


Abstract
Similar to the development of gene set enrichment and gene regulatory network analysis tools over a decade ago, microRNA enrichment tools are currently gaining importance. Building on our experience with the gene set analysis toolkit GeneTrail, we implemented the miRNA Enrichment Analysis and Annotation tool (miEAA). MiEAA is a web-based application that offers a variety of commonly applied statistical tests such as over-representation analysis and miRNA set enrichment analysis, which is similar to Gene Set Enrichment Analysis. Besides the different statistical tests, miEAA also provides rich functionality in terms of miRNA categories. Altogether, over 14 000 miRNA sets have been added, including pathways, diseases, organs and target genes. Importantly, our tool can be applied for miRNA precursors as well as mature miRNAs. To make the tool as useful as possible we additionally implemented supporting tools such as converters between different miRBase versions and converters from miRNA names to precursor names. We evaluated the performance of miEAA on two sets of miRNAs that are affected in lung adenocarcinomas and have been detected by array analysis. The web-based application is freely accessible at: http://www.ccb.uni-saarland.de/mieaa_tool/.
INTRODUCTION
miRNAs are small non-coding RNA molecules of approximately 22 nucleotides length. Since their discovery in Caenorhabditis elegans, they have been established as important regulators of biological and pathological processes (1,2). miRNAs regulate messenger RNAs (mRNAs) in post transcriptional phases by either halting the translation or by degrading the mRNA molecule. In addition to their importance in biological processes, varying miRNA expression levels in specific diseases makes them valuable biomarker candidates, such as for Alzheimer's disease (AD) (3). The overall importance of miRNAs is emphasized by a study of Lewis et al., which estimates that as much as 30% of the human genes are regulated by miRNAs (4). As the amount of data and applications in miRNomics is increasing rapidly, driven by the fast advances in next-generation sequencing (NGS), respective tools supporting the analysis are implemented. These include miRNA prediction tools from NGS reads (5,6), prediction of miRNA targets (7–9), or miRNA enrichment (10,11) and annotation tools (12). Most tools that provide enrichment analyses for miRNAs first convert them to their targets and then perform the analysis on the target genes (13–16). Recently, it has been shown that this approach is biased and leads to inaccurate results (17). To overcome this bias, Godard and van Eyll suggested converting the gene-categories into miRNA-categories and then performing the enrichment analysis directly at the level of miRNAs. Among the most widely used tools implementing this approach is the ‘Tool for annotations of human miRNAs’ (TAM) (18), which is based on over-representation analysis (ORA) and miRNA categories collected from databases such as HMDD (19) or miRBase (20) and additional publications.
Here, we present miEAA which relies on the established statistical framework of the gene set analysis toolkit GeneTrail (21). GeneTrail finds significantly enriched categories for gene sets and annotates them accordingly. Like GeneTrail, miEAA also provides the two most common statistical analyses, ORA and GSEA (gene set enrichment analysis) (22). In contrast to GeneTrail, miEAA is tailored for miRNA input as it supports miRNA precursor names, as well as mature miRNA names. To offer a broad functionality and applicability, we collected about 14 000 different miRNA sets from the literature and various important miRNA databases and integrated them into miEAA. Furthermore, we added tools for the conversion of miRNAs and precursors into different miRBase versions, as well as a converter between miRNA names and precursor names. To exemplify the functionality of miEAA, we analyzed a set of lung adenocarcinoma related miRNAs and also compared the findings to an analysis with TAM. miEAA is designed as a web-based application and is freely accessible at http://www.ccb.uni-saarland.de/mieaa_tool/.
