


Abstract
This article presents W-IQ-TREE, an intuitive and user-friendly web interface and server for IQ-TREE, an efficient phylogenetic software for maximum likelihood analysis. W-IQ-TREE supports multiple sequence types (DNA, protein, codon, binary and morphology) in common alignment formats and a wide range of evolutionary models including mixture and partition models. W-IQ-TREE performs fast model selection, partition scheme finding, efficient tree reconstruction, ultrafast bootstrapping, branch tests, and tree topology tests. All computations are conducted on a dedicated computer cluster and the users receive the results via URL or email. W-IQ-TREE is available at http://iqtree.cibiv.univie.ac.at. It is free and open to all users and there is no login requirement.
INTRODUCTION
IQ-TREE (1), the successor of the TREE-PUZZLE program (2), is an efficient and versatile phylogenetic software for maximum likelihood analysis of large phylogenetic data. IQ-TREE explores the tree space efficiently and often achieves higher likelihoods than RAxML (3) and PhyML (4). Other key features of IQ-TREE are (i) very fast model selection procedure including partition scheme finding (5), (ii) partitioned analysis for phylogenomic data (6), (iii) ultrafast bootstrap approximation (7), and (iv) implementation of several branch tests (8) and (v) tree topology tests (e.g. (9)).
Most phylogenetic software packages (including IQ-TREE) are command line based, and therefore laborious to run for non-experts. Thus, many web applications with intuitive user-interface were developed (e.g. (10,11)).
Here, we present W-IQ-TREE, a user-friendly web application and compute server for phylogenetic analyses with the IQ-TREE software. W-IQ-TREE currently runs on a computer cluster with 32 CPUs, which can be extended depending on the usage. Since its first launch in April 2014 the numbers of users and submitted jobs are steadily increasing (Figure 1). This is most likely attributed to the user-friendly features presented below.
Figure 1.

Number of all W-IQ-TREE jobs per month irrespective of the IP-addresses submitted by external users and number of distinct users per month.
IQ-TREE WEB APPLICATION
W-IQ-TREE was designed to work on all web browsers. It provides a web interface to interact with users and send user requests to the computer cluster, where the actual computation is done with the most recent sequential IQ-TREE version. In the following, we describe important elements of the web interface.
Input data
W-IQ-TREE accepts input alignments in PHYLIP, FASTA, Nexus, Clustal or MSF format. Various sequence data are supported: DNA, amino acids, codons, binary and morphological data. Binary sequences are encoded by 0 and 1 whereas morphological sequences allow 0–9 and A–Z as characters. For phylogenomic alignments, users can supply a partition file defining a partitioning scheme, for example, to specify different genes or to distinguish between codon positions.
Models of sequence evolution
By default, W-IQ-TREE will determine the best-fit substitution model (see below) followed by tree reconstruction. Alternatively, users can specify the substitution model together with models of rate heterogeneity like the discrete Gamma (12) and the FreeRate model (13). IQ-TREE supports a wide range of substitution models including protein mixture models (14,15). An ascertainment bias correction model (16,17) can also be switched on to correct the likelihoods if the alignment does not contain invariable sites (e.g., single nucleotide polymorphism or morphological data).
Model selection
W-IQ-TREE supports a ‘standard’ model selection procedure like jModelTest (18) and ProtTest (19) as well as an extended procedure (i.e. including the FreeRate heterogeneity model). The FreeRate heterogeneity model relaxes the discrete Gamma model by ‘freely’ estimating rates and proportions of the site categories. W-IQ-TREE uses the Bayesian information criterion (20) (default) or the Akaike information criterion (21) to select the best-fit model. For phylogenomic data, W-IQ-TREE determines the best-fit partitioning scheme using a fast implementation of PartitionFinder (5).
IQ-TREE search parameters
IQ-TREE implements a stochastic algorithm to sample local optima in the tree space. To this end, IQ-TREE maintains a set of candidate trees and applies an evolutionary search algorithm to improve the candidate set. This procedure iteratively performs two operations: perturbing a candidate tree and locally optimizing the perturbed tree by nearest neighbor interchange (NNI). They are controlled by two search parameters: 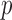 , the perturbation strength, and
, the perturbation strength, and 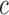 , the number of iterations since the last best tree was found.
