

Summary
Elevated serum IgE has many etiologies including parasitic infection, allergy and asthma, malignancy and immune dysregulation. The hyper IgE syndromes due to mutations in STAT3, DOCK8 and PGM3 are monogenic primary immunodeficiencies that are associated with high IgE, eczema and recurrent infections. All of these primary immunodeficiencies are associated with recurrent pneumonias leading to bronchiectasis; however each has unique features and genetic diagnosis is essential in guiding therapy, discussing family planning and defining prognosis. STAT3 mutated HIES is unique with respect to its multi-organ features and frequent pneumatocele formation. DOCK8 deficiency is characterized by severe cutaneous viral infections and early mortality. PGM3 deficiency is newly defined and is distinct in having neurologic abnormalities and cytopenias.
Introduction
Immunoglobulin E (IgE) is a type of antibody that is linked with host defense against parasitic infection. However, in most countries with a low incidence of parasitic infection, high IgE is more commonly associated with disorders of immune dysregulation such as atopy. There are various pulmonary conditions associated with high serum IgE. IgE is one of the drivers of asthma associated with atopy, which led to the development of omalizumab, an anti-IgE monoclonal antibody, as an asthma therapy(1). High serum IgE is associated with allergic bronchopulmonary aspergillosis (ABPA), which complicates cystic fibrosis as well as asthma; monitoring the fall of IgE can be helpful to assess therapeutic response(2). Peripheral and pulmonary eosinophilia along with high serum IgE can be associated with parasitic infections such as Strongyloides and Ascaris leading to Loeffler’s syndrome. Pulmonary eosinophilia and high serum IgE can also be associated with inflammatory conditions such as eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome), which frequently manifests with pulmonary infiltrates, asthma, and chronic rhinosinusitis(3). Finally, recurrent pneumonias and bronchiectasis with high serum IgE may result from several primary immunodeficiencies.
The hyper IgE syndromes (HIES) are grouped together due to symptoms of eczema, recurrent lung and skin infections, and high serum IgE. Mutations in STAT3 (Signal Transducer and Activator 3), DOCK8 (Dedicator of Cytokinesis 8) and PGM3 (Phosphogucomutase 3) lead to three distinct disease entities that have been classified as HIES(4–9). In this review, we will discuss the clinical features of these primary immunodeficiencies with a particular focus on the pulmonary manifestations as well as discussion of the genetics, pathogenesis and approaches to therapy.
Autosomal Dominant (AD)-HIES (aka Job’s Syndrome)
Job’s syndrome was initially described in two patients with recurrent skin abscesses that were reminiscent of the boils described in the Bible as affecting the prophet Job(10). Several years later, IgE was identified and high serum levels were associated with this syndrome of eczema and recurrent infections(11). Over the next few decades, the clinical spectrum of this syndrome broadened to include many diverse features, the genetics were described, and the understanding of the pathogenesis increased dramatically(12).
Pulmonary Features
Recurrent pyogenic pneumonias caused by Staphylococcus aureus, Streptococcus pneumoniae and Haemophilus influenzae typically begin in the first several years of life(12). The systemic signs of infection are often diminished leading to delayed diagnosis of pneumonia. For instance, S. aureus lobar pneumonia may present with minimal fever, normal peripheral white blood cell count, and fairly normal inflammatory markers. Despite the lack of systemic signs of inflammation, airway inflammation is prominent and mucus production may be especially tenacious (Figure 1). Bronchoscopy may be helpful not only to identify the etiology of infection but to assist in airway clearance. The pyogenic pneumonias typically respond well to appropriate antibiotics, but due to the frequent delay in diagnosis, there may be complications such as empyema, pneumatocele formation, and bronchiectasis. As will be discussed further below, AD-HIES appears to be associated with impaired epithelial repair, which likely explains the high frequency of pneumatoceles and parenchymal abnormalities, encountered in approximately 70% of patients(12).
Figure 1.
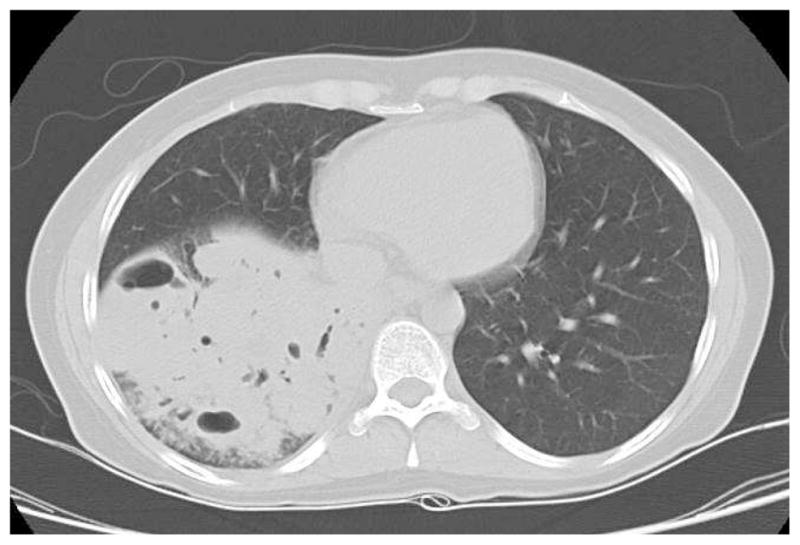
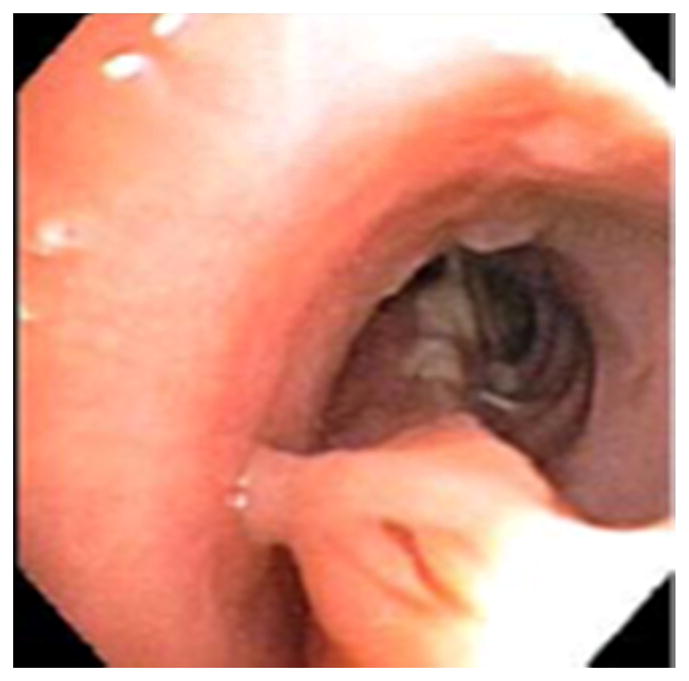
Chest CT demonstrating a lobar infiltrate due to methicillin resistant S. aureus in a 37 year old with AD-HIES (A); bronchoscopy of right bronchus intermedius showed tenacious mucus extending from the right lower lobe (B).
