

Summary
The malaria parasite Plasmodium falciparum dramatically remodels its host red blood cell to enhance its own survival, using a secretory membrane system that it establishes outside its own cell. Cisternal organelles, called Maurer’s clefts, act as a staging point for the forward trafficking of virulence proteins to the red blood cell (RBC) membrane. The Ring-EXported Protein-1 (REX1) is a Maurer’s cleft resident protein. We show that inducible knockdown of REX1 causes stacking of Maurer’s cleft cisternae without disrupting the organization of the knob-associated histidine-rich protein at the RBC membrane. Genetic dissection of the REX1 sequence shows that loss of a repeat sequence domain results in the formation of giant Maurer’s cleft stacks. The stacked Maurer’s clefts are decorated with tether-like structures and retain the ability to dock onto the RBC membrane skeleton. The REX1 mutant parasites show deficient export of the major virulence protein, PfEMP1, to the red blood cell surface and markedly reduced binding to the endothelial cell receptor, CD36. REX1 is predicted to form a largely α-helical structure, with a repetitive charge pattern in the repeat sequence domain, providing potential insights into the role of REX1 in Maurer’s cleft sculpting.
Keywords: Trafficking, Virulence protein, Maurer’s clefts REX1, PfEMP1, KAHRP
Introduction
Despite enormous efforts, malaria remains a disease of global significance. Annually about 600,000 deaths are attributed to infection with Plasmodium falciparum (WHO, 2014, Murray et al., 2012). The particular virulence of P. falciparum is due, in part, to the cytoadherence of infected red blood cells (RBCs) to the walls of post-venule capillaries. Mediated by a protein called P. falciparum erythrocyte membrane protein-1 (PfEMP1) (Scherf et al., 2008, Baruch et al., 1995), cytoadherence enables P. falciparum to avoid splenic clearance, thus permitting a more rapid multiplication rate. An exacerbated inflammatory response to the sequestered parasites is involved in precipitating severe complications, such as coma and under-weight births (White et al., 2013, Beeson et al., 2001, Turner et al., 2013).
Because mature human RBCs lack the machinery for protein synthesis and trafficking, export of Plasmodium virulence proteins requires the assembly of a parasite-derived exomembrane system in the host RBC cytoplasm. For example, PfEMP1, an integral membrane protein, is secreted through the parasite’s endomembrane system, and exported across the parasitophorous vacuole membrane (PVM) in which the parasite resides (Maier et al., 2009). It is trafficked to compartments known as the Maurer’s clefts, and then transferred to the RBC membrane, where it is assembled into knobs (Voigt et al., 2000). Despite its importance, much remains unclear about the trafficking, sorting and fusion machinery that mediates delivery of PfEMP1 to the RBC surface.
The term Maurer’s cleft is, in fact, a misnomer as these parasite-derived structures are flattened cisternae, with no direct opening onto the RBC surface. In the 3D7 strain, they number ~15 per cell, and are roughly disc-shaped, with a diameter of ~500 nm and a thickness of ~40 nm (Hanssen et al., 2008b). They are connected to the RBC membrane via direct cytoskeletal interactions (Cyrklaff et al., 2011) and via distinct tether-like structures (tubes of about ~25 nm by ~200 nm) (Pachlatko et al., 2010, Hanssen et al., 2010). The Maurer’s clefts are thought to function as a sorting compartment; some exported proteins transiently associate with the Maurer’s clefts en route to the RBC cytoskeleton or membrane, while some remain as resident proteins. Resident proteins play important roles in the PfEMP1 trafficking process; and proteins such as the ring exported protein-1 (REX1) (Dixon et al., 2011), the skeleton-binding protein-1 (SBP1) (Maier et al., 2007, Cooke et al., 2006), the membrane-associated histidine-rich protein-1 (MAHRP1) (Spycher et al., 2008), PfEMP1 trafficking protein 1 (PfPTP1) (Rug et al., 2014) and P. falciparum antigen 332 (Glenister et al., 2009) have been shown to be essential for efficient delivery of PfEMP1 to the RBC surface.
REX1 is a peripheral membrane protein that is exported to the RBC cytoplasm in the very early ring stage of infection (~2 h p.i.) (McMillan et al., 2013), where it associates with a population of already formed Maurer’s clefts (Gruring et al., 2012). This 712 amino acid protein possesses a recessed hydrophobic signal sequence (residues 37 – 58), a predicted coiled-coil region (residues 181 – 361), a highly charged repetitive sequence region (residues 362–579) and a C-terminal region (residues 580–712) (Fig. 1). It has no predicted Plasmodium Export Element (PEXEL) and is thus considered to be a PEXEL-Negative Exported Protein (PNEP) (Gruring et al., 2012, Spielmann & Gilberger, 2010).
Fig. 1. Schematics of REX1 and REX1 mutants.
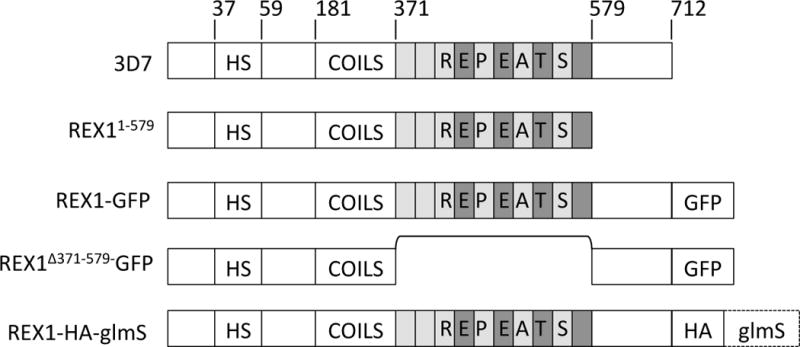
Full length 3D7 REX1 features a recessed hydrophobic signal sequence (HS), a predicted coiled-coil region and a repeat region. In REX11–579 transfectants the C-terminal region is deleted. REX1-GFP and REX1(Δ371–579)-GFP transfectants express REX1 with a C-terminal GFP tag, with and without the repeat region. REX1_KD transfectants have a 3xHA tag and glmS sequence integration into the 3′ untranslated region.
Previously we have shown that attachment of REX1 to the Maurer’s clefts is mediated by a sequence within the predicted coiled-coil domain (Dixon et al., 2008). Interestingly, targeted truncation of REX1 leads to an altered Maurer’s cleft architecture and a significant decrease in PfEMP1 surface exposure (Dixon et al., 2011, Hanssen et al., 2008a). However, complete genetic disruption of REX1 was found to be associated with concomitant deletion of the sub-telomeric region of chromosome 2, with resultant loss of the genes for a number of exported proteins, including the knob-associated histidine-rich protein (KAHRP). KAHRP forms the main structural component of the knobs at the infected RBC surface in which PfEMP1 is concentrated (Horrocks et al., 2005) and promotes PfEMP1 presentation (Crabb et al., 1997). This makes it difficult to distinguish the direct effect of REX1 on PfEMP1 trafficking from an indirect effect due to the loss of knobs and other deleted chromosome 2 genes.
In this work, we have used the inducible ribozyme system (Prommana et al., 2013) to genetically attenuate the expression of REX1 and show that Maurer’s cleft architecture changes can be achieved, independent of loss of the kahrp locus. We have mapped the key domain within REX1 that controls cleft architecture and forward trafficking of PfEMP1. That is, we show that removal of the repeat sequence region (residues 362–579) results in the formation of giant stacked Maurer’s clefts. This is associated with significant loss of PfEMP1 surface delivery and decreased adhesion of infected RBCs. Using sophisticated prediction methods, we gain some insights into the physical nature of REX1 and its role in Maurer’s clefts sculpting.
Results
Knockdown of REX1 expression is associated with Maurer’s cleft reorganization without loss of KAHRP
Genes with important functions in the asexual blood stage are difficult to study using conventional gene disruption strategies due to the haploid nature of the P. falciparum genome. Recently, methods have been introduced to target genes by incorporating a glucosamine-6-phosphate−activated ribozyme (glmS) into their 3′ untranslated region (Fig. S1) (Prommana et al., 2013). In the presence of the cofactor glucosamine-6-phosphate, the glmS ribozyme degrades the mRNA encoding the targeted gene thereby reducing its expression.
The glucosamine (GlcN)-inducible glmS ribozyme and a 3 × HA tag (Fig. 1) were incorporated into the 3′ untranslated region of the REX1 gene by homologous recombination. PCR and DNA sequencing confirmed correct integration of the glmS plasmid. Treatment of ring stage REX1-KD parasites with 2.5 mM GlcN (which is converted to glucosamine-6-phosphate) for 20 h, resulted in efficient (>90%) REX1 knockdown, as quantitated by loss of the HA-tagged REX1 (Fig. 2A). By contrast, the level of a control protein, PfGAPDH, remained unchanged. This indicates that the decline in the REX1 level was not caused by defective or aborted development of parasites. Indeed parasite growth was not affected by the significantly decreased expression of REX1; when monitored over a period of 48 h in the presence of 2.5 mM GlcN, no growth defect was observed in either 3D7 or REX1-KD parasites compared to untreated parasites. Parasitemia levels were 106% of the controls, in both cases (Fig. S2).
Fig. 2. Inducible knockdown of REX1 using the glmS ribozyme system decreases the number of Maurer’s cleft puncta.
