-κB Signaling in Latency Reservoirs of the Host
Numerous in vivo investigations have demonstrated that disruption of NF-κB signaling can impair MHV68 latency establishment in B cells and alter long-term immune control of viral latency (Table 2). LTα-/- mice exhibited increased levels of latency in the spleen that are accompanied by a reduction in T cell activation (Lee et al., 2000). The deletion of CD40, a cell surface molecule that activates canonical and non-canonical NF-κB signaling pathways upon CD40L engagement (Kosaka et al., 1999), prevented germinal center formation and T-dependent class switching (Kawabe et al., 1994). Although latency was established in class-switched B cells in CD40-/- mice at levels comparable to WT mice, reactivation was increased in the spleen and recrudescence was observed in the lungs of the CD40-/- mice (Batten et al., 2000; Willer and Speck, 2005; Kalled, 2006; Frederico et al., 2014b). The absence of MyD88, a molecule that acts downstream of numerous canonical pathway receptors, led to a defect in germinal center formation and a reduction of latency establishment early after infection with MHV68. However, latency ‘recovered’ to WT levels by late times post-infection (Gargano et al., 2008). Interestingly, infection of MyD88-/- mice with the IκBαM-transgenic virus led to an even greater defect in latency establishment, suggesting that MyD88 has additional NF-κB subunit-independent effects on the latency reservoir (Gargano et al., 2008). Germline deletion of a single NF-κB subunit, p50, led to a significant reduction in latency establishment (Krug et al., 2009). The defects in B and T responses to MHV68 infection upon p50 deletion likely underlie the observed recrudescence in the lungs and increased viral load in the spleen at later times post-infection (Krug et al., 2009).
Non-canonical NF-κB signaling is highly active at multiple stages of B cell development, including entry into the germinal center (Batten et al., 2000; Kaisho et al., 2001; Senftleben et al., 2001; Claudio et al., 2002; Vora et al., 2003; Kalled, 2006; Mills et al., 2007). However, the role of the non-canonical pathway in MHV68 latency establishment is not well understood. Deletion of BAFFR, which signals through the non-canonical NF-κB signaling pathway, impaired B cell maturation and germinal center formation (Batten et al., 2000; Kalled, 2006). MHV68 infection of BAFFR-/- mice led to reduced latency establishment and lower titers of virus-specific IgG (Frederico et al., 2014b).
Germline deletion of NF-κB components are associated with altered pathogenesis in the context of a spectrum of immunodeficiencies, demonstrating the importance of lymphocyte development and function in the establishment, and long-term control, of viral latency. However, such complex phenotypes complicate the determination of the cell-intrinsic roles of NF-κB signaling for MHV68 latency. Alternative experimental approaches been used to separate global immune defects from cell intrinsic effects on MHV68 pathogenesis. A recombinant MHV68 engineered to express a dominant negative form of the IκBα protein (MHV68-IκBαM) was used to block canonical NF-κB signaling only in the infected cell. This blockade led to a significant reduction of latency establishment in the lung and splenic B cells of infected mice (Krug et al., 2007). This defect could not be rescued by overexpression of the anti-apoptotic protein Bcl-2, suggesting that blockade of canonical NF-κB signaling led to a defect in latency rather than a defect in cell survival (Krug et al., 2007). The reduction of latency in the spleen was durable and accompanied by a reduction in virally infected B cells of the lungs (Krug et al., 2007). Specific inhibition of genes in the infected cell has also been accomplished using expression of Cre recombinase from the virus (Siegel et al., 2010) and transgenic mice, although this has not been used to probe the role of NF-κB components. The absence of global immune defects and the relative ease of recombinant virus generation makes this an attractive option to tease apart cell intrinsic roles for NF-κB as well as other signaling pathways.
Another method to examine cell-intrinsic roles during pathogenesis while avoiding global immune disruption is the generation of mixed bone marrow chimeric mice. WT stromal cells and a ∼50–80% proportion of WT bone marrow allow for the establishment of a normal immune system while setting the stage for a rigorous competition between a WT and mutant cell-type of interest for supporting viral latency. In a study of mixed bone marrow chimeras, MHV68 failed to establish latency efficiently in the NF-κB1 p50-/- B cells as compared their WT B cell counterparts (Krug et al., 2009). In addition there was a more rapid decline in genome+ cells that lacked p50. The p50-/- B cells suffered from an intrinsic defect in germinal center participation that correlated with the reduction of splenic latency. In addition to the establishment defect in the absence of p50, the persistence of pre-formed infectious virus in the lungs of the mixed bone marrow chimeras long after normal clearance of acute phase replication supports a role for NF-κB transcription factors in the maintenance of latency (Krug et al., 2009).
Abrogation of upstream signaling components also impairs latency. Infection of mixed MyD88-/- and WT bone marrow chimeras revealed a defect in latency establishment in the MyD88-/- B cells that was accompanied by a decrease in activation, germinal center participation, and Ig isotype class switching as compared to WT B cells (Gargano et al., 2008). Infection of mixed CD40-/-/WT chimeras revealed a defect in the long-term maintenance of latency in CD40-/- but not WT cells (Kim et al., 2003). Although the frequency of cells harboring the virus was indistinguishable at 16 dpi, CD40-/- B cells were present at reduced frequencies in the germinal centers and the number of CD40-/- B cells harboring latent virus decreased at late times after infection (Kim et al., 2003). The deterioration in latency observed in the bone marrow chimeras implies that CD40 engagement and recruitment to the germinal center is important for homeostatic maintenance in the memory B cells population (Flano et al., 2003; Kim et al., 2003; Willer and Speck, 2005).
Extrinsic stimulation of latent cells with TLR agonists led to mixed reactivation outcomes. Stimulation of TLRs 3, 4, 5, and 9 induced reactivation from the A20-HE MHV68+ latent cell line (Gargano et al., 2009). In contrast, TLR7 and TLR9 stimulation of the latent S11 cells line led to a NF-κB p65-dependent suppression of reactivation (Haas et al., 2014). Administration of LPS (TLR4), R848 (TLR7), or CpG (TLR9) to infected mice led to heightened reactivation of latent splenocytes in the infected animal (Gargano et al., 2009), and CpG injection led to an increase in the frequency of splenic latency in pups and adult mice (Ptaschinski et al., 2010). The deletion of TLR3, TLR7, or TLR9 or the combination of TLR2 and TLR9 did not alter latency establishment (Gargano et al., 2008, 2009; Haas et al., 2014; Sun et al., 2015), but the TLR9-/- mice were observed to have an increase in reactivation from latency (Haas et al., 2014). Interestingly, ex vivo treatment of splenocytes with BAFF slightly reduced reactivation of MHV68 from WT splenocytes (Williams et al., 2015). NF-κB signaling is likely involved in these phenotypes, but its direct role has not been validated.
Synopsis and Future Mechanistic Investigations of NF-κB Signaling as a Host Determinant of Pathogenesis
Taken together, canonical NF-κB subunit activation is dispensable for MHV68 replication in fibroblasts and immortalized lung epithelial cells in culture, yet critical for control of virus replication in the lung and latency establishment in the spleen (Krug et al., 2007, 2009, 2010). Inhibition of NF-κB signaling via loss of membrane receptors or downstream signaling components impacts latency at different stages of chronic infection that coincide with B cell activation, germinal center differentiation, and the formation of long term memory B cells (Kim et al., 2003; Willer and Speck, 2005; Krug et al., 2007, 2009; Gargano et al., 2008). Albeit less investigated, non-canonical signaling also contributes to B cell functions that are conducive to MHV68 latency (Frederico et al., 2014b). Recent studies have revealed the complexity of latency establishment; there are numerous rounds of infection in multiple cell types and B cell subsets prior to the infection of, and transit through, the germinal center (Gaspar et al., 2011; Frederico et al., 2014a). Most of the studies that examine NF-κB signaling components have not defined the cellular origin of the block in latency establishment. Finally, engagement of TLR receptors influences reactivation from latency, albeit with variable outcomes (Gargano et al., 2009; Haas et al., 2014).
What remains unclear is the mechanism for these defects. Are these phenotypes due to a defect in B cell biology and/or a dysregulation of the viral latency program? We propose that gHVs have usurped the NF-κB signaling that transpires in B cells to promote the viral latency program. Thus, it becomes important to determine if specific viral genes are directly under the control of NF-κB subunits, and if specific NF-κB binding sites or responsive elements in the regulatory regions of viral genes mediate this regulation. This knowledge is critical to target those regions of the viral genome for mutagenesis and characterization in vivo. By this approach, the field can genetically separate the role of NF-κB in B cell biology from its function as a regulator of gHV gene regulation in vivo.
Gammaherpesvirus Modulation of the NF-κB Signaling Pathway
Modulation by EBV and KSHV
The gHVs encode numerous proteins that modulate or activate NF-κB signaling during both latent and lytic infection (Grossmann et al., 2006; Soni et al., 2007; Martin et al., 2008; Raab-Traub, 2012). During latency, EBV encodes two proteins, latent membrane protein 1 and 2 (LMP1/2), that mimic B cell signals that are involved in germinal center reactions and the process of memory B-cell differentiation. The EBV encoded LMP1 protein is a constitutively active CD40 receptor homolog and activates both canonical and non-canonical NF-κB pathways to promote the survival and proliferation of infected B cells (Uchida et al., 1999; Grimm et al., 2005; Le Clorennec et al., 2008; Ersing et al., 2013; Zhao et al., 2014). LMP2A has dual roles in promoting NF-κB signaling via Syk and Lyn kinases and enhances LMP1 signaling by controlling LMP1-interacting cellular TRAF2 transcription (Dawson et al., 2001; Guasparri et al., 2008). When expressed as single transgenes in mice, these molecules can promote B cell proliferation and antibody production, independently of germinal center formation (Rastelli et al., 2008; Minamitani et al., 2015). However, it is not clear what the impact of LMP1 and LMP2A activity are on B cell selection in the normal course of an EBV infection since the B cells of double LMP1/LMP2A transgenic mice were phenotypically normal (Hadinoto et al., 2008; Vrazo et al., 2012). EBV lytic genes may also impact NF-κB signaling. The viral dUTPase encoded by BLLF3 signals through TLR2 in a MyD88-dependant fashion (Ariza et al., 2009). In addition, the EBV protein BGLF4 encoded by ORF36 is a viral kinase that has been shown to phosphorylate cellular UTX to inhibit NF-κB signaling and promote RTA transactivation of lytic reporters (Chang et al., 2012).
Kaposi’s sarcoma-associated herpesvirus encodes multiple factors that activate NF-κB signaling, including the viral-FLICE inhibitor protein (vFLIP), viral G protein-coupled receptor (vGPCR), K1, K15, and ORF75. Cells latently infected with KSHV require activation of NF-κB signaling for survival and lymphomagenesis (Keller et al., 2000; Guasparri et al., 2004). KSHV vFLIP expression during latency drives both canonical and non-canonical NF-κB activation in order to promote cell survival and proliferation, as well as induce alterations in morphology (Chaudhary et al., 1999; Liu et al., 2002; Field et al., 2003; Matta and Chaudhary, 2004; Grossmann et al., 2006; Guasparri et al., 2006). The lytic KSHV vGPCR gene product is constitutively active and drives NF-κB signaling and the expression of proinflammatory chemokines and cytokines (Arvanitakis et al., 1997; Kirshner et al., 1999; Schwarz and Murphy, 2001). The role of K1 as an NF-κB activator is cell-specific; its expression promotes NF-κB signaling in B cells, yet inhibits it in HEK293 cells (Prakash et al., 2002; Konrad et al., 2009). K15 is a transmembrane protein that is expressed at low levels during latency and activates multiple pathways including NF-κB signaling (Brinkmann et al., 2003; Wang et al., 2007). Replacement of EBV LMP2A, which acts as a constitutively active BCR, with K1 and K15 rescues the ability of EBV to establish immortalized LCL lines (Steinbruck et al., 2015). KSHV miR-K1 targets the inhibitor IκBα to maintain NF-κB subunit activation and promote a latent state in PEL cells (Lei et al., 2010). The KSHV ORF75 tegument protein was identified in an in vitro screen for NF-κB activators (Konrad et al., 2009). MHV68 ORF75C and KSHV ORF75 are tegument proteins that have been recently found to activate RIG-I to promote NF-κB signaling (He et al., 2015), and are discussed further below.
NF-κB Signaling Is Usurped by MHV68
During the herpesvirus lytic life cycle, viral gene expression follows an ordered cascade beginning with immediate early (IE), to early (E) and then late (L) genes. The IE protein, RTA is encoded by open reading frame 50 (ORF50) and is a conserved gHV transcription factor (Sun et al., 1998; Lukac et al., 1999; Feederle et al., 2000; Wu et al., 2000; Goodwin et al., 2001). RTA initiates lytic replication or the switch from latency to lytic replication by regulating transcription of E and L lytic genes. For the Rhadinoviridae γ2-herpesviruses, KSHV, RRV, HVS, and MHV68, RTA has been shown to be necessary and sufficient to initiate viral lytic replication during de novo infection (Sun et al., 1998; Feederle et al., 2000; Pavlova et al., 2003; Xu et al., 2005). Moreover, ectopic expression of RTA in quiescently infected cells disrupts latency and induces lytic reactivation (Lukac et al., 1998; Sun et al., 1998; Gradoville et al., 2000; Wu et al., 2000; Goodwin et al., 2001). The Lymphocryptovirus γ1-herpesvirus, EBV, requires the viral transactivator protein ZTA (BZLF1) for lytic replication, and RTA (BRLF1) serves as a critical co-factor (Ragoczy and Miller, 2001).
