


Abstract
tRNA-derived small RNA fragments (tRFs) are one class of small non-coding RNAs derived from transfer RNAs (tRNAs). tRFs play important roles in cellular processes and are involved in multiple cancers. High-throughput small RNA (sRNA) sequencing experiments can detect all the cellular expressed sRNAs, including tRFs. However, distinguishing genuine tRFs from RNA fragments generated by random degradation remains a major challenge. In this study, we developed an integrated web-based computing system, tRF2Cancer, to accurately identify tRFs from sRNA deep-sequencing data and evaluate their expression in multiple cancers. The binomial test was introduced to evaluate whether reads from a small RNA-seq data set represent tRFs or degraded fragments. A classification method was then used to annotate the types of tRFs based on their sites of origin in pre-tRNA or mature tRNA. We applied the pipeline to analyze 10 991 data sets from 32 types of cancers and identified thousands of expressed tRFs. A tool called ‘tRFinCancer’ was developed to facilitate the users to inspect the expression of tRFs across different types of cancers. Another tool called ‘tRFBrowser’ shows both the sites of origin and the distribution of chemical modification sites in tRFs on their source tRNA. The tRF2Cancer web server is available at http://rna.sysu.edu.cn/tRFfinder/.
INTRODUCTION
tRNA-derived small RNA fragments (tRFs) are one group of small non-coding RNAs derived from mature or precursor transfer RNAs (tRNAs) (1,2). tRFs play important roles in the regulation of gene expression (3–5). Their functions include control of viral replication (6), modulation of cell viability and proliferation (7), inhibition of protein translation (8,9) and modulation of cancer progression (10). Recent studies revealed that parental sperm tRNA fragments may influence the metabolism of offspring (11–13).
The earliest findings on the cleavage of tRNAs were observed in bacteria (14), primitive eukaryotes (15) and almost every branch of life (16–22). The fragments are primarily generated by a single cleavage of mature tRNA in or near the anticodon loop when cells respond to stress conditions and are referred to as tRNA halves. At the early stage, they were believed to be the products of degradation and function in the inhibition of translation by depletion of the tRNA pool. However, subsequent findings revealed that tRNAs could generate functional small non-coding RNAs that play important roles in gene expression regulation (1,2,4). These milestone findings revealed that tRNAs have additional functions beyond participating in protein synthesis, thus promoting wide interest in the investigation of tRFs and their functions (1,2,7,8,10,23–41).
tRFs are generated through precise processing of both mature and precursor tRNAs (pre-tRNAs). This process produces at least three types of tRFs, including tRF-5, tRF-3 and tRF-1 (2). tRF-5 and tRF-3 are derived from the 5′ and 3′ ends of mature tRNAs, respectively, and were first observed in LNCaP and C4-2 cells (2). tRF-1 is derived from the 3′ trailer fragment of pre-tRNA transcripts and is tightly correlated with cancer cell proliferation (2,42). Another more recently characterized class of tRFs are those found at the internal region of the mature tRNAs. These tRFs can straddle the anticodon and are termed internal tRFs (i-tRFs) (35). Accumulating evidence has demonstrated that tRFs have important roles in human cancers (8,10,29–31,34–36,43,44), and their aberrant expression in cancers was therefore considered as potential diagnostic biomarkers or even for use in medicine for manipulation of cancer cells (5). The functions of tRFs and their mechanisms influencing cancer processes have become an intriguingly new aspect under investigation.
In recent years, the development of high-throughput sRNA sequencing approaches enabled the detection of sRNAs at unprecedented depth. This approach has been successfully applied to the systematic identification of microRNAs (36). However, many problems associated with the identification of tRFs have been noted (5,45). One of the major challenges involves distinguishing the bona fide tRFs from random degradation fragments in a large pool of sequenced sRNAs. Despite accumulating evidence indicating common features of tRFs, some researchers are still concerned that these fragments could simply represent the by-products of random cleavage of tRNAs (5). While there exist extensive tRNA molecules in cellular total RNAs, their degradation fragments would indeed be present in cells and probably be detected by deep-sequencing technology. In addition, the expression of tRFs is spatiotemporal across different cell types and tissue samples. Therefore, the claim of the discovery of tRFs based on the evidence of detecting sRNAs from tRNA molecules in a single deep sequencing run is untenable and will result in more problems in further functional validation experiments.
Currently, there is no online tool available for biologists, who are unfamiliar with the command-line environments, to efficiently identify tRFs in their own sRNA-sequencing data and compare the results with other public data sets. Therefore, given the increasing amount of small RNA sequencing data available (especially large-scale cancer-related data sets) there is a great need to develop a web-based tool to identify tRFs and explore the expression of tRFs in multiple types of cancers.
To address this need, we developed tRF2Cancer, the first public server for identifying tRFs and their expression in cancers from deep-sequencing data (Figure 1). Our web server is able to accurately identify known and novel types of tRFs using a proven statistical method. In addition, the expression of these tRFs in 10 991 samples from 32 types of cancers is provided. tRF2Cancer is also designed to search for the relationships between tRFs and multiple types of chemical modifications on their source tRNAs. We believe that this web server will help researchers investigate new tRFs and discover their functions and potential use as cancer biomarkers from sRNA deep-sequencing data.
Figure 1.

