

Significance
A cardinal feature of Parkinson’s disease pathology is the aggregation of α-synuclein in ubiquitin-positive inclusions termed Lewy bodies, yet the composition of ubiquitin conjugates in these inclusions and their role in α-synuclein pathobiology remain unclear. Here we demonstrate that α-synuclein inclusions contain K63-linked ubiquitin chains, which are strikingly reduced in dopaminergic neurons. In these neurons, the deubiquitinase Usp8 is present in Lewy bodies, and its content is related inversely to the extent of K63-linked ubiquitination. Our mechanistic studies in vitro and in flies indicate that Usp8 interacts with and deconjugates K63-linked ubiquitin chains on α-synuclein, prolonging its half-life and increasing its toxicity. Thus, Usp8 appears to be a critical factor determining α-synuclein levels that could be targeted for therapies.
Keywords: ubiquitin, Parkinson’s disease, endosome, ubiquitin ligase, neurodegeneration
Abstract
In Parkinson’s disease, misfolded α-synuclein accumulates, often in a ubiquitinated form, in neuronal inclusions termed Lewy bodies. An important outstanding question is whether ubiquitination in Lewy bodies is directly relevant to α-synuclein trafficking or turnover and Parkinson’s pathogenesis. By comparative analysis in human postmortem brains, we found that ubiquitin immunoreactivity in Lewy bodies is largely due to K63-linked ubiquitin chains and markedly reduced in the substantia nigra compared with the neocortex. The ubiquitin staining in cells with Lewy bodies inversely correlated with the content and pathological localization of the deubiquitinase Usp8. Usp8 interacted and partly colocalized with α-synuclein in endosomal membranes and, both in cells and after purification, it deubiquitinated K63-linked chains on α-synuclein. Knockdown of Usp8 in the Drosophila eye reduced α-synuclein levels and α-synuclein–induced eye toxicity. Accordingly, in human cells, Usp8 knockdown increased the lysosomal degradation of α-synuclein. In the dopaminergic neurons of the Drosophila model, unlike knockdown of other deubiquitinases, Usp8 protected from α-synuclein–induced locomotor deficits and cell loss. These findings strongly suggest that removal of K63-linked ubiquitin chains on α-synuclein by Usp8 is a critical mechanism that reduces its lysosomal degradation in dopaminergic neurons and may contribute to α-synuclein accumulation in Lewy body disease.
Parkinson’s disease (PD) is the second most common neurodegenerative disorder, characterized pathologically by neuronal death and the formation of intracellular inclusions termed Lewy bodies (LBs). Although primarily a movement disorder with predilection for the pigmented neurons of the substantia nigra, these neuropathological features are eventually widespread, affecting other areas of the brain, especially the entorhinal and anterior cingulate cortex (1). Misfolded α-synuclein is the major constituent of LBs (2), and the density of cortical LBs correlates with the extent of cognitive dysfunction (3). In familial cases, patients with a triplication of the α-synuclein gene develop dementia at an earlier age than those with duplications (4), whereas in sporadic LB disease, soluble α-synuclein oligomers are increased in patients with dementia in the absence of changes in α-synuclein transcription (5). These findings strongly suggest that the neuronal level of α-synuclein is critical in determining the development of diffuse neurodegeneration with LBs. Conversely, differential expression or activation of enzymes that regulate α-synuclein levels may partly explain the neuronal vulnerability and regional progression of α-synuclein pathology.
Most cellular proteins are selectively targeted for degradation by conjugation to a ubiquitin chain. This modification involves activation of ubiquitin by the enzyme E1, transfer of the reactive ubiquitin to a ubiquitin-conjugating enzyme (E2), and then conjugation by a ubiquitin ligase (E3) to a protein substrate or a preceding ubiquitin to form a ubiquitin chain. Ubiquitin contains seven lysine residues, each of which can be linked to the C terminus of another ubiquitin molecule through an isopeptide bond. Whereas formation of ubiquitin chains in which the ubiquitins are covalently linked through their K48 or K11 residues leads to the degradation of cytosolic proteins by 26S proteasomes, attachment of chains linked through K63 residues to membrane-associated proteins targets them for lysosomal degradation. Both these ubiquitin-dependent degradative processes, as well as macroautophagy, contribute to clearance of α-synuclein (6, 7). For example, in the endosomal process, the ubiquitin ligase Nedd4 forms K63-linked chains on α-synuclein to target it to lysosomes (7). At the proteasome and during endosomal uptake, ubiquitin chains are disassembled by deubiquitinating enzymes (DUBs) so that the ubiquitin molecules can be reused in subsequent rounds of degradation, but this action of DUBs can also serve to prevent the degradation of substrates.
Ubiquitin immunoreactivity is a robust neuropathological hallmark of LBs (8, 9) and a fraction of α-synuclein in LBs is ubiquitinated (10, 11). Therefore, enzymes that catalyze ubiquitin conjugation or deubiquitination may contribute to the cell’s response to α-synuclein accumulation. However, the composition of ubiquitin chains in LBs in different regions of the brain remains unknown. As a consequence, it has been widely assumed that ubiquitin immunoreactivity is a nonspecific modification or a surrogate marker of impaired proteasomal function in PD and other α-synucleinopathies (12). In this study, we revisited this assertion and investigated regional differences in LB ubiquitination and explored its enzymatic basis and significance for α-synuclein–induced toxicity.
Results
Ubiquitination of LBs Involves K63-Linked Ubiquitin Chains and Is Regionally Distinct.
Although it has long been known that LBs can be stained with antibodies against ubiquitin (8, 9), the molecular underpinnings of this modification remain unknown. To address this issue, we performed a comprehensive investigation of the pattern and composition of ubiquitin conjugates in these inclusions across different brain regions using 20 cases of almost pure α-synuclein pathology that were identified in the Thomas Willis Brain Collection (University of Oxford). The cases were characterized in terms of neuropathological staging (1) and LB numbers, as shown in Table S1. Using serial sections, we then quantified the percentage of ubiquitin-positive inclusions relative to α-synuclein immunoreactivity in three brain regions (Fig. 1 A–C) where at least eight LBs were detected [substantia nigra (SN), range 10–119; anterior cingulate (AC), range 8–249; entorhinal cortex (EC), range 11–258]. In the cortex, a high proportion of the inclusions were positive with a pan-ubiquitin antibody with similar percentages detected in the two regions that were examined: AC 69.2% (±5.3%) and EC 62.2% (±4.5%). Unexpectedly, the percentage of LBs that were ubiquitin-positive was lower in the SN (27.3 ± 3.2%; *P < 0.0001, n = 14, one-way ANOVA; Fig. 1 C and D). The low percentage of ubiquitin-positive inclusions in the SN was detected irrespective of the pathological stage or clinical diagnosis, and this fraction was even lower in incidental LB cases than in LB disease (Fig. 1 E), suggesting that ubiquitin immunoreactivity is not related to the extent of cell death in this region. To investigate whether ubiquitin chains that target proteins for degradation are present in LBs, we used linkage-specific ubiquitin antibodies against Lys63 and Lys48. We found only occasional staining of inclusions with anti-K48 ubiquitin antibodies regardless of the epitope retrieval method. In striking contrast, anti-K63 ubiquitin antibodies showed extensive immunoreactivity both in the cortex and SN (Fig. 1A). To confirm that K63-specific antibodies stain the pathological inclusions, we used double-labeling immunofluorescence confocal microscopy and found strong colocalization of the K63-specific and α-synuclein antibodies (Fig. 1B). Quantitative analysis revealed that in the cortex, the percentage of K63-linked ubiquitin-immunoreactive inclusions was much higher (AC 49.0 ± 4.7% and EC 52.5 ± 6.0%) than in the SN (12.8 ± 2.4%; ***P < 0.0001, n = 14, Wilcoxon–Mann–Whitney/Kruskal–Wallis test; Fig. 1D). A similar pattern was seen with the pan-ubiquitin antibody, and there was a highly significant positive correlation between K63-specific and pan-ubiquitin–immunoreactive inclusions (ρ = 0.4990; ***P < 0.0004, Spearman’s rho; Fig. S1A). It should be noted that another, less well recognized inclusion in the SN of patients with PD, the intranuclear Marinesco body, was universally positive with K63-specific antibodies (Fig. S1C), indicating that the regional pattern of LB staining is not due to technical differences in antibody binding between the cortex and SN. Finally, we tested and confirmed the specificity of these antibodies by immunoblotting against single-lysine ubiquitin chains, as shown in Fig. S2. Thus, ubiquitin conjugates in LBs as assessed by immunohistochemistry are to a large extent K63-specific chains with a regionally distinct pattern between the SN and cortex.
Table S1.
Study cohort for neuropathology studies
| Case | Patient demographics | PMI, h | Staging | LB nos. | |||||
| Age, y | Sex, f/m | Clinicopathological diagnosis | Braak PD | Braak AD | SN | AC | EC | ||
| 1 | 87 | m | ILB | 24 | 5 | 1 | 11 | 3* | 4* |
| 2 | 77 | m | ILB | 36 | 4 | 1 | 30 | 2* | 3* |
| 3 | 77 | f | ILB | 34 | N/A | 1 | 10 | 0* | 0* |
| 4 | 82 | m | ILB | 29 | 5 | 1 | 11 | 3* | 1* |
| 5 | 82 | m | PD | 48 | 6 | 1 | 25 | 26 | 22 |
| 6 | 77 | m | PD | 72 | 6 | 1 | 15 | 30 | 25 |
| 7 | 72 | m | PD | 48 | 4 | 1 | 11 | 1* | 3* |
| 8 | 92 | m | PD | 36 | 6 | 1 | 16 | 25 | 15 |
| 9 | 86 | m | PD | 66 | 6 | 1 | 18 | 11 | 11 |
| 10 | 76 | m | PDD/DLB | 72 | 6 | 1 | 21 | 10 | 13 |
| 11 | 80 | f | PDD/DLB | 96 | 6 | 2 | 34 | 72 | 213 |
| 12 | 72 | m | PDD/DLB | 48 | 6 | 2 | 75 | 121 | 71 |
| 13 | 79 | m | PDD/DLB | 96 | 6 | 2 | 85 | 91 | 197 |
| 14 | 76 | m | PDD/DLB | 52 | 6 | 2 | 119 | 249 | 175 |
| 15 | 92 | m | PDD/DLB | 24 | 5 | 1 | 24 | 8 | 22 |
| 16 | 76 | f | PDD/DLB | 96 | 6 | 1 | 53 | 28 | 29 |
| 17 | 72 | m | PDD/DLB | 34 | 6 | 1 | 34 | 73 | 258 |
| 18 | 80 | f | PDD/DLB | 72 | 6 | 2 | 28 | N/A* | 106 |
| 19 | 71 | m | PDD/DLB | 88 | 6 | 1 | 50 | 17 | 31 |
| 20 | 80 | m | PDD/DLB | 37 | 6 | 2 | 10 | 78 | 119 |
Characterization of the 20 cases in terms of neuropathological staging and LB numbers in three brain areas (substantia nigra, anterior cingulate cortex, and entorhinal cortex). Clinicopathological diagnoses: AD, Alzheimer’s disease; DLB, dementia with Lewy bodies; ILB, incidental Lewy body cases; PD, Parkinson’s disease; PDD, Parkinson’s with dementia; PMI, postmortem interval. Staging is according to Braak PD and Braak AD. LB burden is in mean absolute values of quantification triplets, blinded for case and diagnosis. N/A, section not available.
Low LB measures; these cases were excluded from statistical analysis.
Fig. 1.