A number of cellular proteins such as IRF4 and HMGB-1 have been identified as co-factors that synergize with MHV68 RTA to regulate RTA-responsive promoters (Song et al., 2004; O’Flaherty et al., 2014). However, some cellular factors have been reported to inhibit gHV lytic replication by inhibiting RTA expression or activity, thereby promoting latency. For instance, cellular factors such as PARP-1, KSHV RTA-binding protein (K-RBP), and NF-κB can antagonize RTA to inhibit lytic replication (Brown et al., 2003b; Gwack et al., 2003; Yang and Wood, 2007). High levels of NF-κB p65 have a deleterious effect on gHV lytic replication, blocking lytic gene transcription and viral replication (Gutsch et al., 1994; Brown et al., 2003a).
Interestingly, the gHV lytic transactivator proteins inhibit NF-κB signaling during viral replication. For EBV, the major lytic transactivator Zta (BZLF1) inhibits transactivation functions of NF-κB p65 (Morrison and Kenney, 2004). MHV68 and KSHV RTA proteins have been demonstrated to target p65 for degradation (Brown et al., 2003b; Yang et al., 2008; Dong et al., 2012). MHV68 RTA activated in response to MAVS/IKKB leads to ubiquitin-mediated degradation of p65 that is phosphorylated on Ser468 (Dong and Feng, 2011; Dong et al., 2012). MHV68 LANA induces the ubiquitination and degradation of p65, preventing NF-κB-dependent gene expression in 293 cells (Rodrigues et al., 2009). It is not clear if the major consequence of RTA degradation of p65 is to impair NF-κB to promote lytic gene expression or limit the transcription of NF-κB dependent proinflammatory cytokines (Dong et al., 2010, 2012; Dong and Feng, 2011) (Figure 5). Although NF-κB signaling is initially targeted for degradation by viral molecules, NF-κB subunit activation has been observed at late timepoints, from 12 to 24 hpi, during productive replication in fibroblasts (Krug et al., 2007, 2010); the impact of this late stage activation is not known.
FIGURE 5.
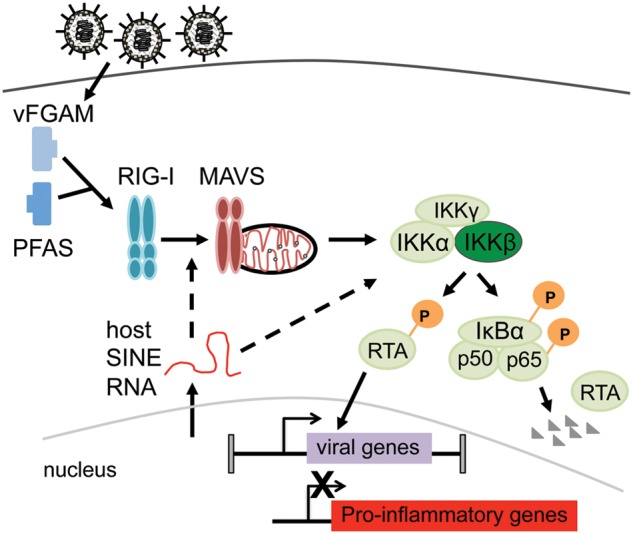
Model for the role of canonical NF-κB signaling in productive infection. Model summarizing recent reports of virus interplay with canonical NF-κB signaling to promote productive infection. Viral tegument protein (vFGAMS/vGAT) encoded by ORF75C and host SINE RNAs induced by infection leads to IKKβ activation and phosphorylation of RTA (Dong and Feng, 2011; He et al., 2015; Karijolich et al., 2015). Phosphorylation of RTA increases transactivation of viral genes (Dong and Feng, 2011). RTA targets NF-κB subunit p65 for degradation to dampen pro-inflammatory gene expression (Dong and Feng, 2011; Dong et al., 2012).
Herpesvirus virions contain a tegument layer between the viral nucleocapsid and the viral envelope. All gHVs encode one to three tegument proteins with an approximate 140 amino acid stretch of homology to a cellular enzyme critical for de novo purine biosynthesis. This enzyme encoded by the Pfas gene, the formyl-glycineamide-phosphoribosyl-amidotransferase (FGARATs/FGAMs) catalyzes the fourth step in the de novo purine biosynthetic pathway (Zhao et al., 2013). The viral homologs herein termed vFGAMs (also referred to as vFGARATs or vGAT) have not preserved the known active sites of the host enzyme and, as such, are not predicted to retain enzymatic FGARAT activity (Gaspar et al., 2008). So far three functions have been described for vFGAMs: ND10 antagonism, facilitation of capsid trafficking in the cytoplasm, and NF-κB activation (Tsai et al., 2015). In a recent study by He et al. (2015), MHV68 vFGAM/vGAT (ORF75C) and KSHV ORF75 were found to recruit host FGARAT/PFAS to deamidate RIG-I. In turn, activated RIG-I complexed with MAVS and resulted in the activation of the upstream NF-κB signaling kinase, IKKβ.
Kaposi’s sarcoma-associated herpesvirus and MHV68 RTA migrate at higher molecular weight than predicted based on their amino acid content, from 64 to 90 kDa for MHV68 and 76 to 110 kDa for KSHV (Lukac et al., 1999; Wu et al., 2001). Phosphatase treatment decreases KSHV RTA mobility to ∼90 kDa suggesting that phosphorylation contributes in large to the change in RTA protein mobility (Lukac et al., 1999). Mass spectrometry analysis of KSHV RTA identified serine/threonine residues that were phosphorylated. Replacement of residues 634 and 636 from serine to alanine impairs KSHV RTA transactivation and reduces reactivation (Tsai et al., 2012). MHV68 RTA is phosphorylated in vitro by the IKKβ kinase at groups of serine/threonine residues S550T552S556 (STS) or T561T562S564 (TTS). Alanine replacement of the residues impairs RTA transactivation and growth of a TTS mutant virus (Dong et al., 2010). Taken together, RTA phosphorylation plays a role in regulating RTA-mediated viral gene expression which impacts gHV lytic replication and reactivation.
Murine gammaherpesvirus 68 RTA phosphorylation by IKKβ enhances RTA transactivation function and promotes RTA ubiquitin-mediated deagration of p65 (Dong et al., 2010, 2012; Dong and Feng, 2011) (Figure 5). Since IKKβ also phosphorylates the NF-κB subunit p65, RTA driven p65 degradation would presumably serve to limit NF-κB-dependent inflammatory cytokine production (Dong et al., 2010; Dong and Feng, 2011; He et al., 2015). An increase in pro-inflammatory cytokine production and lung inflammatory infiltrate was reported for MAVS-/- mice during acute MHV68 infection (Dong and Feng, 2011; Dong et al., 2012). Interestingly, the kinetics of virus replication were also dramatically altered in the lungs in the absence of MAVS. Given that overexpression of p65 impairs MHV68 RTA transactivation (Brown et al., 2003b), the degradation of p65 might further enhance viral lytic gene expression as well.
Karijolich et al. (2015) recently identified another pathway that leads to IKKβ activation upon MHV68 infection. In this study, host short interspersed nuclear elements (SINE) RNAs were induced upon MHV68 infection. SINE RNAs drove IKKβ activation and subsequent phosphorylation of RTA to enhance its transactivation function. MAVS deficiency reduced the activation of IKKβ but did not abrogate it, suggesting that SINEs may activate NF-κB by both MAVS-dependent and independent means (Karijolich et al., 2015). Taken together, upon de novo infection of permissive cell lines, MHV68 activates upstream NF-κB signaling events to drive RTA phosphorylation to enhance lytic gene expression, but impairs downstream NF-κB subunit activation, likely to prevent inflammatory cytokine production (Figure 5).
Evasion of Inflammasome
The inflammasome is an intracellular surveillance system that can respond to both damage-associated and pathogen-associated molecular patterns (DAMPs and PAMPs). By initiating these critical inflammatory responses, the inflammasome plays an important role in innate immune control of numerous pathogens, including double stranded DNA viruses (Rathinam et al., 2010; Sun et al., 2015). The inflammasome is both activated and subverted during human gHV infection in culture (Gregory et al., 2011; Kerur et al., 2011; Haneklaus et al., 2012; Ansari et al., 2013; Singh et al., 2013). Caspase-1 is the inflammasome effector protease that cleaves the proinflammatory cytokines IL-1β and IL-18. Surprisingly, mice deficient in caspase-1 were not impaired for the control of acute replication of MHV68 in the lung or latency in the spleen (Cieniewicz et al., 2015). In contrast to other herpesviruses, infection with 10 infectious MHV68 particles per cell does not activate caspase-1 (Cieniewicz et al., 2015; Sun et al., 2015). The delivery of the ORF64 deubiquitinase as a tegument protein seemingly drives efficient shuttling of intact capsids to the nucleus, shielding the foreign viral genomic DNA from innate sensors that might trigger inflammasome activation upon infection (Sun et al., 2015). In addition, MHV68 infection caused a specific reduction in IL-1β production after extrinsic LPS stimulation or upon co-infection with Salmonella enterica Serovar Typhimurium (Cieniewicz et al., 2015). Interestingly, this impairment occurred at the proIL-1β transcript level without effecting il18 and tnfa mRNA. IL-1β is regulated by NF-κB signaling, thus RTA was suspected to reduce IL-1β transcription via its function to degrade p65. However, the downregulation was independent of the lytic viral transcriptional activator RTA (Cieniewicz et al., 2015). Taken together, MHV68 impairs the inflammasome response by both inhibiting sensing and reducing IL-1β production during the initial stages of infection, likely via different mechanisms involving virion components and RTA-independent IE gene products.
Impact of These Studies
Over the long course of coevolution with their hosts, the gHVs have evolved strategies to subvert host signaling and immunity, culminating in the establishment of a life-long infection that causes little deleterious effect to the host. However, in the context of immunosuppression, these latent infections predispose their host to the development of malignancies. Murine gHV infection of mice serves as a powerful system to investigate the complex interactions of virus and host factors that contribute to pathogenesis in a natural host. Impairment of NF-κB signaling through the use of knockout mice and recombinant viruses has revealed a critical role for this host pathway in the establishment of latency in the host B cell latency reservoir (Table 2). Targeted ablation strategies and experimental determination of the NF-κB-dependent gene expression profile of the virus and host are critical to identify roles intrinsic to the infected cell. With regard to productive replication, de novo infection with MHV68 leads to activation of upstream NF-κB signaling molecules to enhance transactivation of lytic gene expression and promote productive replication (Figure 5). A better understanding of the mechanism of action for the host factors of the infected cell and the molecular interface between the virus and host will identify novel therapeutic approaches to disrupt replication and latency, and ultimately enable the prevention or elimination of virus-driven malignancies.
Author Contributions
All authors contributed to the writing and figure generation for the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Demetra Catalona with assistance in the bibliography. The authors apologize to colleagues whose research was not cited due to space constraints.
Footnotes
Funding. BC was supported by NIAID T32AI007539, AS was supported by NIGMS, NM was supported by NIH AI097875, and LK was supported by ACS Research Scholar grant RSG-11-160-01-MPC, NIH AI097875, and NIH AI110079.