tRF2Cancer workflow.
tRF2Cancer ANALYSIS WORKFLOW
Data sources and gene annotation
Human genome sequences were downloaded from the UCSC bioinformatics websites (version hg19) (46), and human gene annotations were obtained from the UCSC database (47) and Ensembl database (Release 76) (48). Known non-coding RNA sequences were obtained from the Rfam database (Release 12.0) (49). microRNA genes were obtained from the miRBase database (Release 21) (50). The tRNA sequences were downloaded from the GtRNAdb database (Release 2.0) (51), and these data were subject to several pre-processing steps. For precursor tRNA genes, we extracted the sequences including the tRNA genes and 100 bp up- and downstream of the 3′-end of tRNA genes. For mature tRNA sequences, we removed the introns and added CCA to the 3′-end of the tRNA gene sequences. Small RNA sequencing data of cancer samples were retrieved from the Cancer Genome Atlas (TCGA) database (52). A total of 10 991 samples from 32 types of cancers were integrated into tRF2Cancer. Table 1 lists the types and numbers of cancer samples. The tRNA modifications sites were retrieved from RMBase (Release 1.0), a database that contains RNA modifications identified from high-throughput sequencing data sets (53). The secondary structure of tRNA was displayed by forna (54). A javascript-based genome browser, Jbrowse (Release 1.11.6), was used to navigate the modification sites and tRF reads distribution on source tRNAs (55).
Table 1. Cancer types and number of samples used in tRF2Cancer.
| Abbreviation | Cancer Types | Sample Counts |
|---|---|---|
| ACC | Adrenocortical Carcinoma | 80 |
| BLCA | Bladder Urothelial Carcinoma | 437 |
| BRCA | Breast Invasive Carcinoma | 1207 |
| CESC | Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma | 312 |
| CHOL | Cholangiocarcinoma | 45 |
| COAD | Colon Adenocarcinoma | 447 |
| DLBC | Lymphoid Neoplasm Diffuse Large B-cell Lymphoma | 47 |
| ESCA | Esophageal Carcinoma | 200 |
| GBM | Glioblastoma Multiforme | 5 |
| HNSC | Head and Neck Squamous Cell Carcinoma | 569 |
| KICH | Kidney Chromophobe | 91 |
| KIRC | Kidney Renal Clear Cell carcinoma | 616 |
| KIRP | Kidney Renal Papillary Cell Carcinoma | 326 |
| LGG | Brain Lower Grade Glioma | 530 |
| LIHC | Liver Hepatocellular Carcinoma | 425 |
| LUAD | Lung Adenocarcinoma | 567 |
| LUSC | Lung Squamous Cell Carcinoma | 523 |
| MESO | Mesothelioma | 87 |
| OV | Ovarian Serous Cystadenocarcinoma | 499 |
| PAAD | Pancreatic adenocarcinoma | 183 |
| PCPG | Pheochromocytoma and Paraganglioma | 187 |
| PRAD | Prostate Adenocarcinoma | 551 |
| READ | Rectum Adenocarcinoma | 161 |
| SARC | Sarcoma | 263 |
| SKCM | Skin Cutaneous Melanoma | 452 |
| STAD | Stomach Adenocarcinoma | 497 |
| TGCT | Testicular Germ Cell Tumors | 156 |
| THCA | Thyroid Carcinoma | 573 |
| THYM | Thymoma | 126 |
| UCEC | Uterine Corpus Endometrioid Carcinoma | 579 |
| UCS | Uterine Carcinosarcoma | 57 |
| UVM | Uveal Melanoma | 80 |
Searching tRNA-derived small RNAs from deep-sequencing data
Figure 1 presents the workflow of tRF2Cancer. After the pre-processing of the sequencing data, small RNAs were first mapped to the human genome to remove unmapped reads. Aligned reads were then mapped to the known human transcript sequences, including mRNAs, snoRNAs, snRNAs, rRNAs, microRNAs and repeat sequences. Reads that were successfully mapped to those known transcripts were discarded. The remaining reads were then mapped to both precursor tRNA genes and mature tRNA sequences. We use bowtie (version 1 or version 2) (56,57), an ultrafast and exhaustive small RNA mapping program, to perform the sequence alignment between deep-sequencing small RNAs and long transcripts. The alignment mode and parameters can be set by users. The parameters include the number of allowed mismatches (0 by default), the region of tRFs length (16–30 nt by default), and whether indels (insertions and deletions) are allowed (no indels by default). Finally, only those reads that were aligned to the same strand with the source tRNA transcripts were considered as tRNA-derived small RNAs and used for subsequent analysis.
Analyzing the sequenced small RNAs to identify tRFs according to their biogenesis
The major challenge in tRF identification involves distinguishing actual tRFs from random degradation fragments. To solve this problem, we developed tRFfinder to accurately identify tRFs according to their biogenesis. The tRNA precursors (pre-tRNA) first underwent a series of processing to generate mature tRNA molecules, including cleavage of 5′ end (5′ leader) and 3′ end (3′ trailer) by RNase P and tRNaseZ, respectively; addition of 3′ CCA trinucleotide to the acceptor stem; splicing of introns, if necessary; and chemical modification of bases in the nucleus before transportation into the cytoplasm (58). Then, the mature tRNA with a stable structure could be recognized and cut by endonucleases (such as Dicer) to release tRF-5 at the 5′ end and/or tRF-3 at the 3′-end (Figure 2A). Otherwise, the tRNAs are degraded to generate various random sRNAs (Figure 2B). In summary, compared with the randomly degraded fragments, the tRFs possess at least the following three characteristics: (i) remarkable site-specificity; (ii) defined lengths; and (iii) significantly higher abundance. Therefore, if a tRNA generates genuine tRFs, then one can expect that most of the deep-sequencing reads are enriched in one or more of the three following regions: the 5′ end, the 3′ end and the 3′ trailer region of the tRNA (Figure 2A). Furthermore, it is expected that only very few reads do not correspond to these three products. Otherwise, the degradation fragments are distributed randomly on the source tRNA (Figure 2B).
