


Abstract
We present an update to our Galaxy-based web server for processing and visualizing deeply sequenced data. Its core tool set, deepTools, allows users to perform complete bioinformatic workflows ranging from quality controls and normalizations of aligned reads to integrative analyses, including clustering and visualization approaches. Since we first described our deepTools Galaxy server in 2014, we have implemented new solutions for many requests from the community and our users. Here, we introduce significant enhancements and new tools to further improve data visualization and interpretation. deepTools continue to be open to all users and freely available as a web service at deeptools.ie-freiburg.mpg.de. The new deepTools2 suite can be easily deployed within any Galaxy framework via the toolshed repository, and we also provide source code for command line usage under Linux and Mac OS X. A public and documented API for access to deepTools functionality is also available.
INTRODUCTION
The analysis of data from high-throughput DNA sequencing experiments continues to be a major challenge for many researchers. The rapidly increasing diversity of experimental assays using high-throughput sequencing has led to a concomitant increase in the number of analysis packages that allow for insightful visualization and downstream analyses (e.g. ChAsE (1), the ChIP-seq web server (http://ccg.vital-it.ch/chipseq), Genomation (2), Homer (3), ngs.plot (4)). Many of these tools require command line experience and input data, which is already quality controlled and properly normalized. As a result, many research groups still do not have the capacity to process and analyse the data generated with deep sequencing technologies. With this in mind, we developed deepTools, a modular suite of fast and user-friendly tools that we implemented within a publicly accessible Galaxy instance (5). deepTools support a wide range of functions, such as various quality controls, different normalization schemes and genome-wide visualizations. Since the original publication of deepTools, and motivated by frequent requests from biologists and bioinformaticians, we have published 10 releases with numerous performance improvements and expansions of scope. In the process, we rewrote large sections of the code, revised the documentation, and updated our server hardware. We added new ways to process and filter deep sequencing data, provided new tools for quality control and analysis, and updated our documentation and examples. Moreover, we have significantly increased the computational speed (sometimes up to 100-fold), extended automated testing for most components, and created an API allowing others to seamlessly incorporate deepTools’ functionality into their own programs. Here, we present our latest release (deepTools2) and its associated web server, which is based on the well-known Galaxy platform (6). This ensures that users lacking access to specialized resources and servers can still benefit from a simple and flexible framework for reproducible analysis.
POWERFUL WEB SERVER FOR DEEP SEQUENCING ANALYSES
Our deepTools Galaxy server is freely available at http://deeptools.ie-freiburg.mpg.de. The deepTools suite supports four common tasks: (i) quality control, (ii) data processing and normalization, (iii) data integration and (iv) visualization (see Table 1). The package has been designed to take as input files some of the most established formats in the deep sequencing field, such as BAM for aligned reads, bigWig for scores (e.g. normalized read coverages) associated with genomic regions, and BED for coordinates of genome regions. All tools come with a large number of options that can be used to fine-tune analyses or filter input datasets for faster data exploration. The visualizations and output files encompass highly customizable, publication-ready images as well as genome-wide scores in bigWig or bedGraph format and tab-separated summaries, e.g. of pairwise correlation metrics that can readily be used for further downstream analyses.
Table 1. Typical applications of deepTools components and a summary of their main inputs and outputs.