References
- Adler H., Messerle M., Wagner M., Koszinowski U. H. (2000). Cloning and mutagenesis of the murine gammaherpesvirus 68 genome as an infectious bacterial artificial chromosome. J. Virol. 74 6964–6974. 10.1128/JVI.74.15.6964-6974.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfieri C., Birkenbach M., Kieff E. (1991). Early events in epstein-Barr virus infection of human B lymphocytes. Virology 181 595–608. 10.1016/0042-6822(91)90893-G [DOI] [PubMed] [Google Scholar]
- Ambroziak J. A., Blackbourn D. J., Herndier B. G., Glogau R. G., Gullett J. H., Mcdonald A. R., et al. (1995). Herpes-like sequences in HIV-infected and uninfected Kaposi’s sarcoma patients. Science 268 582–583. 10.1126/science.7725108 [DOI] [PubMed] [Google Scholar]
- Ansari M. A., Singh V. V., Dutta S., Veettil M. V., Dutta D., Chikoti L., et al. (2013). Constitutive interferon-inducible protein 16-inflammasome activation during Epstein-Barr virus latency I, II, and III in B and epithelial cells. J. Virol. 87 8606–8623. 10.1128/JVI.00805-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariza M. E., Glaser R., Kaumaya P. T., Jones C., Williams M. V. (2009). The EBV-encoded dUTPase activates NF-kappa B through the TLR2 and MyD88-dependent signaling pathway. J. Immunol. 182 851–859. 10.4049/jimmunol.182.2.851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvanitakis L., Geras-Raaka E., Varma A., Gershengorn M. C., Cesarman E. (1997). Human herpesvirus KSHV encodes a constitutively active G-protein-coupled receptor linked to cell proliferation. Nature 385 347–350. 10.1038/385347a0 [DOI] [PubMed] [Google Scholar]
- Babcock G. J., Decker L. L., Volk M., Thorley-Lawson D. A. (1998). EBV persistence in memory B cells in vivo. Immunity 9 395–404. 10.1016/S1074-7613(00)80622-6 [DOI] [PubMed] [Google Scholar]
- Ballestas M. E., Chatis P. A., Kaye K. M. (1999). Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 284 641–644. 10.1126/science.284.5414.641 [DOI] [PubMed] [Google Scholar]
- Barton E., Mandal P., Speck S. H. (2011). Pathogenesis and host control of gammaherpesviruses: lessons from the mouse. Annu. Rev. Immunol. 29 351–397. 10.1146/annurev-immunol-072710-081639 [DOI] [PubMed] [Google Scholar]
- Basak S., Kim H., Kearns J. D., Tergaonkar V., O’dea E., Werner S. L., et al. (2007). A fourth IkappaB protein within the NF-kappaB signaling module. Cell 128 369–381. 10.1016/j.cell.2006.12.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basak S., Shih V. F., Hoffmann A. (2008). Generation and activation of multiple dimeric transcription factors within the NF-kappaB signaling system. Mol. Cell. Biol. 28 3139–3150. 10.1128/MCB.01469-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batten M., Groom J., Cachero T. G., Qian F., Schneider P., Tschopp J., et al. (2000). Baff Mediates Survival of Peripheral Immature B Lymphocytes. J. Exp. Med. 192 1453–1466. 10.1084/jem.192.10.1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhende P. M., Dickerson S. J., Sun X., Feng W. H., Kenney S. C. (2007). X-box-binding protein 1 activates lytic Epstein-Barr virus gene expression in combination with protein kinase D. J. Virol. 81 7363–7370. 10.1128/JVI.00154-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackman M. A., Flano E., Usherwood E., Woodland D. L. (2000). Murine gamma-herpesvirus-68: a mouse model for infectious mononucleosis? Mol. Med. Today 6 488–490. 10.1016/S1357-4310(00)01813-X [DOI] [PubMed] [Google Scholar]
- Blasdell K., Mccracken C., Morris A., Nash A. A., Begon M., Bennett M., et al. (2003). The wood mouse is a natural host for Murid herpesvirus 4. J. Gen. Virol. 84 111–113. 10.1099/vir.0.18731-0 [DOI] [PubMed] [Google Scholar]
- Blaskovic D., Stancekova M., Svobodova J., Mistrikova J. (1980). Isolation of five strains of herpesviruses from two species of free living small rodents. Acta Virol. 24:468. [PubMed] [Google Scholar]
- Bogerd H. P., Karnowski H. W., Cai X., Shin J., Pohlers M., Cullen B. R. (2010). A mammalian herpesvirus uses noncanonical expression and processing mechanisms to generate viral MicroRNAs. Mol. Cell 37 135–142. 10.1016/j.molcel.2009.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonizzi G., Bebien M., Otero D. C., Johnson-Vroom K. E., Cao Y., Vu D., et al. (2004). Activation of IKKalpha target genes depends on recognition of specific kappaB binding sites by RelB:p52 dimers. EMBO J. 23 4202–4210. 10.1038/sj.emboj.7600391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boshoff C., Schulz T. F., Kennedy M. M., Graham A. K., Fisher C., Thomas A., et al. (1995). Kaposi’s sarcoma-associated herpesvirus infects endothelial and spindle cells. Nat. Med. 1 1274–1278. 10.1038/nm1295-1274 [DOI] [PubMed] [Google Scholar]
- Bowden R. J., Simas J. P., Davis A. J., Efstathiou S. (1997). Murine gammaherpesvirus 68 encodes tRNA-like sequences which are expressed during latency. J. Gen. Virol. 78(Pt 7), 1675–1687. 10.1099/0022-1317-78-7-1675 [DOI] [PubMed] [Google Scholar]
- Brightbill H. D., Jackman J. K., Suto E., Kennedy H., Jones C., III, Chalasani S., et al. (2015). Conditional deletion of NF-kappaB-Inducing kinase (NIK) in adult mice disrupts mature B cell survival and activation. J. Immunol. 195 953–964. 10.4049/jimmunol.1401514 [DOI] [PubMed] [Google Scholar]
- Brinkmann M. M., Glenn M., Rainbow L., Kieser A., Henke-Gendo C., Schulz T. F. (2003). Activation of mitogen-activated protein kinase and NF-kappaB pathways by a Kaposi’s sarcoma-associated herpesvirus K15 membrane protein. J. Virol. 77 9346–9358. 10.1128/JVI.77.17.9346-9358.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown H. J., Song M. J., Deng H., Wu T. T., Cheng G., Sun R. (2003a). NF- B inhibits gammaherpesvirus lytic replication. J. Virol. 77 8532–8540. 10.1128/JVI.77.15.8532-8540.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown H. J., Song M. J., Deng H., Wu T. T., Cheng G., Sun R. (2003b). NF-kappaB inhibits gammaherpesvirus lytic replication. J. Virol. 77 8532–8540. 10.1128/JVI.77.15.8532-8540.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardin R. D., Brooks J. W., Sarawar S. R., Doherty P. C. (1996). Progressive loss of CD8+ T cell-mediated control of a gamma-herpesvirus in the absence of CD4+ T cells. J. Exp. Med. 184 863–871. 10.1084/jem.184.3.863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesarman E. (2014). Gammaherpesviruses and lymphoproliferative disorders. Annu. Rev. Pathol. 9 349–372. 10.1146/annurev-pathol-012513-104656 [DOI] [PubMed] [Google Scholar]
- Chang L. S., Wang J. T., Doong S. L., Lee C. P., Chang C. W., Tsai C. H., et al. (2012). Epstein-Barr virus BGLF4 kinase downregulates NF-kappaB transactivation through phosphorylation of coactivator UXT. J. Virol. 86 12176–12186. 10.1128/JVI.01918-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao B., Frederico B., Stevenson P. G. (2015). B-cell-independent lymphoid tissue infection by a B-cell-tropic rhadinovirus. J. Gen. Virol. 96 2788–2793. 10.1099/vir.0.000188 [DOI] [PubMed] [Google Scholar]
- Chaudhary P. M., Jasmin A., Eby M. T., Hood L. (1999). Modulation of the NF-kappa B pathway by virally encoded death effector domains-containing proteins. Oncogene 18 5738–5746. 10.1038/sj.onc.1202976 [DOI] [PubMed] [Google Scholar]
- Cieniewicz B., Dong Q., Li G., Forrest J. C., Mounce B. C., Tarakanova V. L., et al. (2015). Murine Gammaherpesvirus 68 pathogenesis is independent of Caspase-1 and Caspase-11 in mice and impairs interleukin-1beta production upon extrinsic stimulation in culture. J. Virol. 89 6562–6574. 10.1128/JVI.00658-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claudio E., Brown K., Park S., Wang H., Siebenlist U. (2002). BAFF-induced NEMO-independent processing of NF-kappa B2 in maturing B cells. Nat. Immunol. 3 958–965. 10.1038/ni842 [DOI] [PubMed] [Google Scholar]
- Coleman C. B., Mcgraw J. E., Feldman E. R., Roth A. N., Keyes L. R., Grau K. R., et al. (2014). A gammaherpesvirus Bcl-2 ortholog blocks B cell receptor-mediated apoptosis and promotes the survival of developing B cells in vivo. PLoS Pathog. 10:e1003916 10.1371/journal.ppat.1003916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman C. B., Nealy M. S., Tibbetts S. A. (2010). Immature and transitional B cells are latency reservoirs for a gammaherpesvirus. J. Virol. 84 13045–13052. 10.1128/JVI.01455-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman H. M., De Lima B., Morton V., Stevenson P. G. (2003). Murine gammaherpesvirus 68 lacking thymidine kinase shows severe attenuation of lytic cycle replication in vivo but still establishes latency. J. Virol. 77 2410–2417. 10.1128/JVI.77.4.2410-2417.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins C. M., Boss J. M., Speck S. H. (2009). Identification of infected B-cell populations by using a recombinant murine gammaherpesvirus 68 expressing a fluorescent protein. J. Virol. 83 6484–6493. 10.1128/JVI.00297-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins C. M., Medveczky M. M., Lund T., Medveczky P. G. (2002). The terminal repeats and latency-associated nuclear antigen of herpesvirus saimiri are essential for episomal persistence of the viral genome. J. Gen. Virol. 83 2269–2278. 10.1099/0022-1317-83-9-2269 [DOI] [PubMed] [Google Scholar]
- Collins C. M., Speck S. H. (2014). Expansion of murine gammaherpesvirus latently infected B cells requires T follicular help. PLoS Pathog. 10:e1004106 10.1371/journal.ppat.1004106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins C. M., Speck S. H. (2015). Interleukin 21 signaling in B cells is required for efficient establishment of murine gammaherpesvirus latency. PLoS Pathog. 11:e1004831 10.1371/journal.ppat.1004831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford D. H., Ando I. (1986). EB virus induction is associated with B-cell maturation. Immunology 59 405–409. [PMC free article] [PubMed] [Google Scholar]
- Craxton A., Draves K. E., Gruppi A., Clark E. A. (2005). BAFF regulates B cell survival by downregulating the BH3-only family member Bim via the ERK pathway. J. Exp. Med. 202 1363–1374. 10.1084/jem.20051283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davison A. J. (2002). Evolution of the herpesviruses. Vet. Microbiol. 86 69–88. 10.1016/S0378-1135(01)00492-8 [DOI] [PubMed] [Google Scholar]
- Dawson C. W., George J. H., Blake S. M., Longnecker R., Young L. S. (2001). The Epstein-Barr virus encoded latent membrane protein 2A augments signaling from latent membrane protein 1. Virology 289 192–207. 10.1006/viro.2001.1142 [DOI] [PubMed] [Google Scholar]
- De Silva N. S., Silva K., Anderson M. M., Bhagat G., Klein U. (2016). Impairment of Mature B Cell Maintenance upon combined deletion of the alternative NF-kappaB transcription factors RELB and NF-kappaB2 in B Cells. J. Immunol. 196 2591–2601. 10.4049/jimmunol.1501120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejardin E., Droin N. M., Delhase M., Haas E., Cao Y., Makris C., et al. (2002). The Lymphotoxin-β Receptor induces different patterns of gene expression via Two NF-κB pathways. Immunity 17 525–535. 10.1016/S1074-7613(02)00423-5 [DOI] [PubMed] [Google Scholar]
- Dias P., Giannoni F., Lee L. N., Han D., Yoon S., Yagita H., et al. (2010). CD4 T-cell help programs a change in CD8 T-cell function enabling effective long-term control of murine gammaherpesvirus 68: role of PD-1-PD-L1 interactions. J. Virol. 84 8241–8249. 10.1128/JVI.00784-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebel K. W., Oko L. M., Medina E. M., Niemeyer B. F., Warren C. J., Claypool D. J., et al. (2015). Gammaherpesvirus small noncoding RNAs are bifunctional elements that regulate infection and contribute to virulence in vivo. MBio 6:e01670–14 10.1128/mBio.01670-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty P. C., Christensen J. P., Belz G. T., Stevenson P. G., Sangster M. Y. (2001). Dissecting the host response to a gamma-herpesvirus. Philos. Trans. R. Soc. Lond. B Biol. Sci. 356 581–593. 10.1098/rstb.2000.0786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X., Feng H., Sun Q., Li H., Wu T. T., Sun R., et al. (2010). Murine gamma-herpesvirus 68 hijacks MAVS and IKKbeta to initiate lytic replication. PLoS Pathog. 6:e1001001 10.1371/journal.ppat.1001001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X., Feng P. (2011). Murine gamma herpesvirus 68 hijacks MAVS and IKKbeta to abrogate NFkappaB activation and antiviral cytokine production. PLoS Pathog. 7:e1002336 10.1371/journal.ppat.1002336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X., He Z., Durakoglugil D., Arneson L., Shen Y., Feng P. (2012). Murine gammaherpesvirus 68 evades host cytokine production via replication transactivator-induced RelA degradation. J. Virol. 86 1930–1941. 10.1128/JVI.06127-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunmire S. K., Grimm J. M., Schmeling D. O., Balfour H. H., Jr., Hogquist K. A. (2015). The incubation period of primary epstein-barr virus infection: viral dynamics and immunologic events. PLoS Pathog. 11:e1005286 10.1371/journal.ppat.1005286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutia B. M., Clarke C. J., Allen D. J., Nash A. A. (1997). Pathological changes in the spleens of gamma interferon receptor-deficient mice infected with murine gammaherpesvirus: a role for CD8 T cells. J. Virol. 71 4278–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efstathiou S., Ho Y. M., Hall S., Styles C. J., Scott S. D., Gompels U. A. (1990a). Murine herpesvirus 68 is genetically related to the gammaherpesviruses Epstein-Barr virus and herpesvirus saimiri. J. Gen. Virol. 71(Pt 6), 1365–1372. 10.1099/0022-1317-71-6-1365 [DOI] [PubMed] [Google Scholar]