Figure 2.

Analysis of small RNA sequencing reads for identification of tRNA-derived small RNA fragments (tRFs). (A) After cleavage of transfer RNAs (tRNA) at specific positions, small RNA fragments with consistent characteristics are generated. Each of the small RNA could be detected (with certain probability) by sequencing. After mapping sequencing reads back to the tRNA transcripts, most of the reads are distribute within similar regions with significantly higher frequencies at the tRF-5, tRF-3 and tRF-1 regions. (B) Random degradation of tRNA transcripts produces small RNA fragments. When mapped to the tRNA transcripts, it is expected that these random fragments are uniformly distributed across the entire length of source tRNA, and that features of these fragments (such as sites of origin and frequencies of distribution) are inconsistent with those of bona fide tRFs.
After mapping the small RNA reads to tRNA sequences, tRFfinder first assumes that reads are randomly distributed along the entire tRNA. According to this assumption, we could conclude that, of the entire length of tRNA, the probability (p) of specific nucleotide position in tRNA sequence (either precursor or mature) being mapped by one small-RNA read is
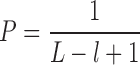 |
(1) |
where L is the length of the tRNA, and l is the length of the small RNA fragment being mapped onto the tRNA.
Based on the assumption of random distribution, the probability of more than k (inclusive) small RNA fragments mapped onto the same position in the tRNA follows the binomial distribution, and the probability of this event is
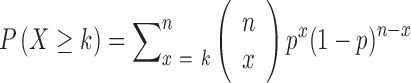 |
(2) |
where k is the observed counts of small RNA fragments mapped onto that specific nucleotide position in the tRNA, and n is the total number of reads mapped onto the entire length of tRNA. Hence, P is the probability that one nucleotide position in the tRNA is included into k or more small RNA fragments as a product of random degradation.
For the real mapping case, if one specific nucleotide in the tRNA sequence corresponds to more than k or more small RNA sequencing reads, but the probability of this event occurring by chance (Equation 2) is less than 1% (by default), then we could conclude that this site contains a significant enrichment of small RNAs (with 99% confidence, by default). Thus, this site is defined as a significant site. If a region in the tRNA sequence contains more than m consecutive significant sites, the region is considered a tRF region, where m is the shortest tRF length set by users (16 by default). After the tRF region is obtained, all the reads in the tRF region compose a stack of reads. The total count of reads in a stack was calculated and is represented by C. The largest read count is represented by c. We then calculated the coverage rate as c/C, if the value is greater than 0.6, the read with the highest count will be chosen as the main tRF and presented in red in the result page (Figure 4C). In addition to the main tRF, we also provide the sequence and the abundance of other reads to the users, since the less abundant tRFs may also have functions (35,59). In the result page, users can sort the reads mappable to tRNA based on start positions, lengths or abundance.
Figure 4.

Overview of the tRF2Cancer result. (A) The summary table of predicted tRFs results. (B) Detailed information regarding tRFs. (C) The display of sequencing reads distribution on source tRNA. (D) The region of tRFs on tRNA secondary structure. (E) The expression of tRFs in 32 types of cancers. (F) The genome browser for tRF profiling and modification site distribution on source tRNA.
Classifying tRFs based on sites of origin in tRNA
The types of tRFs are classified by their sites of origin in pre-tRNA or mature tRNA. tRF2Cancer adopts a 4-category system similar to that of tRFdb (60) with some modifications (Figure 3), as follows: (i) tRF-5, which originates from the first or second base of mature tRNAs; (ii) tRF-3, which originates from the 3′ end of mature tRNA with CCA trinucleotide at 3′-end; (iii) tRF-1, which originates from the beginning of the 3′ end flanking sequences (sequences that are cleaved before maturation of tRNA) with poly-U residues at 3′-end; and (iv) tRF-novel, which does not belong to tRF-5, tRF-3 or tRF-1 and is typically derived from the internal region of the mature tRNA sequence (Figure 3).
Figure 3.

Demonstration of the biogenesis of different types of tRFs. tRF-1 is generated from the 3′-trailer of primary tRNA. tRF-5, -novel and -3 are produced from the 5′-end, internal and 3′-end of mature tRNA, respectively.
Evaluating tRFs expression in different cancers types
We developed tRFinCancer to provide an integrated view of the expression of one selected tRF among 32 types of cancers. We retrieved raw data of small RNA deep-sequencing from public TCGA repositories, and the raw data underwent a series of processing steps, including clipping of 3′-adapters using FASTX-toolkit software (http://hannonlab.cshl.edu/fastx_toolkit/), removal of low-complexity reads and collapsing identical reads together (Figure 1). Then, the remaining reads were used for the identification of tRFs in all of the cancer samples. When users obtain a tRF of interest either by tRFfinder or from literature, tRFinCancer can be used to inspect the expression of this particular tRF in 32 types of cancers. For cancers with multiple samples, we provide the value of mean reads per million (RPM) of all the samples to represent the tRF expression.
The abundance of tRFs is evaluated using their sequencing counts and is normalized to the total count of all reads that mapped to tRNAs to obtain RPM values:
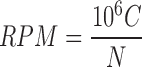 |
(3) |
where C is the sum of reads mapped onto the tRF region, and N is the total number of reads mapped onto all of the tRNA genes.
