

Supplemental Digital Content is Available in the Text.
Twelve-week treatment with mirtazapine (30 mg/d) was effective and safe in controlling fibromyalgia pain in Japanese nonelderly adult patients without coexisting depression.
Keywords: Fibromyalgia, Mirtazapine, Pain treatment, Placebo-controlled study
Abstract
To evaluate the efficacy and safety of mirtazapine in Japanese patients with fibromyalgia (FM), a parallel-group, randomized, double-blind, placebo-controlled phase IIa study was conducted at 57 sites between November 2012 and February 2014. Patients aged 20 to 64 years who met the American College of Rheumatology 1990 diagnostic FM criteria and had stably high pain scores during a placebo run-in period were randomly assigned (1:1) by a computer-generated allocation sequence (block size 4) to receive mirtazapine orally (15 mg/d for 1 week and then 30 mg/d) or matching placebo for 12 weeks. The primary endpoint was change in mean numerical rating scale (NRS) pain score from baseline to endpoint (week 12 or early discontinuation). Of the 430 patients randomized (n = 215 each group), 422 (n = 211 each group) were analyzed for the primary endpoint. At the study endpoint, mirtazapine caused a significantly greater reduction in the mean NRS pain score compared with placebo (difference, 0.44; 95% confidence interval, −0.72 to −0.17; P = 0.0018). The reduction by mirtazapine remained significantly greater compared with placebo from week 6 onward. More patients treated with mirtazapine had their NRS pain score reduced by ≥30% from baseline (45.5% vs 30.8%). Mirtazapine also improved pain-related quality of life assessed by the Japanese version of the Fibromyalgia Impact Questionnaire and the Short-Form 36 Questionnaire. Adverse events were more common with mirtazapine than placebo (68.8% vs 56.7%), including somnolence (32.1% vs 7.4%), weight gain (17.7% vs 0.9%), and increased appetite (11.6% vs 3.3%). In conclusion, mirtazapine was an effective and safe treatment for Japanese patients with FM.
1. Introduction
Fibromyalgia (FM) is a severely disabling condition characterized by widespread, often intense bodily pain. This condition often coexists with sleep disturbances, depression, fatigue, and headaches, which further impair patients' quality of life (QOL).13,14 Estimates from the Japan College of Fibromyalgia Investigation indicate that FM affects more than 2 million Japanese individuals (approximately 1.7% of the Japanese population). FM has a distinct predominance for women (male-to-female ratio 1:4.6) and mainly affects individuals aged between 30 and 70 years.13 Currently, there is no cure for FM and the available treatments are merely symptomatic (eg, analgesics, antidepressants, antiepileptics, nonsteroidal anti-inflammatory drugs, and hypnotics).14,31
In Japan, pregabalin (an antiepileptic drug) and duloxetine (a serotonin–noradrenaline reuptake inhibitor) have been approved for the treatment of FM. In addition to these 2 drugs, selective serotonin reuptake inhibitors and tricyclic antidepressants are commonly used for patients with FM.31 Descending noradrenergic and serotonergic pathways in the spinal cord are involved in the inhibitory control of nociceptive responses in the spinal dorsal horn.31 The above-mentioned antidepressants seem to suppress the abnormal nociceptive responses in FM by activating these descending antinociceptive pathways.
Mirtazapine promotes the release of noradrenaline and serotonin by blocking α2-adrenergic autoreceptors and α2-adrenergic heteroreceptors, respectively. It also enhances serotonin neurotransmission, mainly through 5-HT1A receptors, and blocking postsynaptic 5-HT2A, 5-HT2C, and 5-HT3 receptors. Based on these pharmacological mechanisms, mirtazapine is classified as a noradrenergic and specific serotonergic antidepressant.6 Because mirtazapine increases the extracellular levels of noradrenaline and serotonin (an effect similar to serotonin–noradrenaline reuptake inhibitors and tricyclic antidepressants, albeit via different mechanisms), mirtazapine is expected to have analgesic effects29 and improve sleep disturbances31 in patients with FM. Evidence from several studies on animal models indicates that mirtazapine reduced experimentally induced pain.4,26,27,32 Its anti-FM efficacy in non-Japanese patients has been evaluated in 2 clinical studies. One was an open-label study (n = 26) that provided preliminary evidence for the effects of mirtazapine on FM pain, fatigue, and sleep disturbances.24,25 The other was a randomized placebo-controlled study (n = 40), which showed that the drug significantly relieved FM pain and had a favorable tolerability profile.30 Furthermore, evidence from a randomized placebo-controlled study (n = 281) showed the efficacy and safety of mirtazapine in Japanese patients with depression.11
We previously conducted a double-blind study (JapicCTI-101,176) in Japanese patients with FM and compared the efficacy of mirtazapine (15-45 mg/d) (n = 92) with that of placebo (n = 44). The change in the mean numerical rating scale (NRS) value for strongest pain (the primary endpoint) from baseline to the end of the study was greater in the mirtazapine group than in the placebo group, although the difference was not statistically significant (unpublished data). Therefore, in the present study, we investigated the efficacy and safety of a fixed dose of mirtazapine (30 mg/d). The primary endpoint in the present study was the NRS value for the mean pain. In the present study, we also excluded patients with depressive symptoms, according to the guidelines of the European Medicines Agency,8 to avoid potential confounding effects of depression on the efficacy evaluations.
2. Methods
2.1. Study design
This was a multicenter, parallel-group, randomized, double-blind, placebo-controlled phase IIa study. This study consisted of a 2-week single-blind placebo run-in period (weeks −2 to 0; visits 1-3) and a subsequent 12-week double-blind treatment period (weeks 0-12; visits 3-10) (Fig. 1).
Figure 1.
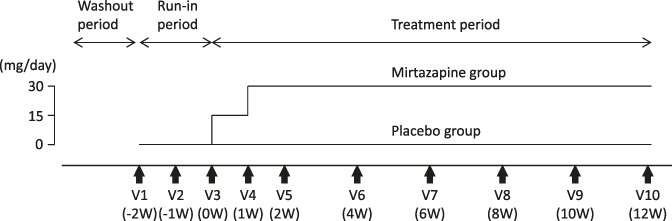
Study design. Consenting patients receiving any prohibited pharmacological or nonpharmacological treatment were allowed to enter the run-in period after discontinuing any previous treatment during the washout period. During the run-in period, all enrolled patients were given placebo in a single-blind manner for 2 weeks to identify and exclude those responsive to placebo. At the end of the run-in period (visit 3), patients who did not respond to placebo were randomly assigned to receive mirtazapine or placebo in the double-blind treatment period.
The study was conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki and the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (IHC), Good Clinical Practice guidelines. The study protocol was reviewed and approved by the institutional review board of each trial site. Before enrollment, all patients gave written informed consent to participation in the study. This study was registered with the Japan Pharmaceutical Information Center's Clinical Trials Information (www.clinicaltrials.jp), under the identifier JapicCTI-122,005, before commencing the study. All endpoints and evaluations were prespecified in the approved study protocol.