Once the parenchyma of the lungs in AD-HIES has been altered by pyogenic pneumonias, the list of infecting microbes expands to include nontuberculous mycobacteria (NTM), molds such as Aspergillus fumigatus, and persistent Gram-negative bacilli such as Pseudomonas (Figure 2). In our series, 16% of our relatively large AD-HIES cohort met the American Thoracic Society/Infectious Disease Society of America (ATS/IDSA) criteria for NTM infection, a prevalence similar to that seen in cystic fibrosis (CF)(13). Molds and Gram negative bacilli cause chronic infection even more commonly. These chronic infections are the cause of significant morbidity and mortality for these individuals as they may cause significant hemoptysis and/or become increasingly resistant to antimicrobial agents with time(14, 15).
Figure 2.
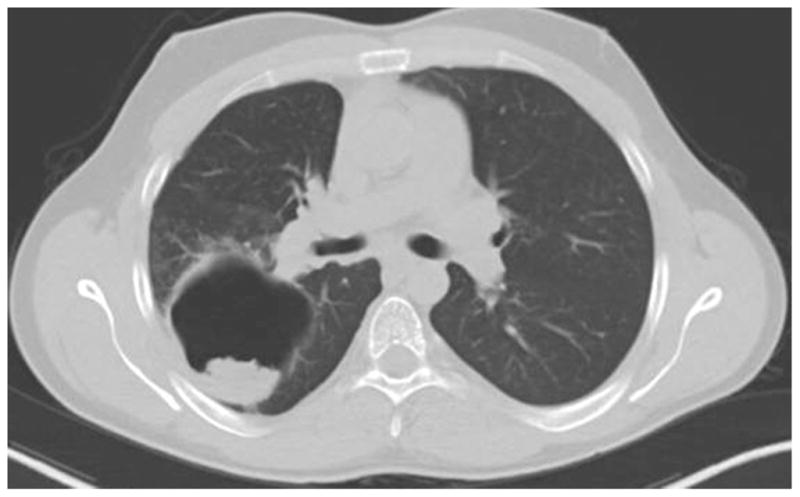
Large pneumatocele with an aspergilloma seen on chest CT of a 23 year old with AD-HIES. The patient had recurrent episodes of hemoptysis that improved with systemic antifungal therapy.
The majority of pulmonary mold infections occur in areas of pre-existing pneumatoceles and bronchiectasis, leading to chronic airway infection(16). Occasionally, however, features of allergic broncho-pulmonary mycosis are seen. This is difficult to diagnose since the serum IgE is high and antigen-specific serologies may be falsely positive to many allergens. The typical imaging findings can lead to this diagnosis and corticosteroid, anti-fungal, or omalizumab treatment initiated (Figure 3). Generally, however, long-term steroid administration is not recommended due to the high incidence of bone fractures (discussed below).
Figure 3.
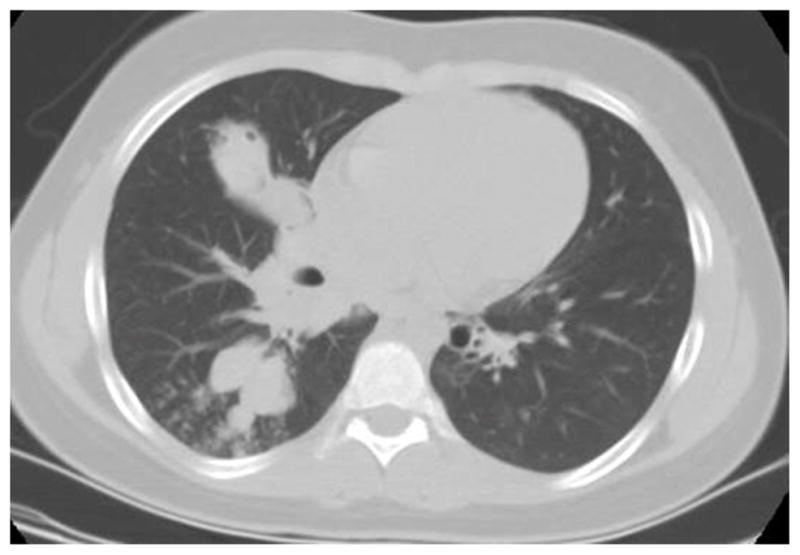
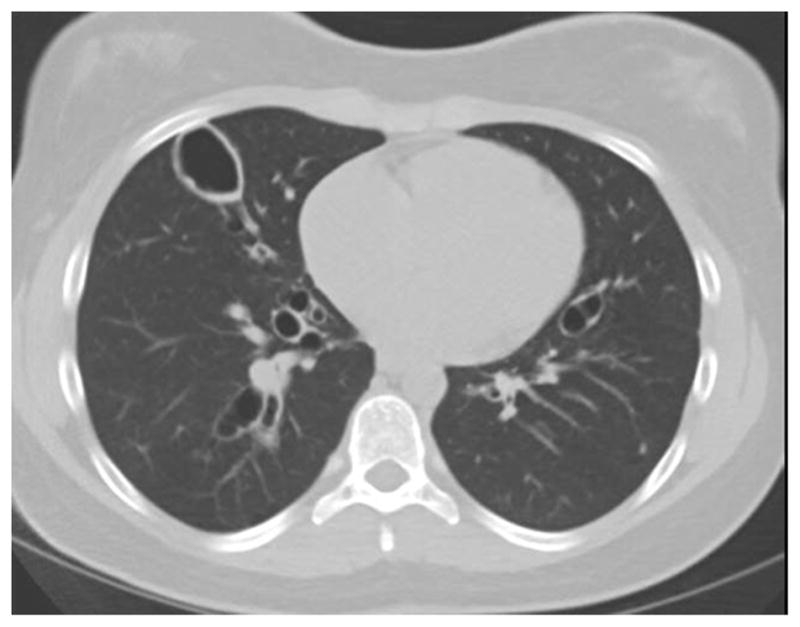
Chest CT of a 15 year old with AD-HIES who presented with increased cough and wheezing (A); CT revealed proximal airway dilation with mucus plugging characteristic of allergic bronchopulmonary Aspergillosis (ABPA) and sputum cultures grew Aspergillus fumigatus. Patient responded to systemic antifungals and corticosteroids, although had persistent airway dilation (B).
Other Clinical Features
The first manifestation of disease is typically a rash, which is present at birth or develops within the first few weeks(17–19). The rash is typically diagnosed as neonatal acne or erythema toxicum neonatorum, and predominates on the head and upper body. If scraped, eosinophils are typically present. The rash evolves into an eczematous dermatitis that is usually driven by S. aureus colonization and chronic infection, and therefore frequently clears if anti-staphylococcal antibiotics are given. Skin abscesses, or boils, frequently appear in the first few years of life, and, similar to the pyogenic pneumonias, often are associated with minimal systemic symptoms such as fever and pain. If drained, though, the abscesses are purulent, and S. aureus predominates. Sinus and ear infections are also common.