Evaluation of different types of mismatches/indels and displaying chemical modification sites on tRFs
tRNAs undergo extensive modifications. Researchers are concerned that the pausing of reverse transcriptase at chemically modified sites in tRNA contributes to the reads detected in deep-sequencing data (61). To avoid this type of error, tRFfinder excluded the tRF candidates whose 3′ ends match exactly to the chemical modification sites on the pre-tRNA. In addition, chemical modifications can lead to reverse transcriptase pausing, which often results in misincorporation of nucleotides, or indels (61–63). tRFfinder uses a scoring scheme to handle different types of mismatches/indels. When the reads are mapped to the tRNA gene sequences and a tRF region is obtained, tRFfinder scores each site of the tRF region according to the following rules (Supplementary Table S1 and Figure S1).
After each site of the tRF region is scored, tRFfinder sums the scores to obtain a total score. To eliminate the effect of length on the total score (the longer the region, usually the higher the total score), the total score is divided by the length of the tRF region to obtain the alignment score for this region. By default, tRFfinder outputs only regions with alignment scores greater than 100 (this threshold can be set by users in the parameter option lists).
For users who want to investigate the chemical modifications on tRFs, we provide ‘tRFBrowser’ for browsing and visualizing the profiling of tRFs and the modification sites distribution on the source tRNAs. We collected modification sites including 5-methylcytosines (m5c), 2′-O-methylations (2′-O-Me), pseudouridine (Ψ) and N6-methyladenosine (m6A). The profiling of tRFs from the 32 types of cancers are also embedded in tRFBrowser. Because there are 10 991 samples in total, tRFBrowser randomly selects 10 samples in each cancer to show at one time. Therefore, users can simultaneously inspect tRF profiling and distribution of modification sites on tRNAs.
IMPLEMENTATION
tRF2Cancer was developed under the Apache/PHP/MySQL environment on the Linux system. The backend was implemented in Perl. The server was equipped with 64 bit 8-core 2.00 GHz Intel Xeon and 12 GB of RAM. The web application was designed for multiple platforms, and is able to run in Google Chrome (17 and later), Firefox (10 and later), Apple Safari (6 and later) and Internet Explorer (9 and later). The method that tRF2Cancer adopts is time efficient. During a test with 11 641 sRNA sequences, running times of approximately 0 m:48 s and 4 m:18 s are required with the loosest filtering criteria and the strictest filtering criteria, respectively.
DATA INPUT
tRF2Cancer offers a user-friendly interface for convenient manipulation. The main input to tRF2Cancer is small RNA deep-sequencing data in the FASTA format. The identical reads must be collapsed together. This should be done by the users before they upload the file to tRF2Cancer. Actually, most of the sequencing company will generate the small RNA sequencing results in the FASTA format, with the identical reads collapsed together. Users can input the sequence data either from a local file or by direct pasting, and tRF2Cancer also supports compressed file (in ZIP, tar.gz or RAR format) as inputs. tRF2Cancer provides additional options for fine-tuning parameters. Users can set the number of allowed mismatches, the mode of mapping (with or without indels), the range of tRFs length and the P-value cutoff before submitting a job. Depending on the number of sRNAs and the size of samples, a typical run may take several minutes to finish after data submission. For notification of job completion, users can provide tRF2Cancer a valid email address.
DATA OUTPUT
A summary table with eight fields is provided in the result page for inspection of predicted tRFs (Figure 4). The fields include tRF type (tRF-5, tRF-3, tRF-1 or tRF-novel), RPM, tRF length, the name of the source tRNA and the positions on source tRNA from which the tRF is derived (Figure 4A). In addition, each button on the result table will lead the users to a special tRF visualization page, which includes visualization of sequencing-read distribution on source tRNA (Figure 4C); the structure of source tRNA, with the highlighted sequence representing the origin of tRFs (Figure 4D); the expression of tRFs in 32 types of cancers (Figure 4E); and the distribution of both tRFs and possible chemical modification sites on the corresponding source tRNA (Figure 4F).
To export the results, users can either copy to clipboard or download the file in Excel format or CSV format. For further retrieval of results, users can bookmark the result page. Each result is assigned a random and unique ID by the server.
EVALUATING tRF2Cancer PERFORMANCE WITH EXPERIMENTALLY VALIDATED DATA AND OTHER TOOLS/ PIPELINES
To evaluate the performance of tRF2Cancer, we collected known tRFs from previous studies. Lee et al. detected 135 tRFs in deep-sequencing data from prostate cancer cell lines (LNCaP and C4-2), 17 of which were validated by Northern hybridization, quantitative RT-PCR and splinted ligation assays (2). We applied tRFfinder on an independent data set downloaded from GEO (GSE79365), which contains 5 samples of prostate cancer cell lines (P69, M12, M2182). Of the 17 known tRF sequences, 15 were present in at least one of the 5 samples. Of these, 13 (86.67%) were successfully recovered by tRFfinder (Supplementary Table S2). To examine the usefulness of the alignment score parameter, we ran tRFfinder with increasing alignment score thresholds to provide insight into the number of validated tRFs retained along with the number of other tRFs reported in relation to the increasing threshold (Supplementary Table S2). Note that an alignment score cut-off of 0 captures all possible predictions.
Next, we performed a comparison between the results of tRFfinder and tDRmapper (45), a tool to identify tRNA-derived RNAs from human small RNA-sequencing data. To avoid system error, we set the same parameters for both tools as much as possible (the same version of GtRNAdb, expression thresholds: 100; and allow for 2 deletions). The result indicated that 46.67% (7 in 15) of the validated tRFs were recaptured by both tRFfinder and tDRmapper (Supplementary Table S3).