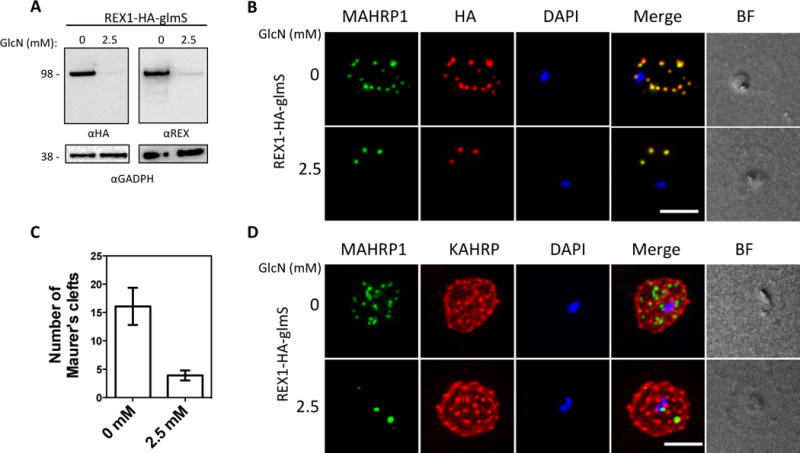
A. Western blots confirming knockdown of expression of REX1 in REX1_KD transfectants probed with anti-HA and anti-REX1. B,D. Immunofluorescence microscopy of paraformaldehyde-fixed infected RBCs probed with Maurer’s cleft marker anti-MAHRP1 (green) and anti-HA or anti-KAHRP (red). Nuclei are stained with DAPI (blue). Scale bar = 3 μm. C. Quantitation of numbers of Maurer’s clefts produced by REX1_KD transfectants in the presence and absence of 2.5 mM GlcN. The mean number of Maurer’s clefts produced per singly nucleated infected RBC was determined by counting SBP1-labelled puncta in at least 10 cells. Error bars = S.D.
Maurer’s clefts can be observed by immunofluorescence microscopy following labelling with antibodies recognizing resident proteins such as MAHRP1 and SBP1 (Fig. 2B, D; S3D). In the absence of GlcN, we observed 16 ± 3 REX1-HA and SBP1-positive puncta in the REX1-KD parasites (Fig. 2C), which is equivalent to the number found in wildtype 3D7 (see Fig. 3C). These puncta correspond to individual Maurer’s cleft cisternae that have a distributed organization within the RBC cytoplasm (Hanssen et al., 2008a).
Fig. 3. Deletion of the repeat region of REX1 decreases the number of Maurer’s cleft puncta.
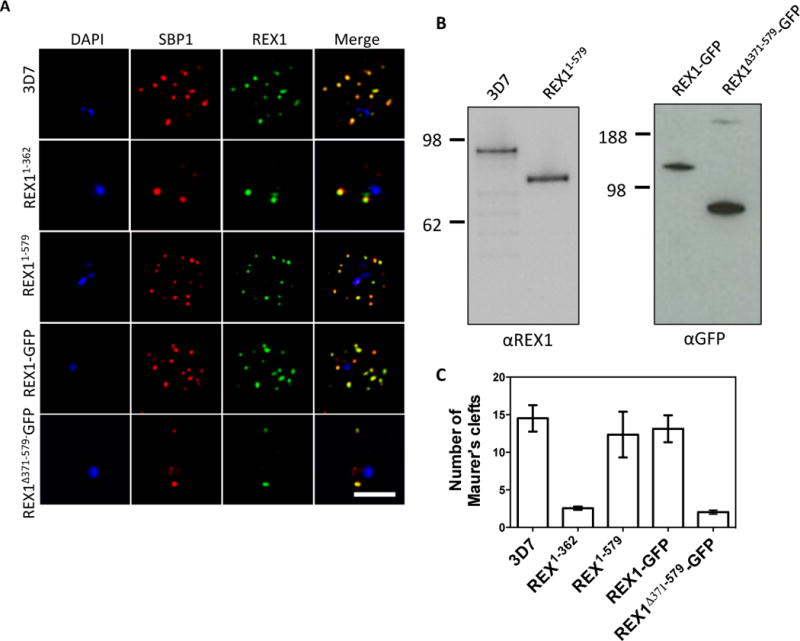
A. Immunofluorescence microscopy of acetone-fixed infected RBCs probed with anti-SBP1 (red) and anti-REX1 (green). Nuclei are stained with DAPI (blue). Scale bar = 3 μm. B. Western blots confirming expression of REX1 in wildtype 3D7 and truncated REX11–579 (probed with anti-REX1), and REX1-GFP and REX1(Δ371–579)-GFP chimeras (probed with anti-GFP). C. Quantitation of numbers of Maurer’s clefts produced by 3D7 and different REX1 transfectants in singly nucleated infected RBCs. Error bars = SD.
Upon exposure to 2.5 mM GlcN, the REX1-KD parasites exhibited a decreased number (4 ± 1) of SBP1-positive puncta (Fig. 2C; S3D) indicating reorganization of Maurer’s cleft architecture. Treatment of 3D7 parasites with 2.5 mM GlcN did not affect the number of Maurer’s clefts puncta (data not shown). Deletion of the rex1 gene has been reported to be associated with concomitant disruption of the chromosome 2 locus, which contains genes encoding KAHRP and other proteins such as P. falciparum Erythrocyte Membrane Proteins-3 (PfEMP3) (Dixon et al., 2011). Interestingly the GlcN-treated REX1-KD parasites retained expression of KAHRP (Fig. 2D) and PfEMP3 (Fig. S3C), indicating that Maurer’s cleft restructuring can occur even in the presence of an intact chromosome 2 locus, and that reduction of REX1 levels does not affect the trafficking of these proteins to the RBC periphery.
The repeat region of REX1 is involved in Maurer’s cleft sculpting
In an effort to identify the region within REX1 that is responsible for maintaining normal Maurer’s cleft architecture, we targeted the locus to modify the REX1 sequence. We found that removal of the C-terminal half of the REX1 protein, using an integration strategy (REX11–362) (see (Dixon et al., 2011); Fig S1) is associated with a dramatic decrease in the number (3 ± 0.2) of SBP1-positive puncta (Fig. 3A,C), similar to that observed with REX1 knock-down parasites. We next generated parasites in which REX1 is truncated at residue 579 (REX11–579; Fig. 1) and used Western analysis to show that a protein of approximately 70 kDa was expressed (Fig. 3B, left panel). These parasites exhibit 12 ± 3 SBP1-labelled puncta (Fig. 3A,C), a Maurer’s cleft profile that more closely resembles wildtype 3D7 (p = 0.54, unpaired t-test). This indicates that the region between amino acids 363 and 579 carries the major determinant that controls Maurer’s cleft architecture.
To further investigate this we generated a REX1 construct in which the repeat sequence domain (371 – 579) was deleted (REX1Δ371–579-GFP; Fig. 1). For this transfectant, we used a GFP-tagged REX1 construct, to facilitate downstream characterization, and compared the mutants with transfectants expressing full-length REX1-GFP and with wild type 3D7. Western analysis confirms that full-length REX1-GFP migrates with an apparent molecular mass of 120 kDa as expected for the chimera, while the REX1Δ371–579-GFP migrates with the expected molecular mass of 87 kDa (Fig. 3B, right panel).
Immunofluorescence microscopy reveals that full-length REX1-GFP is correctly localized at the Maurer’s clefts (Fig. 3A) and that these transfectants exhibit an average number of puncta equivalent to that of the 3D7 parent (13 ± 2; p = 0.59, unpaired t-test; Fig. 3C). By contrast, the REX1Δ371–579-GFP parasites exhibited a striking Maurer’s cleft phenotype, with only 2 ± 0.3 punctate structures observed in the infected RBC cytoplasm (Fig. 3A, C). This is much less than in wildtype 3D7 and the REX1-GFP line (unpaired t-test, p < 0.0001) and even less than the number observed in the REX1 knockdown parasites. These structures were still labelled with SBP1, but appeared larger than the puncta in wildtype parasites.
We performed immunofluorescence microscopy to determine if deletion of the REX1 repeat domain affected the expression or trafficking of other parasite proteins. An anti-KAHRP antiserum gave a roughly homogenous staining pattern at the periphery of RBCs infected with all parasite lines (Fig. S3A). Similarly, a characteristic PfEMP3 profile at the RBC membrane was observed in all parasite lines (Fig. S3B).
Maurer’s cleft cisternae have a diameter that is close to the limit of resolution of conventional light microscopy. Therefore, it was not clear whether the decreased number of puncta in the REX1Δ371–579-GFP line was due to a deficiency in generating Maurer’s clefts or to the stacking of Maurer’s cleft cisternae. We therefore examined these structures using 3D-Structured Illumination Microscopy (SIM), which provides an 8-fold increase in volume resolution (Hanssen et al., 2010, Schermelleh et al., 2008). Infected RBCs were permeabilized with the pore-forming toxin, Equinatoxin II (EqtII) and labelled with an antibody recognizing GFP (Fig. 4A). 3D-SIM reveals giant Maurer’s clefts with an apparently convoluted surface. We also examined the relative organization of REX1Δ371–579-GFP and SBP1 by dual labelled immunofluorescence after fixation and permeabilization (Fig. 4B). A complementary organization of REX1Δ371–579-GFP and SBP1 is evident, consistent with sub-compartmentalization of the Maurer’s cleft lamella.
Fig. 4. REX1(Δ371–579)-GFP parasites exhibit giant Maurer’s clefts.
