

ABSTRACT
Receptor destruction has been considered one of the mechanisms of homologous Sendai virus (SeV) interference. However, direct evidence of receptor destruction upon virus infection and its relevance to interference is missing. To investigate a precise mechanism of homologous interference, we established SeV persistently infected cells. The persistently infected cells inhibited superinfection by homologous SeV but supported replication of human parainfluenza virus 2 (hPIV2) and influenza A virus (IAV). We confirmed that SeV particles could not attach to or penetrate the infected cells and that the hemagglutinin-neuraminidase (HN) protein of SeV was involved in the interference. Lectin blot assays showed that the α2,3-linked sialic acids were specifically reduced in the SeV-infected cells, but the level of α2,6-linked sialic acids had not changed. As infection with IAV removed both α2,3- and α2,6-linked sialic acids, especially α2,3-linked sialic acids, IAV-infected cells inhibited superinfection of SeV. These results provide concrete evidence that destruction of the specific SeV receptor, α2,3-linked sialic acids, is relevant to homologous interference by SeV.
IMPORTANCE Viral interference is a classically observed phenomenon, but the precise mechanism is not clear. Using SeV interference, we provide concrete evidence that reduction of the α2,3-linked sialic acid receptor by the HN of SeV is closely related with viral interference. Since SeV infection resulted in decrease of only α2,3-linked sialic acids, IAV, which also utilized α2,6-linked sialic acids to initiate infection, superinfected the SeV-infected cells. In contrast, SeV could not superinfect the IAV-infected cells because both α2,3- and α2,6-linked sialic acids were removed. These results indicate that receptor destruction critically contributes to viral interference.
INTRODUCTION
Viral interference is a phenomenon in which virus infection confers resistance to the infected cells against subsequent infection by homologous or heterologous viruses. Although interferon is a well-known factor that is associated with interference, other mechanisms have been suggested. Intracellular interference was classically documented by the fact that defective virus interfered with the replication of homologous viruses (1, 2, 3). Shimazu et al. (4) identified the two nucleotide mutations in the leader sequence of Sendai virus (SeV) that was involved in the homologous interference. The two mutations in the leader sequence determined not only protein synthesis but also genome replication of SeV, resulting in dominance between two viruses. Interference at initiation of infection has also been proposed as a major mechanism of viral interference. Kimura et al. (5) indicated that cells infected with temperature-sensitive SeV suppressed the growth of wild-type SeV. Furthermore, they showed that exposure of cells by UV-irradiated SeV exerted the homologous interference. This interference was considered to be due to receptor destruction by neuraminidase (NA) activity of inactivated virions.
SeV is a member of the family of Paramyxoviridae, which includes pathogens of humans and animals, such as human parainfluenza virus type 2 (hPIV2), hPIV3, and Newcastle disease virus (NDV) (6). SeV has an envelope that has two glycoproteins: the hemagglutinin-neuraminidase protein (HN) and the fusion protein (F). The HN protein binds to a sialic acid-containing cellular receptor to initiate infection. The F protein functions during uncoating of the virus by mediating membrane fusion between the viral envelope and the plasma membrane (7). At the late stage of viral replication, NA activity of the HN protein plays a crucial role in generating progeny virions. NA activity removes sialic acid receptors from glycans, preventing binding of the HN protein to a cellular receptor and enabling release of progeny virions. Thus, the HN protein supposedly contributes to viral interference by receptor destruction.
The possible role of HN protein in viral interference has been documented in other paramyxoviruses. The HN protein from hPIV3 expression and NA activity on the cell surface were required to confer resistance to hPIV3 infection (8). Morrison and McGinnes (9) provided evidence that the expression of the HN protein of NDV alone decreased susceptibility of cells to NDV infection. These results indicate that the HN protein and its NA activity are involved in viral interference, and thus it is reasonable to conceive of attachment interference by removal of the receptor. However, evidence that links attachment interference to receptor destruction has not been provided due to a lack of kinetic analysis of receptor molecules in virus-infected cells.
Terminal sialic acids of glycoconjugates are considered to act as receptors for SeV, hPIV2, hPIV3, and NDV. It has been shown that SeV and hPIV2 bind terminal α2,3-linked sialic acids (10, 11, 12, 13). The biological significance of sialic acids as viral receptors was well documented in influenza A viruses (IAVs). Avian and human IAVs preferentially bind to α2,3- and α2,6-linked sialic acids, respectively. The receptor specificity of IAVs is an important factor of host range and tissue tropism (14).
