

Abstract
The protein frustratometer is an energy landscape theory-inspired algorithm that aims at localizing and quantifying the energetic frustration present in protein molecules. Frustration is a useful concept for analyzing proteins’ biological behavior. It compares the energy distributions of the native state with respect to structural decoys. The network of minimally frustrated interactions encompasses the folding core of the molecule. Sites of high local frustration often correlate with functional regions such as binding sites and regions involved in allosteric transitions. We present here an upgraded version of a webserver that measures local frustration. The new implementation that allows the inclusion of electrostatic energy terms, important to the interactions with nucleic acids, is significantly faster than the previous version enabling the analysis of large macromolecular complexes within a user-friendly interface. The webserver is freely available at URL: http://frustratometer.qb.fcen.uba.ar.
INTRODUCTION
Natural protein molecules are highly evolved complex systems (1). Spontaneous folding of individual proteins and recognition between polypeptides leading to well-defined structural ensembles are fundamental concepts in the biology of macromolecules, the specificity of which is explained by the ‘Principle of minimal frustration’ (2). The physical concept of ‘frustration’, the system's inability to simultaneously minimize the competing interaction energy between its components, has lead to multiple developments in the understanding of protein folding and function (for recent reviews see (3,4)). The minimal frustration principle does not rule out that some energetic frustration may be present in a folded protein. Moreover, the remaining frustration may not be a random occurrence but an evolved characteristic, facilitating motion of the protein around its native basin, as well as binding to appropriate partners, and as such the residual frustration is fundamental to protein function (5,6). We have developed theoretical methods for spatially localizing and quantifying the energetic frustration present in native proteins (7,8). These have proven useful in the study of binding interfaces (7), allosteric transitions (9), aggregation and ligand binding (10,11), conformational dynamics (12–14), and have been related with evolutionary patterns (15–18) and disease-related polymorphisms (19). Here, we present an updated version of a web server to perform local frustration calculations. The new server is based on the associative memory, water mediated, structure and energy model (AWSEM)(20). AWSEM provides a transferable, coarse-grained, non-additive force field that is able to predict the native structures of many proteins and protein complexes from sequence information (21). Recently, electrostatic forces have been included in the AWSEM suite and have been shown to play a role in modulating the asperities of the folding and binding landscapes (22). This new server allows for the possibility of analyzing the local frustration that arises by electrostatic interactions and is integrated within the AWSEM suite.
MATERIALS AND METHODS
Input files
The input file is a protein structure model in the standard format of the Protein Data Bank (http://www.rcsb.org). Users can upload a structure file or provide a four-letter code for existing PDB entries. The files are checked for formatting style and if there is more than one amino acid chain in the model, the user is asked to specify which chain(s) to process. A dialog box and an interactive JSmol interface are provided to assist in this process. The user can specify as many chains for analysis as desired. A dialog box is provided to optionally include electrostatics in the calculations. Also, users can optionally provide an e-mail address to receive a notification of job completion. The webserver is free and open to all and there is no login requirement.
Server calculations
The server automatically applies filters that remove hydrogens, heteroatoms and alternative conformations for residues. If the input file contains multiple models, only the first one is analyzed by default. The most common 20 amino acids are taken into account and if backbone or Cβ atoms are missing, they are automatically built into the file using Modeller (http://salilab.org/modeller/). This version of the server is based on the AWSEM suite (20). Long range electrostatic interactions can be included in the calculations. These are modeled using a Debye-Hückel potential that includes both the solvent dielectric effect and the screening of charge-charge interactions by mobile ions in the solvent:
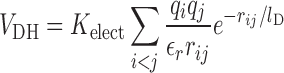 |
(1) |
where qi and qj are the charges of residues i and j that are separated by a distance rij, εr is the dielectric constant of the medium (1 in vacuum) and lD is the Debye–Hückel screening length that takes into account the temperature, the ionic strength of the medium and its dielectric constant. These are set to typical physiological values of T = 25°C, εr = 80 for water I = 0.1 M, and lD = 10 Å. The VDH term can be scaled by a numerical factor εr. The electrostatic constant k is calculated as k = Kelect/εr and represents the electrostatic strength of the system. Smaller values of εr increase the electrostatic strength and vice versa. The user can specify k as input. By default a k = 4.15, corresponding to an aqueous solution, is assumed. Further information can be found in ref (22). In AWSEM only four types of residues are considered to be charged: Arg, Asp, Glu and Lys. To make the results backward-compatible with the previous version, the user is allowed to switch the sequence separation used to calculate the local densities of the amino acids, which is 3 in the new version and 12 in the previous one.