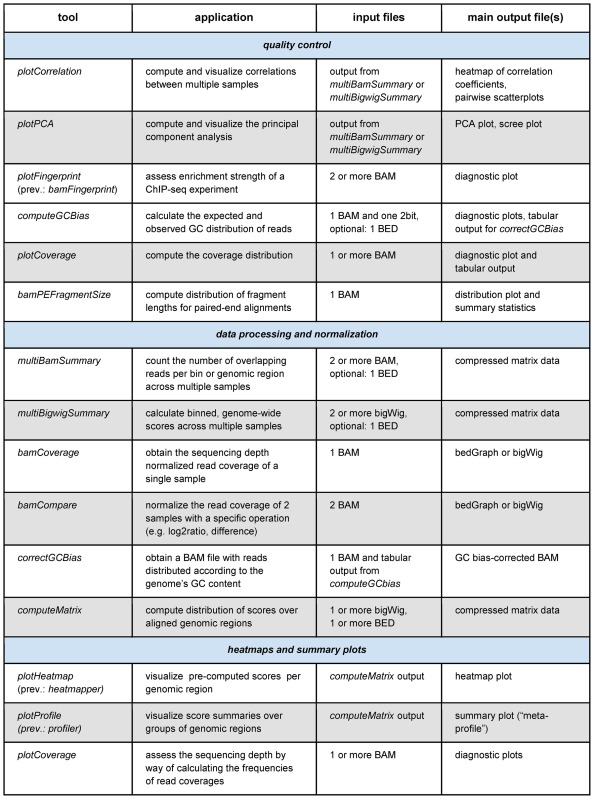
|
Users can upload data from their computers, e.g. via FTP, and can download additional files from the UCSC table browser (7). In addition, we have added more commonly used reference genomes and annotations to the web server's data library. Moreover, comprehensive test data is provided to allow users to easily explore the server's functionality without the need to upload their own data. Registered users can share data, histories and workflows with collaborators and reviewers. To support the increasing number of users, we have made substantial upgrades to our hardware, both in terms of the number of CPUs and available storage.
ADDITIONAL QUALITY CONTROLS
The first step of all analyses is quality control. In addition to the previously established GC-bias detection (computeGCbias) and the ChIP-specific assessment of enrichment (plotFingerprint), we have expanded the diagnostic capabilities of deepTools (Figure 1A). First, we have added a new program, plotCoverage, to inspect the genome-wide distribution of fragment coverage for several samples. This is crucial information for deciding whether the basic goals of the experimental design were met, particularly regarding the desired sequencing depth. For paired-end data, the new program bamPEFragmentSize provides a very sensitive quality check of whether the size distribution of sequenced fragments corresponds to the expectations based on the library preparation. The average fragment size of a sample is also an important parameter for many downstream analyses, such as peak calling (8,9).
Figure 1.

deepTools2 has enhanced features for quality control, data reduction, normalization and visualization of deeply sequenced data. (A) deepTools2 comes with two new quality control tools: plotCoverage, which displays sequencing depth distribution; and plotPCA, which plots the results of principal component analysis (PCA) on BAM or bigWig files. (B) Signal visualization by means of normalized bigWig tracks, produced by deepTools’ bamCompare (ChIP-seq samples) and bamCoverage, which can now handle MNase- and strand-specific RNA-seq samples. In the genes track, the gray color represents untranslated regions and the thin lines represent introns. (C) computeMatrix and plotHeatmap were used to summarize and cluster multiple bigWig scores over genomic intervals. The image shows the resulting k-means clustering of ChIP-seq signals of three histone marks around the transcription start site of genes (with k = 2). Subsequently, the clustered regions were used to plot MNase-seq and CAGE read coverages. In the image, each row corresponds to the same genomic region. (D) plotProfile now offers additional means for visualization. The top panel shows the average signal for different samples and different cluster of regions (left and right plot). In the middle, the same profiles are represented as heatmaps. The bottom panel shows summary profiles where colors correspond to the observed frequency of the signal within each cluster. All data are from the Drosophila melanogaster S2 cell line and are publically available (see Section Accession numbers).
Finally, we have enhanced the capacity for comparative quality control and replicate analysis over multiple samples. Users can now perform principal component analysis (PCA) of multiple samples and generate the corresponding plots with the program plotPCA. This tool takes as input a matrix, in which each column represents a sample-specific vector of fragment counts or any other genome-wide score. It can unveil unexpected patterns, such as batch effects, outliers or sample swaps. In a similar vein, plotCorrelation has been significantly updated to fine-tune the joint analysis of samples and visualization of the correlation structure. It is now also able to produce scatterplots. Previously, the correlation analysis was performed together with the data collection from BAM files by one tool, bamCorrelate. To improve both speed and flexibility, we have replaced this tool and separated the computationally demanding data retrieval from the actual visualization tasks (plotCorrelation and plotPCA). The data collection step can now be done using multiBamSummary or multiBigwigSummary, as described below.