Mucocutaneous candidiasis is another common feature, often manifesting as thrush or nail infections. Endemic fungal infections occur, but may have atypical presentations(20). For instance, histoplasmosis may manifest as a gastrointestinal (GI) tract infection, with minimal pulmonary findings, mimicking inflammatory bowel disease (21–23). Cryptococcus has been reported to cause meningitis, but also isolated GI infection, and Coccidiodes has been associated with meningitis(24–26). Pneumocystis jirovecii pneumonia (PJP) is a rare cause of initial pneumonia in infants(27). Although molds can cause significant pulmonary disease, these are usually airway-based and dissemination is rare.
What makes AD-HIES unique amongst many primary immunodeficiencies is the multi-system involvement with vascular, GI and musculoskeletal manifestations. There is a typical facial appearance that usually manifests during adolescence and includes a prominent forehead, deep-set eyes, a broad nose, and porous skin(12). The skull often has some degree of craniosynostosis. A high arched palate is usually present,. Primary teeth are generally not shed and, if not surgically removed prior to emergence of secondary teeth, dental crowding may be noted.(28). Scoliosis is present in the majority of patients and may be severe enough to require surgical correction. Skeletal fractures can occur following minimal trauma, with or without concurrent osteoporosis. The joints are typically very flexible, and in adulthood, significant arthritis and degenerative spinal disease frequently develop. Middle-sized arterial abnormalities have most frequently been recognized as coronary artery aneurysm and tortuousity, with an increasing prevalence with age(29, 30). Other vascular manifestations include cerebral aneurysm, which can present as subarachnoid hemorrhage(31);and pulmonary hemorrhage, which may be due, in part, to tortuous and dilated bronchial arteries adjacent to areas of chronic airway inflammation. GI manifestations include eosinophilic esophagitis and dysmotility, diverticulosis and, infrequently, GI perforations without clearly associated pathology. The incidence of lymphoma is increased, and there have been several cases of extranodal B cell disease(32).
Although higher than the general population, rates of allergy and anaphylaxis are considerably less common than in other conditions with similarly elevated IgE levels(33). Asthma, while present in some, is generally not that severe or difficult to manage. (Box 1)
Box 1. Distinctive Clinical Features of AD-HIES.
Pneumatoceles with secondary infections
Prolonged bronchopleural fistulae after lung resection
Mucocutaneous Candidiasis
Characteristic facial appearance
Retained primary teeth
Minimal trauma fractures and scoliosis
Less allergies and anaphylaxis than expected for level of serum IgE
Laboratory Findings
As is evident from the name of the syndrome, the most consistent laboratory finding is a high serum IgE. The peak is typically greater than 2000 IU/ml, but in adulthood, the level can decrease and even normalize. The IgE level does not necessarily correlate with disease activity. Eosinophilia is common as well. The complete blood count is frequently normal, but the white blood cell count may be low with a relative neutropenia. The white blood cell count often fails to increase as expected during bacterial infection. Serum immunoglobulins are usually normal, but specific antibody protection is variable. Lymphocyte phenotyping typically reveals low memory T and B lymphocytes, and IL-17 producing T cells (Th17 cells) are very diminished(34–36).
Genetics and Pathogenesis
Dominant negative mutations in STAT3 were identified as the cause of AD-HIES in 2007(4, 5). Sporadic mutations are fairly common, which then can be passed down in an autosomal dominant pattern. The mutations are missense or small in-frame deletions that allow for normal protein expression but diminished function. STAT3 is a key signal transduction molecule for many cytokines including, among others, IL-6, IL-10, IL-21, and IL-17. Upon binding to their receptors, the JAK-STAT pathway is activated and STAT3 dimerizes and enters the nucleus to initiate transcription of many genes involved in immunity, wound healing, oncogenesis, embryogenesis and cell survival. Absence of STAT3 is embryonically lethal in mice.
STAT3 signaling is required for the differentiation of naïve T lymphocytes into Th17 lymphocytes. Shortly after STAT3 mutations were identified as causing AD-HIES, these patients were found to have a lack of differentiation of T cells into CD4+Th17 cells. Th17 lymphocytes secrete IL-17 and IL-22, cytokines that were shown initially in mice and later in humans to be associated with Candida mucosal susceptibility(35, 37). After this discovery in AD-HIES, several other primary immunodeficiencies with Candida susceptibility were noted to have disruption of this pathway. Increased IL-17 and IL-22 activity leads to upregulation of antimicrobial peptides at mucosal borders, which are involved in the killing of Candida. The saliva of STAT3 deficient patients has been shown to be deficient in some of these antimicrobial peptides including beta defensin 2 and histatins(38). However, while the diminished antimicrobial peptides likely explains the Candida susceptibility in AD-HIES, the susceptibility to S. aureus infections remains unclear. The S. aureus infections are also at epithelial borders in AD-HIES, but the lack of recurrent S. aureus infections in other diseases with disruption of this pathway remains puzzling.
Other aspects of the immunodeficiency of AD-HIES are explained by low numbers of memory T and B lymphotyes, with preservation of the naïve lymphocyte subsets. Individuals with AD-HIES typically handle viral infections without complications; however, there is a higher incidence of viral reactivation manifesting as uncomplicated zoster or asymptomatic EBV infection(36). This is thought to be due to the poor maturation of T lymphocytes leading to decreased numbers of memory T lymphocytes. Memory B lymphocyte numbers are low as well, and combined with impaired T follicular helper cell function and IL-21 signaling, likely explain the variable production of specific antibodies by patients with AD-HIES (34, 39).
Several clinical manifestations appear consistent with defective tissue remodeling. Most apparent is the frequent occurrence of pneumatoceles, as well as prolonged bronchopleural fistulae after lung surgery or spontaneous pneumothoraces(40). While normal STAT3 signaling in mouse models is not essential for lung development, it is required for normal repair of bronchiolar and alveolar epithelium after damage(41). With epithelial injury, basal cells are thought to multiply to cover the denuded epithelium and produce IL-6 locally. The subsequent IL6/STAT3 signaling promotes basal cell differentiation into ciliated cells and inhibits secretory cell differentiation by inhibiting Notch 1 expression and up-regulating cilia biogenesis genes such as Mcidas and Foxj1(41). These ciliated and secretory cells are key components of the mucociliary clearance apparatus. Following epithelial damage during infection-driven airway inflammation in AD-HIES, altered regulation of these normal repair mechanisms might partially explain the marked buildup of tenacious secretions, which can firmly adhere to the airway walls (Figure 1). Abnormal tissue remodeling likely also explains the middle sized arterial aneurysms. STAT3 is involved in the regulation of matrix metalloproteinases (MMPs), and plasma levels of MMPs have been found to be abnormal in STAT3 deficient patients compared to controls(42).