In the present study, we generated two recombinant SeVs of which the genomes were identical except for a reporter gene, discriminating infection of each virus and eliminating possible intracellular interference. Using these rSeVs, we investigated factors to determine the homologous interference. Our analysis detected the alteration of sialic acids on the infected-cell surface, demonstrating that destruction of a specific sialic acid was involved in the homologous interference.
MATERIALS AND METHODS
Cells and viruses.
LLC-MK2 cells and Vero cells were maintained in Eagle's minimal essential medium (MEM) containing 10% fetal calf serum (FCS). Madin-Darby canine kidney (MDCK) cells were maintained in MEM containing 5% newborn calf serum. All cells were maintained at 37°C under 5% CO2. IAVs (A/WSN/33 and A/Puerto Rico/8/34; referred to as WSN and PR8, respectively) were provided by Yoko Matsuzaki (Yamagata University) and were propagated in embryonated chicken eggs. hPIV2 (Toshiba strain) was propagated in Vero cells. All virus stocks were stored at −80°C until use.
Antibodies.
Rabbit serum against SeV, anti-HN protein (HN43), and anti-F protein (F881) of SeV monoclonal antibodies (MAbs) were provided by Takemasa Sakaguchi (Hiroshima University) (15).
Plasmid construction.
To construct a plasmid for rescue of recombinant SeVs (rSeVs) carrying reporter genes, enhanced green fluorescent protein (EGFP) and secreted NanoLuc luciferase (sNluc) (Promega) genes were amplified by PCR and then inserted into an MluI site of the FL-5 plasmid, which contains the full-length genome of SeV Z strain with a unique MluI site at the intergenic region between the M and F genes (16). For expression of SeV HN, F, or M proteins, the episomal Epstein-Barr virus-based expression system pEBS-PL was employed (17). Corresponding genes were amplified by PCR and subcloned into NheI and KpnI sites of the pEBS-PL plasmid.
Generation of rSeVs.
Virus recovery was carried out according to Nishio et al. (16) with modifications. Briefly, BSR T7/5 cells (18) were transfected with the full-length cDNA of the SeV genome and pTM1 plasmids (19) encoding the N, P, and L proteins for 48 h and subsequently incubated with medium containing tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-trypsin for a further 24 h. The suspension of transfected cells was injected into 9- to 10-day-old embryonated chicken eggs. The allantoic fluid was harvested and stored at −80°C. rSeV strains carrying the EGFP gene and sNluc were designated rSeV-EGFP and rSeV-sNluc, respectively.
Virus infection.
Virus infection was carried out on a 24-well plate. For multiple cycles of replication (IAV-WSN and hPIV2), cells were inoculated for 60 min at 37°C and then incubated in Dulbecco's modified Eagle medium (DMEM) containing 0.1% FCS. Replication of the inoculated viruses was evaluated by virus titration as described below. rSeV- infected cells were incubated in DMEM without FCS. Infection of rSeVs was confirmed by EGFP expression or detection of sNluc in the medium using the Nano-Glo assay system (Promega).
Establishment of rSeV persistently infected cells.
LLC-MK2 cells were infected with rSeV-EGFP at a multiplicity of infection (MOI) of 30 and incubated with MEM containing 0.5 μg/ml TPCK-trypsin. The remaining cells were expanded after 7 days of incubation and then passaged in a manner similar to normal LLC-MK2 cells. Stocks of the infected cells that kept EGFP expression for 20 passages were designated LLC-MK2_SeV-EGFP and subjected to subsequent experiments.
Virus titration.
The fluorescence focus assay was employed to quantitate rSeVs. Briefly, LLC-MK2 cells or MDCK cells in a 24-well plate were infected with viruses and cultured in DMEM without trypsin for 24 h. Then infected cells detected with anti-SeV rabbit serum and Alexa Fluor 594-conjugated anti-rabbit IgG (H+L) (Invitrogen) were counted. The titer of rSeV-EGFP was determined by counting EGFP-expressing cells. The plaque assay was employed to quantitate IAV-WSN and PR8. Briefly, infected MDCK cells in a 12-well plate were cultured in MEM containing 0.3% bovine serum albumin, 0.5 μg/ml TPCK-trypsin, and 1% agarose for 36 to 48 h until plaques were visible. For hPIV2, the virus titer was determined by the 50% tissue culture infectious dose (TCID50) assay using a 24-well plate. Briefly, Vero cell suspensions were simultaneously mixed with 10-fold serial dilutions of virus in wells of a plate and then incubated in MEM with 10% FCS for 4 to 5 days. Virus infection was confirmed by cytopathic effect, and TCID50 was calculated using the Reed and Muench method (20).