K63-linked ubiquitin conjugates are detected in α-synuclein–positive inclusions and are reduced in the substantia nigra. (A) Schematic view of the studied brain regions and corresponding light microscopy images showing K63-linked ubiquitinated inclusions. [Scale bars, 5 mm (schemes) and 20 mm (images).] (B) Confocal immunofluorescence showing colocalization of K63-linked ubiquitin chains and α-synuclein in Lewy bodies and Lewy neurites in nigral neurons. (C) Quantification of ubiquitin-positive inclusions as a percentage of α-synuclein–positive inclusions (Ub/α-Syn) in serial sections of the AC, EC, and SN; ***P < 0.0001, n = 14. (D) Quantification of K63-linked ubiquitinated inclusions as a percentage of ubiquitin–positive inclusions (K63-Ub/Ub) in serial sections of the AC, EC, and SN; ***P < 0.0001, n = 14. (E) The percentage of ubiquitinated inclusions in nigral neurons was low irrespective of the stage of disease and lower in incidental Lewy body compared with Lewy body disease cases. Ub/Syn, ***P < 0.0001, n = 14; K63/Syn, *P < 0.05, n = 14. AQ, central aqueduct; CA1–4, cornu ammonis 1–4; CC, corpus callosum; CP, corticopontine and pyramidal tracts; DG, dentate gyrus; IC, inferior colliculus; LAT.V., lateral ventricle; Ncl IV, trochlear nucleus; SCP, superior cerebellar peduncle. Error bars correspond to standard error of the mean.
Fig. S1.

(A) Correlation of K63-specific ubiquitin antibody-positive inclusions as a percentage of pan-ubiquitin antibody-positive inclusions in the AC, EC, and SN. Minimal number of analyzed sections of one brain region, n = 14. ρ = 0.4990, *P < 0.0004, Spearman’s rho. (B) Pan-ubiquitin antibody-positive Lewy bodies and Lewy neurites in the substantia nigra. (C) Intranuclear inclusions in a nigral neuron (Marinesco bodies are indicated by an arrowhead) and dystrophic neurites stained with a K63-specific ubiquitin antibody. (Scale bar, 20 µm.)
Fig. S2.
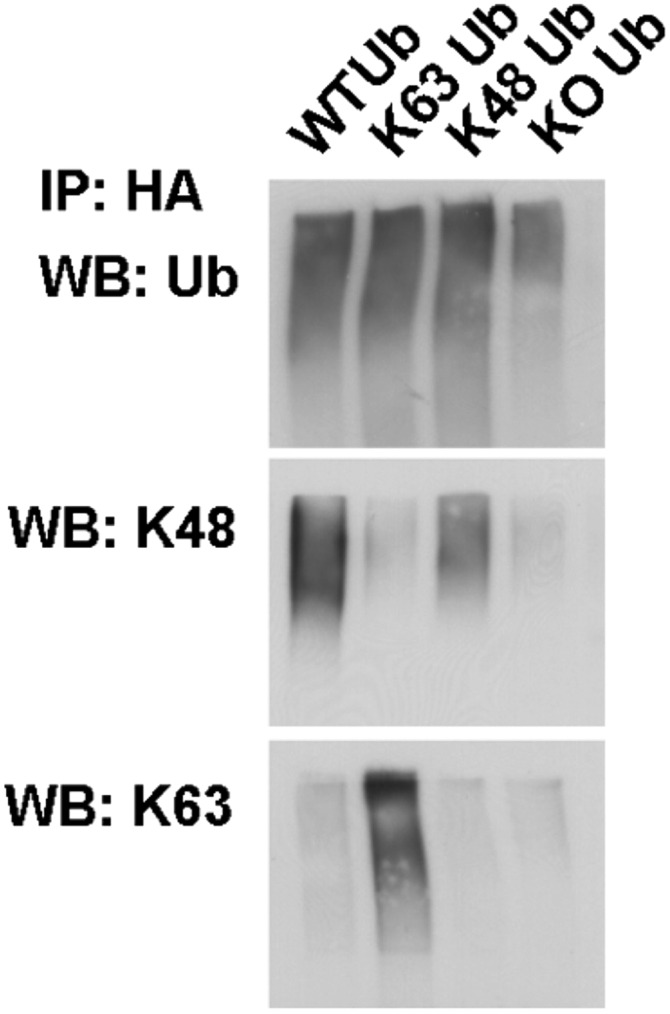
Wild-type or single-lysine HA-tagged ubiquitin was expressed in HEK-293T cells. Ubiquitinated proteins were immunoprecipitated with anti-HA antibodies and blotted with anti–K63-specific or anti–K48-specific antibodies as well as antibodies against pan-ubiquitin. Note that both antibodies and single-lysine ubiquitin constructs have the expected specificity without any cross-reactivity detected.
K63-Linked Ubiquitin Conjugates in LBs Inversely Correlate with Pathological Localization of Usp8.
Inside cells, K63-linked ubiquitin conjugates function in cell signaling, DNA repair, and trafficking proteins to the endosomal or autophagic pathways. The endosomal sorting complex required for transport (ESCRT) components STAM1/2 and HRS help deliver proteins bearing K63-linked chains to the endosomal pathway (13, 14) and, in this process, two DUBs, Usp8 and AMSH, disassemble the ubiquitin chain to release ubiquitin (15–19). Distinct ubiquitin-binding proteins, such as p62 and NRB1, can serve as specific adaptors that promote autophagic degradation of ubiquitinated protein aggregates and organelles (20). We therefore investigated whether differences in the levels of such interactors in the SN and cortex of the same brain or in these regions of healthy and diseased brains could explain the differences in ubiquitin immunoreactivity (Fig. S3). Quantitative immunoblotting revealed a significant increase in Usp8 levels in brain lysates of patients with LB disease compared with healthy controls in the SN (**P = 0.0028, Wilcoxon–Mann–Whitney/Kruskal–Wallis test; Fig. 2 A–C) but not in the cortex. This biochemical finding was corroborated by the detection of a substantial localization of Usp8 but not AMSH in neurons with pathological inclusions (Fig. 2 D–F). Double-labeling immunofluorescence confocal microscopy confirmed that Usp8 was colocalized with the α-synuclein–positive inclusions (Fig. 2G). Importantly, quantitative analysis of Usp8 immunoreactivity showed a striking inverse correlation with either pan-ubiquitin or K63-specific ubiquitin staining: Whereas only a low number of cortical inclusions stained with Usp8 antibodies (AC 14.7 ± 4.0%; EC 16.8 ± 2.8%), many more inclusions were Usp8-positive in the SN (43.9 ± 5.0%; ***P < 0.0001, n = 14; Wilcoxon–Mann–Whitney/Kruskal–Wallis test; Fig. 2H; negative correlation coefficient ρ = −0.5044, ***P < 0.0002; ρ = −0.4186, **P = 0.0038; Spearman’s rho; Fig. 2 I and J). It is noteworthy that the soluble levels of LC3II and p62, two widely used markers of autophagic degradation, did not differ between cases versus controls or between regions (Fig. S3). Thus, contrary to the prevailing views, our data suggest that ubiquitin immunoreactivity in Lewy body disease may represent regional differences in protein deubiquitination and that Usp8 may be the critical determinant of this pattern and therefore important in the pathogenesis of sporadic PD.
Fig. S3.

Targeted screen for proteins that are known to interact with or regulate the assembly of K63-linked ubiquitin chains in the endosomal and autophagic pathways. Total lysates from the SN and AC were compared across regions of the same brain and between healthy and LB-containing regions (n = 8 cases per region). The detection of a significantly increased expression of Usp8 in the SN with LBs despite the use of crude lysates strongly implicates this enzyme in disease pathogenesis. HC, healthy control, **P = 0.0028. Error bars correspond to standard error of the mean.
Fig. 2.

Usp8 expression and localization inversely correlate with ubiquitinated inclusions in α-synucleinopathies. (A) Representative immunoblot showing increased levels of Usp8 but not AMSH relative to the actin loading control in the SN from patients with LB disease; a specific band is indicated by an arrowhead. (B) Quantification of Usp8 protein level in the human brain showed a significant increase in the SN but not AC of patients with Lewy body disease compared with controls HC, healthy control (**P = 0.0028, n = 8). Quantification of AMSH protein levels did not show a significant difference (C). Usp8-positive LBs and Lewy neurites in the AC (D) and SN (E). (Scale bar, 20 µm.) (F) No AMSH staining was seen in nigral LBs, as indicated by the arrowhead. (G) Double immunofluorescence and confocal imaging confirmed the colocalization of Usp8 (red) and α-synuclein (green) in nigral LBs. DAPI indicates nuclear staining in blue. (H) Quantification of Usp8-positive as a percentage of α-synuclein–positive inclusions (Usp8/α-Syn) in serial sections showed a significant increase in the SN compared with cortical areas (AC, EC) (***P = 0.0001, n = 14). (I) Negative correlation between Usp8-positive and ubiquitin-positive inclusions in the SN (***P = 0.0002, ρ = −0.5044). (J) Negative correlation of Usp8-positive and K63-linked ubiquitinated inclusions shown as the ratio K63/Ub (**P = 0.0038, ρ = −0.4186). Error bars correspond to standard error of the mean.
Usp8 Interacts with and Colocalizes with α-Synuclein in Neurons.
Usp8 has pleiotropic effects in the regulation of endosomal trafficking: It directly deubiquitinates endosomal cargo proteins (15–19), but is also implicated in the stability of the ESCRT complexes (21, 22). Because α-synuclein is the most abundant protein in LBs, one possible explanation for our observations in postmortem brains is that the colocalization of Usp8 with α-synuclein in LBs may signify a direct interaction between them. To address this question, we first cotransfected FLAG-tagged wild-type Usp8 or catalytically inactive (Cys786 to Ala) Usp8CA or an empty vector and untagged human α-synuclein in HEK-293T cells. When overexpressed, α-synuclein was immunoprecipitated from lysates of cells expressing wild-type or catalytically inactive Usp8 with anti-FLAG–tagged antibodies but not from lysates of a control expressing an empty vector (Fig. 3A). To test the possibility that monomeric or toxic forms of α-synuclein may bind to and inhibit the activity of Usp8, we assayed for such an effect by using an HA-tagged ubiquitin-bromide (HA-Ub-Br2) probe, which can conjugate to the catalytic cysteine of most DUBs and thus can be used to measure their activity. The amount of HA-Ub–modified Usp8 was not altered by the overexpression of wild-type or PD-related mutant forms of α-synuclein and did not correlate with increasing α-synuclein levels in HEK-293T cells (Fig. 3B). To investigate whether the interaction between Usp8 and α-synuclein may be of physiological relevance, we asked whether they colocalize on endosomal membranes, a known site of action for Usp8. We first confirmed in HEK-293T cells that although Usp8 was primarily cytosolic, it colocalized to a small extent with early but not late or recycling endosomes using either endogenous or transfected (Rab 5, Rab7, Rab11) markers (Fig. S4), in accordance with previous studies that reported a transient association of Usp8 with early sorting endosomes in HeLa cells (15). To investigate whether Usp8 interacts with α-synuclein at specific endosomal compartments, we used bimolecular fluorescence complementation signifying Usp8–α-synuclein interaction (green) with expression of endosomal Rabs (Rab5, Rab7, Rab11) tagged to a red fluorescent protein and assessed the extent of colocalization (yellow). This analysis showed that although the interaction of Usp8 and α-synuclein was detected in the cytosol, puncta of increased signal intensity colocalized principally with Rab5- rather than Rab7- or Rab11-positive endosomes (Fig. 3C). To investigate the normal neuronal localization of these proteins, we asked whether endogenous Usp8 and α-synuclein colocalize in induced pluripotent stem cell (iPSc)-derived human dopaminergic neurons. We first tested and confirmed that these cells express both neuronal (β-III tubulin) and dopaminergic [tyrosine hydroxylase (TH) and G protein-activated inward rectifier potassium channel 2 (GIRK2)] markers (Fig. S5). In these neurons, we found that Usp8 is detected in cytosolic puncta, which partly colocalized with α-synuclein and overlapped to some degree with markers of the early sorting (anti-EEA1) but not the late (anti-LBPA) endosome (Fig. 3D). Collectively, our data demonstrate that α-synuclein directly interacts and colocalizes with Usp8 in cells, including human iPSc-derived dopaminergic neurons, and suggest that this interaction may involve at least transiently association with primarily early (Rab5-positive) endosomes.