Management
Prophylactic antimicrobials to prevent pyogenic pneumonias and early recognition and treatment of infection are essential to try and prevent development of pneumatoceles and bronchiectasis. Trimethoprim/sulfamethoxasole (given once or twice daily) is frequently used due to its long-term tolerability and activity against S. aureus, including most community acquired methicillin resistant strains. Dilute bleach baths or other antiseptics can improve the skin disease remarkably. Prophylactic antifungals, such as fluconazole, are used if chronic or frequent recurrences of Candida infections occur. In areas with a high incidence of endemic mycoses, antifungal prophylaxis such as fluconazole for Coccidioides or itraconazole for Histoplasma should be considered. It is not clear if anti-mold prophylaxis, such as with itraconazole, will prevent colonization and infection of molds inside pneumatoceles, but this can be considered. Immune globulin replacement is frequently given due to the variable specific antibody production, and in one series, reduced the rate of pneumonias by about two thirds(43).
Management of lung disease in the setting of chronic infection with bronchiectasis and pneumatoceles is more difficult, and proving efficacy of therapeutic interventions is limited by the rare nature of the disease. Airway clearance techniques such as nebulized hypertonic saline and secretion clearance devices (e.g. percussive vest, cough assist, or hand-held oscillatory PEP) should generally be considered as in other patients with bronchiectasis. However, it should be noted that in some AD-HIES patients, especially those with extensive bronchiectasis, chronically infected pneumatoceles, or mycetomas, hemoptysis can be a significant cause of morbidity and mortality, likely from abnormal vasculature associated with the chronic infection(14). In addition to optimizing antimicrobial treatment and withholding inhaled airway irritants during acute episodes of hemoptysis, bronchial arterial embolization or surgical resection may be required. Patients should be counseled about bleeding thresholds for seeking emergent evaluation. Minimal trauma fractures are fairly frequent, and include rib fractures with coughing, thus limiting techniques such as percussive vests for some. In our experience, azithromycin appears to be helpful in decreasing the frequency of exacerbations for many with AD-HIES and bronchiectasis. This has the advantage of offering some anti-bacterial prophylaxis as well, and may be given daily for this purpose. Evaluating the QTc interval on EKG is prudent when combining an azole antifungal and azithromycin, especially as these patients may receive intermittent fluoroquinolones as well. When chronic fungal infection of pneumatoceles or bronchiectatic airways is present, long-term administration of a mold-active antifungal, such as posaconazole, seems to minimize spread of infection and associated bleeding. If voriconazole is used, consideration of long-term toxicities such as phototoxicity, skin cancers, and hyperfluorosis is important(44).
Antifungals are not always effective in treating aspergillomas that form inside pneumatoceles. Resection is the generally accepted therapy; however, it has unique challenges in patients with AD-HIES. In a published series of lung surgeries performed in STAT3 deficient patients in the US and Germany, there was a complication rate of approximately 50%, with frequent prolonged bronchopleural fistulae, often persisting for months and causing complicated empyema and at times the need for further lung surgery(40). Prolonged bronchopleural fistulae can complicate pneumonias as well; in one case, prompt resolution occurred after bronchoscopic placement of one-way endobronchial valves(45). Although bone and joint surgeries seem to heal well, complications have been reported after open GI surgeries, suggesting that surgery involving epithelial borders may have more healing complications(46).
Several successful hematopoietic stem cell transplants (HSCT) in patients with AD-HIES have been published, prompting increased consideration of this option (47, 48). The immune defect corrects, but it is unclear how many of the somatic features will improve. There is a suggestion that the bone phenotype may improve. Lung transplantation has only been reported in one patient whose lungs were chronically infected with Mycobacterium abscessus, Asperillus fumigatus and Stenotrophomonas maltophilia entering transplant (Figure 4). Unfortunately, the surgery was complicated and post-transplantation, there were multiple Asperillus infections including supradiaphragmatic abscesses and endobronchial infection of the right sided anastomosis that likely extended into the adjacent hilar and mediastinal space and obstructed the right pulmonary artery(49). The patient died approximately 3 years after lung transplantation with signs of transplant vasculopathy. Post-transplantation, she was maintained on prophylactic antibiotics and immune globulin replacement and did not experience the typical AD-HIES associated recurrent bacterial pneumonias. She did have asymptomatic airway infection due to WU polyomavirus (50).
Figure 4.
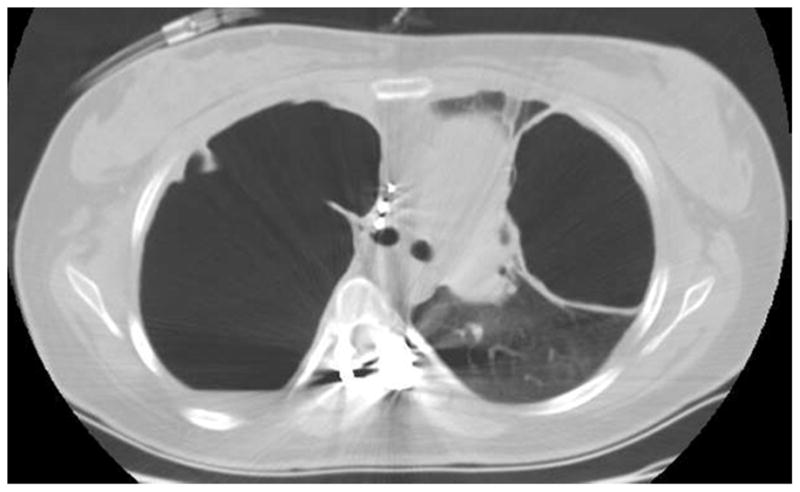
27 year old with AD-HIES and large pneumatoceles chronically infected with Mycobacterium abscessus, Aspergillus fumigatus and Stenotrophomonas maltophilia.
DOCK8 Deficiency
In 2009, homozygous and compound heterozygous mutations in the Dedicator of Cytokinesis 8 (DOCK8) gene were identified in patients previously described as having autosomal recessive HIES(6). Besides the recessive inheritance pattern, several clinical features set DOCK8 deficiency apart from those with AD-HIES, including a lack of the non-immunologic features as well as a higher incidence of viral skin infections and malignancy.
Clinical Features
DOCK8 deficiency typically presents in infancy or early childhood with rash and recurrent sinopulmonary infections(19, 51–53). The eczema ranges from mild to much more severe than is seen in AD-HIES. Atopy is more common in DOCK8 deficiency and significant food allergies and asthma may be present. Viral skin infections are common including recurrent or chronic mucocutaneous herpes simplex virus (HSV), recurrent zoster, and severe and often disfiguring Human Papilloma Virus (HPV) and Molluscum contagiosum infection. Although cutaneous viral infections predominate, there are rare severe systemic viral infections including JC virus-associated progressive multifocal leukoencephalopathy (PML). The long-term prognosis with DOCK8 deficiency is worse than with AD-HIES with only about 50% of affected individuals living beyond 20 years(52). In part, this is due to infection, but this also reflects the tendency to develop malignancy at young ages. Malignancy is frequently driven by chronic viral infections, including HPV-associated squamous cell carcinomas and Epstein Barr Virus (EBV)-associated lymphoma. Non-EBV lymphomas occur as well.
Bronchiectasis is the most common lung finding due to recurrent bacterial and viral pneumonias (Figure 5). Pneumocystis pneumonia occurs as well. Eosinophilic pneumonia has been described(54); however, it is prudent to search thoroughly for an etiologic agent causing the eosinophilic infiltration, and to minimize corticosteroid use as corticosteroid therapy can worsen the cutaneous viral infections and potentially increase the malignancy risk. Asthma is common.