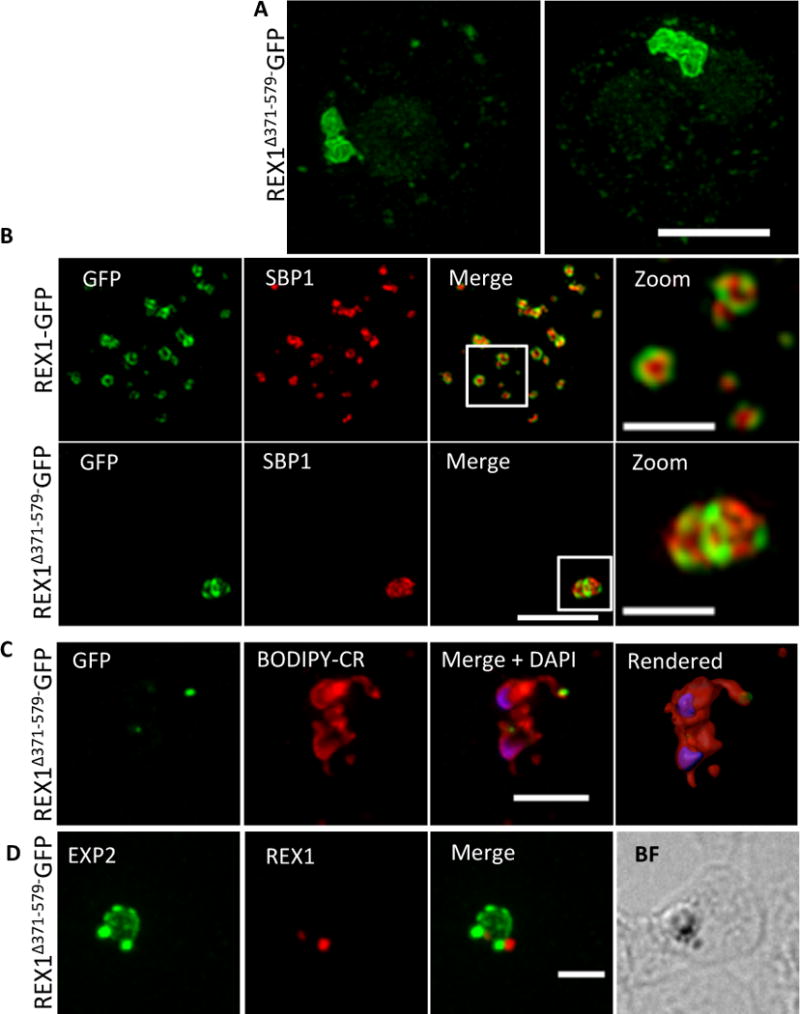
A. REX1(Δ371–579)-GFP-infected RBCs fixed with paraformaldehyde, permeabilized with EqtII and labelled with anti-GFP. B. REX1-GFP and REX1(Δ371–579)-GFP-infected RBCs were fixed with paraformaldehyde/glutaraldehyde and probed with anti-GFP (green) and anti-SBP1 (red). Samples were examined using 3D-SIM. C. REX1(Δ371–579)-GFP-infected RBCs were labelled with BODIPY-ceramide, fixed with paraformaldehyde/glutaraldehyde, labelled with DAPI and imaged using widefield deconvolution microscopy. The right hand panel shows rendering of the surface of the 3D structure using Imaris software. D. REX1(Δ371–579)-GFP-infected RBCs were fixed with paraformaldehyde/glutaraldehyde and probed with anti-EXP2 (green) and anti-REX1 (red). Samples were examined using deconvolution microscopy. Scale bars = 3 μm; zoom bar = 1 μm.
We co-labelled early stage REX1Δ371–579-GFP parasites with the membrane probe, BODIPY-ceramide (Fig. 4C) in an effort to determine the physical organization of the Maurer’s clefts relative to the parasitophorous vacuole membrane (PVM) that surrounds the parasite. The lipid probe BODIPY-ceramide strongly labels membrane structures around the parasite, such as the PVM and the tubolovesicular network (Adisa et al., 2003). As observed in this doubly infected ring stage parasite, the REX1Δ371–579-GFP appears to accumulate at discrete regions of the PVM. This accumulation is also seen in cells labelled with the PVM marker EXP2 (de Koning-Ward et al., 2009) (Fig. 4D) and may represent either nascent Maurer’s clefts budding from the PVM or a stack of Maurer’s clefts that has become tethered to the PVM.
Transmission electron microscopy was performed to examine further the cleft phenotype in the REX1Δ371–579-GFP parasites. The REX1-GFP parasites displayed an unstacked Maurer’s cleft phenotype (Fig. 5A,C) equivalent to that previously reported for the 3D7 strain (Hanssen et al., 2008a). Dramatically, the REX1Δ371–579-GFP parasites showed very large stacks of Maurer’s clefts (Fig. 5B). For stacks cut in transverse section, it was possible to estimate the average number of layers in the stack as 6 ± 2. The average distance between the lamellae was 20 ± 5 nm. The average thickness of the Maurer’s cleft lamellae (lumen and both membranes) was 41 ± 8 and 38 ± 8 nm, respectively, for the REX1-GFP and REX1Δ371–579-GFP lines. These data are consistent with the REX1Δ371–579-GFP parasites producing a similar number of potential cisternae as REX1-GFP parasites but failing to separate them into individual structures.
Fig. 5. Transmission electron microscopy analysis of REX1-GFP and REX1(Δ371–579)-GFP Maurer’s clefts.
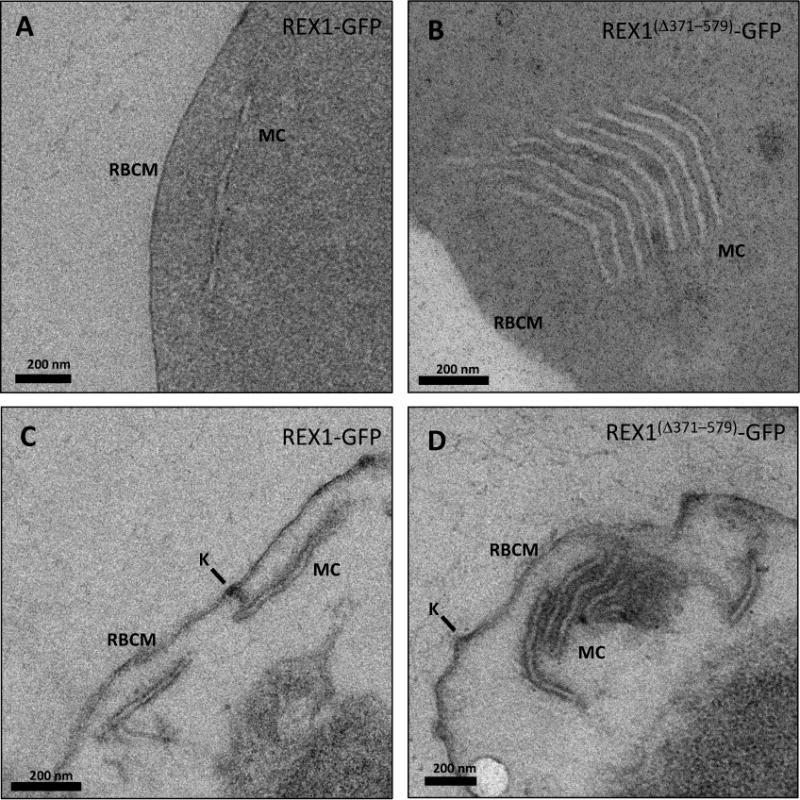
A,B. TEM images of glutaraldehyde-fixed REX1-GFP and REX1(Δ371–579)-GFP-infected RBCs (100 nm sections) revealing unstacked and highly stacked Maurer’s clefts (MC) lamellae. C,D. TEM images of EqtII-permeabilized samples showing single Maurer’s cleft lamellae and stacked lamellae (100 nm sections). Knobs (K) are indicated.
Tether-like structures accumulate on the stacked Maurer’s clefts
In 3D7 parasites, Maurer’s clefts are generated soon after invasion (Gruring et al., 2011, McMillan et al., 2013). During the first ~20 h of intraerythrocytic development, Maurer’s clefts are often mobile within the RBC cytoplasm (McMillan et al., 2013). They then dock onto the RBC membrane in a process that involves tether-like structures as well as direct interactions with a remodeled RBC membrane skeleton (McMillan et al., 2013, Cyrklaff et al., 2011). To determine the organization of the tether-like structures in REX1Δ371–579-GFP parasites, we made use of a previously described marker of the tethers, the membrane-associated histidine-rich protein-2 (MAHRP2) (Pachlatko et al., 2010). In REX1-GFP parasites, immunofluorescence reveals MAHRP2 labelling (Fig. 6A) closely adjacent to the REX1-GFP labelled Maurer’s clefts, as reported previously for wildtype 3D7 (McMillan et al., 2013). For the REX1Δ371–579-GFP parasites, the MAHRP2 labelling partly overlaps with and partly sits outside the single puncta of GFP labelling. To examine this more closely we subjected these samples to 3D-SIM (Fig. 6B). This indicates that MAHRP2 is present in small structures that decorate the Maurer’s cleft stacks.
Fig. 6. Analysis of the Maurer’s clefts ultrastructure and distribution of tethers of REX1(Δ371–579)-GFP parasites.