2.2. Subjects
Japanese male or female patients aged between 20 and 64 years who met the American College of Rheumatology 1990 diagnostic criteria for FM28 were enrolled in the placebo run-in period. To be entered into the subsequent double-blind treatment period, they were required to properly keep a pain diary during the run-in period and to have a visual analogue scale (VAS; 100-mm scale) pain score of ≥40 mm at weeks −2, −1, and 0 and an NRS pain score of ≥4 at weeks −1 and 0. Exclusion criteria were as follows: (1) a VAS pain score reduced by ≥30% at week −1 or 0 compared with week −2 or an NRS pain score reduced by ≥2 at week 0 compared with week −1; (2) patients with refractory FM, which was defined as patients who were simultaneously taking 3 drugs (pregabalin + an antidepressant + another antiepileptic drug) or a narcotic analgesic; (3) any coexisting inflammatory disease (eg, rheumatoid arthritis) as indicated by an erythrocyte sedimentation rate >40 mm/h, C-reactive protein ≥1.0 mg/dL, antinuclear antibody titer ≥320-fold, or rheumatoid factor level >100 IU/mL; (4) presence of pain from any nonorganic disease other than FM; (5) an established diagnosis of chronic fatigue syndrome, sleep apnea syndrome, or restless leg syndrome; (6) any concomitant or previous clinically important disease (eg, uncontrolled autoimmune, hepatic or renal disease) or psychoneurological disorder (eg, schizophrenia, manic depressive psychosis, and epilepsy); (7) patients who met the criteria for major depression episodes according to diagnosis module A of the Mini-International Neuropsychiatric Interview or patients with a total score of ≥6 points for the risk of suicide in module C12,21; (8) National Glycohemoglobin Standardization Program hemoglobin A1c ≥6.9%; (9) use of any monoamine oxidase inhibitor within 14 days before randomization; and (10) previous use of mirtazapine for pain control.
2.3. Treatments
To maintain blinding, mirtazapine 15-mg tablets and placebo tablets with the same appearance as the study drug were used. All subjects enrolled were given placebo tablets for 2 weeks in a single-blind manner during the run-in period. At the end of the run-in period, subjects with confirmed eligibility were randomized in blocks of 4 at 1:1 to receive double-blind treatment with mirtazapine (15 mg/d for 1 week and then at 30 mg/d) or matching placebo for 12 weeks. Both treatments were given orally once daily at bedtime. Subjects and investigators were masked to treatment assignment. The randomization was done by a computer-generated allocation sequence, which was delivered by a telephone randomization service (randomization manager) not involved in subject recruitment or treatment to ensure allocation concealment. The randomization manager securely kept the randomization list and confirmed that the blinding was maintained until the official decoding of the randomization list. The manager also checked and confirmed the similarity between the active and placebo tablets regarding appearance, shape, size, packaging, and labeling.
Subjects were prohibited from using any nonstudy drug (eg, antidepressants, antiepileptics, pregabalin, narcotics, and narcotic analogs) or nonpharmacological treatment (eg, surgery, electroconvulsive therapy, electrostimulation therapy, acupuncture and moxibustion, and nerve block) that might affect pain scoring. Patients using analgesics discontinued these drugs in the washout period, and concomitant analgesics were prohibited during the treatment period. As-needed use of acetaminophen (up to 1.5 g/d), aspirin (up to 300 mg/d), any nonsteroidal anti-inflammatory drug (up to 3 days), and dextromethorphan was permitted. Subjects were allowed to receive a drug to treat any stably controlled concomitant condition without modification of its dosage throughout the treatment period if the drug had been instituted before the start of the run-in period. Subjects were also allowed to receive any nonpharmacological anti-FM treatment, such as physical (eg, exercise and hyperthermia) and psychological therapy (eg, biofeedback treatment and cognitive behavioral therapy), without modification of its protocol, provided that the treatment had been instituted at least 30 days before the start of the run-in period.
2.4. Efficacy evaluations
Each subject self-rated average pain over the last 24 hours on arising everyday using an 11-point NRS (0 = no pain to 10 = worst imaginable pain) and recorded the score in a pain diary. At each of the 9 visits from visits 2 to 10, the investigator determined NRS pain score by averaging the scores assigned by the subject after the previous visit (or the scores assigned on the last 7 days before discontinuing the treatment). At each specified time point, the mean NRS pain score was determined for each treatment group. To assess the impact of treatment on QOL, the Japanese version of Fibromyalgia Impact Questionnaire (JFIQ)20 was administered at weeks 0 (baseline), 4, 8 and 12, and the Japanese Short-form 36 Questionnaire (SF-36) version 210 was administered at baseline and at week 12.
The primary endpoint of this study was change in mean NRS pain score from baseline to week 12 or early discontinuation (endpoint). Secondary efficacy endpoints were change from baseline in mean NRS pain score at other specified time points, percentage of patients with a reduction in the NRS pain score by ≥30% and ≥50% from baseline (30% and 50% responder rate), and changes from baseline to endpoint in JFIQ total and subscale scores (lower score = better QOL) as well as SF-36 summary and subscale scores (higher score = better QOL).
Baseline symptomology was assessed using the Beck Depression Inventory-II for depressive symptoms,12 the Japanese version of the Insomnia Severity Index for insomnia-related symptoms,3,16 and the Japanese version of the Pittsburgh Sleep Quality index for overall sleep quality.7
2.5. Safety evaluation
During the double-blind treatment period, subjects in both groups were asked about their symptoms in an open-ended fashion. Symptoms collected in this way and abnormal physical and laboratory findings observed were reported as adverse events (AEs). AEs other than those considered unrelated to the treatment were regarded as treatment-related AEs. The frequencies of AEs and treatment-related AEs during the treatment period were determined by treatment group.
2.6. Statistical analyses
To determine the target sample size, we assumed that the difference between the 2 groups in the primary endpoint (change in mean NRS pain score from baseline to endpoint) would be 0.7 with a standard deviation of 2.4 based on the results of previous studies.2,5,15,22 Therefore, 200 patients were required in each group to detect this difference with ≥80% power at the 5% (2-sided) level.
The primary efficacy population comprised of subjects who received at least 1 dose of double-blind treatment and had evaluable primary endpoint data at baseline and at 1 or more postbaseline time points. The change in mean NRS pain score from baseline to endpoint (primary endpoint) was compared between the 2 treatment groups using an analysis of covariance (ANCOVA). The ANCOVA model included treatment as a fixed effect and the mean NRS pain score at baseline as a covariate.
Of the secondary efficacy endpoints, changes in the mean NRS pain score from baseline to other specified time points and in JFIQ scores from baseline to endpoint were analyzed by ANCOVA. Between-group comparisons were performed with the Fisher exact test for 30% responder rate and ANCOVA for changes in SF-36 scores from baseline to endpoint.