It is not possible to assess the false positive results of these tRFs predicted by tRFfinder and tDRmapper, given that no experimentally validated database currently available for use as the true positive result; although there is a relational database of tRNA-related fragments called tRFdb, it is also a collection of tRFs predicted from sequencing data. Therefore, tRFdb can only be treated as the data sets with potentially positive tRF records. So we compared the prediction results of tRFfinder and tDRmapper from tRFdb as well as experimentally validated data sets provided by Lee et al. (2). In total, 155 tRF candidates were detected by tRFfinder, 95 of which are not supported by the potentially positive tRF data set. There are 149 tRF candidates detected by tDRmapper, and 110 are not supported by the potentially positive tRF data set. Therefore, the potential false positive rates of tRFfinder and tDRmapper are 61.29% and 73.83%, respectively (Supplementary Table S4). We believe that numerous unsupported sequences will be validated as true tRFs in the future.
Several studies provided strategies for tRF identification (35,45,59,60). We compared our tool with three other studies (Supplementary Table S5). Our pipeline provides more selectable parameters, which makes it flexible for the users to identify and visualize tRFs. Most importantly, tRF2Cancer firstly uses a statistical model to distinguish tRFs from random degradation fragments.
DISCUSSION AND CONCLUSIONS
tRF2Cancer facilitates the identification of tRFs to study their expression in cancers from deep-sequencing data with user-friendly interfaces and time-efficient algorithms. tRF2Cancer provides three useful tools for researchers to investigate tRFs. ‘tRFfinder’ is developed to identify genuine tRF signals from random degradation RNA fragments. One statistical method, the binomial test, is introduced to evaluate the significance of the abundance of sequenced sRNAs distributed on each tRNA. A classification method is subsequently used to annotate the types of tRFs based on their position of origin in pre-tRNA or mature tRNA; the four types of tRFs are tRF-5, tRF-3, tRF-1 and tRF-novel. ‘tRFinCancer’ enables users to inspect the expression of any tRFs in different types of cancers. ‘tRFBrowser’ presents both the sites of origin and the distribution of modification sites of tRFs, including m5C, 2′-O-Me, Ψ and m6A., on their corresponding source tRNA. In addition to cancer samples, tRFfinder can be applied to many samples from different kind of tissue/disease context. However, users may be interested in inspecting the expression of tRFs in multiple types of cancers, even though their samples are obtained from normal tissues or other disease. Therefore, we believed that it will be very useful to combine the three tools and we gave it an integrated name, tRF2Cancer. In conclusion, by integrating 10 991 small RNA sequencing data from 32 types of cancers, we hope our tRF2Cancer will help researchers to investigate the features and functions of tRFs in different types of cancers and discover potential medical applications, such as cancer biomarkers.
In addition to tRFs, tRNA halves are another important type of small RNAs derived from tRNAs, that are typically greater than 32 nt in length. tRFfinder focuses on tRFs identification for the following reasons: First, current small RNA sequencing experiments typically contain reads ranging from 18–30 nt. This is the major length range of tRFs. Although some tRNA halves may exist in the data set, they may not be completely detected. Second, we believe that the detection of tRFs in a single deep sequencing run is not sufficient, so it is very important to inspect their expression in other independent laboratories. The TCGA database is a valuable source that contains approximately 3T raw small RNA reads from 10 991 samples, and these data serve as evidence to support the existence and functionality of tRFs. The small RNAs in the TCGA database are less than 36 nt, which contains abundant tRFs and is very suitable for tRF identification. Therefore, we provided a link from tRFfinder to tRFinCancer, to allow users to easily inspect their results in the TCGA database.
Although deep-sequencing technology can detect almost all RNAs in cells, some fragments may be unobserved in small RNA-seq data sets, due to chemical modifications on some parts of the tRNA. This phenomenon is a known limitation in the field of small RNA investigation, especially of tRF identification. Researchers have made efforts to develop special experimental methods to overcome this limitation. For example, the ARM-seq method developed by Cozen et al. requires the samples to be treated with a dealkylating enzyme during library preparation to remove the tRNAs modifications (61). This will eliminate the interference caused by modifications. Therefore, with the development of experimental methods, we believe tRNA fragments will be sequenced more frequently and be detected by our tRFfinder as well as other tRFs finding tools.
Supplementary Material
Acknowledgments
The authors would like to take this opportunity to thank Prof. Anindya Dutta at the University of Virginia, who kindly provided much useful information to us. We are indebted to Peter Kerpedjiev at University of Vienna for his great help in displaying RNA secondary structure using the forna tool. We thank Ze-Lin Wang, Ke-Ren Zhou, Ye Yang and Yi-Dan Wang for their valuable comments. We appreciate the GEO, TCGA and SRA databases for providing their platforms, and also thank contributors for their valuable public data sets.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [30900820, 31230042, 31370791, 31401975, 31471223, 91440110]; Guangdong Province [S2012010010510, S2013010012457]; The project of Science and Technology New Star in ZhuJiang Guangzhou city [2012J2200025]; The project of Science and Technology of Guangzhou [201504010022]; Fundamental Research Funds for the Central Universities [2011330003161070, 14lgjc18, 15lgpy38]; China Postdoctoral Science Foundation [200902348]; Guangdong Province Key Laboratory of Computational Science and the Guangdong Province Computational Science Innovative Research Team (in part). Funding for open access charge: Ministry of Science and Technology of China, Special Program for Applied Research on Super Computation of the NSFC-Guangdong Joint Fund (the second phase).
Conflict of interest statement. None declared.