FACS analysis.
Suspensions of virus-infected cells or virus protein-expressing cells were prepared by trypsinization. The cells were suspended in 1% FCS–phosphate-buffered saline (PBS)(−) containing antibody and kept on ice for 60 min and washed three times with 1% FCS–PBS(−). Next the cells were incubated with Alexa Fluor 488 goat anti-mouse IgG (H+L) (Life Technologies) or goat anti-rabbit IgG-phycoerythrin (PE) (Santa Cruz Biotechnology). The labeled cells were fixed with 2% paraformaldehyde in PBS(−) in the case of virus-infected cells. The cell suspension was analyzed by fluorescence-activated cell sorting (FACS) using a FACSCalibur (BD Biosciences).
Virus adsorption assay.
Cells cultured in a 6-well plate (1 × 106 cells/well) were inoculated with 2 × 105 focus-forming units (FFU) of rSeV-sNluc. The inoculated cells were incubated on ice for 60 min with tilting every 10 min and washed 5 times with cold PBS or MEM. Then the cells were incubated on ice in cold MEM for 20 min. Total RNA was isolated using Isogen (Nippon Gene) according to the manufacturer's instructions. The sNluc gene was amplified using a Qiagen One-Step reverse transcription (RT)-PCR kit. The following primers were used: secNluc-F (AGATTTCGTTGGGGACTGGC) and secNluc-R (GATCAGGCGCTCGTCGATAA). Basically the RT-PCR condition used was according to the manufacturer's instructions. PCR was carried out with 26 cycles of 94°C for 30 s, 52°C for 30 s, and 72°C for 1 min. The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene was detected as an internal control.
Expression of viral proteins by plasmid transfection.
To establish the cells that express the SeV HN, F, or M proteins, LLC-MK2 cells were transfected with pEBS-SeVHN, SeVF, or SeVM by using Lipofectamine 3000 transfection reagent (Life Technologies). The transfected cells were passaged at least twice with medium containing hygromycin B (0.5 mg/ml), and hygromycin B-resistant cells were obtained. The empty plasmid pEBS-PL was used for the control cells. Three independent transfections were carried out and then were subjected to experiments.
Western blot analysis.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blots were performed according to standard procedures. The cells were lysed in lysis buffer (50 mM Tri-HCl [pH 8.0], 150 mM NaCl, 0.6% octylphenoxypolyethoxyethanol [IGEPAL CA-630], 1 mM EDTA), separated on a 10% Tris-glycine gel, and then transferred to a nitrocellulose membrane. After blocking with 5% skim milk, blots were probed with a primary antibody and then with peroxidase-labeled goat anti-rabbit IgG (H+L) (Vector Laboratories). The probed bands were then visualized by using ImmunoCruz Western blotting luminol reagent (Santa Cruz Biotechnology) according to the manufacturer's instructions. Images were captured with a cooled charge-coupled device (CCD) camera.
Lectin blot analysis.
Cultured cells were suspended by treatment with PBS(−) containing 0.02% EDTA and washed twice with saline. A total of 1 × 105 cells suspended in 100 μl of H2O were placed on ice for 20 min and subsequently clarified by centrifugation at 1,000 × g for 10 min. The clarified lysate was incubated with 0.01% trypsin at 37°C for 30 min. The cell lysate was blotted on a nitrocellulose membrane using the Bio-Dot SF (Bio-Rad) according to the manufacturer's instructions. Blots were blocked with soy milk and then labeled with biotinylated Maackia amurensis lectin II (MALII) or biotinylated Sambucus nigra lectin (SNA) (Vector Laboratories). The labeled dots were detected with the Vectastain ABC system (Vector Laboratories). Visualization and image capture were carried out in the same manner described for Western blot analysis.
RESULTS
SeV infection inhibits superinfection of homologous SeV in LLC-MK2 cells.
To investigate viral interference by an SeV, we generated recombinant SeVs (rSeVs), which carry an EGFP or a secretory luciferase (sNluc) gene between the M and F genes. rSeV-EGFP and rSeV-sNluc have identical genomes, except for their reporter genes, and thus reporter expression can distinguish their infection. These recombinant viruses replicated to similar titers as the wild-type virus (data not shown).