Figure 5.
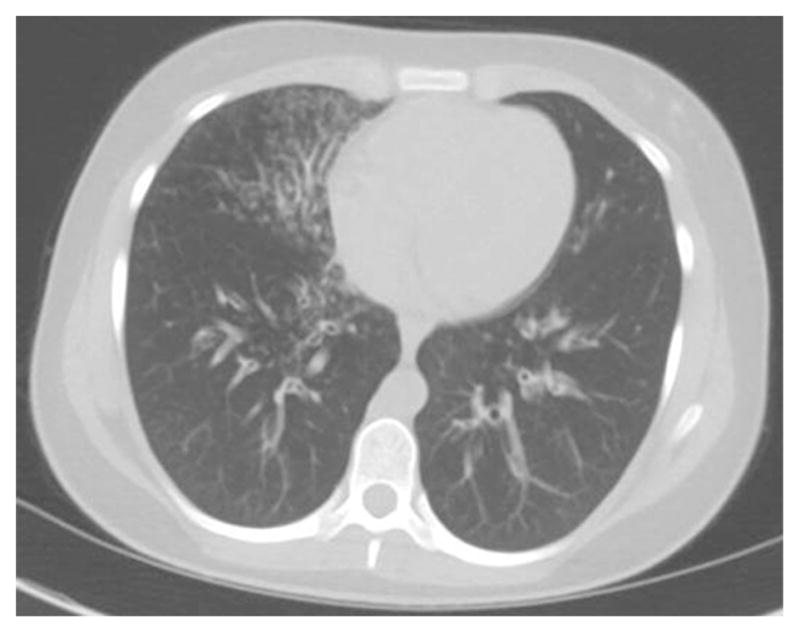
15 year old with DOCK8 deficiency and bronchiectasis after recurrent pneumonias.
Vasculitis and vasculopathy of the cerebral arteries and aorta have been described, the latter rarely resulting in aortic aneurysm and calcification(55). The etiology of most of the vascular defects remains unknown, though one case of zoster vasculitis was described(56). Hepatic disease is also seen in some patients, which particularly affects the biliary system(57). In some but not all cases, this is due to chronic Cryptosporidia infection. (Box 2)
Box 2. Distinctive Features of DOCK8 Deficiency.
Viral cutaneous infections
Malignancies including lymphoma and squamous cell carcinoma
Vasculopathy of aorta with early calcification (infrequent)
Decreased serum IgM
Progressive lymphopenia over time
Laboratory Findings
Serum IgE is usually elevated in DOCK8 deficiency and there typically is eosinophilia, which can be elevated in the range of hyper-eosinophilic syndromes. Serum IgG is usually normal, but specific antibody production is often poor; serum IgM is frequently low and serum IgA is variable(6). There is usually some degree of lymphopenia, particularly of the T lymphocytes, but this is often progressive and so in young children may not be as pronounced. If there has been past EBV infection, chronic EBV viremia is very common.
Genetics and Pathogenesis
Homozygous or compound heterozygous mutations typically lead to lack of DOCK8 expression(6). Diagnosis is by gene sequencing, with comparative genomic hybridization frequently being utilized to detect large deletions. Flow assays to detect DOCK8 protein are being developed to allow for easier screening as the DOCK8 gene is very large and sequencing isn’t widely available.
DOCK8 is involved in actin cytoskeleton organization for cell migration and synapse formation(58–61). Lymphocytes deficient in DOCK8 have trouble migrating through collagen gel matrices, which is thought to be one of the factors in the high incidence of severe cutaneous viral infections(61). DOCK8 is also involved in the survival of T, B and NK cells.
Management
HSCT is the accepted treatment for DOCK8 deficiency due to the high rates of morbidity and mortality at a young age(62–66). DOCK8 is more of a pure immune deficiency than AD-HIES, and therefore HSCT is thought to be curative. Unfortunately, the diagnosis is often not made until end organ damage has occurred, such as bronchiectasis. It is important to optimize airway clearance and minimize chronic infection in preparation for HSCT. Frequently with engraftment there can be worsening of mucus production and airway clearance measures may need to be intensified.
Prophylactic antimicrobials and immune globulin supplementation are the mainstay of therapies until HSCT is performed. TMP/SMX offers both bacterial as well as Pneumocystis prophylaxis. If there is a history of HSV or VZV infection, acyclovir prophylaxis should be considered as well. Interferon-alpha therapy has been used at times to treat recalcitrant viral infections(67, 68) but there are significant side effects such as depression and cytopenias that need to be monitored.
PGM3 Deficiency
Homozygous and compound heterozygous mutations in phosphoglucomutase 3 (PGM3) were identified in 2014 as causing a hyper IgE syndrome phenotype(7–9). PGM3 is a proximal enzyme in the glycosylation pathway to decorate proteins with sugars.
Clinical Features
As PGM3 deficiency is a congenital disorder of glycosylation, many proteins throughout the body with diverse functions are affected leading to multi-system abnormalities. Similar to STAT3 and DOCK8 deficiencies, PGM3 deficiency is associated with eczema, recurrent sinopulmonary infections and high IgE. Asthma and food allergy may be present as well. In contrast to the other syndromes discussed here, neurologic abnormalities such as cognitive and motor delays are usually present. Autoimmunity may be seen as well, such as vasculitis, psoriasis and glomerulonephritis. Hodgkin’s lymphoma has been seen and musculoskeletal abnormalities such as scoliosis and hypotonia have been described(7–9).
Lung disease can be severe with recurrent pneumonias leading to severe bronchiectasis. In one family, one member died in early adulthood due to progressive lung disease, and another had a successful bilateral lung transplant (Figure 6) (7). Allergic bronchopulmonary aspergillosis has been seen as well (Figure 7) (Box 3).
Figure 6.

25 year old with PGM3 deficiency with severe bronchiectasis prior to successful bilateral lung transplantation.
Figure 7.
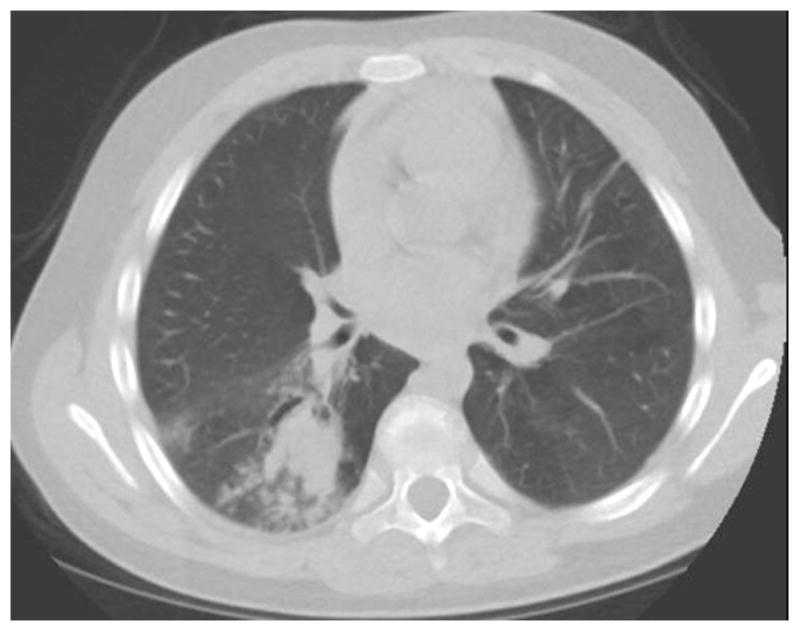
Chest CT of a 13 year old with PGM3 deficiency showing proximal airway dilation with mucus plugging characteristic of ABPA. Sputum cultures grew Aspergillus flavus.