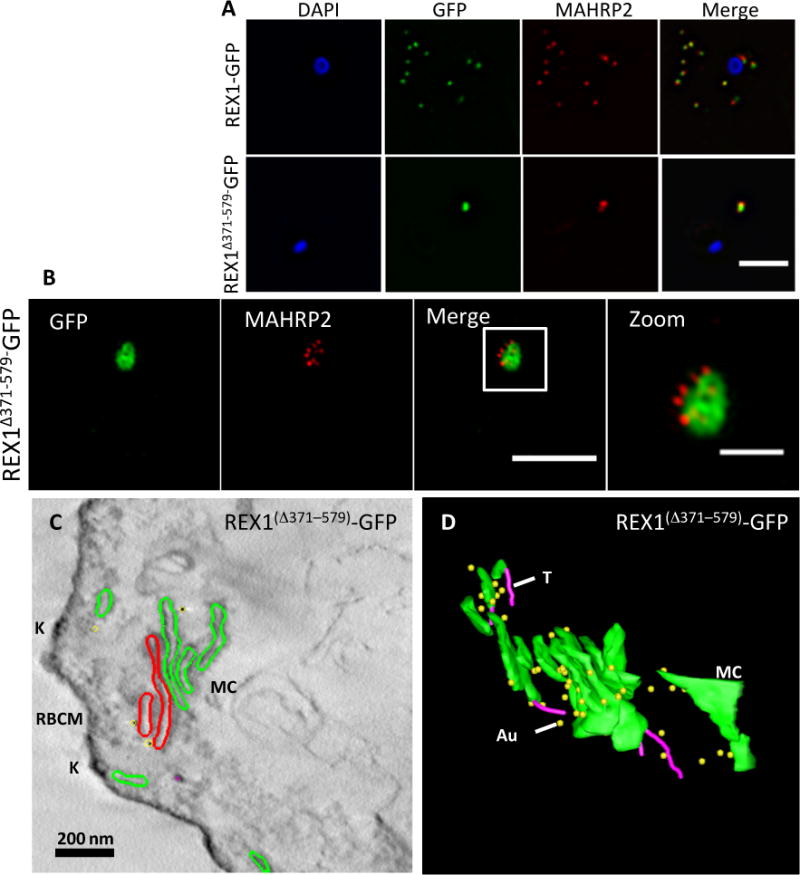
REX1-GFP and REX1(Δ371–579)-GFP transfectants were fixed with paraformaldehyde/glutaraldehyde, permeabilized with Triton X-100, and probed with anti-GFP (green) and anti-MAHRP2 (red). Samples imaged using (A) widefield deconvolution microscopy or (B) 3D-SIM. Scale bars = 3 μm; zoom bar = 1 μm. C. STEM tomogram (600 nm section) of an EqII-permeabilized REX1(Δ371–579)-GFP-infected RBC showing the stacked Maurer’s clefts (MC) layers, and knobs (K) on the RBC membrane (RBCM). The lamella indicated in red share a membrane continuum. D. Rendered STEM tomogram of REX1(Δ371–579)-GFP-infected RBC labelled with anti-GFP antibodies and protein A gold showing Maurer’s clefts (MC, green), tethers (T, magenta) and gold particles (Au, yellow).
To obtain further insights into the Maurer’s cleft architecture, REX1Δ371–579-GFP transfectants were EqtII-permeabilized, then labeled with anti-GFP and protein A-gold. Sections (600 nm) were prepared for scanning transmission electron microscopy (STEM) tomography (Fig. 6C). Rendering of the different features provides a 3D view of the stacked Maurer’s cleft compartments (Fig. 6D). Examination of the STEM tomogram indicated that the some of the Maurer’s cleft layers are connected by a membrane continuum (Fig. 6C, red), indicating a failure to separate the organelle into individual cisternae. It remains possible that the layered structure is more fully inter-connected in regions that are out of the plane of the STEM tomogram. Protein A-gold-labeled anti-GFP is particularly concentrated on the edges of the REX1Δ371–579-GFP Maurer’s cleft cisternae, as previously reported for endogenous REX1 (Hanssen et al., 2008b). Several tubular tether structures with a diameter of ~25 nm are observed in the reconstructions (rendered in magenta). The convoluted nature of the Maurer’s cleft stack is best appreciated by examining a 3D rotation of the rendered tomogram (Video S1).
It is clear that some of the tether-like structures attached to the Maurer’s clefts are not connected through to the RBC membrane; however, given the limited depth of the tomograms, it was difficult to ascertain whether any of the stacked cisternae is directly linked to the RBC membrane. To examine this further we undertook time-lapse imaging of live REX1-GFP and REX1Δ371–579-GFP parasites. As previously reported (McMillan et al., 2013), the REX1-GFP labeled Maurer’s clefts are highly mobile during the early to mid-ring stage of parasite development (Video S2; Fig. S4). The majority of Maurer’s clefts observed in REX1Δ371–579-GFP ring stage-infected RBCs appear to have limited mobility, potentially due to linkage to the parasite surface or trapping in the limited physical space (Video S3; Fig. S4). However, in some REX1Δ371–579-GFP cells the Maurer’s clefts are clearly mobile at the ring stage (Video S4; Fig. S4). The Maurer’s clefts of both REX1-GFP and REX1Δ371–579-GFP immobilize at the trophozoite stage (Fig. S4) consistent with docking onto the RBC membrane.
The repeat region of REX 1 is needed for efficient trafficking of PfEMP1 to the RBC surface
PfEMP1 trafficking requires export into the RBC cytoplasm and transport through the Maurer’s cleft intermediate compartment, prior to insertion into the RBC membrane. We examined the organization of PfEMP1 in wildtype 3D7 and REX1 mutant parasites, using immunofluorescence microscopy on acetone-fixed cells. In agreement with previous studies (Blisnick et al., 2000, Wickham et al., 2001) we observed accumulation of PfEMP1 at the Maurer’s clefts in the 3D7 wildtype parasites, as confirmed by co-labelling with the Maurer’s cleft marker, SBP1 (Fig. 7A). PfEMP1 is also delivered to the Maurer’s clefts in the REX11–579, REX1-GFP and REX1Δ371–579-GFP mutant parasite lines (Fig. 7A) and in the REX-KD parasites (Fig. S3D). Thus despite the aberrant morphology there is no obvious defect in trafficking of PfEMP1 from the parasite to the stacked Maurer’s clefts.
Fig. 7. PfEMP1 surface-exposure and cytoadherence of REX1 transfectants.
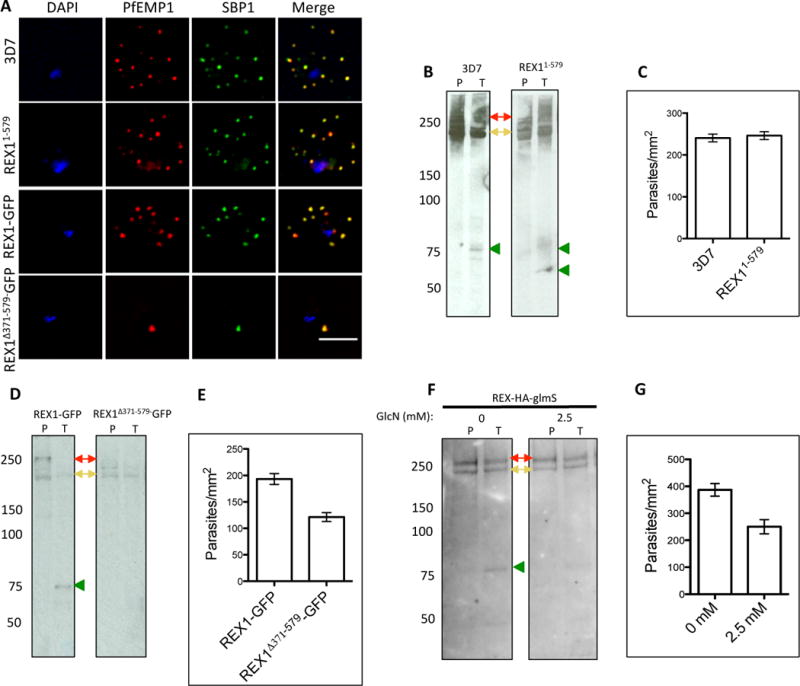
A. Immunofluorescence microscopy of acetone-fixed RBCs infected with 3D7 and REX1 transfectants. Maurer’s clefts were identified by immunolabeling with anti-SBP1 (green). Anti-PfEMP1 antibodies (red) revealed colocation of the virulence protein with Maurer’s clefts. The nuclei are stained with DAPI (blue). Scale bar = 3 μm. B, D, F. Trypsin digestion of surface-exposed PfEMP1 in RBC infected with wildtype and REX11–579 transfectants (B), in REX1-GFP and REX1(Δ371–579)-GFP transfectants (D) and in REX1-HA-GlmS parasites without or with treatment with 2.5 mM GlcN. Full-length PfEMP1 (~270 kDa, red arrows) and a cross-reactive spectrin band (~240 kDa, yellow arrows) are indicated. Trypsin cleavage products (75 kDa) are indicated with green arrowheads. The data are representative of three separate experiments. C, E, G. Adherence of trophozoite-stage infected RBCs to recombinant CD36 under flow conditions (0.1 Pa) ± S.E.M. measured in 10 different areas in each of three separate experiments. REX1(Δ371–579)-GFP binding was significantly lower than REX1-GFP (p < 0.0001, unpaired t-test).