Fig. 3.

Usp8 interaction and colocalization with α-synuclein. (A) Wild-type (Usp8WT) or catalytically inactive (Cys786 to Ala) Usp8 was immunoprecipitated with untagged α-synuclein when expressed in HEK-293T cells. (B) Usp8 activity as measured by binding to HA-Ub-Br2 is not affected by increasing α-synuclein levels or expression of α-synuclein mutants. EV, empty vector; IP, immunoprecipitation; WCL, whole-cell extract. (C) Bimolecular fluorescence complementation signifying Usp8–α-synuclein interaction and expression of endosomal Rabs (Rab5, Rab7, and Rab11) tagged to a red fluorescent protein. Schematic depicting the bimolecular fluorescence complementation assay used in this study. Human Usp8 is tagged to the N-terminal fragment of Venus fluorescent protein (VFP) and α-synuclein is tagged to the C-terminal fragment of VFP. Quantification of the percentage overlap (Manders’ colocalization coefficient) between Usp8 and α-synuclein (green) with the indicated endosomal markers (red); n = 100 cells per condition; ***P < 0.001, one-way ANOVA. (D) Localization of Usp8 in relation to endosomal markers in human iPSc-derived dopaminergic neurons: triple labeling of neurons with TH (for detection of dopaminergic neurons; first column, blue), Usp8 (second column, green), and one of the following: (i) early endosome, EEA1; (ii) late endosome, LBPA; or (iii) α-synuclein (third column, red). Quantification of the percentage overlap (MCC) of Usp8 with the indicated markers in B; n = 50 neurons per condition. (Scale bar, 10 μm.) Error bars correspond to standard error of the mean.
Fig. S4.

(A) Colocalization of Usp8 with various endosomal markers in HEK-293T cells. Cells were either transfected with fluorescent reporters or stained with specific antibodies: (i) early endosome, EEA1; (ii) early endosome, Rab5; (iii) late endosome, Rab7; (iv) late endosome, LBPA; and (v) recycling endosome, Rab11, and also costained for Usp8 (second column, green). (Scale bar, 10 μm.) (B) Quantification of the percentage overlap (MCC) of Usp8 with EEA1 and LBPA (endogenously expressed and stained using antibodies). (C) Quantification of the percentage overlap (MCC) of Usp8 with Rab5, Rab7, and Rab11 (fluorescent reporters transiently expressed). (D) Quantification of the size of EEA1-positive puncta corresponding to early endosomes in HEK-293T cells expressing FLAG-tagged wild-type Usp8 or an empty vector. Early endosome size was quantified by confocal microscopy [LSM 710 (Carl Zeiss)] and ImageJ, with n = 20 cells imaged per condition. The presence of untagged GFP within the vector enabled the identification of transfected cells. Error bars correspond to standard error of the mean.
Fig. S5.

(A) Characterization of dopaminergic neurons derived from healthy control iPS cells. (A, i) Embryoid bodies (EB) were differentiated to neural precursor cells (PREC) and then manually passaged and matured to neurons (NEUR), as described in SI Materials and Methods. (A, ii and iii) Immunostaining for dopaminergic (TH and GIRK2) (ii) and neuronal (β-III tubulin) markers (iii). (Scale bars, 10 µm.) (B) The expression of neuronal markers (TH, Tau, and α-Syn) at the appropriate stage of differentiation was confirmed by immunoblotting.
Usp8 Deubiquitinates α-Synuclein in Human Cells.
To assess whether α-synuclein is a substrate of Usp8, we transiently transfected HA-tagged ubiquitin and FLAG-tagged Usp8 in HEK-293T. The overexpressed wild-type but not catalytically inactive Usp8 caused a marked decrease in the content of ubiquitinated endogenous α-synuclein in HEK-293T cells, as shown by HA pull-down and staining with two different anti–α-synuclein antibodies [Syn1 (Fig. 4A) and C20 (Fig. S6B)]. In addition, by expressing HA-tagged single-lysine ubiquitin, which is capable of only forming K63 or K48 linkages, we found that in human cells, overexpression of wild-type Usp8 deconjugated chains containing both K63 and K48 linkages on α-synuclein but showed preference for K63-linked ubiquitin chains (Fig. 4B). It is noteworthy that on the same immunoblots, total ubiquitination of immunoprecipitated substrates was similar across all of the experimental conditions, suggesting that the activity of Usp8 is selective against certain substrates. To further investigate the site of the Usp8–α-synuclein interaction, we generated a deletion mutant of Usp8 lacking the microtubule interacting and transport (MIT) domain, which was previously shown to mediate its endosomal association (21). Expression of this construct reduced but did not abolish the ability of Usp8 to deubiquitinate α-synuclein in HEK-293T cells (Fig. 4C). In addition, expression of the related DUB Usp7 had less activity against α-synuclein compared with Usp8 (Fig. 4C). Thus, regional differences in LB ubiquitination may be explained, at least partly, by differences in their content of Usp8, which deubiquitinates α-synuclein.
Fig. 4.

Usp8 deconjugates preferentially K63-linked ubiquitin chains on α-synuclein. (A) Expression of Usp8WT caused robust deubiquitination of endogenous α-synuclein compared with expression of Usp8CA or empty vector. Quantification of ubiquitinated α-synuclein in Usp8WT- relative to Usp8CA-expressing cells shown as arbitrary units (AU; *P = 0.0410, n = 3 biological replicates). WB, Western blot. (B) Coexpression of Usp8 with either K63 or K48 single-lysine ubiquitin showed that it deconjugated preferentially K63-linked chains on α-synuclein without any obvious difference in the total amount of ubiquitinated proteins between the immunoprecipitated samples (representative of n = 3 biological replicates). (C) Expression of a deletion mutant of Usp8 lacking the MIT domain required for endosomal localization (Usp8ΔMIT) reduced its activity against K63-linked ubiquitin chains on α-synuclein compared with Usp8WT. Usp7 had reduced activity against α-synuclein compared with Usp8 in cells (n = 5 biological replicates). (D) Recombinant α-synuclein was linked to uniform K63-linked chains in vitro by purified Nedd4 and incubated with the indicated deubiquitinases (50 nM). Robust deubiquitination was observed with Usp7 and Usp8. All seven enzymes were assessed for purity with Coomassie and activity by HA-Ub-Br labeling. Immunoblotting with anti-HA revealed a shift in the molecular mass of labeled DUBs, which indicates covalent binding to HA-Ub-Br. Error bars correspond to standard error of the mean.
Fig. S6.

(A) Ubiquitinated α-synuclein was immunoprecipitated with anti-HA antibodies in M17D cells stably expressing α-synuclein and transiently transfected with HA-Ub. (B) Overexpression of wild-type but not catalytically inactive Usp8 in HEK-293T cells removes ubiquitin chains on α-synuclein, as detected by an antibody against the last 20 amino acids of the protein (C20).
To further assess this model, we reconstituted the deubiquitination of α-synuclein in vitro. Building on our prior finding that the ubiquitin ligase Nedd4 forms K63-linked ubiquitin chains on α-synuclein (7), we compared the abilities of seven diverse cysteine protease DUBs at equimolar concentrations (OTUB1, OTUB2, JosD2, Usp2, Usp5, Usp7, and Usp8) to digest K63-linked ubiquitin chains on α-synuclein. Although all seven DUBs were active in vitro, as evidenced by their binding to HA-ubiquitin-bromide, only Usp8, and the closely related enzyme Usp7, could hydrolyze the K63-linked chains on α-synuclein (Fig. 4D and Fig. S7A).
Fig. S7.

(A) Time course of α-synuclein deubiquitination by recombinant Usp7 and Usp8. K63-linked ubiquitin chains were conjugated on α-synuclein using purified Nedd4 as the E3. An equal amount of ubiquitinated α-synuclein was incubated with each enzyme (50 nM). Aliquots were taken every 10 min, resolved on SDS/PAGE, and immunoblotted with anti–α-synuclein antibody (C20). (B) SH-SY5Y cells were transduced with Usp8 shRNA or Scr shRNA control. Eight days after transduction, the cells were treated for 24 h with 25 μM chloroquine or 12 h with 1 μΜ lactacystin. α-Synuclein levels were analyzed in total lysates after immunoblotting with the C20 antibody and are presented as the ratio to the actin loading control. Error bars correspond to standard error of the mean.
Deubiquitination by Usp8 Regulates α-Synuclein Degradation by the Lysosome.
Deubiquitination by Usp8 was previously reported either to slow the degradation of substrates and promote their recycling (16–19) or facilitate endosomal trafficking and lysosomal degradation (21, 22). Because Usp8 is up-regulated in neurons with LB pathology, we tested the effect of increased Usp8 expression on α-synuclein clearance in the presence of cycloheximide to prevent protein synthesis. Overexpression of Usp8 in HEK-293T cells reduced the rate of clearance of endogenous α-synuclein (Fig. 5A). To test whether these findings on the function of Usp8 in α-synuclein degradation were restricted to these nonneuronal cells or caused only by DUB overexpression, we generated and validated lentiviral particles expressing shRNA against Usp8 in SH-SY5Y neuroblastoma cells, a cell line that expresses dopaminergic markers. Upon transduction, this construct markedly reduced the levels of Usp8 in SH-SY5Y. Strikingly, 7–8 d after transduction with Usp8 shRNA, endogenous α-synuclein levels were reduced by ∼35% (Fig. 5B; P = 0.0031, n = 5) and ubiquitinated α-synuclein was detected by immunoprecipitation (Fig. 5C). The prediction of these experiments is that α-synuclein trafficking to and degradation by the lysosomal pathway are increased when Usp8 is knocked down. To investigate this model, we treated SH-SY5Y cells lacking Usp8 or Scr shRNA controls with chloroquine (50 μM) or lactacystin (5 μM) for 8 h and measured the level of α-synuclein in the cytosol and membrane fractions that are enriched in endosomal/autophagic and lysosomal compartments. We found that at baseline the level of α-synuclein in the cytosolic and membrane fractions was significantly reduced when Usp8 is knocked down, as expected from our data in total lysates. In the membrane fraction that is enriched in endosomes/autophagosomes/lysosomes, increased α-synuclein was detected within 8 h of treatment with chloroquine but not lactacystin (Fig. 5D). Similar results were obtained in whole lysates following a prolonged treatment with 25 μM chloroquine (Fig. S7B). These data indicate that blocking the lysosome but not the proteasome increases α-synuclein in cells lacking Usp8, suggesting that they exhibit accelerated lysosomal degradation of α-synuclein.
Fig. 5.

Usp8 regulates the degradation of α-synuclein by the lysosome. (A) Cycloheximide chase of endogenous α-synuclein showed that its rate of clearance was reduced at 7 h when wild-type Usp8 was expressed in HEK-293T cells (**P = 0.0091, n = 3 biological replicates). (B and C) Lentiviral-mediated shRNA knockdown of Usp8 in SH-SY5Y cells reduced endogenous α-synuclein levels by 35% relative to the actin loading control compared with Scr shRNA controls (**P = 0.0031, n = 5 biological replicates) (B) and increased the amount of ubiquitinated α-synuclein as evidenced by immunoblotting of immunoprecipitated α-synuclein with anti-ubiquitin and anti-K63 antibodies (C). No smear was seen when anti-HA was used as an IgG control. (D) Lysates isolated from Usp8 knockdown and Scr shRNA-treated SH-SY5Y cells were fractionated into cytosolic and membrane fractions and tested for α-synuclein levels at baseline and following 8 h of treatment with either 50 μM chloroquine (CQ) or 5 μM lactacystin (Lact). Accumulation of α-synuclein was observed in the membrane fraction, which is enriched in endosomal/autophagic–lysosomal compartments in Usp8 knockdown cells treated with chloroquine, suggesting that there is accelerated lysosomal degradation of α-synuclein in these cells (*P = 0.019, n = 3). Error bars correspond to standard error of the mean.