REFERENCES
- 1.Li Y., Luo J., Zhou H., Liao J.Y., Ma L.M., Chen Y.Q., Qu L.H. Stress-induced tRNA-derived RNAs: a novel class of small RNAs in the primitive eukaryote Giardia lamblia. Nucleic Acids Res. 2008;36:6048–6055. doi: 10.1093/nar/gkn596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee Y.S., Shibata Y., Malhotra A., Dutta A. A novel class of small RNAs: tRNA-derived RNA fragments (tRFs) Genes Dev. 2009;23:2639–2649. doi: 10.1101/gad.1837609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Y., Zhou H. tRNAs as regulators in gene expression. Sci. China C. 2009;52:245–252. doi: 10.1007/s11427-009-0039-y. [DOI] [PubMed] [Google Scholar]
- 4.Thompson D.M., Parker R. Stressing out over tRNA cleavage. Cell. 2009;138:215–219. doi: 10.1016/j.cell.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 5.Keam S.P., Hutvagner G. tRNA-derived fragments (tRFs): emerging new roles for an ancient RNA in the regulation of gene expression. Life. 2015;5:1638–1651. doi: 10.3390/life5041638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Q., Lee I., Ren J., Ajay S.S., Lee Y.S., Bao X. Identification and functional characterization of tRNA-derived RNA fragments (tRFs) in respiratory syncytial virus infection. Mol. Ther. 2013;21:368–379. doi: 10.1038/mt.2012.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maute R.L., Schneider C., Sumazin P., Holmes A., Califano A., Basso K., Dalla-Favera R. tRNA-derived microRNA modulates proliferation and the DNA damage response and is down-regulated in B cell lymphoma. Proc. Natl. Acad. Sci. U.S.A. 2013;110:1404–1409. doi: 10.1073/pnas.1206761110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sobala A., Hutvagner G. Small RNAs derived from the 5' end of tRNA can inhibit protein translation in human cells. RNA Biol. 2013;10:553–563. doi: 10.4161/rna.24285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gebetsberger J., Zywicki M., Kunzi A., Polacek N. tRNA-derived fragments target the ribosome and function as regulatory non-coding RNA in Haloferax volcanii. Archaea. 2012;2012:260909. doi: 10.1155/2012/260909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goodarzi H., Liu X., Nguyen H.C., Zhang S., Fish L., Tavazoie S.F. Endogenous tRNA-derived fragments suppress breast cancer progression via YBX1 displacement. Cell. 2015;161:790–802. doi: 10.1016/j.cell.2015.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma U., Conine C.C., Shea J.M., Boskovic A., Derr A.G., Bing X.Y., Belleannee C., Kucukural A., Serra R.W., Sun F., et al. Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science. 2016;351:391–396. doi: 10.1126/science.aad6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Q., Yan M., Cao Z., Li X., Zhang Y., Shi J., Feng G.H., Peng H., Zhang X., Zhang Y., et al. Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science. 2016;351:397–400. doi: 10.1126/science.aad7977. [DOI] [PubMed] [Google Scholar]
- 13.Peng H., Shi J., Zhang Y., Zhang H., Liao S., Li W., Lei L., Han C., Ning L., Cao Y., et al. A novel class of tRNA-derived small RNAs extremely enriched in mature mouse sperm. Cell Res. 2012;22:1609–1612. doi: 10.1038/cr.2012.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levitz R., Chapman D., Amitsur M., Green R., Snyder L., Kaufmann G. The optional E.coli prr locus encodes a latent form of phage T4-induced anticodon nuclease. EMBO J. 1990;9:1383–1389. doi: 10.1002/j.1460-2075.1990.tb08253.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee S.R., Collins K. Starvation-induced cleavage of the tRNA anticodon loop in Tetrahymena thermophila. J. Biol. Chem. 2005;280:42744–42749. doi: 10.1074/jbc.M510356200. [DOI] [PubMed] [Google Scholar]
- 16.Haiser H.J., Karginov F.V., Hannon G.J., Elliot M.A. Developmentally regulated cleavage of tRNAs in the bacterium Streptomyces coelicolor. Nucleic Acids Res. 2008;36:732–741. doi: 10.1093/nar/gkm1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jochl C., Rederstorff M., Hertel J., Stadler P.F., Hofacker I.L., Schrettl M., Haas H., Huttenhofer A. Small ncRNA transcriptome analysis from Aspergillus fumigatus suggests a novel mechanism for regulation of protein synthesis. Nucleic Acids Res. 2008;36:2677–2689. doi: 10.1093/nar/gkn123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ogawa T., Tomita K., Ueda T., Watanabe K., Uozumi T., Masaki H. A cytotoxic ribonuclease targeting specific transfer RNA anticodons. Science. 1999;283:2097–2100. doi: 10.1126/science.283.5410.2097. [DOI] [PubMed] [Google Scholar]
- 19.Tomita K., Ogawa T., Uozumi T., Watanabe K., Masaki H. A cytotoxic ribonuclease which specifically cleaves four isoaccepting arginine tRNAs at their anticodon loops. Proc. Natl. Acad. Sci. U.S.A. 2000;97:8278–8283. doi: 10.1073/pnas.140213797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamasaki S., Ivanov P., Hu G.F., Anderson P. Angiogenin cleaves tRNA and promotes stress-induced translational repression. J. Cell Biol. 2009;185:35–42. doi: 10.1083/jcb.200811106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thompson D.M., Lu C., Green P.J., Parker R. tRNA cleavage is a conserved response to oxidative stress in eukaryotes. RNA. 2008;14:2095–2103. doi: 10.1261/rna.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Selitsky S.R., Baran-Gale J., Honda M., Yamane D., Masaki T., Fannin E.E., Guerra B., Shirasaki T., Shimakami T., Kaneko S., et al. Small tRNA-derived RNAs are increased and more abundant than microRNAs in chronic hepatitis B and C. Sci. Rep. 2015;5:7675. doi: 10.1038/srep07675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsieh L.C., Lin S.I., Kuo H.F., Chiou T.J. Abundance of tRNA-derived small RNAs in phosphate-starved Arabidopsis roots. Plant Signal. Behav. 2010;5:537–539. doi: 10.4161/psb.11029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen C.J., liu Q., Zhang Y.C., Qu L.H., Chen Y.Q., Gautheret D. Genome-wide discovery and analysis of microRNAs and other small RNAs from rice embryogenic callus. RNA Biol. 2011;8:538–547. doi: 10.4161/rna.8.3.15199. [DOI] [PubMed] [Google Scholar]