- Efstathiou S., Ho Y. M., Minson A. C. (1990b). Cloning and molecular characterization of the murine herpesvirus 68 genome. J. Gen. Virol. 71(Pt 6), 1355–1364. 10.1099/0022-1317-71-6-1355 [DOI] [PubMed] [Google Scholar]
- Ehtisham S., Sunil-Chandra N. P., Nash A. A. (1993). Pathogenesis of murine gammaherpesvirus infection in mice deficient in CD4 and CD8 T cells. J. Virol. 67 5247–5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ersing I., Bernhardt K., Gewurz B. E. (2013). NF-kappaB and IRF7 pathway activation by epstein-barr virus latent membrane protein 1. Viruses 5 1587–1606. 10.3390/v5061587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans A. G., Moser J. M., Krug L. T., Pozharskaya V., Mora A. L., Speck S. H. (2008). A gammaherpesvirus-secreted activator of Vbeta4+ CD8+ T cells regulates chronic infection and immunopathology. J. Exp. Med. 205 669–684. 10.1084/jem.20071135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feederle R., Kost M., Baumann M., Janz A., Drouet E., Hammerschmidt W., et al. (2000). The epstein-barr virus lytic program is controlled by the co-operative functions of two transactivators. EMBO J. 19 3080–3089. 10.1093/emboj/19.12.3080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman E. R., Kara M., Coleman C. B., Grau K. R., Oko L. M., Krueger B. J., et al. (2014). Virus-encoded microRNAs facilitate gammaherpesvirus latency and pathogenesis in vivo. MBio 5:e00981–14 10.1128/mBio.00981-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman E. R., Kara M., Oko L. M., Grau K. R., Krueger B. J., Zhang J., et al. (2016). A gammaherpesvirus noncoding RNA is essential for hematogenous dissemination and establishment of peripheral latency. mSphere 1:e00105–e00115 10.1128/mSphere.00105-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field N., Low W., Daniels M., Howell S., Daviet L., Boshoff C., et al. (2003). KSHV vFLIP binds to IKK-gamma to activate IKK. J. Cell Sci. 116 3721–3728. 10.1242/jcs.00691 [DOI] [PubMed] [Google Scholar]
- Flano E., Husain S. M., Sample J. T., Woodland D. L., Blackman M. A. (2000). Latent murine gamma-herpesvirus infection is established in activated B cells, dendritic cells, and macrophages. J. Immunol. 165 1074–1081. 10.4049/jimmunol.165.2.1074 [DOI] [PubMed] [Google Scholar]
- Flano E., Jia Q., Moore J., Woodland D. L., Sun R., Blackman M. A. (2005). Early establishment of gamma-herpesvirus latency: implications for immune control. J. Immunol. 174 4972–4978. 10.4049/jimmunol.174.8.4972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flano E., Kim I. J., Moore J., Woodland D. L., Blackman M. A. (2003). Differential gamma-herpesvirus distribution in distinct anatomical locations and cell subsets during persistent infection in mice. J. Immunol. 170 3828–3834. 10.4049/jimmunol.170.7.3828 [DOI] [PubMed] [Google Scholar]
- Flano E., Kim I. J., Woodland D. L., Blackman M. A. (2002a). Gamma-herpesvirus latency is preferentially maintained in splenic germinal center and memory B cells. J. Exp. Med. 196 1363–1372. 10.1084/jem.20020890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flano E., Woodland D. L., Blackman M. A. (1999). Requirement for CD4+ T cells in V beta 4+CD8+ T cell activation associated with latent murine gammaherpesvirus infection. J. Immunol. 163 3403–3408. [PubMed] [Google Scholar]
- Flano E., Woodland D. L., Blackman M. A. (2002b). A mouse model for infectious mononucleosis. Immunol. Res. 25 201–217. 10.1385/IR:25:3:201 [DOI] [PubMed] [Google Scholar]
- Forrest J. C., Speck S. H. (2008). Establishment of B-cell lines latently infected with reactivation-competent murine gammaherpesvirus 68 provides evidence for viral alteration of a DNA damage-signaling cascade. J. Virol. 82 7688–7699. 10.1128/JVI.02689-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francois S., Vidick S., Sarlet M., Desmecht D., Drion P., Stevenson P. G., et al. (2013). Illumination of murine gammaherpesvirus-68 cycle reveals a sexual transmission route from females to males in laboratory mice. PLoS Pathog. 9:e1003292 10.1371/journal.ppat.1003292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederico B., Chao B., Lawler C., May J. S., Stevenson P. G. (2015). Subcapsular sinus macrophages limit acute gammaherpesvirus dissemination. J. Gen. Virol. 96 2314–2327. 10.1099/vir.0.000140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederico B., Chao B., May J. S., Belz G. T., Stevenson P. G. (2014a). A murid gamma-herpesviruses exploits normal splenic immune communication routes for systemic spread. Cell Host Microbe 15 457–470. 10.1016/j.chom.2014.03.010 [DOI] [PubMed] [Google Scholar]
- Frederico B., May J. S., Efstathiou S., Stevenson P. G. (2014b). BAFF receptor deficiency limits gammaherpesvirus infection. J. Virol. 88 3965–3975. 10.1128/JVI.03497-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederico B., Milho R., May J. S., Gillet L., Stevenson P. G. (2012). Myeloid infection links epithelial and B cell tropisms of Murid Herpesvirus-4. PLoS Pathog. 8:e1002935 10.1371/journal.ppat.1002935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman M. L., Lanzer K. G., Cookenham T., Peters B., Sidney J., Wu T. T., et al. (2010). Two kinetic patterns of epitope-specific CD8 T-cell responses following murine gammaherpesvirus 68 infection. J. Virol. 84 2881–2892. 10.1128/JVI.02229-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangappa S., Kapadia S. B., Speck S. H., Virgin H. W. T. (2002). Antibody to a lytic cycle viral protein decreases gammaherpesvirus latency in B-cell-deficient mice. J. Virol. 76 11460–11468. 10.1128/JVI.76.22.11460-11468.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardam S., Sierro F., Basten A., Mackay F., Brink R. (2008). TRAF2 and TRAF3 signal adapters act cooperatively to control the maturation and survival signals delivered to B cells by the BAFF receptor. Immunity 28 391–401. 10.1016/j.immuni.2008.01.009 [DOI] [PubMed] [Google Scholar]
- Gargano L. M., Forrest J. C., Speck S. H. (2009). Signaling through toll-like receptors induces murine gammaherpesvirus 68 reactivation in vivo. J. Virol. 83 1474–1482. 10.1128/JVI.01717-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargano L. M., Moser J. M., Speck S. H. (2008). Role for MyD88 signaling in murine gammaherpesvirus 68 latency. J. Virol. 82 3853–3863. 10.1128/JVI.02577-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar M., Gill M. B., Losing J. B., May J. S., Stevenson P. G. (2008). Multiple functions for ORF75c in murid herpesvirus-4 infection. PLoS ONE 3:e2781 10.1371/journal.pone.0002781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar M., May J. S., Sukla S., Frederico B., Gill M. B., Smith C. M., et al. (2011). Murid herpesvirus-4 exploits dendritic cells to infect B cells. PLoS Pathog 7:e1002346 10.1371/journal.ppat.1002346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiser M. (2002). Morphological aspects of particle uptake by lung phagocytes. Microsc. Res. Tech. 57 512–522. 10.1002/jemt.10105 [DOI] [PubMed] [Google Scholar]
- Giesecke C., Frolich D., Reiter K., Mei H. E., Wirries I., Kuhly R., et al. (2014). Tissue distribution and dependence of responsiveness of human antigen-specific memory B cells. J. Immunol. 192 3091–3100. 10.4049/jimmunol.1302783 [DOI] [PubMed] [Google Scholar]
- Gill M. B., May J. S., Colaco S., Stevenson P. G. (2010). Important role for the murid herpesvirus 4 ribonucleotide reductase large subunit in host colonization via the respiratory tract. J. Virol. 84 10937–10942. 10.1128/JVI.00828-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill M. B., Wright D. E., Smith C. M., May J. S., Stevenson P. G. (2009). Murid herpesvirus-4 lacking thymidine kinase reveals route-dependent requirements for host colonization. J. Gen. Virol. 90 1461–1470. 10.1099/vir.0.010603-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz C. A., Baldwin A. S. (2008). NF-kappaB pathways in the immune system: control of the germinal center reaction. Immunol. Res. 41 233–247. 10.1007/s12026-008-8033-1 [DOI] [PubMed] [Google Scholar]
- Goodwin D. J., Walters M. S., Smith P. G., Thurau M., Fickenscher H., Whitehouse A. (2001). Herpesvirus saimiri open reading frame 50 (Rta) protein reactivates the lytic replication cycle in a persistently infected A549 cell line. J. Virol. 75 4008–4013. 10.1128/JVI.75.8.4008-4013.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelik L., Gilbride K., Dobles M., Kalled S. L., Zandman D., Scott M. L. (2003). Normal B cell homeostasis requires B cell activation factor production by radiation-resistant cells. J. Exp. Med. 198 937–945. 10.1084/jem.20030789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradoville L., Gerlach J., Grogan E., Shedd D., Nikiforow S., Metroka C., et al. (2000). Kaposi’s sarcoma-associated herpesvirus open reading frame 50/Rta protein activates the entire viral lytic cycle in the HH-B2 primary effusion lymphoma cell line. J. Virol. 74 6207–6212. 10.1128/JVI.74.13.6207-6212.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gredmark-Russ S., Cheung E. J., Isaacson M. K., Ploegh H. L., Grotenbreg G. M. (2008). The CD8 T-cell response against murine gammaherpesvirus 68 is directed toward a broad repertoire of epitopes from both early and late antigens. J. Virol. 82 12205–12212. 10.1128/JVI.01463-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory S. M., Davis B. K., West J. A., Taxman D. J., Matsuzawa S., Reed J. C., et al. (2011). Discovery of a viral NLR homolog that inhibits the inflammasome. Science 331 330–334. 10.1126/science.1199478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm T., Schneider S., Naschberger E., Huber J., Guenzi E., Kieser A., et al. (2005). EBV latent membrane protein-1 protects B cells from apoptosis by inhibition of BAX. Blood 105 3263–3269. 10.1182/blood-2004-07-2752 [DOI] [PubMed] [Google Scholar]
- Grossmann C., Podgrabinska S., Skobe M., Ganem D. (2006). Activation of NF-kappaB by the latent vFLIP gene of Kaposi’s sarcoma-associated herpesvirus is required for the spindle shape of virus-infected endothelial cells and contributes to their proinflammatory phenotype. J. Virol. 80 7179–7185. 10.1128/JVI.01603-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guasparri I., Bubman D., Cesarman E. (2008). EBV LMP2A affects LMP1-mediated NF-kappaB signaling and survival of lymphoma cells by regulating TRAF2 expression. Blood 111 3813–3820. 10.1182/blood-2007-03-080309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guasparri I., Keller S. A., Cesarman E. (2004). KSHV vFLIP is essential for the survival of infected lymphoma cells. J. Exp. Med. 199 993–1003. 10.1084/jem.20031467 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Guasparri I., Wu H., Cesarman E. (2006). The KSHV oncoprotein vFLIP contains a TRAF-interacting motif and requires TRAF2 and TRAF3 for signalling. EMBO Rep. 7 114–119. 10.1038/sj.embor.7400580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutsch D. E., Holley-Guthrie E. A., Zhang Q., Stein B., Blanar M. A., Baldwin A. S., et al. (1994). The bZIP transactivator of Epstein-Barr virus, BZLF1, functionally and physically interacts with the p65 subunit of NF-kappa B. Mol. Cell. Biol. 14 1939–1948. 10.1128/MCB.14.3.1939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwack Y., Nakamura H., Lee S. H., Souvlis J., Yustein J. T., Gygi S., et al. (2003). Poly(ADP-ribose) polymerase 1 and Ste20-like kinase hKFC act as transcriptional repressors for gamma-2 herpesvirus lytic replication. Mol. Cell. Biol. 23 8282–8294. 10.1128/MCB.23.22.8282-8294.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas F., Yamauchi K., Murat M., Bernasconi M., Yamanaka N., Speck R. F., et al. (2014). Activation of NF-kappaB via endosomal Toll-like receptor 7 (TLR7) or TLR9 suppresses murine herpesvirus 68 reactivation. J. Virol. 88 10002–10012. 10.1128/JVI.01486-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habison A. C., Beauchemin C., Simas J. P., Usherwood E. J., Kaye K. M. (2012). Murine gammaherpesvirus 68 LANA acts on terminal repeat DNA to mediate episome persistence. J. Virol. 86 11863–11876. 10.1128/JVI.01656-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadinoto V., Shapiro M., Greenough T. C., Sullivan J. L., Luzuriaga K., Thorley-Lawson D. A. (2008). On the dynamics of acute EBV infection and the pathogenesis of infectious mononucleosis. Blood 111 1420–1427. 10.1182/blood-2007-06-093278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haneklaus M., Gerlic M., Kurowska-Stolarska M., Rainey A. A., Pich D., Mcinnes I. B., et al. (2012). Cutting edge: miR-223 and EBV miR-BART15 regulate the NLRP3 inflammasome and IL-1beta production. J. Immunol. 189 3795–3799. 10.4049/jimmunol.1200312 [DOI] [PubMed] [Google Scholar]