IMPLEMENTATION
Workflow
The workflow of our miEAA tool is presented in Figure 1. We differentiate between miRNA precursor names and mature miRNA names as input, because we collected the data specific for those entities if possible. Since our annotations rely on the miRBase v21 names, users can also convert their miRNA/precursor names from earlier miRBase versions to version 21 using an additional tool linked on the miEAA homepage. In addition, users can also convert their miRNAs to precursor names or vice versa. After choosing the input type, the user can upload their test set and then select between ORA or GSEA (gene set enrichment analysis). The ORA also demands a reference/background set that the user can also upload. If this option is skipped, all annotated miRNAs/precursors we collected are used as reference set. If performing a GSEA, the input set of miRNAs/precursors must be sorted by some criterion, e.g. expression level. In a last step, the user can choose the miRNA/precursor categories that they want to analyze with miEAA, as well as set some statistical parameters such as significance threshold and P-value adjustment. After miEAA has finished the computation, the results are illustrated in tabular form on an HTML page.
Figure 1.

Workflow of miEAA. Input for miEAA are either miRNA or precursor names from miRBase version 21. After uploading a plain text file containing these names, the user can chose the enrichment method: ORA or GSEA. Depending on this choice, the user can provide an own reference set for ORA. For GSEA, the input list must be sorted by a meaningful criterion. In a last step, the user can choose the categories that should be analyzed, as well as the P-value significance threshold and adjustment method. After computation, the results are presented in a tabular format on an HTML web site and can also be downloaded as Excel sheet or tab-separated text file.
Integrated resources
Since miEAA is intended to serve as a miRNA annotation and enrichment tool, we collected information from different miRNA-specific tools and databases (miRBase, HMDD2, miRWalk, miRTarBase) (20,23–25) and our own publications (3,26–28). An overview of the collected data for miRNAs and precursors is presented in Table 1. We included only subcategories in miEAA that contained at least two miRNAs or precursors, respectively. For mature miRNAs, our tool offers 10 categories and 13 962 subcategories in total. For precursors, miEAA includes five categories and 792 subcategories. From our own publications, we collected the data sets diseases (3,28), age/gender (26) and immune cells (27). These collected literature miRNAs stem from our own studies, where miRNAs were analyzed as biomarkers in peripheral blood. For the disease category, we provide three data sets per disease: miRNAs found deregulated in this disease (significant P-value), miRNAs significantly upregulated in the disease, miRNAs significantly downregulated in the disease. The immune cell data set comprises the miRNAs we found expressed in at least three individuals for the respective cell type (CD14, CD15, CD19, CD3, CD56). In addition, we assembled the immune cell specific miRNAs showing an expression in at least three individuals of one cell type, but not in the others. For the age- and gender-dependent miRNA set, we provide the miRNAs found generally correlated with age, as well as those that are positively and negatively correlated, and those that are deregulated between male and female, as well as those that are upregulated in male and upregulated in female. From miRBase (version 21), we collected the information about chromosomal location, families, cluster (50 kB) and conserved miRNAs. A conserved miRNA is in our case a miRNA that has the same sequence in at least five different species. The other data sets from miRWalk 2.0 (downloaded in 2015/05), HMDD2 and miRTarbase (Release 4.5: Nov. 1, 2013) were downloaded from their homepage and the miRNA names were converted to the current miRBase v21. For miRWalk we downloaded the data from the ‘validated miRNA-target interactions’ (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2/holistic.html). Afterward, miRNAs were annotated to belong to a category if they were annotated by miRWalk to target at least one gene in that category. The enrichment analysis is performed directly at the level of miRNAs or precursors for all collected categories. The collected data sets can be downloaded from our home page (http://www.ccb.uni-saarland.de/mieaa_tool/downloads/). Additional information about the data sets can also be found in Supplementary Table S1.