Usp8 Knockdown Rescues α-Synuclein–Induced Toxicity in the Drosophila Model.
Our neuropathological and cell-based studies suggest that in α-synucleinopathies, increased deubiquitination by Usp8 in nigral neurons may be harmful, at least partly by increasing α-synuclein levels. We tested this prediction by expressing α-synuclein in the fly eye, which has proven to be a valuable experimental model of α-synuclein–induced toxicity (23, 24). This assay showed that the rough eye phenotype caused by ectopic expression of either human wild-type α-synuclein or A53T mutant α-synuclein was prevented by concomitant knockdown of Usp8 (Fig. 6 A and B). In this model, the level of α-synuclein mRNA measured by quantitative (q)PCR was not lower in single- than in double-transgenic flies, thus excluding a phenotype due to GAL4 dilution in double-transgenic flies (Fig. 6 A and B). In control experiments, we showed that the rough eye phenotype was not observed with the GMR-GAL4 driver alone (Fig. 6C). In addition, we showed that the eye phenotype was not improved when A53T α-synuclein was coexpressed with RNAi against the other endosomal DUB AMSH (Fig. 6C) or the proteasomal DUBs Usp14 or Usp47 (Fig. S8A). In addition, the wild-type α-synuclein phenotype was worsened when coexpressed with an RNAi against the ESCRT I protein Vps28, a downstream interactor in endosomal trafficking (Fig. 6C). Importantly, we also showed that the rough eye phenotype caused by two pathogenic proteins, expanded Ataxin 3 (Fig. 6D) and expanded huntingtin (Fig. S8B), was not rescued by Usp8 knockdown. We confirmed the functional efficiency of Usp8 knockdown by detecting the previously documented wing defect (17) when the RNAi was expressed under the MS1096-GAL4 driver (Fig. S8C).
Fig. 6.

Knockdown of endogenous Usp8 in Drosophila protects against α-synuclein–induced toxicity. (A and B) Overexpression of α-synuclein caused a rough eye phenotype, which was more severe in flies expressing A53T mutant α-synuclein, as detected by SEM. This phenotype was rescued in double-transgenic flies with eye-specific Usp8 knockdown and either wild-type (A) or A53T mutant α-synuclein (B). *P < 0.05. Quantitative PCR did not show a reduction in α-synuclein mRNA levels between single- and double-transgenic lines to explain the phenotype. (C) Knockdown of AMSH did not rescue this phenotype, whereas knockdown of Vps28 made it worse. (D) Usp8 knockdown did not affect the expanded Ataxin 3 phenotype. (E) Representative immunoblot of fractionated lysate (C, cytosol; P, pellet) from A53T mutant α-synuclein flies and flies expressing A53T mutant α-synuclein with Usp8 knockdown (+Usp8 KD). Quantitative band densitometry showed that, relative to the actin loading control, protein levels of either wild-type or A53T mutant monomeric α-synuclein (indicated by an asterisk) were significantly reduced in flies coexpressing Usp8 RNAi (n = 4 biological replicates) in the absence of a reduction in mRNA levels, as shown in A and B. *P < 0.05; ****P < 0.0001. (F) Accelerated loss of climbing ability was seen in transgenic flies expressing human A53T mutant α-synuclein in dopaminergic neurons (ddc-GAL4 driver) with increasing age. The climbing ability of double-transgenic lines expressing A53T α-synuclein with Usp8 knockdown in dopaminergic cells was significantly improved (shown with asterisks) compared with A53T α-synuclein–expressing flies and was similar to the control genotype, ddc-GAL4/+ (*P = 0.0381 for day 10; **P = 0.01 for day 12; *P = 0.0159 for day 14; n = 50 flies per group). (G) Knockdown of JosD2 or AMSH in dopaminergic neurons slightly worsened the A53T mutant α-synuclein phenotype. (H) Expression of A53T mutant α-synuclein but not control constructs in dopaminergic neurons (ddc-GAL4 driver) led to loss of TH-immunoreactive neurons in the PPM1/2 cluster, which was prevented by concomitant Usp8 knockdown (***P < 0.001). NS, nonspecific band. Error bars correspond to standard error of the mean.
Fig. S8.

(A) The rough eye phenotype of A53T α-synuclein was not rescued in flies coexpressing in the eye RNAi to knock down AMSH, Usp14, or Usp47. (B) The rough eye phenotype of pathogenic expanded huntingtin (Htt) was not rescued in the corresponding double-transgenic line expressing RNAi against Usp8 (Htt/Usp8 KD). (C) The functional efficiency of Usp8 knockdown was confirmed by detecting the previously documented wing defect when the RNAi was expressed under the MS1096-GAL4 driver.
To assess the relevance of our finding in SH-SY5Y cells that endogenous Usp8 knockdown reduces α-synuclein levels (Fig. 5B) in the Drosophila model using an alternative RNAi approach, we performed serial fractionation of fly head lysates and measured the intensity of the monomeric α-synuclein band as a ratio to the actin loading control in the cytosolic and pelleted fractions of single- and double-transgenic flies. When Usp8 was knocked down, wild-type or A53T α-synuclein protein level was reduced (Fig. 6E) even though α-synuclein mRNA levels were unchanged (Fig. 6 A and B).
Finally, to corroborate our findings in the eye using an alternative readout of toxicity and to investigate whether similar mechanisms function in dopaminergic neurons, we knocked down Usp8 specifically in these neurons of the Drosophila model of α-synuclein toxicity using the dopamine decarboxylase ddc-GAL4 driver. Strikingly, we found that knockdown of Usp8 (Fig. 6F), but not knockdown of AMSH or JosD2 (Fig. 6G), prevented the age-dependent locomotor defect caused by expression of human A53T mutant α-synuclein in dopaminergic neurons. Interestingly, Usp8 knockdown in dopaminergic cells reduced the loss of the TH-immunoreactive PPM1/2 cluster of dopaminergic neurons (Fig. 6H) that is most vulnerable in the Drosophila α-synuclein model (23–25) and typically correlates with the locomotor deficit.
Discussion
Although the presence of ubiquitin has long been recognized as a characteristic feature of LBs, the origins and nature of LB-associated ubiquitin conjugates have been unclear. The present data indicate that ubiquitination in these inclusions is composed primarily of K63-linked conjugates and is regionally distinct. These findings challenge the long-held view that ubiquitin immunoreactivity represents a nonspecific modification or the end product of impaired proteasome function. A targeted screen for potential interactors involved in the shuttling of K63-linked chains revealed up-regulation and pathological localization of the DUB Usp8, whose content correlated inversely with the extent of LB ubiquitination in diseased brains. To our knowledge, this dramatic pathological staining pattern identifies Usp8 as one of the best LB surrogates in pigmented neurons. Accordingly, transcriptomic analysis in immunolaser-captured, microdissected nigral neurons showed up-regulation of Usp8 mRNA in neurons with LBs (26). Collectively, these data in human pathological specimens firmly establish Usp8 as a critical factor in the pathogenesis of sporadic PD and suggest an important role for K63-linked polyubiquitin chains in α-synuclein degradation. It is noteworthy that K63-linked ubiquitin chains were detected in about half of the ubiquitin-immunoreactive LBs, suggesting that additional linkages, other than K48-linked chains that we excluded, may be present. However, based on a number of previous studies by us and others that identified primarily monoubiquitinated α-synuclein in LB extracts by immunoblotting (10, 11, 27), it is most likely that the remaining immunoreactivity represents singly or multiply monoubiquitinated α-synuclein, which could be the end product of a deubiquitination reaction.
Usp8 shows some selectivity for recombinant K63–ubiquitin linkages in vitro (28, 29). However, compared with other DUBs of the cysteine protease type, Usp8 as well the closely related enzyme Usp7 showed strong capacity to hydrolyze K63-linked ubiquitin on α-synuclein in vitro. In yeast, the Usp8 ortholog Doa4 is an essential DUB for the maintenance of the free ubiquitin pool upon which endosomal trafficking is specifically dependent (30, 31). In mammalian cells, Usp8 regulates the recycling of membrane-associated proteins in a process that involves K63-linked substrate ubiquitination (16–19). Thus, normally in cells, K63-linked ubiquitin chains are continually being hydrolyzed by Usp8, which serves an important function in endosomal–lysosomal trafficking. We and others have previously shown that the ubiquitin ligase Nedd4 and its yeast ortholog Rsp5 conjugate K63-linked chains on α-synuclein, promoting its trafficking by the endosomal route (7, 32, 33), and that α-synuclein is enriched in endosomal compartments (34, 35). At the synapse, Nedd4 is responsible for formation of K63-linked chains that is antagonized by Usp8 (36), and in vitro these enzymes have opposing activities on α-synuclein ubiquitination. Because α-synuclein functions in synaptic vesicle release by transient association with membranes (37), it is likely that these enzymes are critical in the regulation of its physiological function in the brain. Consistent with this notion, lysine at position 96 on α-synuclein, which is the predominant lysine ubiquitinated by Nedd4 in vitro (7), was also identified as the primary ubiquitination site of α-synuclein in rat brain synaptosomes by an antibody-based proteomic analysis (38). Although further work is needed to understand the molecular basis of this regulation, the close proximity of Usp8 to endosomes suggests that its function in α-synuclein turnover may involve reversible endosomal localization, as shown for other substrates (15–19). Accordingly, in cells, Usp8 and α-synuclein interaction occurs at least partly in endosomes, and deconjugation of preferentially K63-linked ubiquitin chains on α-synuclein is enhanced by the N-terminal MIT domain of Usp8 that mediates its endosomal anchoring (21), whereas knockdown of Usp8 accelerates the degradation of α-synuclein by lysosomes. However, the slow turnover rate of α-synuclein and its transient association with membranes differs substantially from the endocytosis of other Usp8 substrates such as transmembrane receptors that are recycled over minutes upon ligand binding, suggesting that the process described here may involve distinct shuttling factors. K63-linked ubiquitin chains also mediate the autophagic degradation of aggregated proteins (13), and their presence in LBs may also signify an attempt to clear misfolded α-synuclein after its aggregation. It is likely that Usp8 is important in the trafficking of both normal as well as misfolded α-synuclein, opposing its clearance by either an endosomal– or autophagic–lysosomal route, respectively. For example, the Nedd4 ortholog in yeast Rsp5, which functions in endosomal trafficking, also regulates ubiquitin-mediated autophagy (39), and aggregated α-synuclein is also ubiquitinated by Nedd4 in vitro (24). Whether additional DUBs such as Usp7 serve a direct role in α-synuclein pathobiology requires further investigation.
Why Usp8 is induced in neurons with LBs and whether this is a maladaptive response to enhance recycling of ubiquitin or other substrates is unclear. It is noteworthy that both in vivo and in cultured cells, Usp8 knockdown reduced total α-synuclein content, even though α-synuclein is degraded by multiple pathways (6, 7, 40). Consequently, impaired processing of α-synuclein by Usp8 could contribute to its pathological accumulation in LBs. As indicated by hereditary cases with α-synuclein multiplications, a small increase of less than 1.5-fold in α-synuclein protein levels is sufficient to cause neurodegeneration with LBs (4). It is thus possible that in disease, induction of Usp8 in neurons with LBs promotes the accumulation and toxicity of α-synuclein. This conclusion is strongly supported by our findings in flies that Usp8 knockdown, but not knockdown of other DUBs, protected against different readouts of α-synuclein toxicity (i.e., rough eye phenotype, locomotor defects, and cell loss) at least partly by reducing α-synuclein levels. Interestingly, Usp8 expression in the normal mouse brain is higher in neurons of the compacta compared with those in the reticulate part of the substantia nigra (41). Given that Usp8 slows the degradation of α-synuclein, this differential expression may account at least in part for the selective vulnerability of the compacta region of the substantia nigra to α-synuclein accumulation and neurodegeneration.