- Hassman L. M., Ellison T. J., Kedes D. H. (2011). KSHV infects a subset of human tonsillar B cells, driving proliferation and plasmablast differentiation. J. Clin. Invest. 121 752–768. 10.1172/JCI44185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S., Zhao J., Song S., He X., Minassian A., Zhou Y., et al. (2015). Viral pseudo-enzymes activate RIG-I via deamidation to evade cytokine production. Mol. Cell 58 134–146. 10.1016/j.molcel.2015.01.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand J. M., Luo Z., Manske M. K., Price-Troska T., Ziesmer S. C., Lin W., et al. (2010). A BAFF-R mutation associated with non-Hodgkin lymphoma alters TRAF recruitment and reveals new insights into BAFF-R signaling. J. Exp. Med. 207 2569–2579. 10.1084/jem.20100857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoagland R. J. (1964). The incubation period of infectious mononucleosis. Am. J. Public Health Nations Health 54 1699–1705. 10.2105/AJPH.54.10.1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A., Leung T. H., Baltimore D. (2003). Genetic analysis of NF-kappaB/Rel transcription factors defines functional specificities. EMBO J. 22 5530–5539. 10.1093/emboj/cdg534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu B. L., Harless S. M., Lindsley R. C., Hilbert D. M., Cancro M. P. (2002). Cutting edge: BLyS enables survival of transitional and mature B cells through distinct mediators. J. Immunol. 168 5993–5996. 10.4049/jimmunol.168.12.5993 [DOI] [PubMed] [Google Scholar]
- Hughes D. J., Kipar A., Leeming G. H., Bennett E., Howarth D., Cummerson J. A., et al. (2011). Chemokine binding protein M3 of murine gammaherpesvirus 68 modulates the host response to infection in a natural host. PLoS Pathog. 7:e1001321 10.1371/journal.ppat.1001321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S., Wu T. T., Tong L. M., Kim K. S., Martinez-Guzman D., Colantonio A. D., et al. (2008). Persistent gammaherpesvirus replication and dynamic interaction with the host in vivo. J. Virol. 82 12498–12509. 10.1128/JVI.01152-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichii M., Oritani K., Kanakura Y. (2014). Early B lymphocyte development: similarities and differences in human and mouse. World J. Stem Cells 6 421–431. 10.4252/wjsc.v6.i4.421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha H. C., Banerjee S., Robertson E. S. (2016). The role of gammaherpesviruses in cancer pathogenesis. Pathogens 5:E18 10.3390/pathogens5010018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaisho T., Takeda K., Tsujimura T., Kawai T., Nomura F., Terada N., et al. (2001). I b kinase is essential for mature B cell development and function. J. Exp. Med. 193 417–426. 10.1084/jem.193.4.417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalled S. L. (2006). Impact of the BAFF/BR3 axis on B cell survival, germinal center maintenance and antibody production. Semin. Immunol. 18 290–296. 10.1016/j.smim.2006.06.002 [DOI] [PubMed] [Google Scholar]
- Karijolich J., Abernathy E., Glaunsinger B. A. (2015). Infection-induced retrotransposon-derived noncoding RNAs enhance herpesviral gene expression via the NF-kappaB pathway. PLoS Pathog. 11:e1005260 10.1371/journal.ppat.1005260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabe T., Naka T., Yoshida K., Tanaka T., Fujiwara H., Suematsu S., et al. (1994). The immune responses in CD40-deficient mice: impaired immunoglobulin class switching and germinal center formation. Immunity 1 167–178. 10.1016/1074-7613(94)90095-7 [DOI] [PubMed] [Google Scholar]
- Keller S. A., Schattner E. J., Cesarman E. (2000). Inhibition of NF-kappaB induces apoptosis of KSHV-infected primary effusion lymphoma cells. Blood 96 2537–2542. [PubMed] [Google Scholar]
- Kerur N., Veettil M. V., Sharma-Walia N., Bottero V., Sadagopan S., Otageri P., et al. (2011). IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to kaposi sarcoma-associated herpesvirus infection. Cell Host Microbe 9 363–375. 10.1016/j.chom.2011.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I. J., Flano E., Woodland D. L., Blackman M. A. (2002). Antibody-mediated control of persistent gamma-herpesvirus infection. J. Immunol. 168 3958–3964. 10.4049/jimmunol.168.8.3958 [DOI] [PubMed] [Google Scholar]
- Kim I. J., Flano E., Woodland D. L., Lund F. E., Randall T. D., Blackman M. A. (2003). Maintenance of long term gamma-herpesvirus B cell latency is dependent on CD40-mediated development of memory B cells. J. Immunol. 171 886–892. 10.4049/jimmunol.171.2.886 [DOI] [PubMed] [Google Scholar]
- Kirshner J. R., Staskus K., Haase A., Lagunoff M., Ganem D. (1999). Expression of the open reading frame 74 (G-protein-coupled receptor) gene of Kaposi’s sarcoma (KS)-associated herpesvirus: implications for KS pathogenesis. J. Virol. 73 6006–6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konrad A., Wies E., Thurau M., Marquardt G., Naschberger E., Hentschel S., et al. (2009). A systems biology approach to identify the combination effects of human herpesvirus 8 genes on NF-kappaB activation. J. Virol. 83 2563–2574. 10.1128/JVI.01512-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka Y., Calderhead D. M., Manning E. M., Hambor J. E., Black A., Geleziunas R., et al. (1999). Activation and regulation of the IkappaB kinase in human B cells by CD40 signaling. Eur. J. Immunol. 29 1353–1362. 10.1002/(SICI)1521-4141(199904)29:04<1353::AID-IMMU1353>3.0.CO;2-2 [DOI] [PubMed] [Google Scholar]
- Krug L. T., Collins C. M., Gargano L. M., Speck S. H. (2009). NF-kappaB p50 plays distinct roles in the establishment and control of murine gammaherpesvirus 68 latency. J. Virol. 83 4732–4748. 10.1128/JVI.xyb00111-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug L. T., Moser J. M., Dickerson S. M., Speck S. H. (2007). Inhibition of NF-kappaB activation in vivo impairs establishment of gammaherpesvirus latency. PLoS Pathog. 3:e11 10.1371/journal.ppat.0030011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug L. T., Torres-Gonzalez E., Qin Q., Sorescu D., Rojas M., Stecenko A., et al. (2010). Inhibition of NF-kappaB signaling reduces virus load and gammaherpesvirus-induced pulmonary fibrosis. Am. J. Pathol. 177 608–621. 10.2353/ajpath.2010.091122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku B., Woo J. S., Liang C., Lee K. H., Hong H. S., E X., et al. (2008). Structural and biochemical bases for the inhibition of autophagy and apoptosis by viral BCL-2 of murine gamma-herpesvirus 68. PLoS Pathog. 4:e25 10.1371/journal.ppat.0040025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunsch C., Ruben S. M., Rosen C. A. (1992). Selection of optimal kappa B/Rel DNA-binding motifs: interaction of both subunits of NF-kappa B with DNA is required for transcriptional activation. Mol. Cell. Biol. 12 4412–4421. 10.1128/MCB.12.10.4412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laichalk L. L., Thorley-Lawson D. A. (2005). Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J. Virol. 79 1296–1307. 10.1128/JVI.79.2.1296-1307.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgren O., Gilbert E. S., Rizzo J. D., Socie G., Banks P. M., Sobocinski K. A., et al. (2009). Risk factors for lymphoproliferative disorders after allogeneic hematopoietic cell transplantation. Blood 113 4992–5001. 10.1182/blood-2008-09-178046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawler C., Milho R., May J. S., Stevenson P. G. (2015). Rhadinovirus host entry by co-operative infection. PLoS Pathog. 11:e1004761 10.1371/journal.ppat.1004761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Clorennec C., Ouk T. S., Youlyouz-Marfak I., Panteix S., Martin C. C., Rastelli J., et al. (2008). Molecular basis of cytotoxicity of Epstein-Barr virus (EBV) latent membrane protein 1 (LMP1) in EBV latency III B cells: LMP1 induces type II ligand-independent autoactivation of CD95/Fas with caspase 8-mediated apoptosis. J. Virol. 82 6721–6733. 10.1128/JVI.02250-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B. J., Santee S., Von Gesjen S., Ware C. F., Sarawar S. R. (2000). Lymphotoxin-alpha -deficient mice can clear a productive infection with murine gammaherpesvirus 68 but fail to develop splenomegaly or lymphocytosis. J. Virol. 74 2786–2792. 10.1128/JVI.74.6.2786-2792.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. S., Groshong S. D., Cool C. D., Kleinschmidt-Demasters B. K., Van Dyk L. F. (2009). Murine gammaherpesvirus 68 infection of IFNgamma unresponsive mice: a small animal model for gammaherpesvirus-associated B-cell lymphoproliferative disease. Cancer Res. 69 5481–5489. 10.1158/0008-5472.CAN-09-0291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. A., Diamond M. E., Yates J. (1999). Genetic evidence that EBNA-1 is needed for efficient, stable latent infection by Epstein-Barr virus. J. Virol. 73 2974–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei X., Bai Z., Ye F., Xie J., Kim C. G., Huang Y., et al. (2010). Regulation of NF-kappaB inhibitor IkappaBalpha and viral replication by a KSHV microRNA. Nat. Cell Biol. 12 193–199. 10.1038/ncb2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentz V. M., Cancro M. P., Nashold F. E., Hayes C. E. (1996). Bcmd governs recruitment of new B cells into the stable peripheral B cell pool in the A/WySnJ mouse. J. Immunol 157 598–606. [PubMed] [Google Scholar]
- Li X., Massa P. E., Hanidu A., Peet G. W., Aro P., Savitt A., et al. (2002). IKKalpha, IKKbeta, and NEMO/IKKgamma are each required for the NF-kappa B-mediated inflammatory response program. J. Biol. Chem. 277 45129–45140. 10.1074/jbc.M205165200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X., Collins C. M., Mendel J. B., Iwakoshi N. N., Speck S. H. (2009). Gammaherpesvirus-driven plasma cell differentiation regulates virus reactivation from latently infected B lymphocytes. PLoS Pathog. 5:e1000677 10.1371/journal.ppat.1000677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X., Paden C. R., Morales F. M., Powers R. P., Jacob J., Speck S. H. (2011). Murine gamma-herpesvirus immortalization of fetal liver-derived B cells requires both the viral cyclin D homolog and latency-associated nuclear antigen. PLoS Pathog 7:e1002220 10.1371/journal.ppat.1002220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao G., Zhang M., Harhaj E. W., Sun S. C. (2004). Regulation of the NF-kappaB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J. Biol. Chem. 279 26243–26250. 10.1074/jbc.M403286200 [DOI] [PubMed] [Google Scholar]
- Lin X., Mu Y., Cunningham E. T., Jr., Marcu K. B., Geleziunas R., Greene W. C. (1998). Molecular determinants of NF-kappaB-inducing kinase action. Mol. Cell. Biol. 18 5899–5907. 10.1128/MCB.18.10.5899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling L., Cao Z., Goeddel D. V. (1998). NF-kappaB-inducing kinase activates IKK-alpha by phosphorylation of Ser-176. Proc. Natl. Acad. Sci. U.S.A. 95 3792–3797. 10.1073/pnas.95.7.3792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litinskiy M. B., Nardelli B., Hilbert D. M., He B., Schaffer A., Casali P., et al. (2002). DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat. Immunol. 3 822–829. 10.1038/ni829 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Liu L., Eby M. T., Rathore N., Sinha S. K., Kumar A., Chaudhary P. M. (2002). The human herpes virus 8-encoded viral FLICE inhibitory protein physically associates with and persistently activates the Ikappa B kinase complex. J. Biol. Chem. 277 13745–13751. 10.1074/jbc.M110480200 [DOI] [PubMed] [Google Scholar]