Table 1. Overview of the integrated categories in miEAA.
| Precursors | |
| Category | Subcategory |
| Families (miRBase (20)) | 151 |
| Clusters (miRBase (20)) | 216 |
| Chromosomal Locations (miRBase (20)) | 24 |
| Pubmed (miRBase (20)) | 106 |
| Diseases (HMDD2 (23)) | 295 |
| Mature miRNAs | |
| Category | Subcategory |
| Diseases (literature (3,28)) | 60 |
| Age/Gender (literature (26)) | 6 |
| Immune cells (literature (27)) | 6 |
| Diseases (mirWalk (24)) | 172 |
| Pathways (miRWalk (24)) | 484 |
| Organs (miRWalk (24)) | 176 |
| Gene Ontology (miRWalk (24)) | 5401 |
| Target genes (miRTarbase (25)) | 7632 |
| Chromosomal Locations (miRBase (20)) | 24 |
| Conserved miRNAs (miRBase (20)) | 1 |
Statistical analysis
Regarding the statistical analysis, miEAA implements the two most common approaches: the miRNA set based ORA and GSEA. ORA calculates the significance of categories for a test set and shows if the specific category is over-represented or under-represented for the test set with respect to a reference set. P-values are computed by applying the Fisher's exact test according to the following formula:
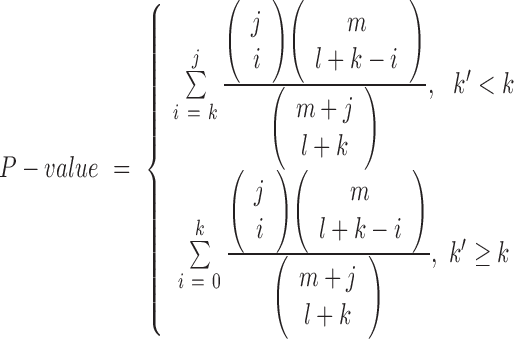 |
Given a test set consisting of miRNAs of which k belong to a certain category C and l do not belong to this category and a reference set of which j miRNAs belong to C and m miRNAs do not belong to this category, we would expect to find 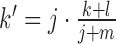 elements in the test set belonging to C by chance. If
elements in the test set belonging to C by chance. If 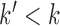 , the considered category is under-represented for the test set, otherwise it is over-represented. A standard significance threshold of 0.05 is applied to check if the computed P-value is statistically significant. In miEAA, the user can adjust this threshold arbitrarily.
, the considered category is under-represented for the test set, otherwise it is over-represented. A standard significance threshold of 0.05 is applied to check if the computed P-value is statistically significant. In miEAA, the user can adjust this threshold arbitrarily.
While ORA relies on a partitioning of miRNAs into test and reference set, GSEA considers only a sorted list of miRNAs/precursors as input. We know that l of these input miRNAs belong to a category C, and m-l do not. While traversing now the sorted input list from top to bottom, the running sum is increased by m-l or decreased by l, whenever we find that the considered miRNA belongs to C or not, respectively. The P-value is computed as the fraction of the number of permutations that have a higher absolute maximum of the running sum as compared to the considered test set. We have implemented a dynamic algorithm, which computes the exact P-value for this unweighted GSEA variant (22).
For both enrichment approaches, the user can set a lower threshold value for the number of miRNAs from the test set that must be contained in the categories. This parameter has no influence on the P-value computation and adjustment and is only applied afterward to reduce the amount of categories displayed in the output. If a category contains less miRNAs from the test set than this threshold, the category is not displayed. Furthermore, we provide the option to adjust P-values for multiple testing by two standard approaches, Bonferroni and Benjamini–Hochberg (29). However, the user can also perform the computation without P-value adjustment.
Results representation
miEAA visualizes the computed results on a clearly arranged web-page in tabular form. In addition, we provide the possibility to download the results as tab-separated text file or as Excel file. The respective results table contains the category, subcategory, P-value, expected and observed number of miRNAs, and the respective miRNAs per subcategory (Figure 2). A red or green arrow in the ORA output represent an optical visualization of over-representation or under-representation, respectively. Furthermore, the results table is freely sortable and filterable. The filters can also be combined, e.g. the user can filter all results having a P-value ‘<0.001’ and an observation value ‘>4.’ In addition, we provide a link to a view where we list for each miRNA its significant subcategories, sorted by the miRNA with the most significant annotations on top (see Supplementary Figure S1). Furthermore, we also collected sequence properties for the miRNAs/precursors in the input list (Supplementary Figure S2), e.g. the minimum free energy (MFE) as computed by RNAfold (2.1.9) from the Vienna package (30). If a miRNA has several precursors, we list all of them.