In summary, we have shown that ubiquitin immunoreactivity in LBs is composed of K63-linked ubiquitin chains and is regionally distinct and have identified the DUB Usp8 as one potentially critical regulator of this pattern. Collectively, our biochemical and in vivo studies suggest that one mechanism by which up-regulation of Usp8 could contribute to PD pathogenesis is by opposing the clearance of α-synuclein. Thus, although Usp8 serves some essential functions, in α-synucleinopathies it may be a potential therapeutic target for small-molecule inhibitors.
Materials and Methods
Staining of Human Brain Sections.
Brain tissue from 20 patients [4 incidental Lewy body (ILB), 5 PD, and 11 PD with dementia (PDD)/Lewy body disease (LBD)] was obtained from the brain bank of John Radcliffe Hospital (Table S1). Research on human tissue was conducted under the ethics approval of the Oxford Brain Bank (Reference: 15/SC/0639, UK South Central-Oxford C Research Ethics Committee) where tissue was collected with informed consent. Tissue was formalin-fixed and paraffin-embedded, and serially sectioned at 7 µm with a sliding microtome. Staining was performed using the Histostain-Plus Detection Kit (Life Technologies) and HISTAR Detection Kit STAR3000C (AbD Serotec), as detailed in SI Materials and Methods. Lewy body pathology was assessed according to the recommendations of the third report of the Dementia with Lewy Bodies Consortium (42) and also by Braak stage (1). For fluorescence double labeling, following dewaxation and rehydration, sections underwent epitope retrieval and preincubation with 10% (vol/vol) goat serum at room temperature for 1 h before incubation with primary antibodies overnight at 4 °C. For double immunofluorescence, Alexa Fluor secondary antibodies (1:500; Life Technologies) at 488 nm (green), 568 nm (red), and 594 nm (far red) were used. Autofluorescence background was blocked by Sudan black, and sections were mounted using fluorescence mounting medium (Dako) containing 1 μg/mL DAPI (Sigma).
Immunoprecipitation.
Cell-lysate supernatants were incubated overnight at 4 °C with anti-HA or anti-FLAG antibodies (Sigma) or 2F12 anti–α-synuclein antibodies. Equilibrated protein G Sepharose beads (P3926; Sigma) were then added for a further 2 h at 4 °C. The beads were washed three times with Tris-buffered saline containing 0.1% Triton X-100, resuspended in NuPAGE Tris-acetate SDS running buffer (Life Technologies) containing NuPAGE Sample Reducing Agent (Life Technologies), and heated at 95 °C for 10 min before loading onto the gel.
Immunoblotting.
Brain samples were homogenized in ice-cold lysis buffer (50 mM Tris⋅HCl, pH 7.4, 150 mM sodium chloride, 1% Nonidet P-40, 0.1% SDS, 1 mM PMSF, 1 mM N-ethylmaleimide; all from Sigma) and centrifuged for 10 min at 15,000 × g at 4 °C. For fractionation of SH-SY5Y, the cells were resuspended in buffer (150 mM NaCl, 10 mM Tris, pH 7.4, complete protease inhibitor mixture, 1 mM PMSF, and 1 mM N-ethylmaleimide) and syringed with a 25-gauge needle 10 times on ice followed by a brief vortexing, and the process was repeated three times with 2-min intervals on ice. Lysates were spun down at 600 × g for 5 min to remove debris, and the supernatants were centrifuged at 100,000 × g for 1 h. The supernatant was designated the cytosolic fraction, and the pellet was resuspended in lysis buffer containing detergents (150 mM NaCl, 10 mM Tris, pH 7.4, complete protease inhibitor mixture, 1 mM PMSF, 1 mM N-ethylmaleimide, 1% Nonidet P-40, 0.1% SDS) and centrifuged at 100,000 × g for 30 min. The supernatant was designated the membranous fraction. The protein content of the resulting supernatants was measured using the BCA assay (Thermo Fisher Scientific). Equal amounts of protein were mixed with NuPAGE Tris-Acetate SDS Running Buffer (Life Technologies) containing NuPAGE Sample Reducing Agent (Life Technologies) and heated at 95 °C for 10 min before being separated by 4–12% SDS/PAGE (NuPAGE Novex Bis-Tris Gels; Life Technologies) and electrotransferred onto a nitrocellulose membrane (Amersham Protran 0.45; GE Healthcare) in transfer buffer [25 mM Tris, 192 mM glycine, 20% (vol/vol) methanol]. After blocking with 4% nonfat powdered milk in PBS containing 0.1% Tween 20 (Sigma) for 1 h, primary antibodies were incubated with the membrane overnight at 4 °C and binding was visualized after washing with peroxidase-labeled anti-rabbit or anti-mouse secondary antibodies (1:10,000; GE Healthcare) and enhanced chemiluminescence (GE Healthcare). Relative quantitation of the protein of interest to the loading control was measured using ImageJ (NIH).
Cell Culture and Transfection.
BE(2)-M17 cells (M17D; ATCC no. CRL-2267) stably expressing α-synuclein and HEK-293T and SH-SY5Y cells, which express α-synuclein endogenously, were maintained at 37 °C and 5% CO2 in DMEM containing 10% (vol/vol) FCS (Sigma) and 1% (vol/vol) penicillin/streptomycin/amphotericin B (Life Technologies). Cells were grown to 60–80% confluency and transfected with plasmid DNA using Lipofectamine 2000 (Life Technologies). Plasmids used and mutagenesis are described in detailed in SI Materials and Methods. Thirty-six hours after transfection, cells were rinsed with PBS and scraped into ice-cold lysis buffer (50 mM Tris⋅HCl, pH 7.4, 150 mM sodium chloride, 1% Nonidet P-40, 0.1% SDS, 1 mM PMSF, 1 mM N-ethylmaleimide, complete protease inhibitor mixture; all from Sigma). Lysates were rotated for 20 min at 4 °C and then centrifuged at 4 °C at 13,000 × g for 5 min. For cycloheximide chase, HEK-293T cells were treated with 20 μg/mL cycloheximide for 7 h before lysis without obvious cell death. For knockdown experiments using lentiviruses expressing shRNA, SH-SY5Y cells were transduced for 24 h and lysed 7–8 d after transduction. For DUB activity measurements, cells were lysed in buffer (50 mM Tris⋅HCl, pH 7.4, 5 mM magnesium chloride, 250 mM sucrose, 1 mM DTT, 2 mM ATP) and an equal amount of total protein was incubated with HA-ubiquitin-Br2, as described previously (29).
Design and Construction of Usp8 or Scrambled shRNA Lentiviral Vectors.
The rat Usp8 sequence (GenBank accession no. NM_001106502.1) was used to generate the Usp8 shRNA (5′-CCGCTCGAGAAAAAAGCTGAGATCTCAAGGCTTTCTTGACAGGAAGA GAAAGCCTTGAGATCTCAGCCAAAACAAGGCTTTTCTCCAAGG-3′), which was predicted to knock down the human protein, as well as a scrambled shRNA (5′-CCGCTCGAGAAAAAA GGCACATTAGGAACCATACATTGACAGGAAGATGTATGGTTCCTAATGTGCCCAAAACAAGGCTTTTCTCCAAGG-3′). A short hairpin expression cassette comprised a sense and an antisense (antisense specific to the target mRNA) strand spaced by a loop sequence, RNA polymerase III transcription termination signal, and XhoI restriction site. A sequence coding for the mouse U6 promoter was inserted upstream of the antisense strand with a SpeI site. A PCR-based method was used to generate double-stranded shRNA expression cassettes using the pSilencer 1.0-U6 expression vector (Life Technologies) as a transcription template and reverse primer encoding the shRNA oligonucleotides. These were subcloned into the lentiviral backbone pRRLsincppt.U6.CMV.EGFP.wpre (43) predigested with SpeI and XhoI, generating the pRRLsincppt.U6.rUsp8shRNA.CMV.EGFP.wpre or pRRLsincppt.U6.scrambledshRNA.CMV.EGFP.wpre construct. The constructs were tested for efficiency in human cell lines and found to be effective in reducing the content of Usp8.
Generation of Human iPSc-Derived Neurons.
Dopaminergic neurons were derived from healthy control iPS cell lines that were previously described (44), as detailed in SI Materials and Methods.
Colocalization Studies.
Four- to 8-wk-old iPSc-derived neurons were plated onto Matrigel (BD Biosciences)-coated glass coverslips and HEK cells were plated onto poly-l-lysine–coated glass coverslips. For Usp8 colocalization with Rab proteins in HEK-293T cells, fluorescently tagged plasmids expressing Rabs were transfected. For the bimolecular fluorescence complementation assay in HEK-293T cells, cells were cotransfected with WT Usp8-VN and WT Syn-VC and fluorescently tagged plasmids expressing Rabs. For localization of endogenous proteins cells were probed with rabbit anti-Usp8 polyclonal antibody (HPA004869; 1:3,000; Sigma), mouse anti–α-synuclein monoclonal antibody (610787; 1:500; BD Biosciences), goat anti-TH polyclonal antibody (101853; 1:400; Abcam), mouse anti-EEA1 monoclonal antibody (610457; 1:500; BD Biosciences), mouse anti-LBPA monoclonal antibody (6C4; 1:200; Echelon Biosciences), and mouse anti-Rab11 monoclonal antibody (A-6; 1:200; Santa Cruz) in PBS and visualized with Alexa 305-, 488-, 546-, or 568-conjugated goat or donkey anti-mouse, -rabbit, or -goat secondary antibodies (1:500; Life Technologies). Confocal imaging is detailed in SI Materials and Methods. To measure the fractional overlap of the Usp8 stain with that of other markers, we have reported the Manders’ colocalization coefficient (MCC), which measures the fraction of one protein that colocalizes with another, poorly measured by the Pearson’s correlation coefficient (45). MCC values range from zero (uncorrelated distributions of two probes with one another) to one (perfect colocalization of two images). No primary antibody controls were included for background subtraction.
Deubiquitination of Ubiquitinated α-Synuclein in Vitro.
Purified α-synuclein was ubiquitinated by Nedd4, as described previously (7). α-Synuclein was incubated with His-Nedd4, His-UbcH5b, His-E1, and ubiquitin in the presence of ATP. After ubiquitination, His-tagged enzymes were removed by incubation with Ni-NTA agarose (Qiagen). The unbound ubiquitinated α-synuclein fraction was used as the substrate of the in vitro deubiquitination reaction. Ubiquitinated α-synuclein was incubated with the indicated DUBs (50 nM) in 50 mM Tris and 2 mM DTT at 37 °C for 90 min. Each sample was analyzed by SDS/PAGE and immunoblotting using an anti–α-synuclein antibody (C20; 1:1,000; Santa Cruz). The recombinant DUBs (OTUB1, OTUB2, JosD2, Usp2 core, Usp5, Usp7, and Usp8 core) used in this study were previously characterized extensively (29), and their activity was confirmed again using the active site-directed probe HA-Ub-Br2, as described previously (29).
Drosophila Genetics.