ENHANCED PROCESSING FLEXIBILITY, SCOPE AND SPEED
Once the general quality controls have been carried out, researchers are faced with the actual processing of data, such as filtering, normalization and format conversions. Due to the sheer size of the aligned reads files, this is a non-trivial and cumbersome aspect of all deep sequencing workflows. Our web server supports this challenging part in two ways: (i) the Galaxy environment automatically keeps track of every analysis step, so that users can always and indefinitely identify the tools and parameters that were used to generate a file; (ii) deepTools can perform combined filtering, normalization and format conversion in a single framework, which has contributed to the appeal of deepTools even outside the web server. An important enhancement in this regard is the general support of all deepTools components for flexible filtering of alignments files (BAM) based on the SAM flag (10), in addition to the optional exclusion of duplicates and reads with low alignment scores. This is designed to prevent accumulation of large and unnecessary intermediate files that often occur when different filtering strategies are compared.
We have also enhanced the capability of deepTools to recognize and process new sequencing read types. For example, deepTools now parses CIGAR strings and spliced-read alignments. This is particularly important for bamCoverage, which can now properly handle spliced reads from strand-specific RNA-seq data and convert them into meaningful coverage tracks (Figure 1B). The same tool was also enhanced to accommodate MNase-seq data, which results in very high-resolution summaries for nucleosome positions.
Frequently, genome-wide data is available from large consortia and data portals, such as IHEC (http://ihec-epigenomes.org), ENCODE (https://www.encodeproject.org) and BLUEPRINT (http://www.blueprint-epigenome.eu). In most cases, the provided data has already been processed and normalized. Since this information is most commonly stored as bigWig files, most deepTools components now have the capacity to handle data stored in bigWig format. Importantly, the processing of bigWig files was sped up by up to 100-fold by using the new Python pyBigWig package (http://dx.doi.org/10.5281/zenodo.45238) and the libBigWig library written in C (http://dx.doi.org/10.5281/zenodo.45278), which were developed simultaneously as side projects. The command line versions of the tools additionally allow users to directly use remotely stored files without downloading them beforehand.
IMPROVED INTEGRATIVE ANALYSES AND VISUALIZATIONS
Following the normalization and aggregation of aligned reads, visualization of the data is a vital part of almost every bioinformatics analysis as it allows users to explore their data and generate hypotheses. In addition, downstream analyses and data interpretation often depend on the integration of multiple samples. One of the major changes for deepTools2 is the capability to process numerous signal (bigWig) and region files (BED) in a joint analysis, particularly for summarizing fragment coverages or other scores across genomic regions. The new tools multiBamSummary and multiBigwigSummary summarize the information from multiple samples into a single score for each region per sample. The resulting matrix can be used for correlation analyses, pairwise scatter plots and PCA (see above), or other downstream analyses.
Similarly, computeMatrix calculates the distribution of scores over selected genomic regions. Typical applications include the computation of signals around promoters or across gene bodies that are scaled to the same length. The primary purpose is to produce a matrix that can later be visualized. One of the most frequently requested features of deepTools was the extension of computeMatrix to allow for the combined analysis of multiple samples. Now, computeMatrix can aggregate regional scores from many different samples into a single matrix for subsequent visualization. Figure 1C illustrates this capability for 5 different signal files (H3K4me3, H3K27me3, H3K36me3, MNase, CAGE). Here, genome-wide scores were provided as five bigWig files, and a single BED file of annotated genes. From these inputs, computeMatrix generated a matrix file that was then visualized using plotHeatmap and plotProfile. Those tools have been extended to produce composite images of multiple heatmaps and summary profiles (Figure 1C and D). They also include options for unsupervised data analysis (k-means and hierarchical clustering), which are most useful if no prior grouping of regions is available. In the example of Figure 1C, two groups of regions were determined by k-means clustering of the signal profiles with k = 2. Alternatively, the user can provide multiple groups of pre-defined regions (multiple BED files) to computeMatrix. The tool will then output a structured matrix in which the rows are grouped according to the pre-defined regions, e.g. different sets of genes. Subsequent visualization corresponds to a supervised analysis, where separate heatmaps and summary profiles are generated for each of the previously defined groups of interest. Both plotHeatmap and plotProfile now also offer many new options for customizing their output (Figure 1D).