Box 3. Distinctive Features of PGM3 Deficiency.
Neurologic abnormalities including cognitive delay
Frequent neutropenia
Autoimmunity such as vasculitis, glomerulonephritis
CD8 lymphopenia
Laboratory Findings
In addition to high serum IgE and frequent eosinophilia, cytopenias are common in PGM3 deficiency. Neutropenia has been fairly common, although not usually associated with sepsis. Lymphopenia is frequent and is predominantly a result of a decrease in CD8 T lymphocytes. , Early onset with a severe combined immunodeficiency-like phenotype also has been described(9). In contrast to AD-HIES, Th17 lymphocytes are increased, which is consistent with the increased finding of autoimmunity. Serum IgA, IgM and IgG are increased, but specific antibody production is variable(7). Diagnosis of PGM3 deficiency is suggested by glycan profiling of urine and serum, with both O- and N-linked glycans being abnormal.
Genetics and Pathogenesis
Hypomorphic homozygous and compound heterozygous mutations have been reported to cause PGM3 deficiency(7–9). Protein is made but is hypofunctional, as protein negative mutations would be embryonic lethal. PGM3 is an early step in many glycosylation pathways, responsible for converting GlcNAc-6-phosphate to GlcNAc-1-phosophate, which then converts to UDP-GlcNAc. Glycosylation is required for adequate function of most proteins. The pathogenesis of the multiple features of this disease is still being investigated.
Management
The management of patients with PGM3 deficiency is still being defined. The described treatments have generally been supportive, including antibiotic prophylaxis, eczema management, and treatment of infections and auto-inflammatory conditions as they arise. However, several children have been described with severe combined immunodeficiency -like presentations and pancytopenia who received early and successful HSCT(9). It remains unclear if HSCT will correct the neurologic and other somatic features. Other congenital disorders of glycosylation have been treated with dietary supplementation to bypass the pathway abnormalities(69). Potential supplementation is being explored in PGM3 deficiency.
Key Points.
The Hyper IgE syndromes (HIES) comprise distinct primary immunodeficiencies characterized by eczema, recurrent skin and lung infections and high serum IgE.
STAT3 deficient HIES patients frequently develop pneumatoceles that become secondarily infected
DOCK8 deficiency is associated with increased viral infections, typically of the skin, and recurrent lung infections that often lead to bronchiectasis.
PGM3 deficiency is a glycosylation disorder characterized by neurologic abnormalities, cytopenias, and significant bronchiectasis.
Acknowledgments
This work was supported in part by the Intramural Research Programs of the NIAID and NHLBI, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schulman ES. Development of a monoclonal anti-immunoglobulin E antibody (omalizumab) for the treatment of allergic respiratory disorders. American journal of respiratory and critical care medicine. 2001;164(8 Pt 2):S6–11. doi: 10.1164/ajrccm.164.supplement_1.2103025. [DOI] [PubMed] [Google Scholar]
- 2.Hogan C, Denning DW. Allergic bronchopulmonary aspergillosis and related allergic syndromes. Seminars in respiratory and critical care medicine. 2011;32(6):682–692. doi: 10.1055/s-0031-1295716. [DOI] [PubMed] [Google Scholar]
- 3.Lally L, Spiera RF. Pulmonary vasculitis. Rheumatic diseases clinics of North America. 2015;41(2):315–331. doi: 10.1016/j.rdc.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 4.Minegishi Y, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007;448(7157):1058–1062. doi: 10.1038/nature06096. [DOI] [PubMed] [Google Scholar]
- 5.Holland SM, et al. STAT3 mutations in the hyper-IgE syndrome. The New England journal of medicine. 2007;357(16):1608–1619. doi: 10.1056/NEJMoa073687. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Q, et al. Combined immunodeficiency associated with DOCK8 mutations. The New England journal of medicine. 2009;361(21):2046–2055. doi: 10.1056/NEJMoa0905506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Y, et al. Autosomal recessive phosphoglucomutase 3 (PGM3) mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, and neurocognitive impairment. The Journal of allergy and clinical immunology. 2014;133(5):1400–1409. 1409 e1401–1405. doi: 10.1016/j.jaci.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sassi A, et al. Hypomorphic homozygous mutations in phosphoglucomutase 3 (PGM3) impair immunity and increase serum IgE levels. The Journal of allergy and clinical immunology. 2014;133(5):1410–1419. 1419 e1411–1413. doi: 10.1016/j.jaci.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stray-Pedersen A, et al. PGM3 mutations cause a congenital disorder of glycosylation with severe immunodeficiency and skeletal dysplasia. American journal of human genetics. 2014;95(1):96–107. doi: 10.1016/j.ajhg.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davis SD, Schaller J, Wedgwood RJ. Job’s Syndrome. Recurrent, “cold”, staphylococcal abscesses. Lancet. 1966;1(7445):1013–1015. doi: 10.1016/s0140-6736(66)90119-x. [DOI] [PubMed] [Google Scholar]
- 11.Buckley RH, Wray BB, Belmaker EZ. Extreme hyperimmunoglobulinemia E and undue susceptibility to infection. Pediatrics. 1972;49(1):59–70. [PubMed] [Google Scholar]
- 12.Grimbacher B, et al. Hyper-IgE syndrome with recurrent infections-- an autosomal dominant multisystem disorder. The New England journal of medicine. 1999;340(9):692–702. doi: 10.1056/NEJM199903043400904. [DOI] [PubMed] [Google Scholar]
- 13.Melia E, et al. Pulmonary nontuberculous mycobacterial infections in hyper-IgE syndrome. The Journal of allergy and clinical immunology. 2009;124(3):617–618. doi: 10.1016/j.jaci.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Freeman AF, et al. Causes of death in hyper-IgE syndrome. The Journal of allergy and clinical immunology. 2007;119(5):1234–1240. doi: 10.1016/j.jaci.2006.12.666. [DOI] [PubMed] [Google Scholar]
- 15.Sowerwine KJ, Holland SM, Freeman AF. Hyper-IgE syndrome update. Annals of the New York Academy of Sciences. 2012;1250:25–32. doi: 10.1111/j.1749-6632.2011.06387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vinh DC, Sugui JA, Hsu AP, Freeman AF, Holland SM. Invasive fungal disease in autosomal-dominant hyper-IgE syndrome. The Journal of allergy and clinical immunology. 2010;125(6):1389–1390. doi: 10.1016/j.jaci.2010.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chamlin SL, et al. Cutaneous manifestations of hyper-IgE syndrome in infants and children. The Journal of pediatrics. 2002;141(4):572–575. doi: 10.1067/mpd.2002.127503. [DOI] [PubMed] [Google Scholar]