The UAS-α-synuclein wild-type (8146) and A53T mutant (8148), UAS-Usp8 RNAi (38982), UAS-Usp14 RNAi (53262), UAS-Usp47 RNAi (44645), and UAS-Ataxin 3 (8150) with expanded polyQ were obtained from the Bloomington Drosophila Stock Center. The UAS-Vps28 RNAi (31894), UAS-JosD2 RNAi (108379), and UAS-AMSH RNAi (108622) were obtained from the Vienna Drosophila Resource Center (VDRC). The GAL4 UAS expression system was used to overexpress these transgenes either in dopaminergic neurons at 25 °C using the ddc-GAL4 driver (kindly provided by A. Lin and G. Miesenbock University of Oxford, Oxford, UK) or specifically in the eye using GMR-GAL4 (provided by I. Davis, University of Oxford, Oxford) at 29 °C. The following genotypes were used: (i) +/+; ddc-GAL4/+, (ii) +/+; ddc-GAL4/UAS-A53T α-synuclein, (iii) UAS-Usp8 RNAi/+; ddc-GAL4/+, (iv) UAS-Usp8 RNAi/+; ddc-GAL4/UAS-A53T α-synuclein, (v) GMR-GAL4/+, (vi) GMR-GAL4/+; UAS-α-synuclein/+, (vii) GMR-GAL4/+; UAS-A53T α-synuclein/+, (viii) GMR-GAL4/UAS-Usp8 RNAi; +/+, (ix) UAS-Usp8 RNAi/GMR-GAL4; UAS-α-synuclein/+, (x) Usp8 RNAi/GMR-GAL4; UAS-A53T α-synuclein/+, (xi) MS1096-GAL4/Y, (xii) MS1096-GAL4/Y; UAS-Usp8 RNAi/+, (xiii) UAS-α-synuclein/GMR-GAL4; UAS-Vps28 RNAi/+, (xiv) UAS-Ataxin 3/GMR-GAL4; UAS-Usp8 RNAi/+, (xv) GMR-GAL4/UAS-Ataxin 3; +/+, (xvi) UAS-AMSH RNAi; ddcGAL4/+, (xvii) UAS-JOSD2 RNAi; ddcGal4/+, (xviii) GMR-GAL4/UAS-AMSH RNAi; +/+, (xix) GMR-GAL4/UAS-AMSH RNAi; UAS-A53T α-synuclein/+, (xx) UAS-JOSD2 RNAi/+; UAS-A53T α-synuclein/ddcGAL4, (xxi) UAS-AMSH RNAi/+; UAS-A53T α-synuclein/ddcGAL4, (xxii) GMR-GAL4/+; UAS-Usp14 RNAi/+, (xxiii) GMR-GAL4/+; UAS-Usp47 RNAi/+, (xxiv) GMR-GAL4/+; UAS-Usp14 RNAi/UAS-A53T α-synuclein, and (xxv) GMR-GAL4/+; UAS-Usp47 RNAi/UAS-A53T α-synuclein. The experiments were repeated in two independently derived transgenic lines and carried out with the experimenter blinded to the sample genotypes throughout the analysis.
Scanning Electron Microscopy of the Drosophila Eye.
Age-matched male flies were fixed in 70% ethanol. The flies were then dehydrated in 100% ethanol, dried, and mounted on SEM stubs. The samples were then sputter-coated with gold and imaged using a JEOL JSM-6390 scanning electron microscope. Eyes were examined for abnormal bristle orientation, ommatidial fusion or pitting, and disorganization of the ommatidial array.
Fractionation of Drosophila Head Lysates.
All steps were performed at 4 °C. For each genotype, five male adult heads were homogenized in lysis buffer (150 mM NaCl, 10 mM Tris, pH 7.4, complete protease inhibitor mixture, 1 mM N-ethylmaleimide). Crude lysates were centrifuged at 4,000 × g for 5 min. Supernatants were then centrifuged at 100,000 × g for 1 h. The resulting supernatant was designated the cytosolic fraction, and the pellet was resuspended in loading buffer.
Climbing Assays.
Locomotor function was assayed using a startle-induced negative geotaxis assay. Ten male flies were placed in each vial and five vials were used per line in each experiment (total 50 flies per line). Each vial was tapped 10 times and the percentage of flies above 6 cm was recorded after 4 s.
Quantitative RT-PCR.
Five fly heads from each line were homogenized for 30 s using a tissue homogenizer, as detailed in SI Materials and Methods.
Confocal Imaging of Fly Brains.
Fly brains were dissected at 25 d posteclosion in ice-cold Schneider’s fly medium and fixed in 4% paraformaldehyde (PFA). They were washed in PBS containing 0.05% Triton X-100 (PBS-T), blocked in goat serum for 1 h, and then incubated for 48 h at 4 °C with monoclonal mouse anti-TH (1:500) followed by PBS-T washes and Alexa 488-coupled goat anti-mouse IgG (1:1,000) for a further 48 h at 4 °C. Stained brains were mounted in ProLong Gold antifade mountant. Z stacks of the brain were obtained on a Zeiss LSM 780 confocal microscope using a 25× objective (1.4× digital zoom) at 1-μm steps and Z-projected. Dopaminergic neuronal clusters were identified and counted.
Statistical Analysis.
The statistical analysis was performed using JMP software (SAS; version 10.0) and Prism (GraphPad). Outliers were identified using an outlier box plot and were excluded from the calculations where observed. All data were examined for distribution, and statistical tests were chosen accordingly. For normally distributed data, a t test or a one-way ANOVA was used. For nonparametric data, the Wilcoxon–Mann–Whitney and Kruskal–Wallis rank-sum tests were performed to assess differences in mRNA or protein levels and for climbing assays. The null hypothesis was rejected at a significance level of P = 0.05. Exponential data were log-converted if reasonable before statistical calculation was performed. Correlations were estimated with a pairwise correlation method for normally distributed data and with Spearman’s rho for nonparametric data. Colocalization was estimated as the fractional overlap of two markers using Manders’ colocalization coefficient.
SI Materials and Methods
Staining of Human Brain Sections.
Brain tissue from 20 patients (4 ILB, 5 PD, and 11 PDD/LBD) was obtained from the brain bank of John Radcliffe Hospital (Table S1). Tissue was formalin-fixed and paraffin-embedded, and serially sectioned at 7 µm with a sliding microtome. Staining was performed using the Histostain-Plus Detection Kit (Life Technologies) and HISTAR Detection Kit STAR3000C (AbD Serotec). Sections were dewaxed and rehydrated, and antigen unmasking was achieved by various retrieval methods. Endogenous peroxidase activity was quenched in 3% (vol/vol) hydrogen peroxide followed by incubation with primary antibodies in Tris-buffered saline containing 0.05% Triton X-100 (TBS-T) at 4 °C overnight. Washes were performed in TBS-T and PBS. The color reaction was developed with diaminobenzidine; counterstaining was done in hematoxylin and eosin. A standard positive control section was stained with every set of sections. Following washes and dehydration, Histo-Clear sections were mounted with glass coverslips.
Lewy body pathology was assessed according to the recommendations of the third report of the Dementia with Lewy Bodies Consortium (42) and also by Braak stage (1). For fluorescence double labeling, following dewaxation and rehydration, sections underwent epitope retrieval and preincubation with 10% goat serum at room temperature for 1 h before incubation with primary antibodies overnight at 4 °C. For double immunofluorescence, Alexa Fluor secondary antibodies (1:500; Life Technologies) at 488 nm (green), 568 nm (red), and 594 nm (far red) were used. Autofluorescence background was blocked by Sudan black and sections were mounted using fluorescence mounting medium (Dako) containing 1 μg/mL DAPI (Sigma).
Immunoprecipitation.
Cell-lysate supernatants were incubated overnight at 4 °C with mouse monoclonal anti-HA (Sigma) and anti-FLAG antibodies (Sigma) and mouse monoclonal anti–α-synuclein antibodies (2F12; a kind gift of D. Selkoe, Harvard Medical School, Boston). Equilibrated protein G Sepharose beads (P3926; Sigma) were then added for a further 2 h at 4 °C. The beads were washed three times with TBS-T, resuspended in 1× NuPAGE LDS sample buffer with NuPAGE Sample Reducing Agent (Life Technologies), and heated at 95 °C for 10 min before loading onto the gel.
Immunoblotting.
Brain samples were homogenized in ice-cold lysis buffer (50 mM Tris⋅HCl, pH 7.4, 150 mM sodium chloride, 1% Nonidet P-40, 0.1% SDS, 1 mM PMSF, 1 mM N-ethylmaleimide; all from Sigma) and centrifuged for 10 min at 15,000 × g at 4 °C. For fractionation of SH-SY5Y, the cells were resuspended in buffer (150 mM NaCl, 10 mM Tris, pH 7.4, complete protease inhibitor mixture, 1 mM PMSF, 1 mM N-ethylmaleimide) and syringed with a 25-gauge needle 10 times on ice followed by brief vortexing and the process was repeated three times with 2-min intervals on ice. Lysates were spun down at 600 × g for 5 min to remove debris and the supernatants were centrifuged at 100,000 × g for 1 h. The supernatant was designated the cytosolic fraction, and the pellet was resuspended in lysis buffer containing detergents (150 mM NaCl, 10 mM Tris, pH 7.4, complete protease inhibitor mixture, 1 mM PMSF, 1 mM N-ethylmaleimide, 1% Nonidet P-40, 0.1% SDS) and centrifuged at 100,000 × g for 30 min. The supernatant was designated the membranous fraction. The protein content of the resulting supernatants was quantified for total protein (BCA assay; Thermo Fisher Scientific). An equal amount of protein was mixed with NuPAGE Tris-Acetate SDS Running Buffer (Life Technologies) containing NuPAGE Sample Reducing Agent (Life Technologies) and heated at 95 °C for 10 min before being separated by 4–12% SDS/PAGE (NuPAGE Novex Bis-Tris Gels; Life Technologies) and electrotransferred onto a nitrocellulose membrane (Amersham Protran 0.45; GE Healthcare) in transfer buffer [25 mM Tris, 192 mM glycine, 20% (vol/vol) methanol]. After blocking with 4% nonfat powdered milk in PBS containing 0.1% Tween 20 (Sigma) for 1 h, primary antibodies were incubated with the membrane overnight at 4 °C and binding was visualized after washing with peroxidase-labeled anti-rabbit or anti-mouse secondary antibodies (1:10,000; GE Healthcare) and enhanced chemiluminescence (GE Healthcare). Relative quantitation of the protein of interest to the loading control was measured using ImageJ.
Cell Culture and Transfection.
BE(2)-M17 (M17D; ATCC no. CRL-2267) stably expressing α-synuclein and HEK-293T cells endogenously expressing α-synuclein were maintained at 37 °C and 5% CO2 in DMEM containing 10% (vol/vol) FCS (Sigma) and 1% (vol/vol) penicillin/streptomycin/amphotericin B (Life Technologies).
Cells were grown to 60–80% confluency and transfected with plasmid DNA using Lipofectamine 2000. Thirty-six hours after transfection, cells were rinsed with PBS and scraped into ice-cold lysis buffer (50 mM Tris⋅HCl, pH 7.4, 150 mM sodium chloride, 1% Nonidet P-40, 0.1% SDS, 1 mM PMSF, 1 mM N-ethylmaleimide, complete protease inhibitor mixture; all from Sigma). Lysates were rotated for 20 min at 4 °C and then centrifuged at 4 °C at 13,000 × g for 5 min. For cycloheximide chase, cells were treated with 20 μg/mL cycloheximide for 7 h before lysis without obvious detection of cell death. For knockdown experiments using lentiviruses expressing shRNA, SH-SY5Y cells were transduced for 24 h and lysed 7 d after transduction.
Design and Construction of Usp8 or Scrambled shRNA Lentiviral Vectors.
The rat Usp8 sequence (accession no. NM_001106502.1) was used to generate the Usp8 shRNA (5′-CCGCTCGAGAAAAAAGCTGAGATCTCAAGGCTTTCTTGACAGGAAGAGAAAGCCTTGAGATCTCAGCCAAAACAAGGCTTTTCTCCAAGG-3′), which was predicted to knock down the human protein as well as a scrambled shRNA (5′-CCGCTCGAGAAAAAAGGCACATTAGGAACCATACATTGACAGGAAGATGTATGGTTCCTAATGTGCCCAAAACAAGGCTTTTCTCCAAGG-3′). A short hairpin expression cassette comprised a sense and an antisense (antisense specific to the target mRNA) strand spaced by a loop sequence, RNA polymerase III transcription termination signal, and XhoI restriction site. A sequence coding for the mouse U6 promoter was inserted upstream of the antisense strand with a SpeI site. A PCR-based method was used to generate double-stranded shRNA expression cassettes using the pSilencer 1.0-U6 expression vector (Life Technologies) as a transcription template and reverse primer encoding the shRNA oligonucleotides. These were subcloned into the lentiviral backbone pRRLsincppt.U6.CMV.EGFP.wpre (43) predigested with SpeI and XhoI, generating the pRRLsincppt.U6.rUsp8shRNA.CMV.EGFP.wpre or pRRLsincppt.U6.scrambledshRNA.CMV.EGFP.wpre construct. The constructs were tested for efficiency in human cell lines and found to be effective in reducing the content of Usp8.