Only a subpopulation of PfEMP1 is delivered to the infected RBC surface (Waterkeyn et al., 2000). This pool of PfEMP1 is oriented with its N-terminal domain exposed at the surface and is thus accessible to cleavage with trypsin (Kriek et al., 2003, Waterkeyn et al., 2000, Gardner et al., 1996). By contrast, intracellular pools of PfEMP1 are protected from protease cleavage. We undertook an analysis of the trypsin accessibility of PfEMP1 in RBCs infected with the different parasite lines. Intact magnet-purified mature stage-infected RBCs were subjected to treatment with PBS (P) or trypsin (T). The Triton-insoluble, SDS-soluble material (representing surface-exposed PfEMP1) was collected and subjected to SDS-PAGE and probed with an antibody recognizing the acidic terminal segment (ATS) domain of PfEMP1. Full length PfEMP1 from 3D7 parasites migrates with an apparent molecular mass of ~270 kDa (Fig. 7B,D,F, red arrows). A cross-reaction of the PfEMP1 antibody with RBC spectrin (at ~250 kDa) is observed in variable intensity in the different samples (Fig. 7B,D,F, yellow arrows). Cleavage products of ~75 kDa were observed in the trypsin-treated 3D7, REX1-GFP and REX1-KD (no GlcN) samples (green arrowheads). A similar cleavage product was observed in the REX11–579 sample as well as an additional product ~60 kDa, likely representing another PfEMP1 variant (Fig. 7B). By contrast, the ~75 kDa cleavage products were not detected in the REX1Δ371–579-GFP parasites (Fig. 7D) and was reduced in the REX1-KD parasites treated with 2.5 mM GlcN (Fig. 7F). This suggests a significant reduction of surface-exposed PfEMP1 in these parasites. By contrast, we observed no obvious difference between the level of full-length Triton X-100-insoluble PfEMP1 in the GlcN-treated and control samples (three experiments). To control for the possibility of cell lysis during trypsin treatment, we used SBP1 as a control. There is no evidence for the SBP1 cleavage product that would be expected if the RBC membrane was breached during the trypsin treatment protocol (see Fig. S5).
To assess the functional consequences of the decreased surface-exposed PfEMP1, parasite-infected RBCs were panned on immobilized CD36, to enrich the population for CD36-binding variants of PfEMP1, and then examined for their ability to adhere to immobilized CD36 under flow at a pressure of 0.1 Pa (Fig. 7C,E,G). Because binding levels differ depending on factors such as CD36 batch, it is important to compare matched pairs of samples strictly in parallel. The 3D7 parasites showed no significant difference in binding compared to REX11–579 (p = 0.68, unpaired t-test). The REX1Δ371–579-GFP mutants bound significantly less efficiently to CD36 (37% decrease; p < 0.001) than REX1-GFP. Similarly, addition of 2.5 mM GlcN to the REX1-KD parasites significantly decreased the binding to CD36 (35% decrease; p < 0.001). This confirms that loss of PfEMP1 surface exposure leads to a significant reduction in adhesion to endothelial ligands. To control for the possibility that treating with GlcN may affect parasite viability, we also measured the CD36 binding of GlcN-treated and untreated 3D7 parent parasites and found no difference (Fig. S6; p = 0.125, unpaired t-test).
REX1 is predicted to be highly α-helical
Sophisticated algorithms for predicting protein structure have been developed recently. For example, I-TASSER (Iterative Threading ASSEmbly Refinement) (http://zhanglab.ccmb.med.umich.edu/I-TASSER/) uses a multiple-threading program (Local Meta-Threading-Server; LOMETS) to identify related structures from the Protein Data Base (PDB), and then constructs full-length models by iterative template fragment assembly simulations (Roy et al., 2010, Zhang, 2008). The REX1 sequence was analyzed using I-TASSER. The resulting secondary structure prediction indicates largely helical structure throughout the protein (Fig. S7A). Of particular note is the repeat region, which is predicted to be helical throughout, with prediction scores of 9, indicating high confidence. The top model generated for REX1 is presented in Fig. S7B and shows a predominantly α-helical structure. The C-score for this model is outside the range specified for high confidence (Roy et al., 2010), and sequence homology between REX1 and the structure used to construct the model is low (<10%). Thus, further work is needed to confirm features of this model. Nevertheless, the prediction of helical structure in the repeat region allows some insight into the possible structural properties of this part of the protein. The repeat sequence domain in the 3D7 strain of P. falciparum takes the form AAABABABAB, where A and B are variants of PQAEKDASKLTTTYDQTKEVK and PQAEKDALAKTENQNGELL, respectively. Analysis of individual repetitive segments reveals alternating negatively and positively charged regions, with a distinct distribution of charged amino acids in the two repeat types (Fig. S7C). These features will result in a repetitive pattern of charge distribution in this area of the protein. The multiple repeat segments may form extended helices, with the proline residues at the start of each repeat potentially inducing kinks or disrupting the extension of the helix.
Discussion
The virulence of P. falciparum is due, in part, to its ability to present PfEMP1 at the RBC surface, which enables sequestration of infected RBCs within the microvasculature of the host. Differences in the severity of malaria-induced pathologies are thought to arise from the expression of different PfEMP1 variants that bind to different endothelial cell receptors or to the same receptor with different affinities. However, differences in the efficiency of trafficking or presentation of PfEMP1 may also contribute to the adhesion phenotype. For example, alterations in Maurer’s cleft architecture may directly or indirectly affect the efficiency of PfEMP1 trafficking to the RBC membrane.
Previous efforts to ablate the rex1 gene resulted in concomitant loss of a subtelomeric region of chromosome 2, encoding KAHRP and other proteins. Because of the inefficiency of conventional knockout strategies in P. falciparum, several months of continuous culturing may be required to retrieve a knockout clone. If a genetic manipulation confers even a small growth disadvantage additional genetic changes may occur that relieve this disadvantage. For example, the loss of the sub-telomeric region of chromosome 2, which encodes several exported proteins that are not needed for survival in culture, is a frequent event during long-term culture.
In this work, we took advantage of a recently developed system for conditional knockdown of P. falciparum genes. We achieved >90% knockdown of REX1 upon addition of GlcN to REX1-KD parasites. This was not associated with a measureable growth defect over the period examined (48 h) but a disadvantage may manifest over a longer period, or upon complete deletion of REX1. Maurer’s cleft reorganization was observed in these REX1-depleted parasites, confirming its important role in maintaining normal organization. No loss of the chromosome 2 proteins, KAHRP and PfEMP3, was observed indicating that it is possible to lose REX1 and maintain the expression of these other exported proteins. Nonetheless, REX1-KD mutants showed defective PfEMP1 surface presentation and defective binding under flow. This confirms that REX1 plays a role in PfEMP1 trafficking that is independent of effects on other exported protein. The level of this defect (~35% lower binding) was less than that reported for complete rex1 deletion (Dixon et al., 2011). This likely indicates that the loss of knobs also contributes to the loss of adhesion in this REX1-deleted line, but may also reflect the fact that a small amount of REX1 is still expressed in the REX1 knockdown. Contrary to previous reports (Knuepfer et al., 2005, Wickham et al., 2001), we did not observe KAHRP or PfEMP3 colocating with Maurer’s clefts proteins. This may be due to stage differences or to the fact that these previous studies used truncated constructs of KAHRP and PfEMP3 which may have affected protein localization.
We were interested to further dissect the region of REX1 that is responsible for Maurer’s cleft sculpting and effective PfEMP1 trafficking. We showed that deletion of 341 amino acids from the C-terminus causes the formation of Maurer’s cleft with an average of 3 stacked cisternae, and is associated with less efficient PfEMP1 trafficking, consistent with previous work (Dixon et al., 2011). By contrast deletion of the C-terminal 133 amino acids had no significant effect on Maurer’s cleft stacking or PfEMP1 trafficking, indicating that the repeat region (371–579 amino acids) contains important functional motifs.
To further investigate the role of this region we generated transfectants in which the repeat sequence domain was deleted. These parasites exhibited a phenotype that is even more severe than the REX11–362 transfectants. The Maurer’s clefts became highly stacked with an average of six layers per stack. Lateral interactions between the cisternal layers membrane were evident, as well as a membrane continuum between some of the layers. Interestingly in early stage parasites the giant Maurer’s clefts were attached to the PVM, which is consistent with the suggestion that Maurer’s cleft lamellae are formed from a single focus. The appearance and dimensions of the individual cisternae were similar to that in wildtype parasites, and Maurer’s cleft markers such as SBP1 were still trafficked to the stacked clefts and located in distinct sub-compartments, as for wildtype 3D7 (McMillan et al., 2013). The clefts were decorated with multiple 25-nm tether-like structures, indicating that this association still occurs; and they still docked onto the RBC membrane in the mature stage of development. The data suggest that the clefts are formed is a similar manner to wildtype Maurer’s clefts, but fail to separate into individual cisternae.