- 25.Wang L., Yu X., Wang H., Lu Y.Z., de Ruiter M., Prins M., He Y.K. A novel class of heat-responsive small RNAs derived from the chloroplast genome of Chinese cabbage (Brassica rapa) BMC Genomics. 2011;12:289. doi: 10.1186/1471-2164-12-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loss-Morais G., Waterhouse P.M., Margis R. Description of plant tRNA-derived RNA fragments (tRFs) associated with argonaute and identification of their putative targets. Biol. Direct. 2013;8:6. doi: 10.1186/1745-6150-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hackenberg M., Huang P.J., Huang C.Y., Shi B.J., Gustafson P., Langridge P. A comprehensive expression profile of microRNAs and other classes of non-coding small RNAs in barley under phosphorous-deficient and -sufficient conditions. DNA Res. 2013;20:109–125. doi: 10.1093/dnares/dss037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karaiskos S., Naqvi A.S., Swanson K.E., Grigoriev A. Age-driven modulation of tRNA-derived fragments in Drosophila and their potential targets. Biol. Direct. 2015;10:51. doi: 10.1186/s13062-015-0081-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Z., Ender C., Meister G., Moore P.S., Chang Y., John B. Extensive terminal and asymmetric processing of small RNAs from rRNAs, snoRNAs, snRNAs, and tRNAs. Nucleic Acids Res. 2012;40:6787–6799. doi: 10.1093/nar/gks307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haussecker D., Huang Y., Lau A., Parameswaran P., Fire A.Z., Kay M.A. Human tRNA-derived small RNAs in the global regulation of RNA silencing. RNA. 2010;16:673–695. doi: 10.1261/rna.2000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cole C., Sobala A., Lu C., Thatcher S.R., Bowman A., Brown J.W., Green P.J., Barton G.J., Hutvagner G. Filtering of deep sequencing data reveals the existence of abundant Dicer-dependent small RNAs derived from tRNAs. RNA. 2009;15:2147–2160. doi: 10.1261/rna.1738409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yeung M.L., Bennasser Y., Watashi K., Le S.Y., Houzet L., Jeang K.T. Pyrosequencing of small non-coding RNAs in HIV-1 infected cells: evidence for the processing of a viral-cellular double-stranded RNA hybrid. Nucleic Acids Res. 2009;37:6575–6586. doi: 10.1093/nar/gkp707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Babiarz J.E., Ruby J.G., Wang Y., Bartel D.P., Blelloch R. Mouse ES cells express endogenous shRNAs, siRNAs, and other Microprocessor-independent, Dicer-dependent small RNAs. Genes Dev. 2008;22:2773–2785. doi: 10.1101/gad.1705308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kawaji H., Nakamura M., Takahashi Y., Sandelin A., Katayama S., Fukuda S., Daub C.O., Kai C., Kawai J., Yasuda J., et al. Hidden layers of human small RNAs. BMC Genomics. 2008;9:157. doi: 10.1186/1471-2164-9-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Telonis A.G., Loher P., Honda S., Jing Y., Palazzo J., Kirino Y., Rigoutsos I. Dissecting tRNA-derived fragment complexities using personalized transcriptomes reveals novel fragment classes and unexpected dependencies. Oncotarget. 2015;6:24797–24822. doi: 10.18632/oncotarget.4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rounge T.B., Furu K., Skotheim R.I., Haugen T.B., Grotmol T., Enerly E. Profiling of the small RNA populations in human testicular germ cell tumors shows global loss of piRNAs. Mol. Cancer. 2015;14:153. doi: 10.1186/s12943-015-0411-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guzman N., Agarwal K., Asthagiri D., Yu L., Saji M., Ringel M.D., Paulaitis M.E. Breast cancer-specific miR signature unique to extracellular vesicles includes ‘microRNA-like’ tRNA fragments. Mol. Cancer Res. 2015;13:891–901. doi: 10.1158/1541-7786.MCR-14-0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vojtech L., Woo S., Hughes S., Levy C., Ballweber L., Sauteraud R.P., Strobl J., Westerberg K., Gottardo R., Tewari M., et al. Exosomes in human semen carry a distinctive repertoire of small non-coding RNAs with potential regulatory functions. Nucleic Acids Res. 2014;42:7290–7304. doi: 10.1093/nar/gku347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hanada T., Weitzer S., Mair B., Bernreuther C., Wainger B.J., Ichida J., Hanada R., Orthofer M., Cronin S.J., Komnenovic V., et al. CLP1 links tRNA metabolism to progressive motor-neuron loss. Nature. 