, the number of iterations since the last best tree was found.
In the default setting, 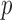 is set to 0.5 (i.e. half of the internal branches are randomly perturbed by NNI) and
is set to 0.5 (i.e. half of the internal branches are randomly perturbed by NNI) and 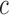 equals 100 (i.e. IQ-TREE stops if no better tree was found within the last 100 iterations). Although this setting was empirically determined to work well (1), it might not hold true for all data sets. For data sets with many sequences, users should specify a higher
equals 100 (i.e. IQ-TREE stops if no better tree was found within the last 100 iterations). Although this setting was empirically determined to work well (1), it might not hold true for all data sets. For data sets with many sequences, users should specify a higher 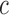 to explore the tree space more extensively. For short sequences a smaller
to explore the tree space more extensively. For short sequences a smaller 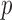 is recommended, whereas for long sequences a larger
is recommended, whereas for long sequences a larger 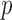 allows for broader sampling of the tree space. It is also recommended conducting multiple IQ-TREE runs using different search parameters.
allows for broader sampling of the tree space. It is also recommended conducting multiple IQ-TREE runs using different search parameters.
Branch support analysis
W-IQ-TREE provides a number of methods to assess the reliability of internal branches: standard bootstrap (22), the SH-aLRT (4), aBayes test (8) and the ultrafast bootstrap (7) (UFBoot).These tests can be combined in a single run. The UFBoot has two parameters that can be set via the web interface: the minimum correlation coefficient (default: 0.99) and the maximum number of iterations (default: 1000). Here, UFBoot computes the Pearson correlation coefficient of two sets of support values during the analysis. UFBoot stops as soon as the maximum number of iterations is reached or if the correlation between the two sets of support values exceeds 0.99, which works for most data sets. When the alignment contains little phylogenetic information, the correlation between the two sets of support values might not exceed 0.99. In such a case, users are advised to increase the maximum number of iterations.
Tree topology evaluation and tests
If users provide a tree file containing several trees in NEWICK format, W-IQ-TREE will compute the log-likelihoods for all given trees. Here, IQ-TREE estimates model parameters (e.g. substitution rates) on a parsimony tree and only optimizes the branch lengths of the user trees to save computation. Moreover, W-IQ-TREE performs several tree topology tests including the KH test (23), the SH test (24), the approximately unbiased (AU) test (9) and the expected likelihood weight (25).
Analysis results
After job submission, W-IQ-TREE provides a URL that allows users to monitor the progress of the job(s). If an email address was provided, W-IQ-TREE automatically sends an email to inform the user that the job is done and where to access the results. Moreover, W-IQ-TREE will display the tree for a quick assessment of the result (Figure 2). The user can download the corresponding tree file in NEWICK, SVG and PDF formats for further analyses. Finally, a command line showing the user-specifications is provided to enable users to repeat the IQ-TREE run on a local computer system. Note that jobs requiring more than 24 CPU hours or >1GB RAM will be stopped if one of the limits is reached. In such cases, users are advised to download the checkpoint file and then resume a standard IQ-TREE run on local machines.
Figure 2.

Screenshot of an example result with W-IQ-TREE for a chordate data set.
AVAILABILITY
W-IQ-TREE is freely accessible at http://iqtree.cibiv.univie.ac.at. The W-IQ-TREE user interface was developed in Javascript using the Sencha framework (http://www.sencha.com), which works on most web browsers and platforms (e.g. Windows, Mac OSX and Linux). The server code was written in PHP to handle and distribute user jobs in the computing cluster. The source code of the W-IQ-TREE is available upon request. Tutorials and extensive documentation are available on the IQ-TREE homepage http://www.cibiv.at/software/iqtree/.
Acknowledgments
The authors thank Robert Happel and Heiko Schmidt for technical supports, Alexandros Stamatakis and two anonymous reviewers for helpful comments and suggestions on the web interface and the manuscript.
FUNDING
Austrian Science Fund (FWF) [I 2805-B29]. Funding for open access charge: FWF [I 2805-B29].
Conflict of interest statement. None declared.