It is evident that the repeat region plays an important role (either direct or indirect) in Maurer’s cleft sculpting and it is interesting to consider possible molecular mechanisms. I-TASSER predicts that REX1 comprises extended regions of α-helix. The repeat region (residues 371–579) is highly charged with an excess of negatively charged amino acids, giving it an overall pI of 4.9. The bioinformatics analysis suggests that the repeat sequence is organized as α-helical segments, each with a distinct distribution of charged amino acids. REX1 accumulates at the edges of the Maurer’s clefts and it is possible that the charged α-helical regions generate a high surface charge that repels individual cisternae away from their neighbors. Alternatively, it is possible that the REX1 functions as a severing protein in a similar manner to members of the dynamin family. These proteins have extended α-helices that form helical “coats” that constrict membranes and promote fission (Morlot et al., 2012, Low et al., 2009). Many Plasmodium proteins contain repeat regions, including SBP1 which has degenerate repeats of the sequence QXXQ in the C-terminal domain, with a pI of 4.25. These repeats are predicted by I-TASSER to form random coil (data not shown). However, it remains possible that other Plasmodium proteins may contribute to the structure of Maurer’s clefts by forming extended helices. It is also possible that deletion of the REX1 repeats alters the 3D structure of the REX1 protein a way that is deleterious to its function.
The REX1Δ371–579 mutants exhibited an intact chromosome 2 locus, a normal distribution of KAHRP and PfEMP3, and normal knob morphology. This suggests that REX1 plays no role in the trafficking of these proteins. Similarly, PfEMP1 appeared to be delivered efficiently to the Maurer’s clefts indicating that REX1 is not involved in this transport step. By contrast, the REX1Δ371–579 mutants (like the GlcN-treated REX1-KD parasites) showed defective PfEMP1 surface presentation and defective binding under flow. It is possible that the repeat domain directly participates in PfEMP1 trafficking, for example by promoting the budding of PfEMP1-containing vesicles from the Maurer’s clefts. Alternatively, deleting the repeat region of REX1 may exert an effect by altering the conformation of the protein. Another possibility is that the stacking of the Maurer’s clefts may compromise lateral associations of individual Maurer’s cleft cisternae with the RBC membrane that are needed for budding of vesicles or packaging of cargo.
Interestingly an analysis of the REX1 sequences available in PlasmoDB indicates that the repeat region is polymorphic (in sequence and length) in different P. falciparum strains. These differences in repeats do not seem to result in the dramatic change in Maurer’s clefts morphology that are observed when this region is deleted. Nonetheless, it is possible that repeat region polymorphisms will affect the efficiency of PfEMP1 trafficking, which could in turn contribute to different levels of cytoadhesion, and thus virulence, of field strains. Further analysis of the role of REX1 in trafficking PfEMP1 may provide ways of interfering with its surface presentation. This could provide an important new strategy to combat this lethal human pathogen.
Experimental Procedures
Parasite culture
P. falciparum parasites were cultured as previously described (Trager & Jensen, 1976). Briefly, parasites (3D7 strain) were cultured in O+ RBC (Australian Red Cross Blood Service) at 5% hematocrit, in RPMI-GlutaMAX™-HEPES (Invitrogen) supplemented with 5% human serum (Australian Red Cross Blood Service) and 0.25% AlbuMAX II (Invitrogen). Synchronization of the ring stages were performed by addition of 5% D-sorbitol (Lambros & Vanderberg, 1979). Mature stage parasite-infected RBCs were purified by magnetic separation (CS columns; Miltenyi Biotec). To maintain knob-positive parasites, cultures were routinely subjected to gelatin flotation (Pasvol et al., 1978). Knockdown of protein expression in parasites containing the glmS riboswitch was achieved by addition of 2.5 mM glucosamine to trophozoite-stage parasites in culture.
Plasmid constructs and P. falciparum transfection
The genomic region of REX1 corresponding to amino acids 1–579 was PCR-amplified from 3D7 gDNA using the REX1579fw (cctaggtgccaactcgaaacttcctgc) and REX1579rv (atcgatatctttttcagcttgagtaag) primers (AvrII and ClaI in bold). The resultant PCR products were directionally cloned into pHH1-DEST to get pHH1-REX1–579. The gene sequence used to generate the REX371–579 parasites was synthesized by GeneScript™. This sequence contained BamHI and PstI sites at the 5′ and 3′ ends of the gene allowing directional cloning into the pEntry-GFP gateway compatible vector (Invitrogen). The pEntry-REX371–579-GFP vector was recombined with the Gateway compatible Destination vector, pHH1-DEST, to yield the final transfection plasmid pHH1-REX371–579.
The riboswitch-inducible knockdown construct was made by PCR amplifying the 3′ region of REX1 with the REX1-glmS-fw (ggatcctgccaactcgaaacttcctgc) and REX1-glmS-rv (ctgcagattaaatacagaactttctag) primers (BamHI and PstI in bold). This PCR product was directionally cloned into the pGLMS-HA plasmid (Elsworth et al., 2014) to get the pGLMS-REX1-HA construct. Transfections were performed as previously described (Deitsch et al., 2001).
Fluorescence microscopy
Fluorescence microscopy was performed on either paraformaldehyde/glutaraldehyde (4%/0.0065%) or acetone-fixed thin blood smears as previously described (Spielmann et al., 2006). The following primary antibodies were used: anti-REX1 (rabbit, 1:2000) (Hawthorne et al., 2004), anti-REX1_N-term (mouse, 1:1000) (Hanssen et al., 2008a), anti-GFP (monoclonal antibody, Roche, 1:500), anti-GFP (rabbit, 1:1000) (Humphries et al., 2005), anti-SBP1 (rabbit, 1:2000) (Cooke et al., 2006), anti-MAHRP2 (rabbit, 1:200) (Pachlatko et al., 2010), anti-PfEMP1 ATS (mAb 1B/98-6H1-1, 1:100) (Maier et al., 2007), anti-KAHRP (mouse, 1:200) and anti-PfEMP3 (mouse, 1:200) (Waterkeyn et al., 2000). Nuclei were visualized by addition of DAPI (1 μg ml−1).
Samples were viewed on a DeltaVision DV Elite Restorative Widefield Deconvolution Imaging System (GE Healthcare/Applied Precision) using a 1000× objective (1.4NA). Samples were excited by 390, 475, 542 or 632 nm LEDs and imaged using band pass filters at 435, 523, 994 or 676 nm. Images are presented as projections of whole cell z-stacks (taken at intervals of 0.2 μm) unless otherwise stated.
Structured Illumination Microscopy (Schermelleh et al., 2008) was performed on a DeltaVision OMX V4 Blaze (GE Healthcare/Applied Precision). Samples were excited using 488, 568 or 642 nm lasers and imaged using band pass filters at 528, 608 and 683 nm with a 60× objective (1.42 NA). Images were processed using the SoftWorx imaging software (Applied Precision) or ImageJ software (NIH).
Immunoblotting
Trophozoite stage parasites were enriched from culture by magnetic separation or Percoll purification as previously described (Dixon et al., 2011). Purified cells were lysed with 0.03% saponin and fractionated into supernatant and pellet fractions as previously described (Dixon et al., 2008). Protein samples were adjusted for equal loading, mixed with 6× SDS loading dye and separated on 4–12% Bis-Tris gels (Life Technologies). Gels were transferred to nitrocellulose membranes and blocked for 1 h in 3.5% skim milk in PBS. The following primary antibodies were used in these study: anti-REX1 (rabbit, 1:2000) (Hawthorne et al., 2004), anti-REX1 (mouse, 1:1000) (Hanssen et al., 2008a), anti-GFP (mAb, Roche; 1:500), anti-GFP (rabbit, 1:1000) (Humphries et al., 2005) and anti-GAPDH (rabbit, 1:1000). Anti-mouse or anti-rabbit horseradish peroxidase-conjugated secondary antibodies (1:25000, Promega) were incubated with the membranes for 1 h at room temperature. All membranes were washed 3 times for 10 min with 0.1% Tween-20 in PBS, following antibody incubations. Washed membranes were incubated with Clarity ECL reagents (Bio-Rad) and visualized on a Las3000 Imager (Fujifilm).
Trypsin cleavage assay
Surface presentation of PfEMP1 was assessed by trypsin cleavage assay as previously described (Dixon et al., 2011). Briefly, purified late stage parasites (~106 cells) were subjected to treatment with either phosphate-buffered saline (PBS) alone or TPCK-treated trypsin (1 mg ml−1; Sigma), for 1 h at 37°C. The reaction was stopped by addition of soybean trypsin inhibitor. Samples were then lysed and solubilized with 1% Triton X-100 on ice for 20 min and pelleted by centrifugation. The resultant pellet was solubilized in 2% SDS and the proteins separated on 3–8% Tris-acetate gels (Life Technologies) and transferred to nitrocellulose. Membranes were blocked for 1 h in 3.5% skim milk in PBS prior to incubation with anti-PfEMP1 ATS (mAb 1B/98-6H1-1, 1:100) (Maier et al., 2007) antibodies for 2 h at room temperature. To determine if intracellular epitopes are exposed during trypsin treatment, intact infected RBCs were exposed to trypsin, then TPCK-treated. The intact infected RBCs were then lysed with EqtII, to release hemoglobin, and the pellets subjected to SDS-PAGE, transferred to nitrocellulose, blocked and probed with anti-SBP1 (BR5) rabbit antibody (1:1000) (Blisnick et al., 2000). Secondary antibodies and detection were performed as described above.
CD36 binding assays and PfEMP1 variant up-selection
Up-selection of parasites on immobilized CD36 was employed prior to binding experiments to maximize surface expression of PfEMP1. Recombinant human CD36 (125 μg ml−1 in PBS) was immobilized on plastic petri dishes (Spycher et al., 2008). Dishes were blocked with 1% BSA in PBS and washed with RPMI-HEPES. Infected RBCs (1% hematocrit, 3% parasitemia) in RPMI-HEPES were added and incubated for 1 h at 37°C. Unbound cells were gently washed from the dish with RPMI-HEPES. Fresh culture media and RBCs were added to the dish and cultures were maintained as described above.