A local frustration index quantifies how much a residue or residue pair contributes to the energy in a given structure compared to what it would contribute in a typical decoy or molten globule configuration. Making numerous mutations and changing local environments lets us assess the mean and variance of the energies of molten globule configurations (decoys) relative to the native. By normalizing the difference between the native and the average decoy by the contribution of the same residue (or pair of residues) to the variance of the energies of the decoys we thereby can get an idea whether that contribution to the energy is typical of what would be expected in a minimally frustrated protein or more like that in a random unevolved heteropolymer. In the case of pairs of residues we ask: how favorable is the actual native pair relative to other possible interactions? To compute the frustration index for interacting pairs of amino acids i, j simultaneous mutations on residues i and j are made. We have proposed two related but complementary ways for localizing frustration at the pairwise contact level. These ways differ in how the set of decoys is constructed. In one choice, the decoy set is made by randomizing only the identities of the interacting amino acids i, j, keeping all other interaction parameters at their native value. This scheme effectively evaluates every possible mutation of the amino acid pair that forms a particular contact in a robustly fixed structure. We call the resulting index the ‘mutational frustration’:
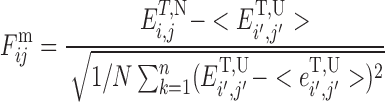 |
(2) |
Here, ET, N is the total energy of the protein in the native configuration and ET, U is the average energy of the decoys. The decoy energy distribution is calculated by randomly selecting amino acid identities from the protein composition and fixing the density ρi and the pairwise distances ri, j to the native conformation. It is worth noting that the energy change upon pair mutation not only comes directly from the particular contact probed but also changes through interactions of each residue with other residues not in the pair, as those contributions may also vary upon mutation. One advantage of the mutational frustration index is that, in principle, this local measure of frustration also could be experimentally determined in the laboratory by combinatorial protein engineering.
A second way of quantifying pairwise local frustration imagines that the residues are not only changed in identity but also can be displaced in location: how favorable is the native interaction between two residues in the native structure relative to other interactions these residues could form in globally different distinct compact structures? The energy variance thus reflects contributions from the energies of molten globule conformations of the same polypeptide chain. For this index, specially suitable for examining alternative tertiary structures, the decoy set involves randomizing not just the identities but also the distances ri, j and densities ρi of the interacting amino acids. Given a protein sequence and structure this strategy compares the energy of each native contact with the distribution of a randomly interacting set.
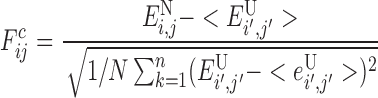 |
(3) |
when 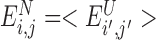 the native energy would not be discriminated from the typical energy of a random interaction in the molten globule and
the native energy would not be discriminated from the typical energy of a random interaction in the molten globule and 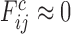 . This scheme effectively evaluates the native pair with respect to a set of structural decoys that might be encountered in the folding process. We call the frustration index computed in this way the ‘configurational frustration’.
. This scheme effectively evaluates the native pair with respect to a set of structural decoys that might be encountered in the folding process. We call the frustration index computed in this way the ‘configurational frustration’.
The jobs are assigned a JobID, organized in a run queue and processed as computational resources become available. A typical run of a 400 residue protein takes ∼2 min of CPU time and a protein complex of 19 chains with 2600 residues takes ∼15 min.
Output files
A results page is generated for each job. A link is provided at the time of data submission and allows the user to bookmark and access the results at a later time. This link also reports the status of the job if it is not finished. The result pages can also be accessed following the link sent by e-mail (if provided) or by specifying the JobID. The server generates several projections of the local frustration calculations (Figure 1). An interactive JSmol interface facilitates inspection of the structures where the minimally frustrated and highly frustrated contacts are highlighted. A contact map is provided in which the frustration index of every contact is shown. Projections of local frustration distributions along the sequence are also provided. If multiple chains were specified, various tabs are available in order to inspect each chain separately. Results can be downloaded by following the link at the bottom of the results page. The download pack includes the input file as processed by the server, image files of the results, scripts to interactively visualize results in PyMOL or VMD programs, the raw tables of the frustration index calculations and an explanatory README file. A user's submitted data are private and are not viewable by anyone other than the user or those given the JobID. The results are accessible for 30 days after completion and permanently deleted afterwards.
Figure 1.

Frustration analysis for the nucleosome complex (Pdb ID: 4z66). The server generates several projections of the local frustration patterns, two of which are shown here. (A) Local frustration pattern mapped to the overall structure of the complex. Minimally frustrated interactions are depicted as green lines, highly frustrated interactions as red lines. The protein backbone is represented as blue ribbon and the DNA backbone as gray cartoon. (B) Local frustration of one of the chains of the complex mapped to the sequence (chain D). The fraction of native contacts in each frustration class around a 5 Å sphere of each Cα is shown. The calculations were performed with different electrostatic constants, k = 0 shown at left and k = 16.6 shown at right. The larger the value for the k parameter, the stronger the effect of the Debye–Hückel term. In the web server, the user can specify k.