To confirm interference by these rSeVs, LLC-MK2 cells were first infected with rSeV-EGFP (MOI of 5) and then superinfected with rSeV-sNluc (MOI of 0.3) at 0 or 48 h after rSeV-EGFP infection. sNluc expression was measured after 24 h of superinfection as an indicator of rSeV-sNluc infection (Fig. 1A). When rSeV-EGFP and rSeV-sNluc simultaneously infected cells (0 h), sNluc expression was almost the same as in cells infected with rSeV-sNluc alone. In contrast, sNluc expression was significantly suppressed in cells preincubated for 48 h with rSeV-EGFP, indicating that rSeV-sNluc infection was apparently inhibited. We confirmed by FACS analysis that a majority of cells preincubated for 48 h with rSeV-EGFP expressed EGFP and SeV antigen (data not shown).
FIG 1.

SeV infection inhibits superinfection of homologous virus. (A) LLC-MK2 cells infected with or without rSeV-EGFP for 0 or 48 h were superinfected with rSeV-sNluc. Culture supernatants were recovered at 24 h after rSeV-sNluc infection, and sNluc activity was measured. The data are shown as mean luciferase counts ± standard deviation (SD) (n = 4). *, P < 0.01, according to Student's t test. (B) EGFP expression in LLC-MK2_SeV-EGFP or LLC-MK2 cells was detected by FACS analysis. The x axis indicates relative fluorescence. (C) sNluc activities at 1 h and 48 h post-rSeV-sNluc infection in LLC-MK2_SeV-EGFP or LLC-MK2 cells were measured. The data are shown as mean luciferase counts ± SD (n = 3). *, P < 0.01, according to Student's t test. (D) LLC-MK2_SeV-EGFP cells infected with or without rSeV-sNluc were incubated for 48 or 72 h. Culture supernatants were treated with trypsin to activate rSeV-EGFP produced from LLC-MK2_SeV-EGFP cells and used for rSeV-EGFP titration. The data are shown as mean FFU titer ± SD (n = 3).
To analyze the mechanism of rSeV-sNluc inhibition, we established persistently rSeV-EGFP-infected LLC-MK2 cells (LLC-MK2_SeV-EGFP). We detected EGFP expression to confirm viral infection, and cell morphology was indistinguishable from that of normal LLC-MK2 cells (data not shown). FACS analysis showed that 96% of all cells were expressing EGFP, indicating that almost all cells were infected with rSeV-EGFP (Fig. 1B). The sNluc production in LLC-MK2_SeV-EGFP cells superinfected with rSeV-sNluc at an MOI of 0.2 (4 × 104 FFU) was significantly hampered at 48 h postinfection compared to that in LLC-MK2 cells (Fig. 1C). Since infected cells were incubated in a medium without trypsin, a 1/104 decrease of luciferase activity in LLC-MK2_SeV-EGFP cells against LLC-MK2 cells indicated that the infection of rSeV-sNluc was intensely inhibited in LLC-MK2_SeV-EGFP cells. Production of rSeV-EGFP particles from the LLC-MK2_SeV-EGFP cells was not affected by superinfection with rSeV-sNluc at an MOI of 3 (Fig. 1D), indicating that intracellular interference was unlikely to be involved. These results suggested that rSeV-sNluc infection was inhibited at an early step in LLC-MK2_SeV-EGFP cells.
To investigate the susceptibility of the LLC-MK2_SeV-EGFP cells to other viruses, we infected the cells with hPIV2 or IAV-WSN, which are known to utilize sialic acids to initiate infection (Fig. 2A and B). To avoid the effect of trypsin treatment of the LLC-MK2_SeV-EGFP cells, we used IAV-WSN because its growth does not depend on trypsin (21). Although the growth of hPIV2 in the LLC-MK2_SeV-EGFP cells was less efficient than that in normal LLC-MK2 cells (at 48 and 72 h), the LLC-MK2_SeV-EGFP cells supported hPIV2 propagation. IAV-WSN propagation in the LLC-MK2_SeV-EGFP cells was identical to that in normal LLC-MK2 cells. These results indicated that the LLC-MK2_SeV-EGFP cells inhibited virus infection, specifically by rSeV-sNluc.
FIG 2.