Figure 2.

Example of an ORA for miRNA input. This screenshot visualizes an example, where we analyzed a set of miRNAs with ORA as enrichment method. The output has a tabular format containing the category (e.g. target genes from miRTarbase), subcategories (e.g. a certain target gene), the P-value, the miRNAs from the test set that are contained in the subcategory, the type of enrichment, the number of miRNAs that we would expect to find and the number of miRNAs that we actually observed.
Test data for enrichment analysis
For testing the utility of our tool, we downloaded a publicly available data set from GEO (GSE48414). This data set contains array data of lung adenocarcinoma patients and normal controls and has been published by Bjaanæs et al. in 2014 (31). We downloaded the raw data of the Agilent arrays (miRBase v16) and extracted the gTotalProbeSignal, which is the average of all the background corrected signals for each replicated probe. These values were summed up to calculate the total expression value for each miRNA per sample. Normalization was applied by using the quantile normalization implemented in the preprocessCore package of the programming language R. Finally, we performed a log2 transformation of the data. For computing the differentially expressed miRNAs, we used 20 lung cancer samples and 20 matched controls. We computed the median fold changes, Wilcoxon–Mann–Whitney P-values and AUC values for all miRNAs that showed at least expression in half of the samples of one group (434 miRNAs, Supplementary Table S2). After conversion of the miRBase v16 miRNAs into v21, 423 miRNAs remained. We sorted this list by their descending AUC values and used this list of 423 miRNAs for miRNA set enrichment analysis. As a second example, we extracted miRNAs with a fold change of more than 1.5 in tumor samples compared to normal samples and having a significant adjusted two-tailed P-value (< 0.05) in the Wilcoxon–Mann–Whitney test. This list contained 49 miRNAs and was further converted into precursors with our miRNA-precursor conversion tool. For this conversion, we allowed non-unique mappings, which resulted in a set of 55 precursors.
RESULTS AND DISCUSSION
As mentioned in the Introduction, many enrichment tools are already available, at least for miRNA target genes. Some popular tools such as DIANA miRPath (16), miTalos (32) and miRTar (15) work on predicted or validated targets of miRNAs, which has been shown to be biased (17). In addition, they mostly provide only one or the other standard enrichment analysis or have some restrictions on the input size (e.g. 100 miRNAs for DIANA miRPath). Some other tools such as miSEA (10) or miTEA (11) provide alternative approaches, but they require some specialized input. The input for miTEA is a list of ranked genes and then it identifies the miRNAs that are significantly associated with these genes. miSEA requires the upload of a control and treatment file with expression data and seems to work on a mixture of miRNA names and precursor names from the look of the example files. It is not obvious how or if a conversion of miRNAs to precursors is done. Other tools as miRNApath (13) and CORNA (33) are only available as R package and are not easily applicable for nonexpert users. Therefore, we provide in this study primarily a comparison of our tool to TAM (18), which is the most similar in functionality.
As a first use case, we explored a set of 49 miRNAs we found significantly upregulated (FC > 1.5, adjusted P-value < 0.05) when comparing 20 lung cancer samples and 20 matched controls (Supplementary Table S2). With these miRNAs we performed an ORA with miEAA using the default parameters and uploading the human miRNAs spotted on the Agilent array as a reference set. This analysis resulted in 59 significant categories (Supplementary Table S3). Among the most significant categories, we found for example the target genes HOXB5, KLF11, ZEB1, RASSF2 and PTPRD. The HOX genes encode transcription factors that regulate the embryonic morphogenesis and have previously been described to be deregulated in lung cancer (34). KLF11 is also a transcription factor and tumor suppressor, which plays an important role in TGF-β induced growth inhibition in pancreatic cells (35). ZEB1 and ZEB2 are key regulators of the epithelial to mesenchymal transition, a process which contributes in cancer to the formation of metastases, and have also been described in lung cancer (36–39). Loss of RASSF2 is associated with an enhanced tumorigenicity of lung cancer cells (40). PTPRD is a candidate for a tumor suppressor gene in lung cancer (41). Besides these target genes, we also found the pathways ‘EGF EGFR signaling’ and ‘small cell lung cancer’ significantly enriched.