Descriptive summaries of data for each group were provided for demographic, disease, and other baseline variables potentially predictive of treatment response. Data for each variable were categorized before the official decoding of the randomization list.
The safety analyses included all subjects that received at least 1 dose of double-blind treatment. The AEs and treatment-related AEs reported during the double-blind treatment period were summarized by group in terms of frequency, timing, and severity. The frequencies of AEs and treatment-related AEs were compared between the 2 groups using the Fisher exact test.
All statistical analyses were performed using SAS Version 9.3 for Windows (SAS Institute, Cary, NC). Data were considered significant if the 2-sided P value was less than 0.05.
3. Results
3.1. Disposition of subjects
From November 2012 to February 2014, 514 patients were enrolled in the placebo run-in period at one of the 57 Japanese tertiary care hospitals. Of these, 84 were excluded before entering the double-blind treatment period, leaving 430 patients randomized to receive mirtazapine (n = 215) or placebo (n = 215) (see the online-only Figure, available online as Supplemental Digital Content at http://links.lww.com/PAIN/A300). All the randomized patients received the respective treatments assigned. Of the randomized patients, 422 were included in the primary efficacy population. The reasons for excluding the 8 patients from the efficacy analysis set were (1) lack of evaluable on-treatment primary endpoint data (mirtazapine, n = 4; placebo, n = 1), (2) placebo responder (placebo, n = 2), and (3) prior treatment with mirtazapine for pain control (placebo, n = 1). Whether each subject was included in the primary efficacy population was determined in a case conference before officially decoding the randomization list (ie, before unblinding).
During follow-up, treatment was discontinued in 25 patients (11.6%) randomized to mirtazapine and in 23 patients (10.7%) randomized to placebo. In both groups, the most frequent reason for discontinuation was withdrawal of consent (n = 9 in each group), followed by any AE (mirtazapine, n = 6, which includes 1 patient with a serious AE [SAE]; placebo, n = 5). Other reasons for treatment discontinuation were noncompliance with medication (mirtazapine, n = 3; placebo, n = 1), ineligibility (placebo, n = 3), worsening of FM (mirtazapine, n = 1), use of any prohibited drug (placebo, n = 1), worsening of the Mini-International Neuropsychiatric Interview Module C–Suicidal ideation score (mirtazapine, n = 1), and miscellaneous (mirtazapine, n = 5; placebo, n = 4). Three patients in the mirtazapine group and 2 in the placebo group were lost to follow-up.
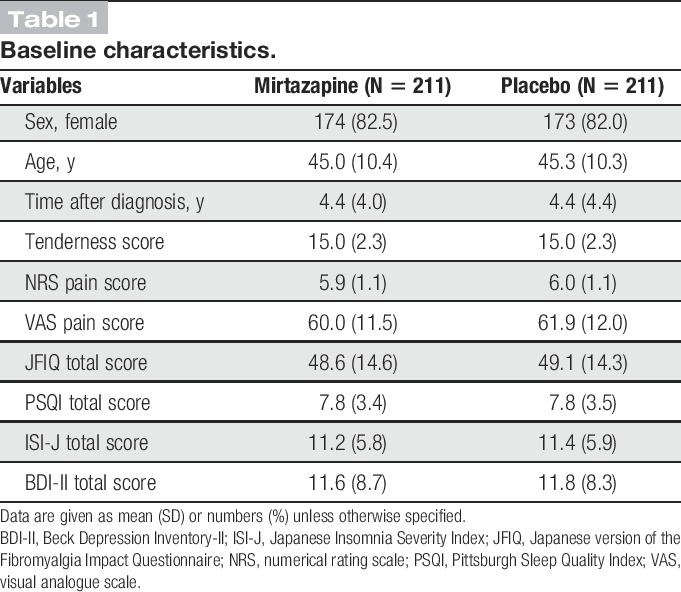
3.2. Baseline characteristics
In the efficacy population, the 2 groups were well balanced with regard to clinical characteristics at baseline (Table 1). Women represented 82.5% of those assigned to the mirtazapine group and 82.0% of those to the placebo group. The respective groups had mean ages of 45.0 and 45.3 years and mean NRS pain scores of 5.9 and 6.0 at baseline. The blinding was securely kept from randomization until official decoding.
Table 1.
Baseline characteristics.
3.3. Efficacy
3.3.1. Effects on NRS pain scores
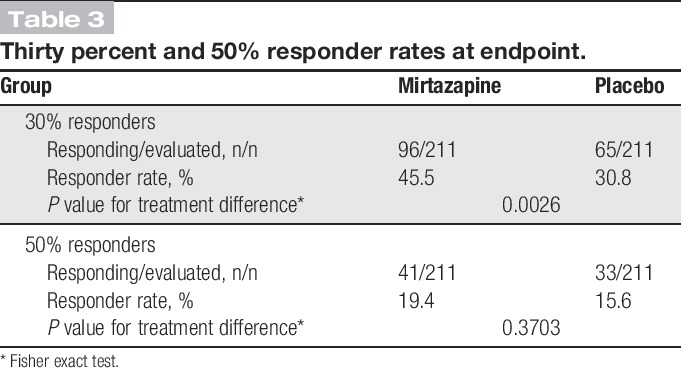
The mean NRS pain score was reduced from baseline to endpoint by 1.61 in the mirtazapine group compared with 1.17 in the placebo group (P = 0.0018; Table 2). The reduction of the score remained greater with mirtazapine than with placebo from week 2 until the end of treatment. The difference between the 2 groups increased gradually from week 2 and remained constant (0.41-0.47) from week 8 onward. The difference was statistically significant at weeks 6 (P = 0.0192), 8 (P = 0.0192), 10 (P = 0.0036), and 12 (P = 0.0013; Fig. 2). The mirtazapine group also had a higher 30% responder rate than the placebo group (45.5% [96/211] vs 30.8% [65/211], P = 0.0026; Table 3). The 50% responder rate was not significantly different between the mirtazapine and placebo groups.
Table 2.
Change in mean numerical rating scale pain score from baseline to endpoint.
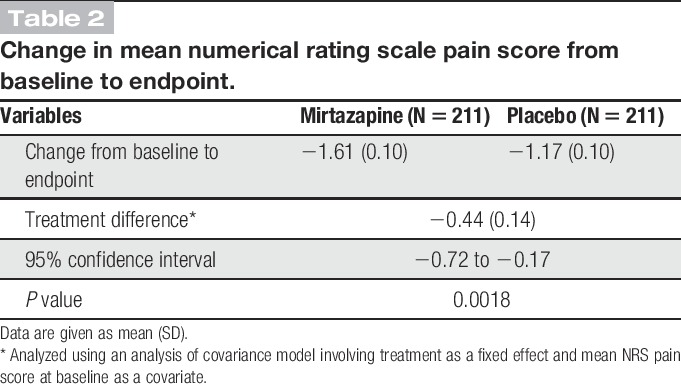
Figure 2.