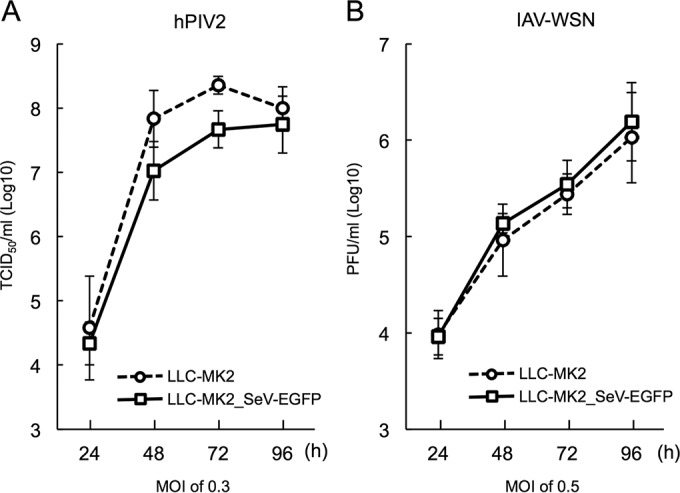
LLC-MK2_SeV-EGFP cells support hPIV2 and IAV propagation. The growth kinetics of hPIV2 and IAV-WSN was studied in LLC-MK2 (circles) or LLC-MK2_SeV-EGFP (squares) cells. (A) hPIV2 titers at each time point were measured by TCID50 assay in Vero cells. (B) IAV-WSN titers were measured by plaque assay in MDCK cells. The data are shown as mean virus titer ± SD (n = 3).
Inhibition of rSeV-sNluc infection depends on expression of the SeV HN protein.
Since rSeV-sNluc infection of the LLC-MK2_SeV-EGFP cells did not affect replication of rSeV-EGFP, rSeV-sNluc infection was apparently inhibited at an early stage. We thus examined the attachment of rSeV-sNluc to the LLC-MK2_SeV-EGFP cells. SeV-sNluc was adsorbed on the LLC-MK2_SeV-EGFP cells for 60 min on ice, and then total cellular RNA was isolated to detect sNluc RNA by RT-PCR. The amount of sNluc PCR product in the LLC-MK2_SeV-EGFP cells was dramatically reduced compared to that in normal LLC-MK2 cells, indicating that attachment of rSeV-sNluc on LLC-MK2_SeV-EGFP cells was inefficient (Fig. 3A).
FIG 3.
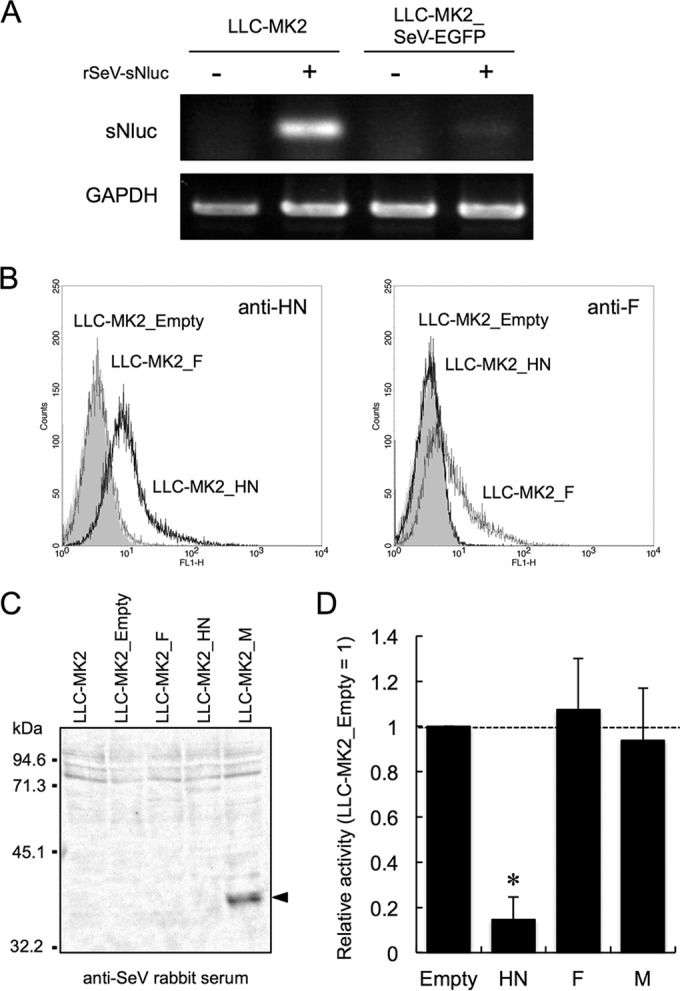
The SeV HN protein is responsible for inhibition of homologous virus. (A) LLC-MK2_SeV-EGFP cells or LLC-MK2 cells were incubated with rSeV-sNluc for 60 min on ice. Total RNA was extracted from the incubated cells after 5 washes with a medium. The rSeV-sNluc attachment was evaluated by detection of the sNluc gene using RT-PCR. (B) LLC-MK2 cells were transfected with a plasmid for expression of SeV HN or F protein. Transfected cells resistant to hygromycin B were tested for protein expression by FACS analysis using the monoclonal antibody to SeV HN or F protein. LLC-MK2_Empty was generated by introduction of the EBS-PL vector and used as control cells. The x axis indicates relative fluorescence. (C) Expression of SeV M protein was detected by Western blot analysis using anti-SeV rabbit serum. (D) LLC-MK2 cells expressing the SeV proteins were infected with rSeV-sNluc. At 24 h after rSeV-sNluc infection, sNluc production was measured as described in the legend to Fig. 1A. The sNluc activity in LLC-MK2_Empty was set to 1. The data are shown as mean relative sNluc activity ± SD (n = 6). *, P < 0.05, according to a one-way ANOVA followed by a Dunnett's test.