Plasmids.
Usp8 cDNA was amplified from FLAG-HA-Usp8 in pDEST_Tet_ON_CMV_N_FLAG_HA_PGK_puro (Addgene; plasmid 22608) using forward (FLAG in italics) 5′-CGATCAGCTAGCCACCATGGACTACAAGGATGACGATGACAAGATGCCTGCTGTGGCTTCAGTTCCTAAAGAACTCTACCTCAGTTCTTC-3′ and reverse 5′-GTCTCTGGATCCTTATGTGGCTACATCAGTTACTCGTGGTC-3′ primers. The Usp8 MIT deletion mutant was amplified from FLAG-HA-Usp8 in pDEST_Tet_ON_CMV_N_FLAG_HA_PGK_puro using forward 5′-CGATCAGCTAGCTCTGCCACCATGGACTACTATGAAGAAGCTGAAGTCCGGAAAAAACTT-3′ and the reverse primer listed above. The products were digested with BamHI and NheI (underlined) and subcloned into the corresponding digests of pIRES2-EGFP (Clontech). The Cys786-to-Ala mutation was introduced using the QuikChange site-directed mutagenesis protocol (Stratagene). HA-ubiquitin, HA-K63–only lysine ubiquitin, and HA-K48–only lysine ubiquitin were purchased from Addgene (plasmids 17608, 17606, and 17605, respectively). Human α-synuclein in pIRES2-EGFP was a kind gift of M. G. Spillantini, University of Cambridge, Cambridge, UK. Human Usp7 was cloned in pcDNA3.1. Fluorescently tagged plasmids expressing Rabs [mRFP-Rab5 (plasmid 14437), dsRed-Rab7 (plasmid 12661), and dsRed-Rab11 (plasmid 12679)] were purchased from Addgene.
Generation of Human iPSc-Derived Neurons.
Dopaminergic neurons were derived from healthy control iPS cell lines that were previously described (39). For neural induction, embryoid bodies generated from feeder-free iPS cells were subjected to dual-Smad inhibition using noggin (200 ng/mL), SB431542 (10 µM), and ChiR 99021 (0.7 µM). Neural precursor cells were ventralized using sonic hedgehog (C24II; 200 ng/mL) and expanded in growth medium containing fibroblast growth factor 8a (100 ng/mL) and brain-derived neurotrophic factor (BDNF; 20 ng/mL). After manual passaging of neural precursor cells, neurons were matured in the presence of glial-derived neurotrophic factor (20 ng/mL), BDNF (20 ng/mL), ascorbic acid (200 µM), cAMP (0.5 mM), and laminin (1 µg/mL) for 2–8 wk.
Colocalization Studies.
Four- to 8-wk-old neurons were plated onto Matrigel (BD Biosciences)-coated glass coverslips and HEK cells were plated onto poly-l-lysine–coated glass coverslips. For Usp8 colocalization with Rab proteins in HEK-293T cells, fluorescently tagged plasmids expressing Rabs were transfected. For the bifluorescence complementation assay in HEK-293T cells, cells were cotransfected with WT Usp8-VN and WT Syn-VC and fluorescently tagged plasmids expressing Rabs. Cells were probed with rabbit anti-Usp8 polyclonal antibody (HPA004869; 1:3,000; Sigma), mouse anti–α-synuclein monoclonal antibody (610787; 1:500; BD Biosciences), goat anti-TH polyclonal antibody (101853; 1:400; Abcam), mouse anti-EEA1 monoclonal antibody (610457; 1:500; BD Biosciences), mouse anti-LBPA monoclonal antibody (6C4; 1:200; Echelon Biosciences), and mouse anti-Rab11 monoclonal antibody (A-6; 1:200; Santa Cruz) in PBS and visualized with Alexa 305-, 488-, 546-, or 568-conjugated goat or donkey anti-mouse, -rabbit, or -goat secondary antibodies (1:500; Life Technologies). Coverslips were mounted onto glass slides with fluorescence mounting medium (Dako) containing 1 μg/mL DAPI (Sigma). Confocal images were acquired using an LSM 710 (Carl Zeiss) microscope (63× objective) and colocalization analysis was performed using the associated Zen 2009 software (Carl Zeiss). To measure the fractional overlap of the Usp8 stain with that of other markers, we have reported the Manders’ colocalization coefficient (MCC), which measures the fraction of one protein that colocalizes with another, poorly measured by Pearson’s correlation coefficient (40). MCC values range from zero (uncorrelated distributions of two probes with one another) to one (perfect colocalization of two images). No primary antibody controls were included for background subtraction.
Deubiquitination of Ubiquitinated α-Synuclein in Vitro.
Purified α-synuclein was ubiquitinated by Nedd4, as described previously (7). α-Synuclein was incubated with His-Nedd4, His-UbcH5b, His-E1, and ubiquitin in the presence of ATP. After ubiquitination, His-tagged enzymes were removed by incubation with Ni-NTA agarose (Qiagen). The unbound ubiquitinated α-synuclein fraction was used as the substrate of the in vitro deubiquitination reaction. Ubiquitinated α-synuclein was incubated with the indicated DUBs (50 nM) in 50 mM Tris and 2 mM DTT at 37 °C for 90 min. Each sample was analyzed by SDS/PAGE and immunoblotting using an anti–α-synuclein antibody (C20; 1:1,000; Santa Cruz). The recombinant DUBs (OTUB1, OTUB2, JosD2, Usp2 core, Usp5, Usp7, and Usp8 core) used in this study were previously characterized extensively (29), and their activity was confirmed again using the active site-directed probe HA-Ub-Br2, as described previously (29).
Drosophila Genetics.
The UAS-α-synuclein wild-type (8146) and A53T mutant (8148), UAS-Usp8 RNAi (38982), and UAS-Ataxin 3 (8150) with expanded polyQ were obtained from the Bloomington Drosophila Stock Center. The UAS-Vps28 RNAi (31894) was obtained from the VDRC. The GAL4 UAS expression system was used to overexpress these transgenes either in dopaminergic neurons at 25 °C using the ddc-GAL4 driver (kindly provided by A. Lin and G. Miesenbock, University of Oxford, Oxford) or specifically in the eye using GMR-GAL4 (provided by I. Davis, University of Oxford, Oxford) at 29 °C. The following genotypes were used: (i) +/+; ddc-GAL4/+, (ii) +/+; ddc-GAL4/UAS-A53T α-synuclein, (iii) UAS-Usp8 RNAi/+; ddc-GAL4/+, (iv) UAS-Usp8 RNAi/+; ddc-GAL4/UAS-A53T α-synuclein, (v) GMR-GAL4/+, (vi) GMR-GAL4/+; UAS-α-synuclein/+, (vii) GMR-GAL4/+; UAS-A53T α-synuclein/+, (viii) GMR-GAL4/UAS-Usp8 RNAi; +/+, (ix) UAS-Usp8 RNAi/GMR-GAL4; UAS-α-synuclein/+, (x) Usp8 RNAi/GMR-GAL4; UAS-A53T α-synuclein/+, (xi) MS1096-GAL4/Y, (xii) MS1096-GAL4/Y; UAS-Usp8 RNAi/+, (xiii) UAS-α-synuclein/GMR-GAL4; UAS-Vps28 RNAi/+, (xiv) UAS-Ataxin 3/GMR-GAL4; UAS-Usp8 RNAi/+, (xv) GMR-GAL4/UAS-Ataxin 3; +/+, (xvi) UAS-AMSH RNAi; ddcGAL4/+, (xvii) UAS-JOSD2 RNAi; ddcGal4/+, (xviii) GMR-GAL4/UAS-AMSH RNAi; +/+, (xix) GMR-GAL4/UAS-AMSH RNAi; UAS-A53T α-synuclein/+, (xx) UAS-JOSD2 RNAi/+; UAS-A53T α-synuclein/ddcGAL4, (xxi) UAS-AMSH RNAi/+; UAS-A53T α-synuclein/ddcGAL4, (xxii) GMR-GAL4/+; UAS-Usp14 RNAi/+, (xxiii) GMR-GAL4/+; UAS-Usp47 RNAi/+, (xxiv) GMR-GAL4/+; UAS-Usp14 RNAi/UAS-A53T α-synuclein, and (xxv) GMR-GAL4/+; UAS-Usp47 RNAi/UAS-A53T α-synuclein. The experiments were repeated in two independently derived transgenic lines and carried out with the experimenter blinded to the sample genotypes throughout the analysis.
Scanning Electron Microscopy of the Drosophila Eye.
Age-matched male flies were fixed in 70% ethanol. The flies were then dehydrated in 100% ethanol, dried, and mounted on SEM stubs. The samples were then sputter-coated with gold and imaged using a JEOL JSM-6390 scanning electron microscope. Eyes were examined for abnormal bristle orientation, ommatidial fusion or pitting, and disorganization of the ommatidial array.
Fractionation of Drosophila Head Lysates.
All steps were performed at 4 °C. For each genotype, five male adult heads were homogenized in lysis buffer (150 mM NaCl, 10 mM Tris, pH 7.4, complete protease inhibitor mixture, 1 mM N-ethylmaleimide). Crude lysates were centrifuged at 4,000 × g for 5 min. Supernatants were then centrifuged at 100,000 × g for 1 h. The resulting supernatant was designated the cytosolic fraction, and the pellet was resuspended in loading buffer.
Climbing Assays.
Locomotor function was assayed using a startle-induced negative geotaxis assay. Ten male flies were placed in each vial and five vials were used per line in each experiment (total 50 flies per line). Each vial was tapped 10 times and the percentage of flies above 6 cm was recorded after 4 s.
Quantitative RT-PCR.
Five fly heads from each line were homogenized for 30 s using a tissue homogenizer. RNA was extracted (Qiagen; RNeasy Mini Kit) and reverse-transcribed (Qiagen; QuantiTect Reverse Transcription Kit) according to the manufacturer’s instructions. Real-time qPCR was performed using Roche LightCycler 480 II and LightCycler 480 SYBR Green I Master. The primers used were 5′-GGCCAAGGAGGGAGTTGTGGC-3′ and 5′-TGCTGTCACACCCGTCACCA-3′ for α-synuclein and 5′-TGCTAAGCTGTCGCACAAATGGC-3′ and 5′-CGATCCGTAACCGATGTTGGGC-3′ for rp49, which was used for normalization.
Confocal Imaging of Fly Brains.
Fly brains were dissected at 25 d posteclosion in ice-cold Schneider’s fly medium and fixed in 4% PFA. They were washed in PBS-T, blocked in goat serum for 1 h, and then incubated for 48 h at 4 °C with monoclonal mouse anti-TH (1:500) followed by PBS-T washes and Alexa 488-coupled goat anti-mouse IgG (1:1,000) for a further 48 h at 4 °C. Stained brains were mounted in ProLong Gold antifade mountant. Z stacks of the brain were obtained on a Zeiss LSM 780 confocal microscope using a 25× objective (1.4× digital zoom) at 1-μm steps and Z-projected. Dopaminergic neuronal clusters were identified and counted.
Statistical Analysis.