To compare our tool to TAM, we performed the analysis with this tool for the same set of 49 miRNAs with the following parameters: annotation set version 2, FDR adjustment, same reference set as above. For these data, the TAM analysis finds in total 14 categories significantly enriched: Learned Helplessness, mir-8 family, Pain, Carcinoma, Cell cycle related, carbohydrate metabolism, Glomerulonephritis, Breast Neoplasms, hsa-mir-200a cluster, hsa-mir-182 cluster, Nephrosclerosis, Carcinoma Renal Cell, Carcinoma Spindle Cell, Cholangiocarcinoma, which seem to be rather unspecific and not necessarily related to lung cancer. The different annotation and data handling can explain the differences in the results between TAM and miEAA. TAM converts the input into precursor names by cutting off the -3p/-5p/* ending of the name and renaming ‘miR’ into ‘mir.’ Thus, if data sets contain two mature miRNAs of the same precursor, these would be merged, introducing a potential bias. Furthermore, this way of conversion may cause challenges if mature miRNAs stem from different precursors, e.g. in our data set of 49 miRNAs, we have four cases, where we have several potential precursors. As an example, the miRNA hsa-miR-9-3p can stem from the precursors hsa-mir-9-1, hsa-mir-9-2 or hsa-mir-9-3, the miRNA hsa-miR-7-5p from the precursors hsa-mir-7-1, hsa-mir-7-2 or hsa-mir-7-3. Another problem we noticed is that TAM sometimes does not recognize official precursor names. When using hsa-let-7f-1 and hsa-let-7f-2 as input, it seems that TAM does not recognize them correctly, although it provides annotations when hsa-let-7f is used as input. Therefore, when using TAM these conversion steps have to be kept in mind and users should take care on the conversion before uploading the data. To facilitate mapping tasks we implemented conversion tools between different miRBase versions, as well as a tool that converts miRNA names to precursor names and vice versa. These supporting tools can be accessed from the miEAA homepage. To show that there are still many differences between miEAA and TAM, even if we provide TAM with correctly converted precursors and although these tools have an overlap of annotation resources, we converted the 49 miRNAs into their corresponding 55 precursors, as well as the reference set, and repeated the above analysis with the precursor input for both tools. While TAM still finds the same 14 categories significant, miEAA finds 77 categories significantly enriched. Of course, there is now an overlap of the findings of miEAA and TAM, but miEAA finds also categories such as ‘carcinoma, non-small-cell lung’ and ‘lung neoplasms’ significantly enriched (Supplementary Table S4).
In contrast to ORA, GSEA is a threshold free approach that does not require a reference set as background distribution. The above described analysis is a frequently applied procedure for extracting miRNAs from array experiments that are upregulated according to an arbitrary threshold. The results of the ORA vary largely on this chosen threshold. To overcome this issue, a GSEA can be performed by sorting the expressed miRNAs on the array, e.g., by their AUC values or fold changes. As mentioned in the Methods section, we sorted the list of 423 miRNA from the same lung cancer study by their AUC values and performed a GSEA using the default settings in miEAA. In total, this analysis yielded 148 significant categories, with most hits in pathways (69), Gene Ontology terms (34), diseases (21) and targets (11) (Supplementary Table S5). Among the pathways, we find some that are directly associated with specific cancers (Non small cell lung cancer, Melanoma, Glioma, Prostate cancer) or are known to be often influenced in cancer development and progression (ErbB signaling pathway, p53 pathway, Focal adhesion, Jak STAT signaling pathway, p38 MAPK Signaling Pathway, PI3 kinase pathway, Apoptosis, Wnt signaling pathway, Adherens junction). The GO terms having the most annotated miRNAs are involved in transcription processes. In this context, we also find transcription factors among the significant targets (HOXB5, TCF7L1, KLF11). Another interesting finding is that most of the miRNAs that are downregulated in the tumor tissue are conserved miRNAs.