- Loder B. F., Mutschler B., Ray R. J., Paige C. J., Sideras P., Torres R., et al. (1999). B cell development in the spleen takes place in discrete steps and is determined by the quality of B cell receptor-derived signals. J. Exp. Med. 190 75–90. 10.1084/jem.190.1.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas P. C., Yonezumi M., Inohara N., Mcallister-Lucas L. M., Abazeed M. E., Chen F. F., et al. (2001). Bcl10 and MALT1, independent targets of chromosomal translocation in malt lymphoma, cooperate in a novel NF-kappa B signaling pathway. J. Biol. Chem. 276 19012–19019. 10.1074/jbc.M009984200 [DOI] [PubMed] [Google Scholar]
- Lukac D. M., Kirshner J. R., Ganem D. (1999). Transcriptional activation by the product of open reading frame 50 of Kaposi’s sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J. Virol. 73 9348–9361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukac D. M., Renne R., Kirshner J. R., Ganem D. (1998). Reactivation of Kaposi’s sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 252 304–312. 10.1006/viro.1998.9486 [DOI] [PubMed] [Google Scholar]
- Malnati M. S., Dagna L., Ponzoni M., Lusso P. (2003). Human herpesvirus 8 (HHV-8/KSHV) and hematologic malignancies. Rev. Clin. Exp. Hematol. 7 375–405. [PubMed] [Google Scholar]
- Marques S., Efstathiou S., Smith K. G., Haury M., Simas J. P. (2003). Selective gene expression of latent murine gammaherpesvirus 68 in B lymphocytes. J. Virol. 77 7308–7318. 10.1128/JVI.77.13.7308-7318.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D., Galisteo R., Ji Y., Montaner S., Gutkind J. S. (2008). An NF-kappaB gene expression signature contributes to Kaposi’s sarcoma virus vGPCR-induced direct and paracrine neoplasia. Oncogene 27 1844–1852. 10.1038/sj.onc.1210817 [DOI] [PubMed] [Google Scholar]
- Matar C. G., Rangaswamy U. S., Wakeman B. S., Iwakoshi N., Speck S. H. (2014). Murine gammaherpesvirus 68 reactivation from B cells requires IRF4 but not XBP-1. J. Virol. 88 11600–11610. 10.1128/JVI.01876-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima A., Kaisho T., Rennert P. D., Nakano H., Kurosawa K., Uchida D., et al. (2001). Essential role of nuclear factor (NF)-kappa B-Inducing kinase and inhibitor of kappa b (I b) kinase in Nf-kappa b activation through lymphotoxin receptor, but not through tumor necrosis factor receptor I. J. Exp. Med. 193 631–636. 10.1084/jem.193.5.631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta H., Chaudhary P. M. (2004). Activation of alternative NF-kappa B pathway by human herpes virus 8-encoded Fas-associated death domain-like IL-1 beta-converting enzyme inhibitory protein (vFLIP). Proc. Natl. Acad. Sci. U.S.A. 101 9399–9404. 10.1073/pnas.0308016101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthias P., Rolink A. G. (2005). Transcriptional networks in developing and mature B cells. Nat. Rev. Immunol. 5 497–508. 10.1038/nri1633 [DOI] [PubMed] [Google Scholar]
- May J. S., Walker J., Colaco S., Stevenson P. G. (2005). The murine gammaherpesvirus 68 ORF27 gene product contributes to intercellular viral spread. J. Virol. 79 5059–5068. 10.1128/JVI.79.8.5059-5068.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan K. B., Gangappa S., Speck S. H., Virgin H. W. T. (2006). Antibody-independent control of gamma-herpesvirus latency via B cell induction of anti-viral T cell responses. PLoS Pathog. 2:e58 10.1371/journal.ppat.0020058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedev A. E., Espevik T., Ranges G., Sundan A. (1996). Distinct roles of the two tumor necrosis factor (TNF) receptors in modulating TNF and lymphotoxin alpha effects. J. Biol. Chem. 271 9778–9784. 10.1074/jbc.271.16.9778 [DOI] [PubMed] [Google Scholar]
- Milho R., Frederico B., Efstathiou S., Stevenson P. G. (2012). A heparan-dependent herpesvirus targets the olfactory neuroepithelium for host entry. PLoS Pathog. 8:e1002986 10.1371/journal.ppat.1002986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milho R., Gill M. B., May J. S., Colaco S., Stevenson P. G. (2011). In vivo function of the murid herpesvirus-4 ribonucleotide reductase small subunit. J. Gen. Virol. 92 1550–1560. 10.1099/vir.0.031542-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milho R., Smith C. M., Marques S., Alenquer M., May J. S., Gillet L., et al. (2009). In vivo imaging of murid herpesvirus-4 infection. J. Gen. Virol. 90 21–32. 10.1099/vir.0.006569-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills D. M., Bonizzi G., Karin M., Rickert R. C. (2007). Regulation of late B cell differentiation by intrinsic IKKalpha-dependent signals. Proc. Natl. Acad. Sci. U.S.A. 104 6359–6364. 10.1073/pnas.0700296104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamitani T., Yasui T., Ma Y., Zhou H., Okuzaki D., Tsai C. Y., et al. (2015). Evasion of affinity-based selection in germinal centers by Epstein-Barr virus LMP2A. Proc. Natl. Acad. Sci. U.S.A. 112 11612–11617. 10.1073/pnas.1514484112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minkah N., Macaluso M., Oldenburg D. G., Paden C. R., White D. W., Mcbride K. M., et al. (2015). Absence of the uracil DNA glycosylase of murine gammaherpesvirus 68 impairs replication and delays the establishment of latency in vivo. J. Virol. 89 3366–3379. 10.1128/JVI.03111-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita E. M., Yang B., Babcock G. J., Thorley-Lawson D. A. (1997). Identification of the site of Epstein-Barr virus persistence in vivo as a resting B cell. J. Virol. 71 4882–4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita E. M., Yang B., Lam K. M., Crawford D. H., Thorley-Lawson D. A. (1995). A novel form of Epstein-Barr virus latency in normal B cells in vivo. Cell 80 593–601. 10.1016/0092-8674(95)90513-8 [DOI] [PubMed] [Google Scholar]
- Molloy M. J., Zhang W., Usherwood E. J. (2011). Suppressive CD8+ T cells arise in the absence of CD4 help and compromise control of persistent virus. J. Immunol. 186 6218–6226. 10.4049/jimmunol.1003812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorman N. J., Lin C. Y., Speck S. H. (2004). Identification of candidate gammaherpesvirus 68 genes required for virus replication by signature-tagged transposon mutagenesis. J. Virol. 78 10282–10290. 10.1128/JVI.78.19.10282-10290.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora A. L., Woods C. R., Garcia A., Xu J., Rojas M., Speck S. H., et al. (2005). Lung infection with gamma-herpesvirus induces progressive pulmonary fibrosis in Th2-biased mice. Am. J. Physiol. Lung. Cell Mol. Physiol. 289 L711–L721. 10.1152/ajplung.00007.2005 [DOI] [PubMed] [Google Scholar]
- Morrison T. E., Kenney S. C. (2004). BZLF1, an Epstein-Barr virus immediate-early protein, induces p65 nuclear translocation while inhibiting p65 transcriptional function. Virology 328 219–232. 10.1016/j.virol.2004.07.020 [DOI] [PubMed] [Google Scholar]
- Moser J. M., Farrell M. L., Krug L. T., Upton J. W., Speck S. H. (2006). A gammaherpesvirus 68 gene 50 null mutant establishes long-term latency in the lung but fails to vaccinate against a wild-type virus challenge. J. Virol. 80 1592–1598. 10.1128/JVI.80.3.1592-1598.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser J. M., Upton J. W., Gray K. S., Speck S. H. (2005). Ex vivo stimulation of B cells latently infected with gammaherpesvirus 68 triggers reactivation from latency. J. Virol. 79 5227–5231. 10.1128/JVI.79.8.5227-5231.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata T. (2014). Regulation of Epstein-Barr virus reactivation from latency. Microbiol. Immunol. 58 307–317. 10.1111/1348-0421.12155 [DOI] [PubMed] [Google Scholar]
- Nardelli B., Belvedere O., Roschke V., Moore P. A., Olsen H. S., Migone T. S., et al. (2001). Synthesis and release of B-lymphocyte stimulator from myeloid cells. Blood 97 198–204. 10.1182/blood.V97.1.198 [DOI] [PubMed] [Google Scholar]
- Nash A. A., Sunil-Chandra N. P. (1994). Interactions of the murine gammaherpesvirus with the immune system. Curr. Opin. Immunol. 6 560–563. 10.1016/0952-7915(94)90141-4 [DOI] [PubMed] [Google Scholar]
- Nealy M. S., Coleman C. B., Li H., Tibbetts S. A. (2010). Use of a virus-encoded enzymatic marker reveals that a stable fraction of memory B cells expresses latency-associated nuclear antigen throughout chronic gammaherpesvirus infection. J. Virol. 84 7523–7534. 10.1128/JVI.02572-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeckinghaus A., Ghosh S. (2009). The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 1:a000034 10.1101/cshperspect.a000034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Flaherty B. M., Soni T., Wakeman B. S., Speck S. H. (2014). The murine gammaherpesvirus immediate-early Rta synergizes with IRF4, targeting expression of the viral M1 superantigen to plasma cells. PLoS Pathog. 10:e1004302 10.1371/journal.ppat.1004302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otipoby K. L., Sasaki Y., Schmidt-Supprian M., Patke A., Gareus R., Pasparakis M., et al. (2008). BAFF activates Akt and Erk through BAFF-R in an IKK1-dependent manner in primary mouse B cells. Proc. Natl. Acad. Sci. U.S.A. 105 12435–12438. 10.1073/pnas.0805460105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S., Buck M. D., Desai C., Zhang X., Loginicheva E., Martinez J., et al. (2016). Autophagy genes enhance murine gammaherpesvirus 68 reactivation from latency by preventing virus-induced systemic inflammation. Cell Host Microbe 19 91–101. 10.1016/j.chom.2015.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker B. D., Bankier A., Satchwell S., Barrell B., Farrell P. J. (1990). Sequence and transcription of Raji Epstein-Barr virus DNA spanning the B95-8 deletion region. Virology 179 339–346. 10.1016/0042-6822(90)90302-8 [DOI] [PubMed] [Google Scholar]
- Pasparakis M., Luedde T., Schmidt-Supprian M. (2006). Dissection of the NF-kappaB signalling cascade in transgenic and knockout mice. Cell Death. Differ. 13 861–872. 10.1038/sj.cdd.4401870 [DOI] [PubMed] [Google Scholar]
- Pasparakis M., Schmidt-Supprian M., Rajewsky K. (2002). I B kinase signaling is essential for maintenance of mature B Cells. J. Exp. Med. 196 743–752. 10.1084/jem.20020907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patke A., Mecklenbrauker I., Erdjument-Bromage H., Tempst P., Tarakhovsky A. (2006). BAFF controls B cell metabolic fitness through a PKC beta- and Akt-dependent mechanism. J. Exp. Med. 203 2551–2562. 10.1084/jem.20060990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova I. V., Virgin H. W., Speck S. H. (2003). Disruption of gammaherpesvirus 68 Gene 50 demonstrates that Rta is essential for virus replication. J. Virol. 77 5731–5739. 10.1128/JVI.77.10.5731-5739.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins N. D., Schmid R. M., Duckett C. S., Leung K., Rice N. R., Nabel G. J. (1992). Distinct combinations of NF-kappa B subunits determine the specificity of transcriptional activation. Proc. Natl. Acad. Sci. U.S.A. 89 1529–1533. 10.1073/pnas.89.5.1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer S., Sewer A., Lagos-Quintana M., Sheridan R., Sander C., Grasser F. A., et al. (2005). Identification of microRNAs of the herpesvirus family. Nat. Methods 2 269–276. 10.1038/nmeth746 [DOI] [PubMed] [Google Scholar]
- Pfeffer S., Zavolan M., Grasser F. A., Chien M., Russo J. J., Ju J., et al. (2004). Identification of virus-encoded microRNAs. Science 304 734–736. 10.1126/science.1096781 [DOI] [PubMed] [Google Scholar]
- Prakash O., Tang Z. Y., Peng X., Coleman R., Gill J., Farr G., et al. (2002). Tumorigenesis and aberrant signaling in transgenic mice expressing the human herpesvirus-8 K1 gene. J. Natl. Cancer Inst. 94 926–935. 10.1093/jnci/94.12.926 [DOI] [PubMed] [Google Scholar]
- Ptaschinski C., Wilmore J., Fiore N., Rochford R. (2010). In vivo activation of toll-like receptor-9 induces an age-dependent abortive lytic cycle reactivation of murine gammaherpesvirus-68. Viral Immunol. 23 547–555. 10.1089/vim.2010.0055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullen S. S., Labadia M. E., Ingraham R. H., Mcwhirter S. M., Everdeen D. S., Alber T., et al. (1999). High-affinity interactions of tumor necrosis factor receptor-associated factors (TRAFs) and CD40 require TRAF trimerization and CD40 multimerization. Biochemistry 38 10168–10177. 10.1021/bi9909905 [DOI] [PubMed] [Google Scholar]
- Pullen S. S., Miller H. G., Everdeen D. S., Dang T. T., Crute J. J., Kehry M. R. (1998). CD40-tumor necrosis factor receptor-associated factor (TRAF) interactions: regulation of CD40 signaling through multiple TRAF binding sites and TRAF hetero-oligomerization. Biochemistry 37 11836–11845. 10.1021/bi981067q [DOI] [PubMed] [Google Scholar]