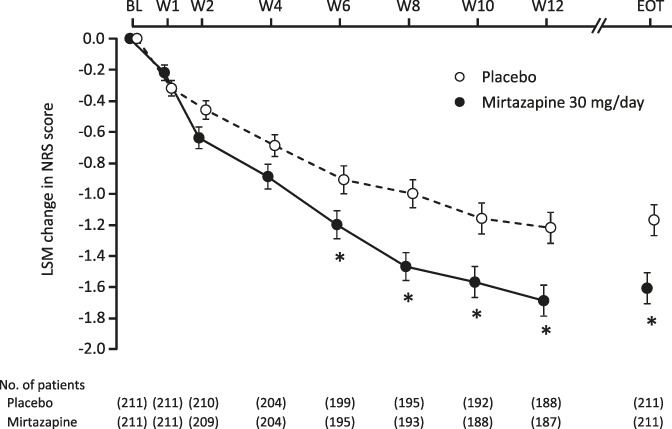
Changes in mean NRS pain score over time. *P < 0.05 (each time point, mixed-effects model repeated measures approach; EOT, ANCOVA). ANCOVA, analysis of covariance; BL, baseline; EOT, end of treatment; LSM, least square mean; NRS, numerical rating scale; W, week.
Table 3.
Thirty percent and 50% responder rates at endpoint.
3.3.2. Effects on QOL scores
The JFIQ total score was reduced from baseline to endpoint by 12.93 in the mirtazapine group compared with 9.29 in the placebo group (P = 0.0097). Figure 3 shows the changes from baseline in JFIQ total score at specified time points. The reduction of the score remained greater with mirtazapine than with placebo at all postbaseline times, with the difference being statistically significant at weeks 8 (P = 0.0042) and 12 (P = 0.0032). Significantly greater reductions with mirtazapine vs placebo were also observed in JFIQ subscales of job ability (P = 0.0259), pain (P = 0.0003), anxiety (P = 0.0475), and depression (P = 0.0020).
Figure 3.
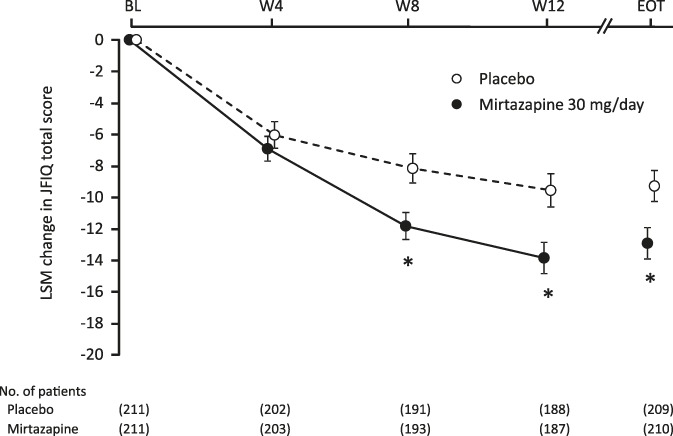
Changes in JFIQ total score over time. *P < 0.05 (each time point, mixed-effects model repeated measures approach; EOT, ANCOVA). ANCOVA, analysis of covariance; BL, baseline; EOT, end of treatment; JFIQ, the Japanese version of Fibromyalgia Impact Questionnaire; LSM, least square mean; W, week.
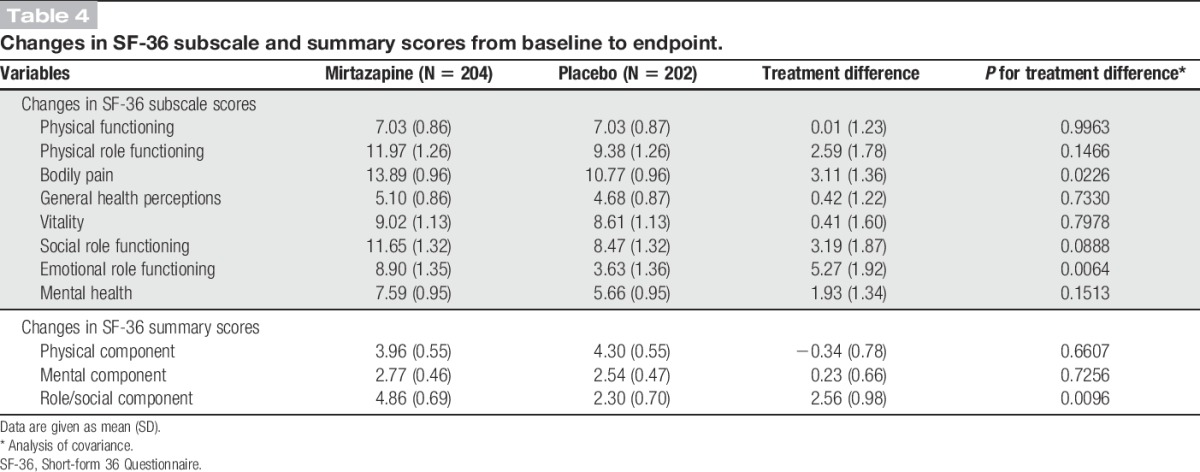
Compared with placebo, mirtazapine increased the scores on the SF-36 subscales of bodily pain (P = 0.0226) and emotional role functioning (P = 0.0064) to a significantly greater extent. Of the 3 SF-36 component summary scores, the role/social component score increased significantly more in mirtazapine recipients than in placebo recipients (P = 0.0096; Table 4).
Table 4.
Changes in SF-36 subscale and summary scores from baseline to endpoint.
3.4. Safety
The safety population comprised 430 patients (n = 215 in each group) who received at least 1 dose of double-blind treatment. In this population, the mean (range) duration of double-blind treatment was 77.4 (1, 91) days for mirtazapine and 78.8 (4, 91) days for placebo.
Table 5 summarizes the AEs reported during the double-blind treatment. A total of 340 AEs occurred in 148 patients treated with mirtazapine and 199 AEs in 122 patients receiving placebo. The incidence of AEs was 68.8% (148/215) with mirtazapine compared with 56.7% (122/215) with placebo (P = 0.0125). The AEs led to treatment discontinuation in 6 patients treated with mirtazapine (including 3 with any treatment-related AE) and 5 patients receiving placebo. As shown in Figure 4, the AEs reported in ≥5% of the patients treated with mirtazapine were somnolence (32.1%, n = 69), weight gain (17.7%, n = 38), nasopharyngitis (12.1%, n = 26), increased appetite (11.6%, n = 25), elevated alanine aminotransferase (ALT) (11.2%, n = 24), elevated gamma-glutamyl transferase (GGT) (9.3%, n = 20), and elevated aspartate aminotransferase (AST) (5.1%, n = 11). In the placebo group, nasopharyngitis (14.9%, n = 32) and somnolence (7.4%, n = 16) were reported in ≥5% of patients. Somnolence was reported in 69 patients after mirtazapine treatment, and it occurred on days 1 to 14 in 53 patients (76.8%) with all episodes being of mild or moderate intensity. Of the 25 patients who reported increased appetite, 15 patients (60.0%) gained weight. The mean body weight for the mirtazapine group was serially increased from baseline by 2.1% at week 4, by 3.0% at week 8, and by 3.7% at week 12.