Binding assays under constant flow conditions were performed in culture chambers (iBIDI µ-Slide I) loaded with recombinant human CD36 (125 μg ml−1 in PBS) and incubated overnight at 4°C. Prior to use the chambers were blocked in 1% BSA in PBS for 1 h at 37°C. Binding assays were performed on trophozoites >24 h post-invasion, synchronized to a 6–8 h window. Parasites (1% hematocrit, 3% parasitemia) were resuspended in bicarbonate-free RPMI-HEPES and flowed through the chambers at 0.1 Pa using a programmable Harvard Elite 11 Syringe Pump. All assays were performed at 37°C and were visualized on a DeltaVision DV Elite Restorative Widefield Deconvolution Imaging System (Applied Precision) using a 60× objective. Parasites were flowed through the chamber for 5 min, prior to washing chamber for 5 min in bicarbonate-free RPMI-HEPES under constant flow conditions. The number of bound cells per field was counted for 10 randomly chosen fields.
Electron microscopy and immunolabeling
Trophozoite stage parasites were harvested by magnetic purification and fixed in either 2.5% glutaraldehyde, or in paraformaldehyde/glutaraldehyde (2%/0.0075%) for immuno-EM. Labelling was performed by permeabilizing with ~40 μg ml−1 Equinatoxin II for 6 min, washing and fixing again with 2% paraformaldehyde. Cells were then washed and incubated with anti-GFP (mAb, Roche; 1:50) in 3% BSA in PBS, washed and incubated with gold-labelled protein A (6 nm Aurion). Cells were embedded in agarose, fixed in 2% OsO4 for 1 h, washed twice in H2O and dehydrated in an ethanol series, then acetone, and then infiltrated with Procure epoxy resin for 2 × 12 h at 60°C. Resin was polymerized with BDMA for at least 48 h at 60°C before thin and thick (100 nm and 600 nm) sections were cut. Sections were placed on 100 h copper grids, stained with uranyl acetate and lead citrate and observed on either a Tecnai Spirit (thin sections) or Tecnai F30 (STEM).
Supplementary Material
Acknowledgments
We thank Courtney Zlatic for technical support, and Dr Megan Dearnley and Dr Aaron Jex, for useful discussions. We thank Prof Brian Cooke (Monash University), Prof Mike Ryan (Monash University), Catherine Braun-Breton (Université Montpellier), Dr Tania de Koning-Ward (Deaking University) and Prof Alan Cowman (Walter & Eliza Hall Institute) for generously providing antibodies. We thank Paul Gilson and Brendan Elsworth (Burnet Institute) for providing the glmS plasmid. Microscopy was performed at the Melbourne Advanced Microscopy Facility and the Biological Optical Microscopy Platform, University of Melbourne. This work was supported by grants from the Australian Research Council and the Australian National Health & Medical Research Council. LT is an ARC Australian Professorial Fellow. MDWG is an ARC Future Fellow.
References
- Adisa A, Rug M, Klonis N, Foley M, Cowman AF, Tilley L. The signal sequence of exported protein-1 directs the green fluorescent protein to the parasitophorous vacuole of transfected malaria parasites. J Biol Chem. 2003;278:6532–6542. doi: 10.1074/jbc.M207039200. [DOI] [PubMed] [Google Scholar]
- Baruch DI, Pasloske BL, Singh HB, Bi X, Ma XC, Feldman M, Taraschi TF, Howard RJ. Cloning the P. falciparum gene encoding PfEMP1, a malarial variant antigen and adherence receptor on the surface of parasitized human erythrocytes. Cell. 1995;82:77–87. doi: 10.1016/0092-8674(95)90054-3. [DOI] [PubMed] [Google Scholar]
- Beeson JG, Reeder JC, Rogerson SJ, Brown GV. Parasite adhesion and immune evasion in placental malaria. Trends Parasitol. 2001;17:331–337. doi: 10.1016/s1471-4922(01)01917-1. [DOI] [PubMed] [Google Scholar]
- Blisnick T, Morales Betoulle ME, Barale J, Uzureau P, Berry L, Desroses S, Fujioka H, Mattei D, Braun Breton C. Pfsbp1, a Maurer’s cleft Plasmodium falciparum protein, is associated with the erythrocyte skeleton. Mol Biochem Parasitol. 2000;111:107–121. doi: 10.1016/s0166-6851(00)00301-7. [DOI] [PubMed] [Google Scholar]
- Cooke BM, Buckingham DW, Glenister FK, Fernandez KM, Bannister LH, Marti M, Mohandas N, Coppel RL. A Maurer’s cleft-associated protein is essential for expression of the major malaria virulence antigen on the surface of infected red blood cells. J Cell Biol. 2006;172:899–908. doi: 10.1083/jcb.200509122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabb BS, Cooke BM, Reeder JC, Waller RF, Caruana SR, Davern KM, Wickham ME, Brown GV, Coppel RL, Cowman AF. Targeted gene disruption shows that knobs enable malaria-infected red cells to cytoadhere under physiological shear stress. Cell. 1997;89:287–296. doi: 10.1016/s0092-8674(00)80207-x. [DOI] [PubMed] [Google Scholar]
- Cyrklaff M, Sanchez CP, Kilian N, Bisseye C, Simpore J, Frischknecht F, Lanzer M. Hemoglobins S and C interfere with actin remodeling in Plasmodium falciparum-infected erythrocytes. Science. 2011;334:1283–1286. doi: 10.1126/science.1213775. [DOI] [PubMed] [Google Scholar]
- de Koning-Ward TF, Gilson PR, Boddey JA, Rug M, Smith BJ, Papenfuss AT, Sanders PR, Lundie RJ, Maier AG, Cowman AF, Crabb BS. A newly discovered protein export machine in malaria parasites. Nature. 2009;459:945–949. doi: 10.1038/nature08104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deitsch K, Driskill C, Wellems T. Transformation of malaria parasites by the spontaneous uptake and expression of DNA from human erythrocytes. Nucleic Acids Res. 2001;29:850–853. doi: 10.1093/nar/29.3.850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon MWA, Hawthorne PL, Spielmann T, Anderson KL, Trenholme KR, Gardiner DL. Targeting of the ring exported protein 1 to the Maurer’s clefts is mediated by a two-phase process. Traffic. 2008;9:1316–1326. doi: 10.1111/j.1600-0854.2008.00768.x. [DOI] [PubMed] [Google Scholar]
- Dixon MWA, Kenny S, McMillan PJ, Hanssen E, Trenholme KR, Gardiner DL, Tilley L. Genetic ablation of a Maurer’s cleft protein prevents assembly of the Plasmodium falciparum virulence complex. Molecular microbiology. 2011;81:982–993. doi: 10.1111/j.1365-2958.2011.07740.x. [DOI] [PubMed] [Google Scholar]
- Elsworth B, Matthews K, Nie CQ, Kalanon M, Charnaud SC, Sanders PR, Chisholm SA, Counihan NA, Shaw PJ, Pino P, Chan JA, Azevedo MF, Rogerson SJ, Beeson JG, Crabb BS, Gilson PR, de Koning-Ward TF. PTEX is an essential nexus for protein export in malaria parasites. Nature. 2014;511:587–591. doi: 10.1038/nature13555. [DOI] [PubMed] [Google Scholar]
- Gardner JP, Pinches RA, Roberts DJ, Newbold CI. Variant antigens and endothelial receptor adhesion in Plasmodium falciparum. Proc Natl Acad Sci U S A. 1996;93:3503–3508. doi: 10.1073/pnas.93.8.3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenister FK, Fernandez KM, Kats LM, Hanssen E, Mohandas N, Coppel RL, Cooke BM. Functional alteration of red blood cells by a megadalton protein of Plasmodium falciparum. Blood. 2009;113:919–928. doi: 10.1182/blood-2008-05-157735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruring C, Heiber A, Kruse F, Flemming S, Franci G, Colombo SF, Fasana E, Schoeler H, Borgese N, Stunnenberg HG, Przyborski JM, Gilberger TW, Spielmann T. Uncovering common principles in protein export of malaria parasites. Cell Host Microbe. 2012;12:717–729. doi: 10.1016/j.chom.2012.09.010. [DOI] [PubMed] [Google Scholar]
- Gruring C, Heiber A, Kruse F, Ungefehr J, Gilberger TW, Spielmann T. Development and host cell modifications of Plasmodium falciparum blood stages in four dimensions. Nat Commun. 2011;2:165. doi: 10.1038/ncomms1169. [DOI] [PubMed] [Google Scholar]
- Hanssen E, Carlton P, Deed S, Klonis N, Sedat J, DeRisi J, Tilley L. Whole cell imaging reveals novel modular features of the exomembrane system of the malaria parasite, Plasmodium falciparum. Int J Parasitol. 2010;40:123–134. doi: 10.1016/j.ijpara.2009.09.004. [DOI] [PubMed] [Google Scholar]
- Hanssen E, Hawthorne P, Dixon MW, Trenholme KR, McMillan PJ, Spielmann T, Gardiner DL, Tilley L. Targeted mutagenesis of the ring-exported protein-1 of Plasmodium falciparum disrupts the architecture of Maurer’s cleft organelles. Mol Microbiol. 2008a;69:938–953. doi: 10.1111/j.1365-2958.2008.06329.x. [DOI] [PubMed] [Google Scholar]