- 18.Eberting CL, Davis J, Puck JM, Holland SM, Turner ML. Dermatitis and the newborn rash of hyper-IgE syndrome. Archives of dermatology. 2004;140(9):1119–1125. doi: 10.1001/archderm.140.9.1119. [DOI] [PubMed] [Google Scholar]
- 19.Chu EY, et al. Cutaneous manifestations of DOCK8 deficiency syndrome. Archives of dermatology. 2012;148(1):79–84. doi: 10.1001/archdermatol.2011.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Odio CD, et al. Endemic mycoses in patients with STAT3-mutated hyper-IgE (Job) syndrome. The Journal of allergy and clinical immunology. 2015;136(5):1411–1413. e1412. doi: 10.1016/j.jaci.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steiner SJ, Kleiman MB, Corkins MR, Christenson JC, Wheat LJ. Ileocecal histoplasmosis simulating Crohn disease in a patient with hyperimmunoglobulin E syndrome. The Pediatric infectious disease journal. 2009;28(8):744–746. doi: 10.1097/INF.0b013e31819b65e0. [DOI] [PubMed] [Google Scholar]
- 22.Cappell MS, Manzione NC. Recurrent colonic histoplasmosis after standard therapy with amphotericin B in a patient with Job’s syndrome. The American journal of gastroenterology. 1991;86(1):119–120. [PubMed] [Google Scholar]
- 23.Alberti-Flor JJ, Granda A. Ileocecal histoplasmosis mimicking Crohn’s disease in a patient with Job’s syndrome. Digestion. 1986;33(3):176–180. doi: 10.1159/000199290. [DOI] [PubMed] [Google Scholar]
- 24.Hutto JO, Bryan CS, Greene FL, White CJ, Gallin JI. Cryptococcosis of the colon resembling Crohn’s disease in a patient with the hyperimmunoglobulinemia E-recurrent infection (Job’s) syndrome. Gastroenterology. 1988;94(3):808–812. doi: 10.1016/0016-5085(88)90257-0. [DOI] [PubMed] [Google Scholar]
- 25.Jacobs DH, et al. Esophageal cryptococcosis in a patient with the hyperimmunoglobulin E-recurrent infection (Job’s) syndrome. Gastroenterology. 1984;87(1):201–203. [PubMed] [Google Scholar]
- 26.Powers AE, et al. Coccidioides immitis meningitis in a patient with hyperimmunoglobulin E syndrome due to a novel mutation in signal transducer and activator of transcription. The Pediatric infectious disease journal. 2009;28(7):664–666. doi: 10.1097/INF.0b013e31819866ec. [DOI] [PubMed] [Google Scholar]
- 27.Freeman AF, et al. Pneumocystis jiroveci infection in patients with hyper-immunoglobulin E syndrome. Pediatrics. 2006;118(4):e1271–1275. doi: 10.1542/peds.2006-0311. [DOI] [PubMed] [Google Scholar]
- 28.Freeman AF, Domingo DL, Holland SM. Hyper IgE (Job’s) syndrome: a primary immune deficiency with oral manifestations. Oral diseases. 2009;15(1):2–7. doi: 10.1111/j.1601-0825.2008.01463.x. [DOI] [PubMed] [Google Scholar]
- 29.Chandesris MO, et al. Frequent and widespread vascular abnormalities in human signal transducer and activator of transcription 3 deficiency. Circulation. Cardiovascular genetics. 2012;5(1):25–34. doi: 10.1161/CIRCGENETICS.111.961235. [DOI] [PubMed] [Google Scholar]
- 30.Freeman AF, et al. Coronary artery abnormalities in Hyper-IgE syndrome. Journal of clinical immunology. 2011;31(3):338–345. doi: 10.1007/s10875-011-9515-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fathi AR, Vortmeyer A, Holland SM, Pluta RM. Intracranial aneurysms associated with hyperimmunoglobulinaemia E (Job) syndrome: report of two cases. Journal of neurology, neurosurgery, and psychiatry. 2011;82(6):704–706. doi: 10.1136/jnnp.2009.198283. [DOI] [PubMed] [Google Scholar]
- 32.Leonard GD, et al. Non-Hodgkin’s lymphoma in Job’s syndrome: a case report and literature review. Leukemia & lymphoma. 2004;45(12):2521–2525. doi: 10.1080/10428190400004463. [DOI] [PubMed] [Google Scholar]
- 33.Siegel AM, et al. Diminished allergic disease in patients with STAT3 mutations reveals a role for STAT3 signaling in mast cell degranulation. The Journal of allergy and clinical immunology. 2013;132(6):1388–1396. doi: 10.1016/j.jaci.2013.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Speckmann C, et al. Reduced memory B cells in patients with hyper IgE syndrome. Clinical immunology. 2008;129(3):448–454. doi: 10.1016/j.clim.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 35.Renner ED, et al. Novel signal transducer and activator of transcription 3 (STAT3) mutations, reduced T(H)17 cell numbers, and variably defective STAT3 phosphorylation in hyper-IgE syndrome. The Journal of allergy and clinical immunology. 2008;122(1):181–187. doi: 10.1016/j.jaci.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Siegel AM, et al. A critical role for STAT3 transcription factor signaling in the development and maintenance of human T cell memory. Immunity. 2011;35(5):806–818. doi: 10.1016/j.immuni.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Milner JD, et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. 2008;452(7188):773–776. doi: 10.1038/nature06764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Conti HR, et al. New mechanism of oral immunity to mucosal candidiasis in hyper-IgE syndrome. Mucosal immunology. 2011;4(4):448–455. doi: 10.1038/mi.2011.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma CS, et al. Functional STAT3 deficiency compromises the generation of human T follicular helper cells. Blood. 2012;119(17):3997–4008. doi: 10.1182/blood-2011-11-392985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Freeman AF, et al. Lung parenchyma surgery in autosomal dominant hyper-IgE syndrome. Journal of clinical immunology. 2013;33(5):896–902. doi: 10.1007/s10875-013-9890-5. [DOI] [PubMed] [Google Scholar]
- 41.Tadokoro T, et al. IL-6/STAT3 promotes regeneration of airway ciliated cells from basal stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(35):E3641–3649. doi: 10.1073/pnas.1409781111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sekhsaria V, et al. Plasma metalloproteinase levels are dysregulated in signal transducer and activator of transcription 3 mutated hyper-IgE syndrome. The Journal of allergy and clinical immunology. 2011;128(5):1124–1127. doi: 10.1016/j.jaci.2011.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chandesris MO, et al. Autosomal dominant STAT3 deficiency and hyper-IgE syndrome: molecular, cellular, and clinical features from a French national survey. Medicine. 2012;91(4):e1–19. doi: 10.1097/MD.0b013e31825f95b9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goyal RK. Voriconazole-associated phototoxic dermatoses and skin cancer. Expert review of anti-infective therapy. 2015;13(12):1537–1546. doi: 10.1586/14787210.2015.1102053. [DOI] [PubMed] [Google Scholar]