- Raab-Traub N. (2012). Novel mechanisms of EBV-induced oncogenesis. Curr. Opin. Virol. 2 453–458. 10.1016/j.coviro.2012.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragoczy T., Miller G. (2001). Autostimulation of the Epstein-Barr virus BRLF1 promoter is mediated through consensus Sp1 and Sp3 binding sites. J. Virol. 75 5240–5251. 10.1128/JVI.75.11.5240-5251.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman Z. S., Rao S. P., Kalled S. L., Manser T. (2003). Normal induction but attenuated progression of germinal center responses in BAFF and BAFF-R signaling-deficient mice. J. Exp. Med. 198 1157–1169. 10.1084/jem.20030495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajcani J., Blaskovic D., Svobodova J., Ciampor F., Huckova D., Stanekova D. (1985). Pathogenesis of acute and persistent murine herpesvirus infection in mice. Acta Virol. 29 51–60. [PubMed] [Google Scholar]
- Raslova H., Berebbi M., Rajcani J., Sarasin A., Matis J., Kudelova M. (2001). Susceptibility of mouse mammary glands to murine gammaherpesvirus 72 (MHV-72) infection: evidence of MHV-72 transmission via breast milk. Microb. Pathog. 31 47–58. 10.1006/mpat.2001.0441 [DOI] [PubMed] [Google Scholar]
- Rastelli J., Homig-Holzel C., Seagal J., Muller W., Hermann A. C., Rajewsky K., et al. (2008). LMP1 signaling can replace CD40 signaling in B cells in vivo and has unique features of inducing class-switch recombination to IgG1. Blood 111 1448–1455. 10.1182/blood-2007-10-117655 [DOI] [PubMed] [Google Scholar]
- Rasul A. E., Nagy N., Sohlberg E., Adori M., Claesson H. E., Klein G., et al. (2012). Simultaneous detection of the two main proliferation driving EBV encoded proteins, EBNA-2 and LMP-1 in single B cells. J. Immunol. Methods 385 60–70. 10.1016/j.jim.2012.08.008 [DOI] [PubMed] [Google Scholar]
- Rathinam V. A. K., Jiang Z. Z., Waggoner S. N., Sharma S., Cole L. E., Waggoner L., et al. (2010). The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 11 395–403. 10.1038/ni.1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razani B., Zarnegar B., Ytterberg A. J., Shiba T., Dempsey P. W., Ware C. F., et al. (2010). Negative feedback in noncanonical NF-kappaB signaling modulates NIK stability through IKKalpha-mediated phosphorylation. Sci. Signal. 3:ra41 10.1126/scisignal.2000778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese T. A., Wakeman B. S., Choi H. S., Hufford M. M., Huang S. C., Zhang X., et al. (2014). Coinfection. Helminth infection reactivates latent gamma-herpesvirus via cytokine competition at a viral promoter. Science 345 573–577. 10.1126/science.1254517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese T. A., Xia J., Johnson L. S., Zhou X., Zhang W., Virgin H. W. (2010). Identification of novel microRNA-like molecules generated from herpesvirus and host tRNA transcripts. J. Virol. 84 10344–10353. 10.1128/JVI.00707-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regnier C. H., Song H. Y., Gao X., Goeddel D. V., Cao Z., Rothe M. (1997). Identification and characterization of an IkappaB kinase. Cell 90 373–383. 10.1016/S0092-8674(00)80344-X [DOI] [PubMed] [Google Scholar]
- Rodrigues L., Filipe J., Seldon M. P., Fonseca L., Anrather J., Soares M. P., et al. (2009). Termination of NF-kappaB activity through a gammaherpesvirus protein that assembles an EC5S ubiquitin-ligase. EMBO J. 28 1283–1295. 10.1038/emboj.2009.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa G. T., Gillet L., Smith C. M., De Lima B. D., Stevenson P. G. (2007). IgG fc receptors provide an alternative infection route for murine gamma-herpesvirus-68. PLoS ONE 2:e560 10.1371/journal.pone.0000560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo J. J., Bohenzky R. A., Chien M. C., Chen J., Yan M., Maddalena D., et al. (1996). Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. U.S.A. 93 14862–14867. 10.1073/pnas.93.25.14862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarawar S. R., Lee B. J., Reiter S. K., Schoenberger S. P. (2001). Stimulation via CD40 can substitute for CD4 T cell function in preventing reactivation of a latent herpesvirus. Proc. Natl. Acad. Sci. U.S.A. 98 6325–6329. 10.1073/pnas.101136898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz M., Murphy P. M. (2001). Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor constitutively activates NF-κB and induces proinflammatory cytokine and chemokine production via a C-Terminal signaling determinant. J. Immunol. 167 505–513. 10.4049/jimmunol.167.1.505 [DOI] [PubMed] [Google Scholar]
- Senftleben U., Cao Y., Xiao G., Greten F. R., Krahn G., Bonizzi G., et al. (2001). Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science 293 1495–1499. 10.1126/science.1062677 [DOI] [PubMed] [Google Scholar]
- Shinohara H., Yasuda T., Aiba Y., Sanjo H., Hamadate M., Watarai H., et al. (2005). PKC beta regulates BCR-mediated IKK activation by facilitating the interaction between TAK1 and CARMA1. J. Exp. Med. 202 1423–1431. 10.1084/jem.20051591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel A. M., Herskowitz J. H., Speck S. H. (2008). The MHV68 M2 protein drives IL-10 dependent B cell proliferation and differentiation. PLoS Pathog. 4:e1000039 10.1371/journal.ppat.1000039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel A. M., Rangaswamy U. S., Napier R. J., Speck S. H. (2010). Blimp-1-dependent plasma cell differentiation is required for efficient maintenance of murine gammaherpesvirus latency and antiviral antibody responses. J. Virol. 84 674–685. 10.1128/JVI.01306-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard E. P., Engels E. A. (2010). Cancer as a cause of death among people with AIDS in the United States. Clin. Infect. Dis. 51 957–962. 10.1086/656416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard E. P., Pfeiffer R. M., Engels E. A. (2011). Cumulative incidence of cancer among individuals with acquired immunodeficiency syndrome in the United States. Cancer 117 1089–1096. 10.1002/cncr.25547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simas J. P., Efstathiou S. (1998). Murine gammaherpesvirus 68: a model for the study of gammaherpesvirus pathogenesis. Trends Microbiol. 6 276–282. 10.1016/S0966-842X(98)01306-7 [DOI] [PubMed] [Google Scholar]
- Singh V. V., Kerur N., Bottero V., Dutta S., Chakraborty S., Ansari M. A., et al. (2013). Kaposi’s sarcoma-associated herpesvirus latency in endothelial and B cells activates gamma interferon-inducible protein 16-mediated inflammasomes. J. Virol. 87 4417–4431. 10.1128/JVI.03282-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S., Colbert C. L., Becker N., Wei Y., Levine B. (2008). Molecular basis of the regulation of Beclin 1-dependent autophagy by the gamma-herpesvirus 68 Bcl-2 homolog M11. Autophagy 4 989–997. 10.4161/auto.6803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sixbey J. W., Nedrud J. G., Raab-Traub N., Hanes R. A., Pagano J. S. (1984). Epstein-Barr virus replication in oropharyngeal epithelial cells. N. Engl. J. Med. 310 1225–1230. 10.1056/NEJM198405103101905 [DOI] [PubMed] [Google Scholar]
- Sixbey J. W., Vesterinen E. H., Nedrud J. G., Raab-Traub N., Walton L. A., Pagano J. S. (1983). Replication of Epstein-Barr virus in human epithelial cells infected in vitro. Nature 306 480–483. 10.1038/306480a0 [DOI] [PubMed] [Google Scholar]
- Solan N. J., Miyoshi H., Carmona E. M., Bren G. D., Paya C. V. (2002). RelB cellular regulation and transcriptional activity are regulated by p100. J. Biol. Chem. 277 1405–1418. 10.1074/jbc.M109619200 [DOI] [PubMed] [Google Scholar]
- Song M. J., Hwang S., Wong W., Round J., Martinez-Guzman D., Turpaz Y., et al. (2004). The DNA architectural protein HMGB1 facilitates RTA-mediated viral gene expression in gamma-2 herpesviruses. J. Virol. 78 12940–12950. 10.1128/JVI.78.23.12940-12950.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M. J., Hwang S., Wong W. H., Wu T. T., Lee S., Liao H. I., et al. (2005). Identification of viral genes essential for replication of murine gamma-herpesvirus 68 using signature-tagged mutagenesis. Proc. Natl. Acad. Sci. U.S.A. 102 3805–3810. 10.1073/pnas.0404521102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soni V., Cahir-Mcfarland E., Kieff E. (2007). LMP1 TRAFficking activates growth and survival pathways. Adv. Exp. Med. Biol. 597 173–187. 10.1007/978-0-387-70630-6_14 [DOI] [PubMed] [Google Scholar]
- Speck S. H., Ganem D. (2010). Viral latency and its regulation: lessons from the gamma-herpesviruses. Cell Host Microbe 8 100–115. 10.1016/j.chom.2010.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbruck L., Gustems M., Medele S., Schulz T. F., Lutter D., Hammerschmidt W. (2015). K1 and K15 of kaposi’s sarcoma-associated herpesvirus are partial functional homologues of latent membrane Protein 2A of Epstein-Barr Virus. J. Virol. 89 7248–7261. 10.1128/JVI.00839-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiniger B., Barth P., Herbst B., Hartnell A., Crocker P. R. (1997). The species-specific structure of microanatomical compartments in the human spleen: strongly sialoadhesin-positive macrophages occur in the perifollicular zone, but not in the marginal zone. Immunology 92 307–316. 10.1046/j.1365-2567.1997.00328.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson P. G., Belz G. T., Altman J. D., Doherty P. C. (1999a). Changing patterns of dominance in the CD8+ T cell response during acute and persistent murine gamma-herpesvirus infection. Eur. J. Immunol. 29 1059–1067. 10.1002/(SICI)1521-4141(199904)29:04<1059::AID-IMMU1059>3.0.CO;2-L [DOI] [PubMed] [Google Scholar]
- Stevenson P. G., Cardin R. D., Christensen J. P., Doherty P. C. (1999b). Immunological control of a murine gammaherpesvirus independent of CD8+ T cells. J. Gen. Virol. 80(Pt 2), 477–483. 10.1099/0022-1317-80-2-477 [DOI] [PubMed] [Google Scholar]
- Stevenson P. G., Doherty P. C. (1998). Kinetic analysis of the specific host response to a murine gammaherpesvirus. J. Virol. 72 943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiglincova V., Chalupkova A., Hrabovska Z., Cipkova J., Wagnerova M., Mistrikova J. (2011). Vertical transmission of murine gammaherpesvirus 68 in mice. Acta Virol. 55 55–59. 10.4149/av_2011_01_55 [DOI] [PubMed] [Google Scholar]
- Suarez A. L., van Dyk L. F. (2008). Endothelial cells support persistent gammaherpesvirus 68 infection. PLoS Pathog. 4:e1000152 10.1371/journal.ppat.1000152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C., Schattgen S. A., Pisitkun P., Jorgensen J. P., Hilterbrand A. T., Wang L. J., et al. (2015). Evasion of innate cytosolic DNA sensing by a gammaherpesvirus facilitates establishment of latent infection. J. Immunol. 194 1819–1831. 10.4049/jimmunol.1402495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C. C., Thorley-Lawson D. A. (2007). Plasma cell-specific transcription factor XBP-1s binds to and transactivates the Epstein-Barr virus BZLF1 promoter. J. Virol. 81 13566–13577. 10.1128/JVI.01055-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun R., Lin S. F., Gradoville L., Yuan Y., Zhu F., Miller G. (1998). A viral gene that activates lytic cycle expression of Kaposi’s sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. U.S.A. 95 10866–10871. 10.1073/pnas.95.18.10866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S. C. (2012). The noncanonical NF-kappaB pathway. Immunol. Rev. 246 125–140. 10.1111/j.1600-065X.2011.01088.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunil-Chandra N. P., Arno J., Fazakerley J., Nash A. A. (1994). Lymphoproliferative disease in mice infected with murine gammaherpesvirus 68. Am. J. Pathol. 145 818–826. [PMC free article] [PubMed] [Google Scholar]
- Sunil-Chandra N. P., Efstathiou S., Arno J., Nash A. A. (1992a). Virological and pathological features of mice infected with murine gamma-herpesvirus 68. J. Gen. Virol. 73(Pt 9), 2347–2356. 10.1099/0022-1317-73-9-2347 [DOI] [PubMed] [Google Scholar]
- Sunil-Chandra N. P., Efstathiou S., Nash A. A. (1992b). Murine gammaherpesvirus 68 establishes a latent infection in mouse B lymphocytes in vivo. J. Gen. Virol. 73(Pt 12), 3275–3279. 10.1099/0022-1317-73-12-3275 [DOI] [PubMed] [Google Scholar]
- Tan C. S., Frederico B., Stevenson P. G. (2014). Herpesvirus delivery to the murine respiratory tract. J. Virol. Methods 206 105–114. 10.1016/j.jviromet.2014.06.003 [DOI] [PubMed] [Google Scholar]
- Tarakanova V. L., Kreisel F., White D. W., Virgin H. W. T. (2008). Murine gammaherpesvirus 68 genes both induce and suppress lymphoproliferative disease. J. Virol. 82 1034–1039. 10.1128/JVI.01426-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarakanova V. L., Suarez F., Tibbetts S. A., Jacoby M. A., Weck K. E., Hess J. L., et al. (2005). Murine gammaherpesvirus 68 infection is associated with lymphoproliferative disease and lymphoma in BALB beta2 microglobulin-deficient mice. J. Virol. 79 14668–14679. 10.1128/JVI.79.23.14668-14679.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telfer S., Bennett M., Carslake D., Helyar S., Begon M. (2007). The dynamics of murid gammaherpesvirus 4 within wild, sympatric populations of bank voles and wood mice. J. Wildl. Dis. 43 32–39. 10.7589/0090-3558-43.1.32 [DOI] [PubMed] [Google Scholar]