Neuraminidase activity of the SeV HN protein cleaves sialic acids at the termini of oligosaccharides. Therefore, the HN protein is likely to be responsible for inhibition of rSeV-sNluc superinfection. To test this hypothesis, we established LLC-MK2 cells expressing SeV HN, F, or M protein, all of which are known to associate with the cellular plasma membrane, and susceptibility to rSeV-sNluc was analyzed. Expression of the HN and F proteins was confirmed by FACS analysis using monoclonal antibodies (Fig. 3B). M protein expression was detected by Western blotting (Fig. 3C). Upon rSeV-sNluc infection, LLC-MK2_F, LLC-MK2_M, and LLC-MK2_Empty produced the same level of sNluc. However, sNluc production was significantly decreased in LLC-MK2_HN, indicating that the HN protein alone conferred resistance to rSeV-sNluc superinfection (Fig. 3D).
Resistance to rSeV-sNluc infection is due to removal of α2,3-linked sialic acids.
α2,3-Sialic acids linked to the cell surface glycoconjugates is considered a receptor for SeV (10, 11, 12). We analyzed the sialic acid population on the surface of LLC-MK2_SeV-EGFP cells by lectin blot analysis. Cell lysates prepared under hypotonic conditions were blotted on a membrane. MALII and SNA lectins were used to detect α2,3-linked and α2,6-linked sialic acids, respectively. We show the result of the lectin blot detected by SNA from 25 μl of lysate because SNA signals of 100 and 50 μl lysates indicated plateau levels. Reactivity of MALII to the LLC-MK2_SeV-EGFP lysate was obviously decreased compared to that in the normal LLC-MK2 lysate (Fig. 4A). In contrast, SNA showed the same reactivity between the LLC-MK2_SeV-EGFP and normal LLC-MK2 lysates (Fig. 4B). Inhibition of rSeV-sNluc attachment to the cells was thus related to the reduction of cell surface α2,3-linked sialic acids.
FIG 4.