RESULTS
The frustratometer algorithm compares the energetic contribution of the extra stabilization energy from a given pair of amino acids in the native protein to the statistics of the energies that is calculated by placing different types of residues in the same native location (mutational frustration index) or by creating a different local environment for the interacting pair (configurational frustration index). If there is a sufficient additional stabilization for an individual native pair as normalized by the typical energy fluctuation, the local interaction is called ‘minimally frustrated’ and is highlighted in green. If the stabilization of the native pair lies in the middle of the distribution of alternatives, the interaction is considered ‘neutral’. On the other hand, if the native pair is sufficiently destabilizing compared with the other possibilities, the interaction is ‘highly frustrated’ and is highlighted in red. The server also provides the calculations of a ‘single residue level frustration’. In this case the decoy set is made randomizing the identity of every single amino acid, keeping all other interaction parameters at their native value. This scheme effectively evaluates every possible mutation at every site in for a well-defined structure. Details of the method and the energy functions can be found in reference (7). For discussions on the interpretation of the results the reader is referred to (5,8) and the server documentation pages.
Electrostatic interactions are able to modulate the stability and folding landscapes of some proteins. The new version of the server introduces the possibility of analyzing the effect of electrostatics on the local frustration patterns. By introducing Debye-Hückel potentials that mimic long-range electrostatic forces into AWSEM, the effects of electrostatics on the landscapes of several proteins were recently analyzed (22). Binding partners can guide their mutual approach with electrostatic interactions prior to making physical contact. Sometimes long range electrostatic interactions not only provide charge–charge stabilization that funnels the landscape for binding but also help steer the docking of intrinsically disordered proteins before structural formation is completed. For other systems, long-range electrostatic interactions cause frustration in the landscape and would seem to impede protein dimer formation. This frustration is often localized in regions that are targeted specifically to other charged partners, such as the surfaces of DNA molecules (Figure 1). Presumably the landscapes of multimeric protein-DNA assemblies are funneled by alleviating this frustration (23).
CONCLUDING REMARKS
The Frustratometer 2 web server is an improved version of an earlier development (8). The new implementation runs within the AWSEM engine, opening the possibility of analyzing together the results of folding simulations and local frustration analysis. Also, this implementation allows for the inclusion of long-range electrostatic energy terms in the calculations. The inclusion of this energy terms have been shown to play a role in binding, in particular DNA-binding proteins, were electrostatics causes frustration in the DNA-binding region favoring its binding with DNA (22,23). The new server runs ∼30 times faster than the previous version, allowing for the analysis of large protein complexes. In this case, the server provides users with projections of the local frustration patterns of the individual chains as well as the overall complex. The server is supported by a Documentation section, including a quick-start guide and illustrative screenshots. A gallery of frustratographs with examples is also hosted at the site, together with fully interactive outputs that are linked from the help pages. We also host a FAQs section and personalized support, provided via e-mail request.
Acknowledgments
We thank Rocío Espada for her contribution to the preparation of the site, and various users of the Frustratometer webserver for helpful comments and suggestions on how to improve the service.
FUNDING
Consejo de Investigaciones Científicas y Técnicas (CONICET); Agencia Nacional de Promoción Científica y Tecnológica [PICT2012/01647 to D.U.F., PPG Grant P01GM071862 to P.G.W.] from the National Institute of General Medical Sciences; D.R. Bullard-Welch Chair at Rice University [C-0016 to P.G.W.]. Funding for open access charge: Agencia Nacional de Promoción Científica y Tecnológica.
Conflict of interest statement. None declared.