- Thompson M. P., Kurzrock R. (2004). Epstein-Barr virus and cancer. Clin. Cancer Res. 10 803–821. 10.1158/1078-0432.CCR-0670-3 [DOI] [PubMed] [Google Scholar]
- Tibbetts S. A., Mcclellan J. S., Gangappa S., Speck S. H., Virgin H. W. T. (2003). Effective vaccination against long-term gammaherpesvirus latency. J. Virol. 77 2522–2529. 10.1128/JVI.77.7.4469.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibbetts S. A., Suarez F., Steed A. L., Simmons J. A., Virgin H. W. T. (2006). A gamma-herpesvirus deficient in replication establishes chronic infection in vivo and is impervious to restriction by adaptive immune cells. Virology 353 210–219. 10.1016/j.virol.2006.05.020 [DOI] [PubMed] [Google Scholar]
- Tibbetts S. A., Van Dyk L. F., Speck S. H., Virgin H. W. T. (2002). Immune control of the number and reactivation phenotype of cells latently infected with a gammaherpesvirus. J. Virol. 76 7125–7132. 10.1128/JVI.76.14.7125-7132.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischer B. K., Smith G. A., Osterrieder N. (2010). En passant mutagenesis: a two step markerless red recombination system. Methods Mol. Biol. 634 421–430. 10.1007/978-1-60761-652-8_30 [DOI] [PubMed] [Google Scholar]
- Tripp R. A., Hamilton-Easton A. M., Cardin R. D., Nguyen P., Behm F. G., Woodland D. L., et al. (1997). Pathogenesis of an infectious mononucleosis-like disease induced by a murine gamma-herpesvirus: role for a viral superantigen? J. Exp. Med. 185 1641–1650. 10.1084/jem.185.9.1641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai K., Messick T. E., Lieberman P. M. (2015). Disruption of host antiviral resistances by gammaherpesvirus tegument proteins with homology to the FGARAT purine biosynthesis enzyme. Curr. Opin. Virol. 14 30–40. 10.1016/j.coviro.2015.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai W. H., Wang P. W., Lin S. Y., Wu I. L., Ko Y. C., Chen Y. L., et al. (2012). Ser-634 and Ser-636 of Kaposi’s sarcoma-associated herpesvirus RTA are involved in transactivation and are potential Cdk9 phosphorylation sites. Front. Microbiol. 3:60 10.3389/fmicb.2012.00060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse E., Kwong Y. L. (2015). Epstein Barr virus-associated lymphoproliferative diseases: the virus as a therapeutic target. Exp. Mol. Med. 47 e136 10.1038/emm.2014.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida J., Yasui T., Takaoka-Shichijo Y., Muraoka M., Kulwichit W., Raab-Traub N., et al. (1999). Mimicry of CD40 signals by Epstein-Barr virus LMP1 in B lymphocyte responses. Science 286 300–303. 10.1126/science.286.5438.300 [DOI] [PubMed] [Google Scholar]
- Upton J. W., Speck S. H. (2006). Evidence for CDK-dependent and CDK-independent functions of the murine gammaherpesvirus 68 v-cyclin. J. Virol. 80 11946–11959. 10.1128/JVI.01722-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban M. B., Baeuerle P. A. (1991). The role of the p50 and p65 subunits of NF-kappa B in the recognition of cognate sequences. New Biol. 3 279–288. [PubMed] [Google Scholar]
- Usherwood E. J., Ross A. J., Allen D. J., Nash A. A. (1996a). Murine gammaherpesvirus-induced splenomegaly: a critical role for CD4 T cells. J. Gen. Virol. 77(Pt 4), 627–630. 10.1099/0022-1317-77-4-627 [DOI] [PubMed] [Google Scholar]
- Usherwood E. J., Stewart J. P., Nash A. A. (1996b). Characterization of tumor cell lines derived from murine gammaherpesvirus-68-infected mice. J. Virol. 70 6516–6518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usherwood E. J., Stewart J. P., Robertson K., Allen D. J., Nash A. A. (1996c). Absence of splenic latency in murine gammaherpesvirus 68-infected B cell-deficient mice. J. Gen. Virol. 77(Pt 11), 2819–2825. 10.1099/0022-1317-77-11-2819 [DOI] [PubMed] [Google Scholar]
- Vallabhapurapu S., Karin M. (2009). Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 27 693–733. 10.1146/annurev.immunol.021908.132641 [DOI] [PubMed] [Google Scholar]
- van Dyk L. F., Hess J. L., Katz J. D., Jacoby M., Speck S. H., Virgin H. I. (1999). The murine gammaherpesvirus 68 v-cyclin gene is an oncogene that promotes cell cycle progression in primary lymphocytes. J. Virol. 73 5110–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dyk L. F., Virgin H. W., Speck S. H. (2003). Maintenance of gammaherpesvirus latency requires viral cyclin in the absence of B lymphocytes. J. Virol. 77 5118–5126. 10.1128/JVI.77.9.5118-5126.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varfolomeev E., Blankenship J. W., Wayson S. M., Fedorova A. V., Kayagaki N., Garg P., et al. (2007). IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 131 669–681. 10.1016/j.cell.2007.10.030 [DOI] [PubMed] [Google Scholar]
- Verma S. C., Robertson E. S. (2003). Molecular biology and pathogenesis of Kaposi sarcoma-associated herpesvirus. FEMS Microbiol. Lett. 222 155–163. 10.1016/S0378-1097(03)00261-1 [DOI] [PubMed] [Google Scholar]
- Verzijl D., Fitzsimons C. P., Van Dijk M., Stewart J. P., Timmerman H., Smit M. J., et al. (2004). Differential activation of murine herpesvirus 68- and Kaposi’s sarcoma-associated herpesvirus-encoded ORF74 G protein-coupled receptors by human and murine chemokines. J. Virol. 78 3343–3351. 10.1128/JVI.78.7.3343-3351.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgin H. W. T., Latreille P., Wamsley P., Hallsworth K., Weck K. E., Dal Canto A. J., et al. (1997). Complete sequence and genomic analysis of murine gammaherpesvirus 68. J. Virol. 71 5894–5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgin H. W. T., Presti R. M., Li X. Y., Liu C., Speck S. H. (1999). Three distinct regions of the murine gammaherpesvirus 68 genome are transcriptionally active in latently infected mice. J. Virol. 73 2321–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vora K. A., Wang L. C., Rao S. P., Liu Z. Y., Majeau G. R., Cutler A. H., et al. (2003). Cutting edge: germinal centers formed in the absence of B cell-activating factor belonging to the TNF family exhibit impaired maturation and function. J. Immunol. 171 547–551. 10.4049/jimmunol.171.2.547 [DOI] [PubMed] [Google Scholar]
- Vrazo A. C., Chauchard M., Raab-Traub N., Longnecker R. (2012). Epstein-Barr virus LMP2A reduces hyperactivation induced by LMP1 to restore normal B cell phenotype in transgenic mice. PLoS Pathog. 8:e1002662 10.1371/journal.ppat.1002662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeling M. N., Roy D. J., Nash A. A., Stewart J. P. (2001). Characterization of the murine gammaherpesvirus 68 ORF74 product: a novel oncogenic G protein-coupled receptor. J. Gen. Virol. 82 1187–1197. 10.1099/0022-1317-82-5-1187 [DOI] [PubMed] [Google Scholar]
- Wang C., Deng L., Hong M., Akkaraju G. R., Inoue J., Chen Z. J. J. (2001). TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412 346–351. 10.1038/35084276 [DOI] [PubMed] [Google Scholar]
- Wang L., Brinkmann M. M., Pietrek M., Ottinger M., Dittrich-Breiholz O., Kracht M., et al. (2007). Functional characterization of the M-type K15-encoded membrane protein of Kaposi’s sarcoma-associated herpesvirus. J. Gen. Virol. 88 1698–1707. 10.1099/vir.0.82807-0 [DOI] [PubMed] [Google Scholar]
- Weck K. E., Barkon M. L., Yoo L. I., Speck S. H., Virgin H. I. (1996). Mature B cells are required for acute splenic infection, but not for establishment of latency, by murine gammaherpesvirus 68. J. Virol. 70 6775–6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weck K. E., Kim S. S., Virgin H. I., Speck S. H. (1999a). B cells regulate murine gammaherpesvirus 68 latency. J. Virol. 73 4651–4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weck K. E., Kim S. S., Virgin H. I., Speck S. H. (1999b). Macrophages are the major reservoir of latent murine gammaherpesvirus 68 in peritoneal cells. J. Virol. 73 3273–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willer D. O., Speck S. H. (2003). Long-term latent murine Gammaherpesvirus 68 infection is preferentially found within the surface immunoglobulin D-negative subset of splenic B cells in vivo. J. Virol. 77 8310–8321. 10.1128/JVI.77.15.8310-8321.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willer D. O., Speck S. H. (2005). Establishment and maintenance of long-term murine gammaherpesvirus 68 latency in B cells in the absence of CD40. J. Virol. 79 2891–2899. 10.1128/JVI.79.5.2891-2899.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams L. M., Niemeyer B. F., Franklin D. S., Clambey E. T., Van Dyk L. F. (2015). A conserved gammaherpesvirus cyclin specifically bypasses host p18(INK4c) to promote reactivation from latency. J. Virol. 89 10821–10831. 10.1128/JVI.00891-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson S. J., Tsao E. H., Webb B. L., Ye H., Dalton-Griffin L., Tsantoulas C., et al. (2007). X box binding protein XBP-1s transactivates the Kaposi’s sarcoma-associated herpesvirus (KSHV) ORF50 promoter, linking plasma cell differentiation to KSHV reactivation from latency. J. Virol. 81 13578–13586. 10.1128/JVI.01663-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodland R. T., Fox C. J., Schmidt M. R., Hammerman P. S., Opferman J. T., Korsmeyer S. J., et al. (2008). Multiple signaling pathways promote B lymphocyte stimulator dependent B-cell growth and survival. Blood 111 750–760. 10.1182/blood-2007-03-077222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T. T., Tong L., Rickabaugh T., Speck S., Sun R. (2001). Function of Rta is essential for lytic replication of murine gammaherpesvirus 68. J. Virol. 75 9262–9273. 10.1128/JVI.75.19.9262-9273.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T. T., Usherwood E. J., Stewart J. P., Nash A. A., Sun R. (2000). Rta of murine gammaherpesvirus 68 reactivates the complete lytic cycle from latency. J. Virol. 74 3659–3667. 10.1128/JVI.74.8.3659-3667.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao G., Harhaj E. W., Sun S. C. (2001). NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol. Cell 7 401–409. 10.1016/S1097-2765(01)00187-3 [DOI] [PubMed] [Google Scholar]
- Xiaofei X., Hwang S., Oh S., Lee J. S., Jeong J. H., Gwack Y., et al. (2009). Viral Bcl-2-mediated evasion of autophagy aids chronic infection of gammaherpesvirus 68. PLoS Pathog. 5:e1000609 10.1371/journal.ppat.1000609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y., Aucoin D. P., Huete A. R., Cei S. A., Hanson L. J., Pari G. S. (2005). A Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 ORF50 deletion mutant is defective for reactivation of latent virus and DNA replication. J. Virol. 79 3479–3487. 10.1128/JVI.79.6.3479-3487.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Wood C. (2007). The transcriptional repressor K-RBP modulates RTA-mediated transactivation and lytic replication of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 81 6294–6306. 10.1128/JVI.02648-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Yan Z., Wood C. (2008). Kaposi’s sarcoma-associated herpesvirus transactivator RTA promotes degradation of the repressors to regulate viral lytic replication. J. Virol. 82 3590–3603. 10.1128/JVI.02229-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates J., Guan N. (1991). Epstein-Barr virus-derived plasmids replicate only once per cell cycle and are not amplified after entry into cells. J. Virol. 65 483–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F., Feng J., Harada J. N., Chanda S. K., Kenney S. C., Sun R. (2007). B cell terminal differentiation factor XBP-1 induces reactivation of Kaposi’s sarcoma-associated herpesvirus. FEBS Lett. 581 3485–3488. 10.1016/j.febslet.2007.06.056 [DOI] [PubMed] [Google Scholar]
- Zhao B., Barrera L. A., Ersing I., Willox B., Schmidt S. C., Greenfeld H., et al. (2014). The NF-kappaB genomic landscape in lymphoblastoid B cells. Cell Rep. 8 1595–1606. 10.1016/j.celrep.2014.07.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H., French J. B., Fang Y., Benkovic S. J. (2013). The purinosome, a multi-protein complex involved in the de novo biosynthesis of purines in humans. Chem. Commun. (Camb) 49 4444–4452. 10.1039/c3cc41437j [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J. Y., Strehle M., Frohn A., Kremmer E., Hofig K. P., Meister G., et al. (2010). Identification and analysis of expression of novel microRNAs of murine gammaherpesvirus 68. J. Virol. 84 10266–10275. 10.1128/JVI.01119-10 [DOI] [PMC free article] [PubMed] [Google Scholar]