- Hanssen E, Sougrat R, Frankland S, Deed S, Klonis N, Lippincott-Schwartz J, Tilley L. Electron tomography of the Maurer’s cleft organelles of Plasmodium falciparum-infected erythrocytes reveals novel structural features. Mol Microbiol. 2008b;67:703–718. doi: 10.1111/j.1365-2958.2007.06063.x. [DOI] [PubMed] [Google Scholar]
- Hawthorne PL, Trenholme KR, Skinner-Adams TS, Spielmann T, Fischer K, Dixon MWA, Ortega MR, Anderson KL, Kemp DJ, Gardiner DL. A novel Plasmodium falciparum ring stage protein, REX, is located in Maurer’s clefts. Molecular and Biochemical Parasitology. 2004;136:181–189. doi: 10.1016/j.molbiopara.2004.03.013. [DOI] [PubMed] [Google Scholar]
- Horrocks P, Pinches RA, Chakravorty SJ, Papakrivos J, Christodoulou Z, Kyes SA, Urban BC, Ferguson DJ, Newbold CI. PfEMP1 expression is reduced on the surface of knobless Plasmodium falciparum infected erythrocytes. J Cell Sci. 2005;118:2507–2518. doi: 10.1242/jcs.02381. [DOI] [PubMed] [Google Scholar]
- Humphries AD, Streimann IC, Stojanovski D, Johnston AJ, Yano M, Hoogenraad NJ, Ryan MT. Dissection of the mitochondrial import and assembly pathway for human Tom40. J Biol Chem. 2005;280:11535–11543. doi: 10.1074/jbc.M413816200. [DOI] [PubMed] [Google Scholar]
- Knuepfer E, Rug M, Klonis N, Tilley L, Cowman AF. Trafficking determinants for PfEMP3 export and assembly under the Plasmodium falciparum-infected red blood cell membrane. Mol Microbiol. 2005;58:1039–1053. doi: 10.1111/j.1365-2958.2005.04895.x. [DOI] [PubMed] [Google Scholar]
- Kriek N, Tilley L, Horrocks P, Pinches R, Elford BC, Ferguson DJ, Lingelbach K, Newbold CI. Characterization of the pathway for transport of the cytoadherence-mediating protein, PfEMP1, to the host cell surface in malaria parasite-infected erythrocytes. Mol Microbiol. 2003;50:1215–1227. doi: 10.1046/j.1365-2958.2003.03784.x. [DOI] [PubMed] [Google Scholar]
- Lambros C, Vanderberg JP. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol. 1979;65:418–420. [PubMed] [Google Scholar]
- Low HH, Sachse C, Amos LA, Lowe J. Structure of a bacterial dynamin-like protein lipid tube provides a mechanism for assembly and membrane curving. Cell. 2009;139:1342–1352. doi: 10.1016/j.cell.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier AG, Cooke BM, Cowman AF, Tilley L. Malaria parasite proteins that remodel the host erythrocyte. Nat Rev Microbiol. 2009;7:341–354. doi: 10.1038/nrmicro2110. [DOI] [PubMed] [Google Scholar]
- Maier AG, Rug M, O’Neill M T, Beeson JG, Marti M, Reeder J, Cowman AF. Skeleton-binding protein 1 functions at the parasitophorous vacuole membrane to traffic PfEMP1 to the Plasmodium falciparum-infected erythrocyte surface. Blood. 2007;109:1289–1297. doi: 10.1182/blood-2006-08-043364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan PJ, Millet C, Batinovic S, Maiorca M, Hanssen E, Kenny S, Muhle RA, Melcher M, Fidock DA, Smith JD, Dixon MW, Tilley L. Spatial and temporal mapping of the PfEMP1 export pathway in Plasmodium falciparum. Cell Microbiol. 2013;15:1401–1418. doi: 10.1111/cmi.12125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morlot S, Galli V, Klein M, Chiaruttini N, Manzi J, Humbert F, Dinis L, Lenz M, Cappello G, Roux A. Membrane shape at the edge of the dynamin helix sets location and duration of the fission reaction. Cell. 2012;151:619–629. doi: 10.1016/j.cell.2012.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray CJL, Rosenfeld LC, Lim SS, Andrews KG, Foreman KJ, Haring D, Fullman N, Naghavi M, Lozano R, Lopez AD. Global malaria mortality between 1980 and 2010: a systematic analysis. The Lancet. 2012;379:413–431. doi: 10.1016/S0140-6736(12)60034-8. [DOI] [PubMed] [Google Scholar]
- Pachlatko E, Rusch S, Muller A, Hemphill A, Tilley L, Hanssen E, Beck HP. MAHRP2, an exported protein of Plasmodium falciparum, is an essential component of Maurer’s cleft tethers. Molecular Microbiology. 2010;77:1136–1152. doi: 10.1111/j.1365-2958.2010.07278.x. [DOI] [PubMed] [Google Scholar]
- Pasvol G, Wilson RJ, Smalley ME, Brown J. Separation of viable schizont-infected red cells of Plasmodium falciparum from human blood. Ann Trop Med Parasitol. 1978;72:87–88. doi: 10.1080/00034983.1978.11719283. [DOI] [PubMed] [Google Scholar]
- Prommana P, Uthaipibull C, Wongsombat C, Kamchonwongpaisan S, Yuthavong Y, Knuepfer E, Holder AA, Shaw PJ. Inducible knockdown of Plasmodium gene expression using the glmS ribozyme. PLoS One. 2013;8:e73783. doi: 10.1371/journal.pone.0073783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rug M, Cyrklaff M, Mikkonen A, Lemgruber L, Kuelzer S, Sanchez CP, Thompson J, Hanssen E, O’Neill M, Langer C, Lanzer M, Frischknecht F, Maier AG, Cowman AF. Export of virulence proteins by malaria-infected erythrocytes involves remodeling of host actin cytoskeleton. Blood. 2014;124:3459–3468. doi: 10.1182/blood-2014-06-583054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherf A, Lopez-Rubio JJ, Riviere L. Antigenic variation in Plasmodium falciparum. Annu Rev Microbiol. 2008;62:445–470. doi: 10.1146/annurev.micro.61.080706.093134. [DOI] [PubMed] [Google Scholar]
- Schermelleh L, Carlton PM, Haase S, Shao L, Winoto L, Kner P, Burke B, Cardoso MC, Agard DA, Gustafsson MG, Leonhardt H, Sedat JW. Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science. 2008;320:1332–1336. doi: 10.1126/science.1156947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielmann T, Gilberger TW. Protein export in malaria parasites: do multiple export motifs add up to multiple export pathways? Trends Parasitol. 2010;26:6–10. doi: 10.1016/j.pt.2009.10.001. [DOI] [PubMed] [Google Scholar]
- Spielmann T, Hawthorne PL, Dixon MW, Hannemann M, Klotz K, Kemp DJ, Klonis N, Tilley L, Trenholme KR, Gardiner DL. A cluster of ring stage-specific genes linked to a locus implicated in cytoadherence in Plasmodium falciparum codes for PEXEL-negative and PEXEL-positive proteins exported into the host cell. Mol Biol Cell. 2006;17:3613–3624. doi: 10.1091/mbc.E06-04-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spycher C, Rug M, Pachlatko E, Hanssen E, Ferguson D, Cowman AF, Tilley L, Beck HP. The Maurer’s cleft protein MAHRP1 is essential for trafficking of PfEMP1 to the surface of Plasmodium falciparum-infected erythrocytes. Mol Microbiol. 2008;68:1300–1314. doi: 10.1111/j.1365-2958.2008.06235.x. [DOI] [PubMed] [Google Scholar]
- Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- Turner L, Lavstsen T, Berger SS, Wang CW, Petersen JE, Avril M, Brazier AJ, Freeth J, Jespersen JS, Nielsen MA, Magistrado P, Lusingu J, Smith JD, Higgins MK, Theander TG. Severe malaria is associated with parasite binding to endothelial protein C receptor. Nature. 2013;498:502–505. doi: 10.1038/nature12216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt S, Hanspal M, LeRoy PJ, Zhao PS, Oh SS, Chishti AH, Liu SC. The cytoadherence ligand Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1) binds to the P. falciparum knob-associated histidine-rich protein (KAHRP) by electrostatic interactions. Mol Biochem Parasitol. 2000;110:423–428. doi: 10.1016/s0166-6851(00)00281-4. [DOI] [PubMed] [Google Scholar]
- Waterkeyn JG, Wickham ME, Davern KM, Cooke BM, Coppel RL, Reeder JC, Culvenor JG, Waller RF, Cowman AF. Targeted mutagenesis of Plasmodium falciparum erythrocyte membrane protein 3 PfEMP3) disrupts cytoadherence of malaria-infected red blood cells. EMBO J. 2000;19:2813–2823. doi: 10.1093/emboj/19.12.2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White NJ, Pukrittayakamee S, Hien TT, Faiz MA, Mokuolu OA, Dondorp AM. Malaria. Lancet. 2013 doi: 10.1016/S0140-6736(13)60024-0. [DOI] [PubMed] [Google Scholar]
- WHO. World malaria report 2014 World Health Organization 2014 [Google Scholar]
- Wickham ME, Rug M, Ralph SA, Klonis N, McFadden GI, Tilley L, Cowman AF. Trafficking and assembly of the cytoadherence complex in Plasmodium falciparum-infected human erythrocytes. The EMBO journal. 2001;20:5636–5649. doi: 10.1093/emboj/20.20.5636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.