Table 5.
Overview of adverse events.
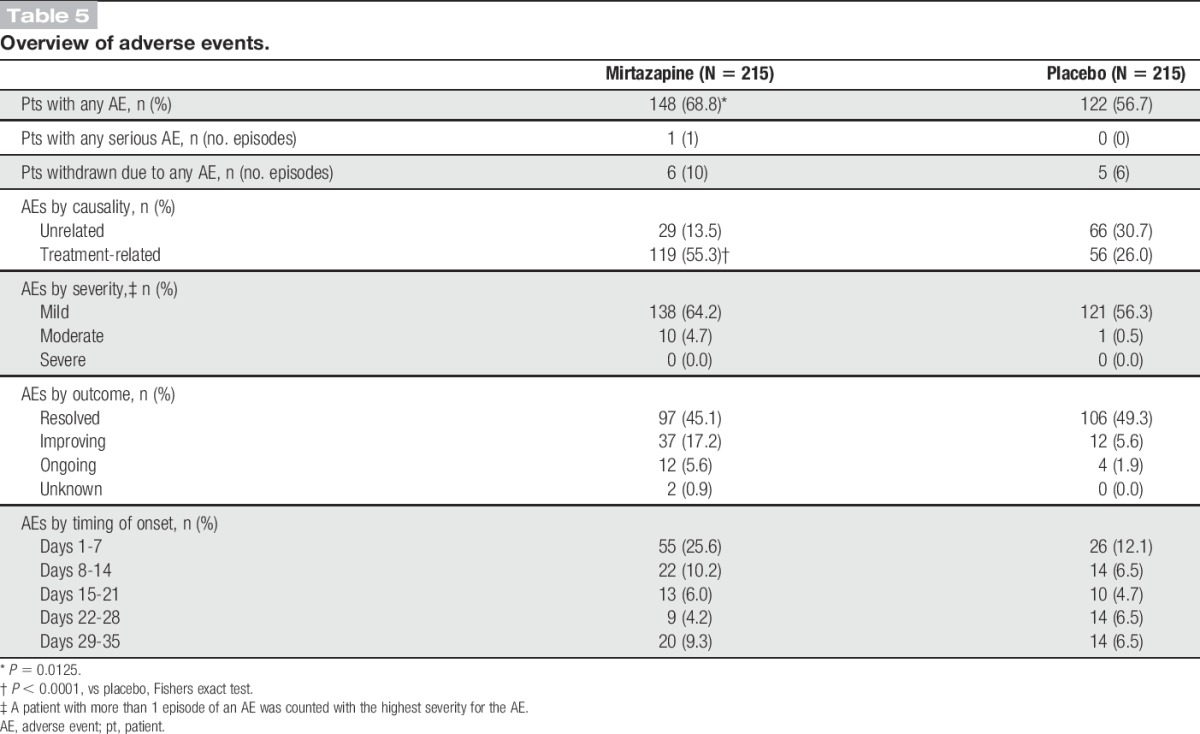
Figure 4.
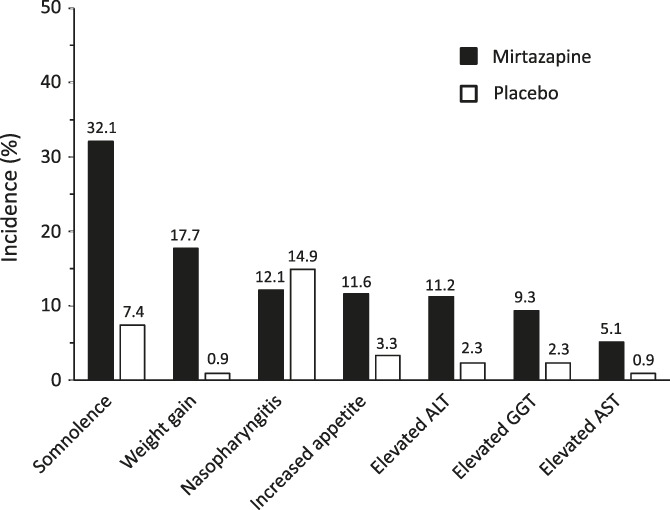
Adverse events reported in ≥5% of patients treated with mirtazapine. ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT, gamma-glutamyl transferase.
There were 248 treatment-related AEs in 119 patients treated with mirtazapine and 76 treatment-related AEs in 56 patients receiving placebo. The incidence of treatment-related AEs was 55.3% (119/215) with mirtazapine compared with 26.0% (56/215) with placebo (P < 0.0001). Among patients treated with mirtazapine, treatment-related AEs reported with a frequency ≥5% were somnolence, weight gain, nasopharyngitis, increased appetite, elevated ALT, elevated GGT, and elevated AST.
No deaths occurred in either group. Intestinal obstruction was reported in 1 patient treated with mirtazapine. This was the only SAE, which was considered unrelated to the study drug.
No severe AEs were reported. There were 12 moderate AEs (somnolence, intestinal obstruction, malaise, epicondylitis, foot fracture, ligament sprain, contusion, meniscus lesion, arthritis, and dizziness) in 10 patients in the mirtazapine group and 1 moderate AE (gastroenteritis) in 1 patient in the placebo group. All other AEs were of mild intensity. Six moderate AEs (somnolence, malaise, arthritis, and dizziness) reported in 5 patients in the mirtazapine group were considered treatment-related, whereas there were no moderate treatment-related AEs in the placebo group. All other treatment-related AEs were of mild intensity.
Of the AEs with a frequency ≥5% in the mirtazapine group, somnolence and increased appetite generally occurred in the early phase of treatment (on days 1-7 and on days 8-21), whereas weight gain and elevated liver enzymes generally occurred in later phases of treatment (on days 29-35 and on days 85 onward). Somnolence, weight gain, and elevated GGT tended to be longlasting with their most frequent duration being ≥85 days.
Fourteen AEs (malaise, elevated ALT, weight gain, somnolence, dry throat, nasal dryness, and alopecia) in 12 patients treated with mirtazapine and 7 AEs (elevated ALT, elevated AST, elevated blood lactate dehydrogenase, elevated GGT, osteoarthritis, and neuralgia) in 4 placebo recipients persistent at the last follow-up. In either group, no AEs resulted in death and patients recovered without sequelae.