2013;495:474–480. doi: 10.1038/nature11923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nolte-'t Hoen E.N., Buermans H.P., Waasdorp M., Stoorvogel W., Wauben M.H., t Hoen P.A. Deep sequencing of RNA from immune cell-derived vesicles uncovers the selective incorporation of small non-coding RNA biotypes with potential regulatory functions. Nucleic Acids Res. 2012;40:9272–9285. doi: 10.1093/nar/gks658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liao J.Y., Guo Y.H., Zheng L.L., Li Y., Xu W.L., Zhang Y.C., Zhou H., Lun Z.R., Ayala F.J., Qu L.H. Both endo-siRNAs and tRNA-derived small RNAs are involved in the differentiation of primitive eukaryote Giardia lamblia. Proc. Natl. Acad. Sci. U.S.A. 2014;111:14159–14164. doi: 10.1073/pnas.1414394111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumar P., Anaya J., Mudunuri S.B., Dutta A. Meta-analysis of tRNA derived RNA fragments reveals that they are evolutionarily conserved and associate with AGO proteins to recognize specific RNA targets. BMC Biol. 2014;12:78. doi: 10.1186/s12915-014-0078-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liao J.Y., Ma L.M., Guo Y.H., Zhang Y.C., Zhou H., Shao P., Chen Y.Q., Qu L.H. Deep sequencing of human nuclear and cytoplasmic small RNAs reveals an unexpectedly complex subcellular distribution of miRNAs and tRNA 3' trailers. PloS One. 2010;5:e10563. doi: 10.1371/journal.pone.0010563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keam S.P., Young P.E., McCorkindale A.L., Dang T.H., Clancy J.L., Humphreys D.T., Preiss T., Hutvagner G., Martin D.I., Cropley J.E., et al. The human Piwi protein Hiwi2 associates with tRNA-derived piRNAs in somatic cells. Nucleic Acids Res. 2014;42:8984–8995. doi: 10.1093/nar/gku620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Selitsky S.R., Sethupathy P. tDRmapper: challenges and solutions to mapping, naming, and quantifying tRNA-derived RNAs from human small RNA-sequencing data. BMC Bioinformatics. 2015;16:354. doi: 10.1186/s12859-015-0800-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meyer L.R., Zweig A.S., Hinrichs A.S., Karolchik D., Kuhn R.M., Wong M., Sloan C.A., Rosenbloom K.R., Roe G., Rhead B., et al. The UCSC Genome Browser database: extensions and updates 2013. Nucleic Acids Res. 2013;41:D64–D69. doi: 10.1093/nar/gks1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Karolchik D., Baertsch R., Diekhans M., Furey T.S., Hinrichs A., Lu Y.T., Roskin K.M., Schwartz M., Sugnet C.W., Thomas D.J., et al. The UCSC genome browser database. Nucleic Acids Res. 2003;31:51–54. doi: 10.1093/nar/gkg129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yates A., Akanni W., Amode M.R., Barrell D., Billis K., Carvalho-Silva D., Cummins C., Clapham P., Fitzgerald S., Gil L., et al. Ensembl 2016. Nucleic Acids Res. 2016;44:D710–D716. doi: 10.1093/nar/gkv1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nawrocki E.P., Burge S.W., Bateman A., Daub J., Eberhardt R.Y., Eddy S.R., Floden E.W., Gardner P.P., Jones T.A., Tate J., et al. Rfam 12.0: updates to the RNA families database. Nucleic Acids Res. 2015;43:D130–D137. doi: 10.1093/nar/gku1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kozomara A., Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42:D68–D73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chan P.P., Lowe T.M. GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 2009;37:D93–D97. doi: 10.1093/nar/gkn787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cancer Genome Atlas Research, N. Weinstein J.N., Collisson E.A., Mills G.B., Shaw K.R., Ozenberger B.A., Ellrott K., Shmulevich I., Sander C., Stuart J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun W.J., Li J.H., Liu S., Wu J., Zhou H., Qu L.H., Yang J.H. RMBase: a resource for decoding the landscape of RNA modifications from high-throughput sequencing data. Nucleic Acids Res. 2016;44:D259–D265. doi: 10.1093/nar/gkv1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kerpedjiev P., Hammer S., Hofacker I.L. Forna (force-directed RNA): Simple and effective online RNA secondary structure diagrams. Bioinformatics. 2015;31:3377–3379. doi: 10.1093/bioinformatics/btv372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Skinner M.E., Uzilov A.V., Stein L.D., Mungall C.J., Holmes I.H. JBrowse: a next-generation genome browser. Genome Res. 2009;19:1630–1638. doi: 10.1101/gr.094607.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abbott J.A., Francklyn C.S., Robey-Bond S.M. Transfer RNA and human disease. Front. Genet. 2014;5:158. doi: 10.3389/fgene.2014.00158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Telonis A.G., Loher P., Kirino Y., Rigoutsos I. Consequential considerations when mapping tRNA fragments. BMC Bioinformatics. 2016;17:123. doi: 10.1186/s12859-016-0921-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kumar P., Mudunuri S.B., Anaya J., Dutta A. tRFdb: a database for transfer RNA fragments. Nucleic Acids Res. 2015;43:D141–D145. doi: 10.1093/nar/gku1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cozen A.E., Quartley E., Holmes A.D., Hrabeta-Robinson E., Phizicky E.M., Lowe T.M. ARM-seq: AlkB-facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments. Nat. Methods. 2015;12:879–884. doi: 10.1038/nmeth.3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dominissini D., Nachtergaele S., Moshitch-Moshkovitz S., Peer E., Kol N., Ben-Haim M.S., Dai Q., Di Segni A., Salmon-Divon M., Clark W.C., et al. The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature. 2016;530:441–446. doi: 10.1038/nature16998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li X., Xiong X., Wang K., Wang L., Shu X., Ma S., Yi C. Transcriptome-wide mapping reveals reversible and dynamic N-methyladenosine methylome. Nat. Chem. Biol. 2016;12:311–316. doi: 10.1038/nchembio.2040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}