IMPLEMENTATION AND ACCESS
Our deepTools web server has been implemented within the Galaxy framework (6). The software has been virtualized as a Docker container, which currently has access to 24 cores, 141GB memory and more than 8TB of disk storage. Our web server offers all deepTools’ functionality for free and without registration. Users with more challenging requirements can access deepTools through multiple additional means:
Toolshed
For already existing local Galaxy instances, deepTools can be easily installed through the Galaxy toolshed (11).
Docker Image
For those wishing to use deepTools locally within the Galaxy framework, we have significantly simplified the way in which a deepTools Galaxy server can be deployed. Through incorporating everything in a single Docker container, users can now install a deepTools Galaxy web server on a desktop machine with a single command. Further details are available in our online documentation (http://deeptools.readthedocs.org/en/latest/content/installation.html).
Stand alone
deepTools can be installed through the Python package index (PyPi, https://pypi.python.org/pypi/deepTools), through GitHub (https://github.com/fidelram/deepTools) and in the bioconda channel of Anaconda (https://anaconda.org/bioconda/deeptools). More information can be found at http://deeptools.readthedocs.org/en/latest/content/installation.html.
API
Finally, deepTools are now fully available as a Python package. Numerous functions, such as the function for summarizing read counts from a BAM file per genomic region (countReadsPerBin), can easily be accessed in any Python script that imports the deepTools package. Usage details can be found at http://deeptools.readthedocs.org/en/latest/content/api.html.
EXTENSIVE TESTING, DOCUMENTATION AND COMMUNITY SUPPORT
deepTools have greatly benefited from transparent development as they are hosted on github.org, where users and collaborators can directly post issues, fork their own versions, and follow the changes to the code. All tools contain comprehensive and automatic tests that evaluate proper functioning after any modification of the code. We continuously update our documentation, and respond to questions sent to our mailing list (deeptools@googlegroups.com) and through the Galaxy bug report system. For this major update, we have reorganized the documentation with updated examples of tools usage, step-by-step protocols and FAQs. Also, detailed usage descriptions have been added to the Galaxy wrappers. To further improve our documentation, we migrated it to readthedocs.org, where we can offer versioned documentation and an easy way to export it as a composite PDF file. The most up-to-date documentation can now always be found at http://deeptools.readthedocs.org/en/latest/.
DISCUSSION AND OUTLOOK
The adoption of deep sequencing in many laboratories has created the need for accessible, efficient and transparent methods to process the data directly by the researchers. However, the ever-increasing throughput of deep sequencing technologies requires sophisticated bioinformatics solutions that are not always available to every lab. Via the deepTools web server, researchers can access a set of easy to use, yet powerful programs that cover quality control, normalization, integration and visualization of the data. Since deepTools employ a high level of parallelization for the computationally most expensive tasks, they are well suited to work with a large number of samples emerging from large-scale data production centers (12,13) or single-cell sequencing (14). The Galaxy framework is frequently used for establishing reproducible and standardized analysis workflows, even for groups where bioinformatics support is otherwise scarce. We have also found it to be a user-friendly environment that most biomedical scientists are comfortable with. This makes it a versatile platform for training workshops, as well as for sharing data and workflows among collaborators.
The modular design of deepTools has allowed us to significantly expand the scope of the analyses compared to its first release. More functionality can be added in the future, and since the software development is completely open and transparent, it will benefit from contributions from the wider community to incorporate new standards and best practices in this rapidly evolving field.
ACCESSION NUMBERS
The datasets used in Figure 1 were: H3K4me3 and H3K26me3 from GEO:GSE41440 (15), H3K36me3 from GEO:GSE27679 (16), MNase-seq from GEO:GSE58821 (17), CAGE from GEO:GSE52884 (18).
Acknowledgments
The authors would like to thank all users of deepTools for their constructive feedback and suggestions. We would further like to thank specific users for their valuable input: Peter Ebert, Sebastian Preissl and Ralf Gilsbach. We would also like to thank Abdullah Sahyoun for insightful discussions.
FUNDING
German Research Foundation [SFB 992, Project Z01]; German Epigenome Programme DEEP [01KU1216G]. Source of Open Access funding: own funds.