In the mirtazapine group, the hematocrit, neutrophil count, and total bilirubin value were significantly lower than baseline, whereas the lymphocyte count, ALT, AST, alkaline phosphatase, GGT, creatinine, and chloride were significantly higher than baseline at all specified time points. However, all these changes were of little clinical significance. The mean body weight for this group increased serially from baseline by 1.20 kg at week 4, by 1.68 kg at week 8, and by 2.08 kg at week 12.
Although abnormal liver function test results were observed in both groups, the changes in these tests were classified as mild in severity.
4. Discussion
In this double-blind, placebo-controlled study, we evaluated the efficacy of mirtazapine (30 mg/d) for the treatment of Japanese patients with FM by assessing its analgesic effect and its impact on their QOL. Therefore, the efficacy analysis set used in this study was considered an appropriate representation of the Japanese FM population with regard to male-to-female ratio (1:4.8) but was younger than the general FM patient population (51.5 [16.9] years).13
In this study, we observed a significantly greater reduction of mean NRS pain score from baseline to the end of treatment with mirtazapine compared with placebo. The difference in the effect size between mirtazapine and placebo in this study was similar to that of pregabalin (−0.44)18 and duloxetine (−0.38),17 which are approved for FM in Japan, and is therefore considered clinically meaningful. The reduction of mean NRS pain score with mirtazapine was significantly greater than that with placebo from week 6 onward. Moreover, mirtazapine significantly improved the QOL of patients with FM compared with placebo in terms of JFIQ total and subscale scores for job ability, pain, anxiety, and depression as well as SF-36 role/social component and subscale scores for bodily pain and emotional role functioning. The improvement of QOL observed in pain-related subscales/components after treatment with mirtazapine supports its analgesic effect as assessed by NRS pain score. Antidepressants are often used to manage chronic pain associated with various conditions and exert an analgesic effect for chronic pain independent of their antidepressant effect.9 The anti-FM efficacy of mirtazapine observed also seemed to be independent of its antidepressant activity and its ability to improve sleep because this study did not include patients with depression, which often coexists with FM. Additionally, the subjects of this study generally had fair quality of sleep at baseline as assessed by the Japanese Sleep Impairment Index and the Pittsburgh Sleep Quality Index.
In a placebo-controlled pilot study, Yeephu et al.30 found that there were no significant between-group differences in most efficacy endpoints between the placebo (n = 13) and mirtazapine 15 mg/d (n = 13) or 30 mg/d (n = 14) groups; however, the authors reported that pain VAS, total FIQ, and Jenkins sleep scale scores significantly improved from baseline in each group, particularly the 30 mg/d mirtazapine group. These findings support the results of our study.
Our previous placebo-controlled study suggested that mirtazapine might relieve pain in Japanese patients with FM but failed to show its superiority over placebo (JapicCTI-101,176; unpublished data). Thus, we planned the current larger study (n = 200 per group planned), which randomly assigned Japanese patients with FM at 1:1 to receive either mirtazapine or placebo to increase the sensitivity for detecting the treatment effect and the precision for estimating the effect size. Additionally, to minimize the placebo effect during double-blind treatment, we included a longer placebo run-in period to identify and exclude subjects responsive to placebo. Assuming that the expected treatment difference in mean NRS pain score was 0.7 (SD, 2.4) with placebo, as had been assumed for pregabalin in evaluating its anti-FM efficacy,2,5,15,22 the planned sample size would provide a power ≥80% for detecting the expected effect of mirtazapine. The duration of the double-blind treatment was 12 weeks according to the European Medical Agency Guideline recommendations to continue maintenance dose therapy for 12 weeks with a drug intended for neuropathic pain.8 This also falls within the range of treatment durations of pregabalin or duloxetine (8-15 weeks) used to evaluate their efficacy for FM.2,5,15,18,22,23
In this study, the difference between mirtazapine and placebo with regard to change from baseline in mean NRS pain score increased gradually from week 2 and remained constant from week 8 onward. This pattern of response indicates a slower onset of analgesic effect for mirtazapine than for pregabalin2,5,15,18,22 or duloxetine.1,23 In contrast, the treatment difference with regard to change from baseline in JFIQ total score increased serially from weeks 4 to 12. As the JFIQ is a validated Japanese version of the FIQ and it is a condition-specific instrument that allows a comprehensive assessment of the impact of FM on the patient's health, this finding indicates that mirtazapine further suppressed disease activity of FM from weeks 8 to 12.19 Mirtazapine was also more effective than placebo in improving health-related QOL as assessed by the SF-36. Taken together, mirtazapine 30 mg/d administered for 12 weeks was shown to be an effective treatment for FM.
With regard to safety, the AE profile of mirtazapine observed in this study was similar to that reported in the double-blind placebo-controlled study of 12-week treatment with 15, 30, or 45 mg/d of mirtazapine in Japanese patients with depression (Study 001),11 with somnolence being the most frequent AE in both studies. The AEs in our study were also similar to those of the earlier randomized study of mirtazapine in patients with FM,30 the most common of which were increased appetite and weight gain. In the present study, somnolence of mild or moderate intensity generally occurred in the early phase of treatment (on days 1-14 in 76.8% patients). Weight gain, a specific safety concern with mirtazapine, and increased appetite, a well-known cause of weight gain (60.0% of the mirtazapine recipients with increased appetite gained weight), were more frequent than in Study 001 (17.7 vs 7.1% and 11.6 vs 4.3%, respectively). The mean body weight of patients treated with mirtazapine increased serially from baseline (by 2.1% at week 4 and by 3.7% at week 12) but to a similar extent to that in Study 001 (by 2.3% at week 6). All episodes of weight gain and increased appetite reported in this study were of mild intensity and considered to have little influence on the subjects' health. Elevated liver enzymes, another category of common AEs with mirtazapine, occurred as often in this study as in Study 001 (elevated ALT [13.8%], elevated AST [8.6%] and elevated GGT [7.6%]). All episodes of elevated liver enzymes reported in this study were of mild intensity and considered to have little influence on the subjects' health.