Summarizing our analysis of the lung cancer data set of Bjaanæs et al., we showed that the ORA as well as GSEA in miEAA yielded interesting results and highlighted some pathways and targets that may be influenced in lung cancer development.
CONCLUSION
The development of miRNA enrichment tools gain rapidly on importance. First solutions are already available, including the TAM tool, which offers ORA for miRNA precursors. Here, we presented miEAA, a comprehensive miRNA enrichment tool in terms of statistical tests and miRNA/precursor categories. While the TAM tool contains 362 categories only for precursors, miEAA includes over 14 000 categories, defined both for precursors and mature miRNAs. In addition, we provide supporting tools for the conversion of precursor and miRNA names into different miRBase versions and for the mapping of precursors to miRNAs or vice versa. The correct mapping and handling of precursors or miRNAs is an important step, often neglected in currently available tools.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Saarland University. Funding for open access charge: Saarland University.
Conflict of interest statement. None declared.
REFERENCES
- 1.Keller A., Leidinger P., Vogel B., Backes C., ElSharawy A., Galata V., Müller S., Marquart S., Schrauder M., Strick R., et al. miRNAs can be generally associated with human pathologies as exemplified for miR-144*. BMC Med. 2014;12:224. doi: 10.1186/s12916-014-0224-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 3.Leidinger P., Backes C., Deutscher S., Schmitt K., Mueller S.C., Frese K., Haas J., Ruprecht K., Paul F., Stahler C., et al. A blood based 12-miRNA signature of Alzheimer disease patients. Genome Biol. 2013;14:R78. doi: 10.1186/gb-2013-14-7-r78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lewis B.P., Burge C.B., Bartel D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 5.Friedlander M.R., Chen W., Adamidi C., Maaskola J., Einspanier R., Knespel S., Rajewsky N. Discovering microRNAs from deep sequencing data using miRDeep. Nat. Biotech. 2008;26:407–415. doi: 10.1038/nbt1394. [DOI] [PubMed] [Google Scholar]
- 6.Hackenberg M., Rodriguez-Ezpeleta N., Aransay A.M. miRanalyzer: an update on the detection and analysis of microRNAs in high-throughput sequencing experiments. Nucleic Acids Res. 2011;39:W132–W138. doi: 10.1093/nar/gkr247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Enright A.J., John B., Gaul U., Tuschl T., Sander C., Marks D.S. MicroRNA targets in Drosophila. Genome Biol. 2004;5:R1. doi: 10.1186/gb-2003-5-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lewis B.P., Shih I.H., Jones-Rhoades M.W., Bartel D.P., Burge C.B. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 9.Xiao F., Zuo Z., Cai G., Kang S., Gao X., Li T. miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res. 2009;37:D105–D110. doi: 10.1093/nar/gkn851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corapcioglu M.E., Ogul H. miSEA: microRNA set enrichment analysis. Biosystems. 2015;134:37–42. doi: 10.1016/j.biosystems.2015.05.004. [DOI] [PubMed] [Google Scholar]
- 11.Steinfeld I., Navon R., Ach R., Yakhini Z. miRNA target enrichment analysis reveals directly active miRNAs in health and disease. Nucleic Acids Res. 2013;41:e45. doi: 10.1093/nar/gks1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang D., Wang J., Lu M., Song F., Cui Q. Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics. 2010;26:1644–1650. doi: 10.1093/bioinformatics/btq241. [DOI] [PubMed] [Google Scholar]