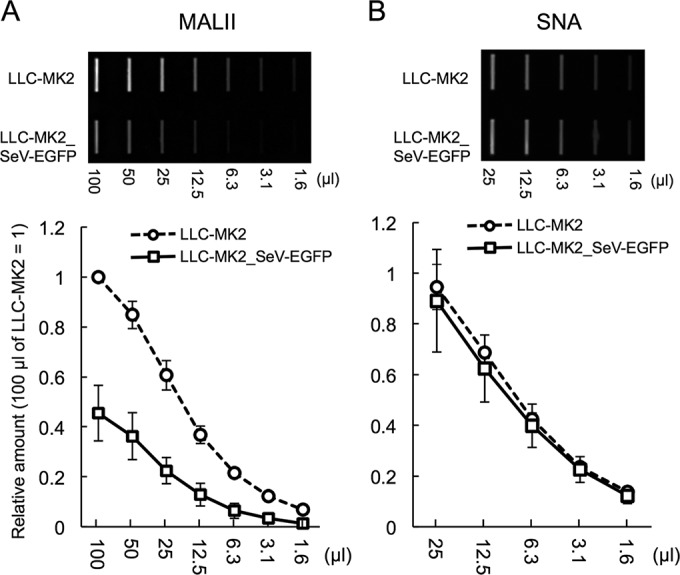
α2,3-linked sialic acids were specifically removed on LLC-MK2_SeV-EGFP. A total of 1 × 105 cells per 100 μl were lysed under a hypotonic condition. A 2-fold serial dilution of the lysates was prepared and spotted on the membrane. The spotted membrane was incubated with biotinylated lectins: MALII for α2,3-linked sialic acids (A) or SNA for α2,6-linked sialic acids (B). The density of the spots was quantified using ImageJ. The density of the normal LLC-MK2 cells (100 μl) was set to 1. The data are shown as the mean of three independent experiments ± SD.
To confirm the plausible role of α2,3-linked sialic acids in SeV interference, we analyzed the sialic acid population of the cells infected with rSeV-EGFP, IAV-PR8, or hPIV2 (Fig. 5A and B). Infection of rSeV-EGFP significantly decreased α2,3-linked sialic acids (Fig. 5A) on LLC-MK2 cells. hPIV2 infection decreased α2,3-linked sialic acids less efficiently. Interestingly, infection of rSeV-EGFP or hPIV2 increased α2,6-linked sialic acids on the cells to the same degree (Fig. 5B). IAV-PR8 infection decreased both α2,3- and α2,6-linked sialic acids most efficiently among the viruses. Since the IAV-PR8 infection removed α2,3-linked sialic acids more efficiently than rSeV-EGFP infection, we examined superinfection of rSeV-sNluc of the IAV-PR8-infected cells (Fig. 5C). When rSeV-sNluc simultaneously infected cells (0 h), sNluc expression was not affected by IAV-PR8. Preincubation of cells with IAV-PR8 for 24 and 48 h significantly suppressed sNluc expression. These results indicated that the amount of α2,3-linked sialic acids was a crucial factor that determined rSeV-sNluc infection.
FIG 5.
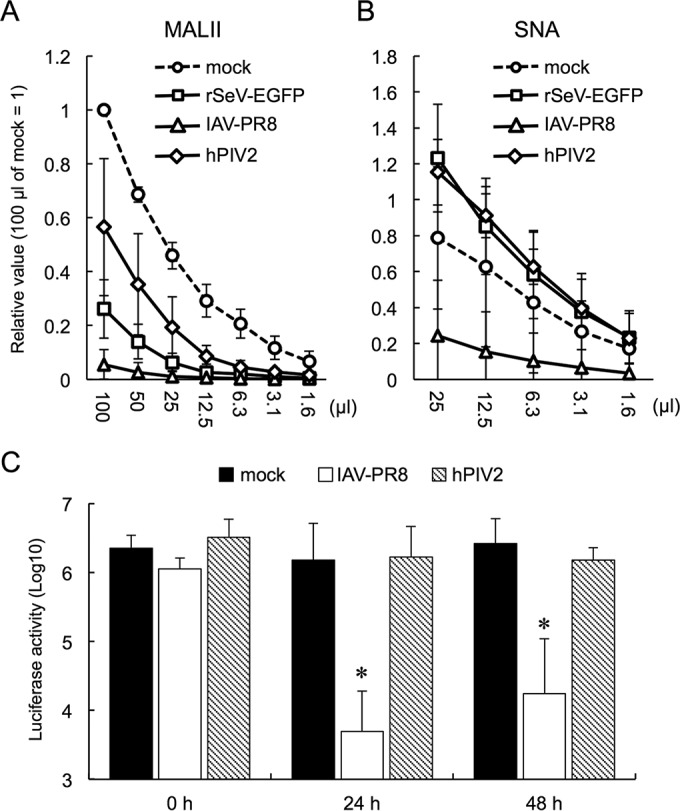
Removal of α2,3-linked sialic acids by influenza A virus inhibited rSeV-sNluc infection. (A and B) LLC-MK2 cells were infected with rSeV-EGFP, IAV-PR8, or hPIV2 at an MOI of 5 and incubated for 48 h. Preparation of cell lysates and lectin blot were the same as for Fig. 4A and B. (C) LLC-MK2 cells incubated with rSeV-EGFP, IAV-PR8, or hPIV2 for 0, 24, or 48 h were superinfected with rSeV-sNluc. Culture supernatants were recovered at 24 h after rSeV-sNluc infection, and sNluc activity was measured. Mock-infected cells were also incubated for the same periods and then infected with rSeV-sNluc. The data are shown as the mean of three independent experiments ± SD. *, P < 0.05 compared to mock infected, according to a one-way ANOVA followed by a Dunnett's test.