The statistical analysis was performed using JMP software (SAS; version 10.0) and Prism (GraphPad). Outliers were identified using an outlier box plot and were excluded from the calculations where observed. All data were examined for distribution, and statistical tests were chosen accordingly. For normally distributed data, a t test or a one-way ANOVA was used. For nonparametric data, the Wilcoxon–Mann–Whitney and Kruskal–Wallis rank-sum tests were performed to assess differences in mRNA or protein levels and for climbing assays. The null hypothesis was rejected at a significance level of P = 0.05. Exponential data were log-converted if reasonable before statistical calculation was performed. Correlations were estimated with a pairwise correlation method for normally distributed data and with Spearman’s rho for nonparametric data. Colocalization was estimated as the fractional overlap of two markers using the Manders’ colocalization coefficient.
Acknowledgments
We thank S. Cowley for providing the control iPS cell lines, M. G. Spillantini for the α-synuclein cDNA, L. Parkkinen for assistance with the selection of neuropathological cases, D. Selkoe for the 2F12 antibody, F. Rinaldi for the lentiviral backbone, and the Bloomington Drosophila Stock Center and VDRC, as well as A. Lin, G. Miesenbock, and I. Davis, for Drosophila stocks. This work was supported by a Wellcome Trust Intermediate Clinical Fellowship, Wellcome-Beit Award, and National institute for Health Research (NIHR) Oxford Biomedical Research Centre (G.K.T.). The Oxford Brain Bank is supported by Alzheimer’s Brain Bank UK, Medical Research Council, and NIHR Oxford Biomedical Research Centre.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1523597113/-/DCSupplemental.
References
- 1.Braak H, et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24(2):197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 2.Spillantini MG, et al. Alpha-synuclein in Lewy bodies. Nature. 1997;388(6645):839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 3.Schneider JA, et al. Cognitive impairment, decline and fluctuations in older community-dwelling subjects with Lewy bodies. Brain. 2012;135(Pt 10):3005–3014. doi: 10.1093/brain/aws234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ross OA, et al. Genomic investigation of alpha-synuclein multiplication and parkinsonism. Ann Neurol. 2008;63(6):743–750. doi: 10.1002/ana.21380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wirdefeldt K, et al. Expression of alpha-synuclein in the human brain: Relation to Lewy body disease. Brain Res Mol Brain Res. 2001;92(1–2):58–65. doi: 10.1016/s0169-328x(01)00150-4. [DOI] [PubMed] [Google Scholar]
- 6.Tofaris GK, Layfield R, Spillantini MG. Alpha-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett. 2001;509(1):22–26. doi: 10.1016/s0014-5793(01)03115-5. [DOI] [PubMed] [Google Scholar]
- 7.Tofaris GK, et al. Ubiquitin ligase Nedd4 promotes alpha-synuclein degradation by the endosomal-lysosomal pathway. Proc Natl Acad Sci USA. 2011;108(41):17004–17009. doi: 10.1073/pnas.1109356108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuzuhara S, Mori H, Izumiyama N, Yoshimura M, Ihara Y. Lewy bodies are ubiquitinated. A light and electron microscopic immunocytochemical study. Acta Neuropathol. 1988;75(4):345–353. doi: 10.1007/BF00687787. [DOI] [PubMed] [Google Scholar]
- 9.Lowe J, et al. Ubiquitin is a common factor in intermediate filament inclusion bodies of diverse type in man, including those of Parkinson’s disease, Pick’s disease, and Alzheimer’s disease, as well as Rosenthal fibres in cerebellar astrocytomas, cytoplasmic bodies in muscle, and mallory bodies in alcoholic liver disease. J Pathol. 1988;155(1):9–15. doi: 10.1002/path.1711550105. [DOI] [PubMed] [Google Scholar]
- 10.Tofaris GK, Razzaq A, Ghetti B, Lilley KS, Spillantini MG. Ubiquitination of alpha-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function. J Biol Chem. 2003;278(45):44405–44411. doi: 10.1074/jbc.M308041200. [DOI] [PubMed] [Google Scholar]
- 11.Anderson JP, et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem. 2006;281(40):29739–29752. doi: 10.1074/jbc.M600933200. [DOI] [PubMed] [Google Scholar]
- 12.Schmidt M, Finley D. Regulation of proteasome activity in health and disease. Biochim Biophys Acta. 2014;1843(1):13–25. doi: 10.1016/j.bbamcr.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nathan JA, Kim HT, Ting L, Gygi SP, Goldberg AL. Why do cellular proteins linked to K63-polyubiquitin chains not associate with proteasomes? EMBO J. 2013;32(4):552–565. doi: 10.1038/emboj.2012.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wollert T, Hurley JH. Molecular mechanism of multivesicular body biogenesis by ESCRT complexes. Nature. 2010;464(7290):864–869. doi: 10.1038/nature08849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Urbé S, et al. Control of growth factor receptor dynamics by reversible ubiquitination. Biochem Soc Trans. 2006;34(Pt 5):754–756. doi: 10.1042/BST0340754. [DOI] [PubMed] [Google Scholar]
- 16.Mizuno E, et al. Regulation of epidermal growth factor receptor down-regulation by UBPY-mediated deubiquitination at endosomes. Mol Biol Cell. 2005;16(11):5163–5174. doi: 10.1091/mbc.E05-06-0560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mukai A, et al. Balanced ubiquitylation and deubiquitylation of Frizzled regulate cellular responsiveness to Wg/Wnt. EMBO J. 2010;29(13):2114–2125. doi: 10.1038/emboj.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reincke M, et al. Mutations in the deubiquitinase gene USP8 cause Cushing’s disease. Nat Genet. 2015;47(1):31–38. doi: 10.1038/ng.3166. [DOI] [PubMed] [Google Scholar]
- 19.Ma ZY, et al. Recurrent gain-of-function USP8 mutations in Cushing’s disease. Cell Res. 2015;25(3):306–317. doi: 10.1038/cr.2015.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kirkin V, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009;33(4):505–516. doi: 10.1016/j.molcel.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 21.Row PE, et al. The MIT domain of UBPY constitutes a CHMP binding and endosomal localization signal required for efficient epidermal growth factor receptor degradation. J Biol Chem. 2007;282(42):30929–30937. doi: 10.1074/jbc.M704009200. [DOI] [PubMed] [Google Scholar]
- 22.Ali N, et al. Recruitment of UBPY and ESCRT exchange drive HD-PTP-dependent sorting of EGFR to the MVB. Curr Biol. 2013;23(6):453–461. doi: 10.1016/j.cub.2013.02.033. [DOI] [PubMed] [Google Scholar]
- 23.Chen L, et al. Tyrosine and serine phosphorylation of alpha-synuclein have opposing effects on neurotoxicity and soluble oligomer formation. J Clin Invest. 2009;119(11):3257–3265. doi: 10.1172/JCI39088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davies SE, et al. Enhanced ubiquitin-dependent degradation by Nedd4 protects against α-synuclein accumulation and toxicity in animal models of Parkinson’s disease. Neurobiol Dis. 2014;64:79–87. doi: 10.1016/j.nbd.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science. 2002;295(5556):865–868. doi: 10.1126/science.1067389. [DOI] [PubMed] [Google Scholar]
- 26.Lu L, et al. Gene expression profiling of Lewy body-bearing neurons in Parkinson’s disease. Exp Neurol. 2005;195(1):27–39. doi: 10.1016/j.expneurol.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 27.Hasegawa M, et al. Phosphorylated alpha-synuclein is ubiquitinated in alpha-synucleinopathy lesions. J Biol Chem. 2002;277(50):49071–49076. doi: 10.1074/jbc.M208046200. [DOI] [PubMed] [Google Scholar]
- 28.Ritorto MS, et al. Screening of DUB activity and specificity by MALDI-TOF mass spectrometry. Nat Commun. 2014;5:4763. doi: 10.1038/ncomms5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McGouran JF, Gaertner SR, Altun M, Kramer HB, Kessler BM. Deubiquitinating enzyme specificity for ubiquitin chain topology profiled by di-ubiquitin activity probes. Chem Biol. 2013;20(12):1447–1455. doi: 10.1016/j.chembiol.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Swaminathan S, Amerik AY, Hochstrasser M. The Doa4 deubiquitinating enzyme is required for ubiquitin homeostasis in yeast. Mol Biol Cell. 1999;10(8):2583–2594. doi: 10.1091/mbc.10.8.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dupré S, Haguenauer-Tsapis R. Deubiquitination step in the endocytic pathway of yeast plasma membrane proteins: Crucial role of Doa4p ubiquitin isopeptidase. Mol Cell Biol. 2001;21(14):4482–4494. doi: 10.1128/MCB.21.14.4482-4494.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sugeno N, et al. Lys-63-linked ubiquitination by E3 ubiquitin ligase Nedd4-1 facilitates endosomal sequestration of internalized α-synuclein. J Biol Chem. 2014;289(26):18137–18151. doi: 10.1074/jbc.M113.529461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wijayanti I, Watanabe D, Oshiro S, Takagi H. Isolation and functional analysis of yeast ubiquitin ligase Rsp5 variants that alleviate the toxicity of human α-synuclein. J Biochem. 2015;157(4):251–260. doi: 10.1093/jb/mvu069. [DOI] [PubMed] [Google Scholar]
- 34.Boassa D, et al. Mapping the subcellular distribution of α-synuclein in neurons using genetically encoded probes for correlated light and electron microscopy: Implications for Parkinson’s disease pathogenesis. J Neurosci. 2013;33(6):2605–2615. doi: 10.1523/JNEUROSCI.2898-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hasegawa T, et al. The AAA-ATPase VPS4 regulates extracellular secretion and lysosomal targeting of α-synuclein. PLoS One. 2011;6(12):e29460. doi: 10.1371/journal.pone.0029460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scudder SL, et al. Synaptic strength is bidirectionally controlled by opposing activity-dependent regulation of Nedd4-1 and USP8. J Neurosci. 2014;34(50):16637–16649. doi: 10.1523/JNEUROSCI.2452-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perrett RM, Alexopoulou Z, Tofaris GK. The endosomal pathway in Parkinson’s disease. Mol Cell Neurosci. 2015;66(Pt A):21–28. doi: 10.1016/j.mcn.2015.02.009. [DOI] [PubMed] [Google Scholar]
- 38.Na CH, et al. Synaptic protein ubiquitination in rat brain revealed by antibody-based ubiquitome analysis. J Proteome Res. 2012;11(9):4722–4732. doi: 10.1021/pr300536k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu K, Psakhye I, Jentsch S. Autophagic clearance of polyQ proteins mediated by ubiquitin-Atg8 adaptors of the conserved CUET protein family. Cell. 2014;158(3):549–563. doi: 10.1016/j.cell.2014.05.048. [DOI] [PubMed] [Google Scholar]
- 40.Tofaris GK. Lysosome-dependent pathways as a unifying theme in Parkinson’s disease. Mov Disord. 2012;27(11):1364–1369. doi: 10.1002/mds.25136. [DOI] [PubMed] [Google Scholar]
- 41.Bruzzone F, Vallarino M, Berruti G, Angelini C. Expression of the deubiquitinating enzyme mUBPy in the mouse brain. Brain Res. 2008;1195:56–66. doi: 10.1016/j.brainres.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 42.McKeith IG, et al. Consortium on DLB Diagnosis and management of dementia with Lewy bodies: Third report of the DLB Consortium. Neurology. 2005;65(12):1863–1872. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- 43.Rinaldi F, et al. Cross-regulation of Connexin43 and β-catenin influences differentiation of human neural progenitor cells. Cell Death Dis. 2014;5:e1017. doi: 10.1038/cddis.2013.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hartfield EM, et al. Physiological characterisation of human iPS-derived dopaminergic neurons. PLoS One. 2014;9(2):e87388. doi: 10.1371/journal.pone.0087388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dunn KW, Kamocka MM, McDonald JH. A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol. 2011;300(4):C723–C742. doi: 10.1152/ajpcell.00462.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]