REFERENCES
- 1.Frauenfelder H. Proteins: paradigms of complexity. Proc. Natl. Acad. Sci. U.S.A. 2002;99(Suppl. 1):2479–2480. doi: 10.1073/pnas.012579999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bryngelson J.D., Wolynes P.G. Spin glasses and the statistical mechanics of protein folding. Proc. Natl. Acad. Sci. U.S.A. 1987;84:7524–7528. doi: 10.1073/pnas.84.21.7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wolynes P.G. Evolution, energy landscapes and the paradoxes of protein folding. Biochimie. 2015;119:218–230. doi: 10.1016/j.biochi.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schafer N.P., Kim B.L., Zheng W., Wolynes P.G. Learning To Fold Proteins Using Energy Landscape Theory. Isr. J. Chem. 2014;54:1311–1337. doi: 10.1002/ijch.201300145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferreiro D.U., Komives E.A., Wolynes P.G. Frustration in biomolecules. Q. Rev. Biophys. 2014;47:285–363. doi: 10.1017/S0033583514000092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gershenson A., Gierasch L.M., Pastore A., Radford S.E. Energy landscapes of functional proteins are inherently risky. Nat. Chem. Biol. 2014;10:884–891. doi: 10.1038/nchembio.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferreiro D.U., Hegler J.A., Komives E.A., Wolynes P.G. Localizing frustration in native proteins and protein assemblies. Proc. Natl. Acad. Sci. U.S.A. 2007;104:19819–19824. doi: 10.1073/pnas.0709915104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jenik M., Parra R.G., Radusky L.G., Turjanski A., Wolynes P.G., Ferreiro D.U. Protein frustratometer: a tool to localize energetic frustration in protein molecules. Nucleic Acids Res. 2012;40:W348–W351. doi: 10.1093/nar/gks447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferreiro D.U., Hegler J.A., Komives E.A., Wolynes P.G. On the role of frustration in the energy landscapes of allosteric proteins. Proc. Natl. Acad. Sci. U.S.A. 2011;108:3499–3503. doi: 10.1073/pnas.1018980108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gianni S., Camilloni C., Giri R., Toto A., Bonetti D., Morrone A., Sormanni P., Brunori M., Vendruscolo M. Understanding the frustration arising from the competition between function, misfolding, and aggregation in a globular protein. Proc. Natl. Acad. Sci. U.S.A. 2014;111:14141–14146. doi: 10.1073/pnas.1405233111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Das A., Plotkin S.S. SOD1 exhibits allosteric frustration to facilitate metal binding affinity. Proc. Natl. Acad. Sci. U.S.A. 2013;110:3871–3876. doi: 10.1073/pnas.1216597110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sutto L., Lätzer J., Hegler J.A., Ferreiro D.U., Wolynes P.G. Consequences of localized frustration for the folding mechanism of the IM7 protein. Proc. Natl. Acad. Sci. U.S.A. 2007;104:19825–19830. doi: 10.1073/pnas.0709922104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fuglestad B., Gasper P.M., McCammon J.A., Markwick P. R.L., Komives E.A. Correlated motions and residual frustration in thrombin. J. Phys. Chem. B. 2013;117:12857–12863. doi: 10.1021/jp402107u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Truong H.H., Kim B.L., Schafer N.P., Wolynes P.G. Funneling and frustration in the energy landscapes of some designed and simplified proteins. J. Chem. Phys. 2013;139:121908. doi: 10.1063/1.4813504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tripathi S., Waxham M.N., Cheung M.S., Liu Y. Lessons in Protein Design from Combined Evolution and Conformational Dynamics. Sci. Rep. 2015;5:14259. doi: 10.1038/srep14259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galen S.C., Natarajan C., Moriyama H., Weber R.E., Fago A., Benham P.M., Chavez A.N., Cheviron Z.A., Storz J.F., Witt C.C. Contribution of a mutational hot spot to hemoglobin adaptation in high-altitude Andean house wrens. Proc. Natl. Acad. Sci. U.S.A. 2015;112:13958–13963. doi: 10.1073/pnas.1507300112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abriata L.A., Salverda M. L.M., Tomatis P.E. Sequence-function-stability relationships in proteins from datasets of functionally annotated variants: the case of TEM beta-lactamases. FEBS Lett. 2012;586:3330–3335. doi: 10.1016/j.febslet.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 18.Parra R.G., Espada R., Verstraete N., Ferreiro D.U. Structural and energetic characterization of the ankyrin repeat protein family. PLoS Comput. Biol. 2015;11:e1004659. doi: 10.1371/journal.pcbi.1004659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dixit A., Verkhivker G.M. The energy landscape analysis of cancer mutations in protein kinases. PLoS One. 2011;6:e26071. doi: 10.1371/journal.pone.0026071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davtyan A., Schafer N.P., Zheng W., Clementi C., Wolynes P.G., Papoian G.A. AWSEM-MD: protein structure prediction using coarse-grained physical potentials and bioinformatically based local structure biasing. J. Phys. Chem. B. 2012;116:8494–8503. doi: 10.1021/jp212541y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng W., Schafer N.P., Davtyan A., Papoian G.A., Wolynes P.G. Predictive energy landscapes for protein-protein association. Proc. Natl. Acad. Sci. U.S.A. 2012;109:19244–19249. doi: 10.1073/pnas.1216215109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsai M.-Y., Zheng W., Balamurugan D., Schafer N.P., Kim B.L., Cheung M.S., Wolynes P.G. Electrostatics, structure prediction, and the energy landscapes for protein folding and binding. Protein Sci. 2016;25:255–269. doi: 10.1002/pro.2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Potoyan D.A., Zheng W., Komives E.A., Wolynes P.G. Molecular stripping in the NF-kappaB/IkappaB/DNA genetic regulatory network. Proc. Natl. Acad. Sci. U.S.A. 2016;113:110–115. doi: 10.1073/pnas.1520483112. [DOI] [PMC free article] [PubMed] [Google Scholar]