Intestinal obstruction in 1 patient treated with mirtazapine was the only SAE reported in this study. Because this patient had previously developed intestinal obstruction, the reported AE was considered to indicate spontaneous recurrence of the previous event, and thus, it was deemed as unrelated to the study drug by the investigator. There were no severe AEs. Additionally, aside from 13 moderate AEs (mirtazapine, n = 12; placebo, n = 1), all AEs were of mild intensity. Fourteen AEs in the mirtazapine group and 7 AEs in the placebo group persisted up to the last follow-up, and the outcome of 2 AEs in the mirtazapine group was unknown. The investigator completed the follow-up of these events after confirming the safety of each subject. Thus, the results of this study raise no new safety concerns related to mirtazapine treatment for FM. The most common AEs observed in Japanese patients with FM treated with mirtazapine in this study were consistent in the nature and frequency with those reported in the Japanese patients with depression treated in Study 001.11
This study has several limitations. First, the study excluded FM patients aged ≥65 years to ensure the subjects' safety and also excluded FM patients with concomitant depression to assess the analgesic effect of mirtazapine independent of its antidepressant effect. Therefore, the study population was not an accurate representation of the Japanese FM population seen in routine clinical practice. Second, this study was overly short in duration (12 weeks) to provide evidence to support the long-term efficacy and safety of mirtazapine in the management of FM. Third, responders during the 2-week placebo run-in period were excluded from this study, which might limit external validity. However, this was deemed clinically appropriate because patients with a valid response while on their current therapy are likely to continue their therapy than add a new drug to their treatment regimen. Fourth, this study was conducted in an exploratory manner to examine the efficacy and safety of a single dose of mirtazapine in Japanese patients with FM, so the results might not reflect those obtained with other doses or in other settings. Hence, the findings from this study need further confirmation.
In summary, the present double-blind placebo-controlled study showed that mirtazapine (30 mg/d) was effective and safe in the treatment of FM in Japanese patients meeting the American College of Rheumatology 1990 diagnostic criteria for FM.28 Compared with placebo, mirtazapine produced a significantly greater analgesic effect and significantly greater improvements in QOL in terms of job ability, anxiety, and role/social functioning. Of note, mirtazapine was found to be effective in controlling FM pain even in patients without coexisting depression, indicating the independence of this drug's anti-FM efficacy from its antidepressant effect. The drug was tolerated well in Japanese patients with FM, having a safety profile similar to that reported in Japanese patients with depression. A further confirmatory study should be designed to establish its benefit for the treatment of FM.
Conflict of interest statement
The authors have no conflicts of interest to declare.
K. Miki, M. Murakami, H. Oka, K. Osada received honorarium from Meiji Seika Pharma Co, Ltd. K. Onozawa and S. Yoshida are employees of Meiji Seika Pharma Co, Ltd.
Trial registration: Japan Pharmaceutical Information Center's Clinical Trials Information (www.clinicaltrials.jp), identifier JapicCTI-122005.
Funded by Meiji Seika Pharma Co, Ltd.
Acknowledgements
We are also grateful to all investigators who participated in this trial: Kanzo Amano, Shinichi Aoki, Chikafumi Arita, Kohei Azuma, Akemi Eto, Toru Fujigaki, Yasushi Fukushima, Yasuo Fukuuchi, Takeshi Hasunuma, Kotaro Hatta, Tomomi Himeno, Hidehiko Honda, Takehiko Ichikawa, Hiroshi Inoue, Mikichika Inoue, Sadahiko Kameda, Yoichiro Kashiwagi, Mariko Kawate, Hyeteok Kim, Masamiki Kimura, Tomomasa Kimura, Keizo Kobayashi, Norihiko Koido, Yoshinobu Koyama, Hiroaki Matsuno, Keita Miyanishi, Akiko Miyazawa, Akihito Mizutani, Masato Moriguchi, Mami Morimoto, Yuko Morita, Yasuhiko Munakata, Kenichi Murase, Kiyoshi Murayama, Shiro Nakayama, Kenya Nishioka, Masanari Omata, Hirofumi Oosaki, Takashi Ooura, Kenmei Sakata, Yuki Sekiguchi, Kazutoshi Seto, Satoe Shimoda, Yoshiki Shiohira, Eishi Shirasawa, Toyohiko Sugimoto, Toshiyuki Sugiura, Fumihiko Takeda, Nobuo Takubo, Fusazo Urano, Yasuyuki Watanabe, Shinichi Yamaguchi, Toshiaki Yamamura, Ryoichi Yamazaki, Kuninobu Yasuda. The study was conducted and is published under the sponsorship of Meiji Seika Pharma Co, Ltd. Additionally, the authors thank Springer Healthcare Communications, Mary Richardson and Nicholas D. Smith for technical editing and English-language editing of the manuscript before submission, which was funded by Meiji Seika Pharma Co, Ltd.
Appendix A. Supplemental Digital Content
Supplemental Digital Content associated with this article can be found online at http://links.lww.com/PAIN/A300.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.painjournalonline.com).
References
- [1].Arnold LM, Rosen A, Pritchett YL, D'Souza DN, Goldstein DJ, Iyengar S, Wernicke JF. A randomized, double-blind, placebo-controlled trial of duloxetine in the treatment of women with fibromyalgia with or without major depressive disorder. PAIN 2005;119:5–15. [DOI] [PubMed] [Google Scholar]
- [2].Arnold LM, Russell IJ, Diri EW, Duan WR, Young JP, Jr, Sharma U, Martin SA, Barrett JA, Haig GA. 14-week, randomized, double-blinded, placebo-controlled monotherapy trial of pregabalin in patients with fibromyalgia. J Pain 2008;9:792–805. [DOI] [PubMed] [Google Scholar]
- [3].Bastien CH, Vallières A, Morin CM. Validation of the insomnia severity index as an outcome measure for insomnia research. Sleep 2001;2:297–307. [DOI] [PubMed] [Google Scholar]
- [4].Bomholt SF, Mikkelsen JD, Blackburn-Munro G. Antinociceptive effects of the antidepressants amitriptyline, duloxetine, mirtazapine and citalopram in animal models of acute, persistent and neuropathic pain. Neuropharmacology 2005;48:252–63. [DOI] [PubMed] [Google Scholar]
- [5].Crofford LJ, Rowbotham MC, Mease PJ, Russell IJ, Dworkin RH, Corbin AE, Young JP, Jr, LaMoreaux LK, Martin SA, Sharma U. Pregabalin for the treatment of fibromyalgia syndrome: results of a randomized, double-blind, placebo-controlled trial. Arthritis Rheum 2005;52:1264–73. [DOI] [PubMed] [Google Scholar]