Conflict of interest statement. None declared.
REFERENCES
- 1.Younesy H., Nielsen C.B., Möller T., Alder O., Cullum R., Lorincz M.C., Karimi M.M., Jones S.J.M. An interactive analysis and exploration tool for epigenomic data. Comput. Graph. Forum. 2013;32:91–100. [Google Scholar]
- 2.Akalin A., Franke V., Vlahoviček K., Mason C.E., Schübeler D. Genomation: a toolkit to summarize, annotate and visualize genomic intervals. Bioinformatics. 2015;31:1127–1129. doi: 10.1093/bioinformatics/btu775. [DOI] [PubMed] [Google Scholar]
- 3.Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shen L., Shao N., Liu X., Nestler E. ngs.plot: quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics. 2014;15:284. doi: 10.1186/1471-2164-15-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramírez F., Dündar F., Diehl S., Grüning B.A., Manke T. deepTools: a flexible platform for exploring deep-sequencing data. 2014;42:W187–W191. doi: 10.1093/nar/gku365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hillman-Jackson J., Clements D., Blankenberg D., Taylor J., Nekrutenko A.Galaxy Team. Using Galaxy to perform large-scale interactive data analyses. Curr. Protoc. Bioinformatics. 2012 doi: 10.1002/0471250953.bi1005s38. Chapter 10, Unit 10.5–10.5.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Speir M.L., Zweig A.S., Rosenbloom K.R., Raney B.J., Paten B., Nejad P., Lee B.T., Learned K., Karolchik D., Hinrichs A.S., et al. The UCSC Genome Browser database: 2016 update. Nucleic Acids Res. 2016;44:D717–D725. doi: 10.1093/nar/gkv1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feng J., Liu T., Qin B., Zhang Y., Liu X.S. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 2012;7:1728–1740. doi: 10.1038/nprot.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Landt S.G., Marinov G.K., Kundaje A., Kheradpour P., Pauli F., Batzoglou S., Bernstein B.E., Bickel P., Brown J.B., Cayting P., et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blankenberg D., Von Kuster G., Bouvier E., Baker D., Afgan E., Stoler N., Taylor J., Nekrutenko A., Galaxy Team Dissemination of scientific software with Galaxy ToolShed. Genome Biol. 2014;15:403. doi: 10.1186/gb4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bernstein B.E., Birney E., Dunham I., Green E.D., Gunter C., Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Consortium R.E., Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klein A.M., Mazutis L., Akartuna I., Tallapragada N., Veres A., Li V., Peshkin L., Weitz D.A., Kirschner M.W. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell. 2015;161:1187–1201. doi: 10.1016/j.cell.2015.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herz H.-M., Mohan M., Garruss A.S., Liang K., Takahashi Y.-H., Mickey K., Voets O., Verrijzer C.P., Shilatifard A. Enhancer-associated H3K4 monomethylation by Trithorax-related, the Drosophila homolog of mammalian Mll3/Mll4. Genes Dev. 2012;26:2604–2620. doi: 10.1101/gad.201327.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Y., Negre N., Li Q., Mieczkowska J.O., Slattery M., Liu T., Zhang Y., Kim T.-K., He H.H., Zieba J., et al. Systematic evaluation of factors influencing ChIP-seq fidelity. Nat. Methods. 2012;9:609–614. doi: 10.1038/nmeth.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramírez F., Lingg T., Toscano S., Lam K.C., Georgiev P., Chung H.-R., Lajoie B.R., de Wit E., Zhan Y., de Laat W., et al. High-affinity sites form an interaction network to facilitate spreading of the MSL complex across the X chromosome in Drosophila. Mol Cell. 2015;60:146–162. doi: 10.1016/j.molcel.2015.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liang J., Lacroix L., Gamot A., Cuddapah S., Queille S., Lhoumaud P., Lepetit P., Martin P.G.P., Vogelmann J., Court F., et al. Chromatin immunoprecipitation indirect peaks highlight long-range interactions of insulator proteins and Pol II pausing. Mol. Cell. 2014;53:672–681. doi: 10.1016/j.molcel.2013.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]