- 13.Cogswell J.P., Ward J., Taylor I.A., Waters M., Shi Y., Cannon B., Kelnar K., Kemppainen J., Brown D., Chen C., et al. Identification of miRNA changes in Alzheimer's disease brain and CSF yields putative biomarkers and insights into disease pathways. J. Alzheimers Dis. 2008;14:27–41. doi: 10.3233/jad-2008-14103. [DOI] [PubMed] [Google Scholar]
- 14.Lu T.P., Lee C.Y., Tsai M.H., Chiu Y.C., Hsiao C.K., Lai L.C., Chuang E.Y. miRSystem: an integrated system for characterizing enriched functions and pathways of microRNA targets. PLoS One. 2012;7:e42390. doi: 10.1371/journal.pone.0042390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsu J.B., Chiu C.M., Hsu S.D., Huang W.Y., Chien C.H., Lee T.Y., Huang H.D. miRTar: an integrated system for identifying miRNA-target interactions in human. BMC Bioinformatics. 2011;12:300. doi: 10.1186/1471-2105-12-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vlachos I.S., Kostoulas N., Vergoulis T., Georgakilas G., Reczko M., Maragkakis M., Paraskevopoulou M.D., Prionidis K., Dalamagas T., Hatzigeorgiou A.G. DIANA miRPath v.2.0: investigating the combinatorial effect of microRNAs in pathways. Nucleic Acids Res. 2012;40:W498–W504. doi: 10.1093/nar/gks494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Godard P., van Eyll J. Pathway analysis from lists of microRNAs: common pitfalls and alternative strategy. Nucleic Acids Res. 2015;43:3490–3497. doi: 10.1093/nar/gkv249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu M., Shi B., Wang J., Cao Q., Cui Q. TAM: a method for enrichment and depletion analysis of a microRNA category in a list of microRNAs. BMC Bioinformatics. 2010;11:419. doi: 10.1186/1471-2105-11-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu M., Zhang Q., Deng M., Miao J., Guo Y., Gao W., Cui Q. An analysis of human microRNA and disease associations. PLoS One. 2008;3:e3420. doi: 10.1371/journal.pone.0003420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Griffiths-Jones S., Grocock R.J., van Dongen S., Bateman A., Enright A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–D144. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Backes C., Keller A., Kuentzer J., Kneissl B., Comtesse N., Elnakady Y.A., Müller R., Meese E., Lenhof H.-P. GeneTrail—advanced gene set enrichment analysis. Nucleic Acids Res. 2007;35:W186–W192. doi: 10.1093/nar/gkm323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keller A., Backes C., Lenhof H.-P. Computation of significance scores of unweighted Gene Set Enrichment Analyses. BMC Bioinformatics. 2007;8:290. doi: 10.1186/1471-2105-8-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y., Qiu C., Tu J., Geng B., Yang J., Jiang T., Cui Q. HMDD v2.0: a database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2014;42:D1070–D1074. doi: 10.1093/nar/gkt1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dweep H., Sticht C., Pandey P., Gretz N. miRWalk–database: prediction of possible miRNA binding sites by "walking" the genes of three genomes. J. Biomed. Inform. 2011;44:839–847. doi: 10.1016/j.jbi.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 25.Hsu S.D., Tseng Y.T., Shrestha S., Lin Y.L., Khaleel A., Chou C.H., Chu C.F., Huang H.Y., Lin C.M., Ho S.Y., et al. miRTarBase update 2014: an information resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2014;42:D78–D85. doi: 10.1093/nar/gkt1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meder B., Backes C., Haas J., Leidinger P., Stahler C., Grossmann T., Vogel B., Frese K., Giannitsis E., Katus H.A., et al. Influence of the Confounding Factors Age and Sex on MicroRNA Profiles from Peripheral Blood. Clin. Chem. 2014;60:1200–1208. doi: 10.1373/clinchem.2014.224238. [DOI] [PubMed] [Google Scholar]