REFERENCES
- 1.Nguyen L.T., Schmidt H.A., von Haeseler A., Minh B.Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015;32:268–274. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schmidt H.A., Strimmer K., Vingron M., von Haeseler A. TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics. 2002;18:502–504. doi: 10.1093/bioinformatics/18.3.502. [DOI] [PubMed] [Google Scholar]
- 3.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guindon S., Dufayard J.F., Lefort V., Anisimova M., Hordijk W., Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 5.Lanfear R., Calcott B., Ho S.Y., Guindon S. Partitionfinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 2012;29:1695–1701. doi: 10.1093/molbev/mss020. [DOI] [PubMed] [Google Scholar]
- 6.Chernomor O., Minh B.Q., von Haeseler A. Consequences of common topological rearrangements for partition trees in phylogenomic inference. J. Comput. Biol. 2015;22:1129–1142. doi: 10.1089/cmb.2015.0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Minh B.Q., Nguyen M.A., von Haeseler A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013;30:1188–1195. doi: 10.1093/molbev/mst024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anisimova M., Gil M., Dufayard J.F., Dessimoz C., Gascuel O. Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood-based approximation schemes. Syst. Biol. 2011;60:685–699. doi: 10.1093/sysbio/syr041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shimodaira H. An approximately unbiased test of phylogenetic tree selection. Syst. Biol. 2002;51:492–508. doi: 10.1080/10635150290069913. [DOI] [PubMed] [Google Scholar]
- 10.Boc A., Diallo A.B., Makarenkov V. T-REX: a web server for inferring, validating and visualizing phylogenetic trees and networks. Nucleic Acids Res. 2012;40:W573–W579. doi: 10.1093/nar/gks485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guindon S., Lethiec F., Duroux P., Gascuel O. PHYML Online—a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res. 2005;33:W557–W559. doi: 10.1093/nar/gki352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang Z. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J. Mol. Evol. 1994;39:306–314. doi: 10.1007/BF00160154. [DOI] [PubMed] [Google Scholar]
- 13.Soubrier J., Steel M., Lee M.S., Der Sarkissian C., Guindon S., Ho S.Y., Cooper A. The influence of rate heterogeneity among sites on the time dependence of molecular rates. Mol. Biol. Evol. 2012;29:3345–3358. doi: 10.1093/molbev/mss140. [DOI] [PubMed] [Google Scholar]
- 14.Le S.Q., Dang C.C., Gascuel O. Modeling protein evolution with several amino acid replacement matrices depending on site rates. Mol. Biol. Evol. 2012;29:2921–2936. doi: 10.1093/molbev/mss112. [DOI] [PubMed] [Google Scholar]
- 15.Wang H.C., Li K., Susko E., Roger A.J. A class frequency mixture model that adjusts for site-specific amino acid frequencies and improves inference of protein phylogeny. BMC Evol. Biol. 2008;8:331. doi: 10.1186/1471-2148-8-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leache A.D., Banbury B.L., Felsenstein J., de Oca A.N.M., Stamatakis A. Short tree, long tree, right tree, wrong tree: new acquisition bias corrections for inferring SNP phylogenies. Syst. Biol. 2015;64:1032–1047. doi: 10.1093/sysbio/syv053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lewis P.O. A likelihood approach to estimating phylogeny from discrete morphological character data. Syst. Biol. 2001;50:913–925. doi: 10.1080/106351501753462876. [DOI] [PubMed] [Google Scholar]
- 18.Darriba D., Taboada G.L., Doallo R., Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods. 2012;9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Darriba D., Taboada G.L., Doallo R., Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011;27:1164–1165. doi: 10.1093/bioinformatics/btr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwarz G. Estimating the dimension of a model. Ann. Statist. 1978;6:461–464. [Google Scholar]
- 21.Akaike H. A new look at the statistical model identification. IEEE Trans. Autom. Control. 1974;19:716–723. [Google Scholar]
- 22.Felsenstein J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- 23.Kishino H., Hasegawa M. Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in hominoidea. J. Mol. Evol. 1989;29:170–179. doi: 10.1007/BF02100115. [DOI] [PubMed] [Google Scholar]
- 24.Shimodaira H., Hasegawa M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol. Biol. Evol. 1999;16:1114–1116. [Google Scholar]
- 25.Strimmer K., Rambaut A. Inferring confidence sets of possibly misspecified gene trees. Proc. Biol. Sci. 2002;269:137–142. doi: 10.1098/rspb.2001.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]