- [6].de Boer T. The pharmacologic profile of mirtazapine. J Clin Psychiatry 1996;57(suppl 4):19–25. [PubMed] [Google Scholar]
- [7].Doi Y, Minowa M, Uchiyama M, Okawa M, Kim K, Shibui K, Kamei Y. Psychometric assessment of subjective sleep quality using the Japanese version of the Pittsburgh Sleep Quality Index (PSQI-J) in psychiatric disordered and control subjects. Psychiatry Res 2000;97:165–72. [DOI] [PubMed] [Google Scholar]
- [8].European Medicines Agency. Guideline on clinical Medicinal Products intended for the Treatment of Neuropathic Pain. 2007: Cpmp/ewp/252/03 Rev.1. Available at http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003478.pdf. [Google Scholar]
- [9].Fishbain D. Evidence-based data on pain relief with antidepressants. Ann Med 2000;32:305–16. [DOI] [PubMed] [Google Scholar]
- [10].Fukuhara S, Suzukamo Y. Manual of SF-36v2 Japanese version [in Japanese]. Kyoto, Japan: Institute for Health Outcomes & Process Evaluation Research, 2004. [Google Scholar]
- [11].Kinoshita T. Double-blind placebo controlled study of mirtazapine, a novel antidepressant, in Japanese patients with depression or depressive state. Jpn J Clin Psychopharmacol 2009;12:289–306. [Google Scholar]
- [12].Kojima M, Furukawa TA, Takahashi H, Kawai M, Nagaya T, Tokudome S. Cross-cultural validation of the Beck Depression Inventory-II in Japan. Psychiatry Res 2002;110:291–9. [DOI] [PubMed] [Google Scholar]
- [13].Matsumoto Y. Epidemiology of fibromyalgia. Pharma Medica 2006;24:35–9. [Google Scholar]
- [14].Matsumoto Y. Medical treatments for fibromyalgia from the perspective of rheumatology. In: Nishioka K, editor. Handbook of fibromyalgia. Tokyo, Japan: Japan Medical Journal, 2007. p. 109–20. [Google Scholar]
- [15].Mease PJ, Russell IJ, Arnold LM, Florian H, Young JP, Jr, Martin SA, Sharma U. A randomized, double-blind, placebo-controlled, phase III trial of pregabalin in the treatment of patients with fibromyalgia. J Rheumatol 2008;35:502–14. [PubMed] [Google Scholar]
- [16].Munezawa T, Morin CM, Inoue Y, Nedate K. Development of the Japanese version of the insomnia severity index (ISI-J) [in Japanese]. Seishinka Chiryōgaku 2009;24:219–25. [Google Scholar]
- [17].Murakami M, Osada K, Mizuno H, Ochiai T, Alev L, Nishioka K. A randomized, double-blind, placebo-controlled phase III trial of duloxetine in Japanese fibromyalgia patients. Arthritis Res Ther 2015;17:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ohta H, Oka H, Usui C, Ohkura M, Suzuki M, Nishioka K. A randomized, double-blind, multicenter, placebo-controlled phase III trial to evaluate the efficacy and safety of pregabalin in Japanese patients with fibromyalgia. Arthritis Res Ther 2012;14:R217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Osada K, Oka H, Isomura T, Nakamura I, Tominaga K, Takahashi S, Kojima A. Development of the Japanese version of the Fibromyalgia Impact Questionnaire (JFIQ): psychometric assessment of reliability and validity. Int J Rheum Dis 2011;14:74–80. [DOI] [PubMed] [Google Scholar]
- [20].Osada K, Tominaga K, Oka H, Nishioka K, Isomura T, Nakamura I, Takahashi S, Kojima A. Development of the Japanese version of the Fibromyalgia Impact Questionnaire (FIQ): translation and linguistic validation. Clin Rheumatol Relat Res 2008;20:19–28. [Google Scholar]
- [21].Otsubo T, Tanaka K, Koda R, Shinoda J, Sano N, Tanaka S, Aoyama H, Mimura M, Kamijima K. Reliability and validity of Japanese version of the Mini-International Neuropsychiatric Interview. Psychiatry Clin Neurosci 2005;59:517–26. [DOI] [PubMed] [Google Scholar]
- [22].Pauer L, Winkelmann A, Arsenault P, Jespersen A, Whelan L, Atkinson G, Leon T, Zeiher B; A0081100 Investigators. An international, randomized, double-blind, placebo-controlled, phase III trial of pregabalin monotherapy in treatment of patients with fibromyalgia. J Rheumatol 2011;38:2643–52. [DOI] [PubMed] [Google Scholar]
- [23].Russell IJ, Mease PJ, Smith TR, Kajdasz DK, Wohlreich MM, Detke MJ, Walker DJ, Chappell AS, Arnold LM. Efficacy and safety of duloxetine for treatment of fibromyalgia in patients with or without major depressive disorder: results from a 6-month, randomized, double-blind, placebo-controlled, fixed-dose trial. PAIN 2008;136:432–44. [DOI] [PubMed] [Google Scholar]
- [24].Samboroski W, Leżańska-Szpera M, Rybakowski JK. Effects of antidepressant mirtazapine on fibromyalgia symptoms. Rocz Akad Med Bialymst 2004;49:265–9. [PubMed] [Google Scholar]
- [25].Samboroski W, Leżańska-Szpera M, Rybakowski JK. Open trial of mirtazapine in patients with fibromyalgia. Pharmacopsychiatry 2004;37:168–70. [DOI] [PubMed] [Google Scholar]
- [26].Schreiber S, Bleich A, Pick CG. Venlafaxine and mirtazapine: different mechanisms of antidepressant action, common opioid-mediated antinociceptive effects—a possible opioid involvement in severe depression? J Mol Neurosci 2002;18:143–9. [DOI] [PubMed] [Google Scholar]
- [27].Schreiber S, Rigai T, Katz Y, Pick CG. The antinociceptive effect of mirtazapine in mice is mediated through serotonergic, noradrenergic and opioid mechanisms. Brain Res Bull 2002;58:601–5. [DOI] [PubMed] [Google Scholar]
- [28].Wolfe F, Smythe HA, Yunus MB, Bennett RM, Bombardier C, Goldenberg DL, Tugwell P, Campbell SM, Abeles M, Clark P, Fam AG, Farber SJ, Fiechtner JJ, Franklin CM, Gatter RA, Hamaty D, Lessard J, Lichtbroun AS, Masi AT, McCain GA, Reynolds WJ, Romano TJ, Russell IJ, Sheon RP. The American College of Rheumatology 1990 criteria for the classification of fibromyalgia. Report of the multicenter criteria committee. Arthritis Rheum 1990;33:160–72. [DOI] [PubMed] [Google Scholar]
- [29].Yamauchi M, Imanishi T, Koyama T. A combination of mirtazapine and milnacipran augments the extracellular levels of monoamines in the rat brain. Neuropharmacology 2012;62:2278–87. [DOI] [PubMed] [Google Scholar]
- [30].Yeephu S, Suthisisang C, Suttiruksa S, Prateepavanich P, Limampai P, Russell IJ. Efficacy and safety of mirtazapine in fibromyalgia syndrome patients: a randomized placebo-controlled pilot study. Ann Pharmacother 2013;47:921–32. [DOI] [PubMed] [Google Scholar]
- [31].Yoshimura M, Furue H. Mechanisms for the anti-nociceptive actions of the descending noradrenergic and serotonergic systems in the spinal cord. J Pharmacol Sci 2006;101:107–17. [DOI] [PubMed] [Google Scholar]
- [32].Zhu J, Wei X, Feng X, Song J, Hu Y, Xu J. Repeated administration of mirtazapine inhibits development of hyperalgesia/allodynia and activation of NF-kappaB in a rat model of neuropathic pain. Neurosci Lett 2008;433:33–7. [DOI] [PubMed] [Google Scholar]