- 45.Olivier KN, Barnhart L, Brown DT, Sartain NN, Kwong KF, Freeman AF. Management Of Persistent Bronchopleural Fistula In Job’s Syndrome. Am J Respir Crit Care Med - Conference. 2013;187:A5870. [Google Scholar]
- 46.Langan RC, et al. Safety of major abdominal surgical procedures in patients with hyperimmunoglobulinemia E (Job’s syndrome): a changing paradigm? Journal of gastrointestinal surgery : official journal of the Society for Surgery of the Alimentary Tract. 2013;17(5):1009–1014. doi: 10.1007/s11605-012-2077-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goussetis E, et al. Successful bone marrow transplantation in a pediatric patient with chronic myeloid leukemia from a HLA-identical sibling selected by preimplantation HLA testing. Pediatric blood & cancer. 2011;57(2):345–347. doi: 10.1002/pbc.23007. [DOI] [PubMed] [Google Scholar]
- 48.Patel NC, Gallagher JL, Torgerson TR, Gilman AL. Successful haploidentical donor hematopoietic stem cell transplant and restoration of STAT3 function in an adolescent with autosomal dominant hyper-IgE syndrome. Journal of clinical immunology. 2015;35(5):479–485. doi: 10.1007/s10875-015-0167-z. [DOI] [PubMed] [Google Scholar]
- 49.Kim T, Jancel T, Kumar P, Freeman AF. Drug-drug interaction between isavuconazole and tacrolimus: a case report indicating the need for tacrolimus drug-level monitoring. Journal of clinical pharmacy and therapeutics. 2015 doi: 10.1111/jcpt.12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Siebrasse EA, et al. WU polyomavirus in respiratory epithelial cells from lung transplant patient with Job syndrome. Emerging infectious diseases. 2015;21(1):103–106. doi: 10.3201/eid2101.140855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Q, Davis JC, Dove CG, Su HC. Genetic, clinical, and laboratory markers for DOCK8 immunodeficiency syndrome. Disease markers. 2010;29(3–4):131–139. doi: 10.3233/DMA-2010-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aydin SE, et al. DOCK8 deficiency: clinical and immunological phenotype and treatment options - a review of 136 patients. Journal of clinical immunology. 2015;35(2):189–198. doi: 10.1007/s10875-014-0126-0. [DOI] [PubMed] [Google Scholar]
- 53.Engelhardt KR, et al. The extended clinical phenotype of 64 patients with dedicator of cytokinesis 8 deficiency. The Journal of allergy and clinical immunology. 2015;136(2):402–412. doi: 10.1016/j.jaci.2014.12.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tsuge I, et al. Acute eosinophilic pneumonia occurring in a dedicator of cytokinesis 8 (DOCK8) deficient patient. Pediatric pulmonology. 2014;49(3):E52–55. doi: 10.1002/ppul.22814. [DOI] [PubMed] [Google Scholar]
- 55.Al Mutairi M, et al. Grave aortic aneurysmal dilatation in DOCK8 deficiency. Modern rheumatology / the Japan Rheumatism Association. 2014;24(4):690–693. doi: 10.3109/14397595.2013.874735. [DOI] [PubMed] [Google Scholar]
- 56.Sabry A, et al. Vaccine strain varicella-zoster virus-induced central nervous system vasculopathy as the presenting feature of DOCK8 deficiency. The Journal of allergy and clinical immunology. 2014;133(4):1225–1227. doi: 10.1016/j.jaci.2013.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alsum Z, et al. Clinical, immunological and molecular characterization of DOCK8 and DOCK8-like deficient patients: single center experience of twenty-five patients. Journal of clinical immunology. 2013;33(1):55–67. doi: 10.1007/s10875-012-9769-x. [DOI] [PubMed] [Google Scholar]
- 58.McGhee SA, Chatila TA. DOCK8 immune deficiency as a model for primary cytoskeletal dysfunction. Disease markers. 2010;29(3–4):151–156. doi: 10.3233/DMA-2010-0740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mizesko MC, et al. Defective actin accumulation impairs human natural killer cell function in patients with dedicator of cytokinesis 8 deficiency. The Journal of allergy and clinical immunology. 2013;131(3):840–848. doi: 10.1016/j.jaci.2012.12.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Randall KL, et al. Dock8 mutations cripple B cell immunological synapses, germinal centers and long-lived antibody production. Nature immunology. 2009;10(12):1283–1291. doi: 10.1038/ni.1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Q, et al. DOCK8 regulates lymphocyte shape integrity for skin antiviral immunity. The Journal of experimental medicine. 2014;211(13):2549–2566. doi: 10.1084/jem.20141307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Barlogis V, et al. Successful allogeneic hematopoietic stem cell transplantation for DOCK8 deficiency. The Journal of allergy and clinical immunology. 2011;128(2):420–422. e422. doi: 10.1016/j.jaci.2011.03.025. [DOI] [PubMed] [Google Scholar]
- 63.Bittner TC, et al. Successful long-term correction of autosomal recessive hyper-IgE syndrome due to DOCK8 deficiency by hematopoietic stem cell transplantation. Klinische Padiatrie. 2010;222(6):351–355. doi: 10.1055/s-0030-1265135. [DOI] [PubMed] [Google Scholar]
- 64.Boztug H, et al. Clinical and immunological correction of DOCK8 deficiency by allogeneic hematopoietic stem cell transplantation following a reduced toxicity conditioning regimen. Pediatric hematology and oncology. 2012;29(7):585–594. doi: 10.3109/08880018.2012.714844. [DOI] [PubMed] [Google Scholar]
- 65.Cuellar-Rodriguez J, et al. Matched related and unrelated donor hematopoietic stem cell transplantation for DOCK8 deficiency. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2015;21(6):1037–1045. doi: 10.1016/j.bbmt.2015.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Metin A, et al. Successful bone marrow transplantation for DOCK8 deficient hyper IgE syndrome. Pediatric transplantation. 2012;16(4):398–399. doi: 10.1111/j.1399-3046.2011.01641.x. [DOI] [PubMed] [Google Scholar]
- 67.Al-Zahrani D, et al. Successful interferon-alpha 2b therapy for unremitting warts in a patient with DOCK8 deficiency. Clinical immunology. 2014;153(1):104–108. doi: 10.1016/j.clim.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Keles S, et al. Plasmacytoid dendritic cell depletion in DOCK8 deficiency: rescue of severe herpetic infections with IFN-alpha 2b therapy. The Journal of allergy and clinical immunology. 2014;133(6):1753–1755. e1753. doi: 10.1016/j.jaci.2014.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tegtmeyer LC, et al. Multiple phenotypes in phosphoglucomutase 1 deficiency. The New England journal of medicine. 2014;370(6):533–542. doi: 10.1056/NEJMoa1206605. [DOI] [PMC free article] [PubMed] [Google Scholar]