- 27.Leidinger P., Backes C., Meder B., Meese E., Keller A. The human miRNA repertoire of different blood compounds. BMC Genomics. 2014;15:474. doi: 10.1186/1471-2164-15-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keller A., Leidinger P., Bauer A., Elsharawy A., Haas J., Backes C., Wendschlag A., Giese N., Tjaden C., Ott K., et al. Toward the blood-borne miRNome of human diseases. Nat. Methods. 2011;8:841–843. doi: 10.1038/nmeth.1682. [DOI] [PubMed] [Google Scholar]
- 29.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B. Methodol. 1995;57:289–300. [Google Scholar]
- 30.Lorenz R., Bernhart S.H., Honer Zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011;6:26. doi: 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bjaanaes M.M., Halvorsen A.R., Solberg S., Jorgensen L., Dragani T.A., Galvan A., Colombo F., Anderlini M., Pastorino U., Kure E., et al. Unique microRNA-profiles in EGFR-mutated lung adenocarcinomas. Int. J. Cancer. 2014;135:1812–1821. doi: 10.1002/ijc.28828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Preusse M., Theis F.J., Mueller N.S. miTALOS v2: Analyzing Tissue Specific microRNA Function. PLoS One. 2016;11:e0151771. doi: 10.1371/journal.pone.0151771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu X., Watson M. CORNA: testing gene lists for regulation by microRNAs. Bioinformatics. 2009;25:832–833. doi: 10.1093/bioinformatics/btp059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abe M., Hamada J., Takahashi O., Takahashi Y., Tada M., Miyamoto M., Morikawa T., Kondo S., Moriuchi T. Disordered expression of HOX genes in human non-small cell lung cancer. Oncol. Rep. 2006;15:797–802. [PubMed] [Google Scholar]
- 35.Buck A., Buchholz M., Wagner M., Adler G., Gress T., Ellenrieder V. The tumor suppressor KLF11 mediates a novel mechanism in transforming growth factor beta-induced growth inhibition that is inactivated in pancreatic cancer. Mol. Cancer Res. 2006;4:861–872. doi: 10.1158/1541-7786.MCR-06-0081. [DOI] [PubMed] [Google Scholar]
- 36.Gemmill R.M., Roche J., Potiron V.A., Nasarre P., Mitas M., Coldren C.D., Helfrich B.A., Garrett-Mayer E., Bunn P.A., Drabkin H.A. ZEB1-responsive genes in non-small cell lung cancer. Cancer Lett. 2011;300:66–78. doi: 10.1016/j.canlet.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Y.J., Ping C., Tang J., Zhang W. MicroRNA-455 suppresses non-small cell lung cancer through targeting ZEB1. Cell Biol. Int. 2016 doi: 10.1002/cbin.10584. doi:10.1002/cbin.10584. [DOI] [PubMed] [Google Scholar]
- 38.Yoshida T., Song L., Bai Y., Kinose F., Li J., Ohaegbulam K.C., Munoz-Antonia T., Qu X., Eschrich S., Uramoto H., et al. ZEB1 Mediates Acquired Resistance to the Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors in Non-Small Cell Lung Cancer. PLoS One. 2016;11:e0147344. doi: 10.1371/journal.pone.0147344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiao A., Sui M., Zhang L., Sun P., Geng D., Zhang W., Wang X., Li J. MicroRNA-200c inhibits the metastasis of non-small cell lung cancer cells by targeting ZEB2, an epithelial-mesenchymal transition regulator. Mol. Med. Rep. 2016;13:3349–3355. doi: 10.3892/mmr.2016.4901. [DOI] [PubMed] [Google Scholar]
- 40.Clark J., Freeman J., Donninger H. Loss of RASSF2 Enhances Tumorigencity of Lung Cancer Cells and Confers Resistance to Chemotherapy. Mol. Biol. Int. 2012;2012:705948. doi: 10.1155/2012/705948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kohno T., Otsuka A., Girard L., Sato M., Iwakawa R., Ogiwara H., Sanchez-Cespedes M., Minna J.D., Yokota J. A catalog of genes homozygously deleted in human lung cancer and the candidacy of PTPRD as a tumor suppressor gene. Genes Chromosomes Cancer. 2010;49:342–352. doi: 10.1002/gcc.20746. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.