DISCUSSION
Since the previous reports indicated that the SeV HN protein and its neuraminidase activity were associated with interference (5, 22), receptor destruction was a feasible mechanism to explain the interference. However, a direct relationship between receptor destruction and the interference had not been shown. In this study, we analyzed interference with homologous virus infection by SeV persistently infected cells. We showed that the LLC-MK2_SeV-EGFP cells suppressed SeV attachment and that the HN protein of SeV was involved in the interference. Analysis of sialic acids using lectins revealed that sialic acids α2,3 linked to glycoconjugates, which are considered a receptor for SeV (10, 11, 12), were specifically reduced in the LLC-MK2_SeV-EGFP cells. Taken together, our results provide evidence that the removal of α2,3-linked sialic acids by the HN protein of SeV is responsible for the interference with homologous virus infection by SeV.
It is widely recognized that a defective virus is produced from a complete virus by a high multiplicity of infection (23). Defective virus infection is one of the mechanisms to maintain persistent infection of SeV (24, 25). Since the LLC-MK2_SeV-EGFP cells were established by infection at an MOI of 30, there is a possibility that defective viruses were involved in the establishment of the LLC-MK2_SeV-EGFP cells. Defective virus infection is also known to abrogate viral gene expression and homologous interference (1, 26). If defective viruses generated from the rSeV-EGFP inhibit replication of rSeV-sNluc, replication of rSeV-EGFP should be similarly affected. However, the EGFP intensity of the LLC-MK2_SeV-EGFP cells (Fig. 1B, LLC-MK2_SeV-EGFP) was detected between 102 and 103 (relative fluorescence) and at almost comparable levels, indicating that replication of rSeV-EGFP in the LLC-MK2_SeV-EGFP cells was not affected. Kingsbury and Portner (3) suggested that a high multiplicity of infection was not related to generation of defective viruses in SeV. Therefore, we excluded interference by defective viruses in the LLC-MK2_SeV-EGFP cells superinfected with rSeV-sNluc.
Eguchi et al. (27) reported that SeV infection in LLC-MK2 cells persistently infected with temperature-sensitive SeV was suppressed due to a defect in SeV-mediated fusion. They also indicated that the persistently infected cells bound SeV particles as efficiently as uninfected cells. This is in contrast to our result that SeV particles were inefficiently detected in the LLC-MK2_SeV-EGFP cells (Fig. 3A). This dissimilar result is presumably attributed to the temperature-sensitive phenotype of SeV. Eguchi et al. used SeV cl.151 (28), a temperature-sensitive strain of SeV, to establish the persistently infected cells. Cells persistently infected with SeV cl.151 showed defective expression of the HN protein at the nonpermissive temperature. Therefore, it is conceivable that a receptor for SeV was still available in SeV cl.151 persistently infected cells. In contrast, the LLC-MK2_SeV-EGFP cells were generated by infection of the rSeV-EGFP, of which the phenotype was not temperature sensitive, and production of virus polypeptides and virus particles was not affected by culture temperature (data not shown), indicating that the HN protein was produced constitutively, and thus a receptor for SeV was removed.
Interference of SeV infection was established in the LLC-MK2_SeV-EGFP cells, in which α2,3-linked sialic acids were reduced to approximately 50% of the level in the uninfected cells (Fig. 4A). Interestingly, even though hPIV2 infection removed approximately 50% of α2,3-linked sialic acids, rSeV-sNluc could infect the hPIV2-preinfected cells (Fig. 5A and C). Since SeV recognizes specific gangliosides as cellular receptors (10, 11, 12), these results suggest that the substrate specificity of the hPIV2 neuraminidase differs from the receptor specificity of SeV HN. Baumann and Neubert (22) suggested that the neuraminidase-deficient HN mutants kept the complex of the HN and receptor. Thus, analysis of the complex may permit receptor identification in more detail.
The results of this study are consistent with the characterization of receptor determinants for SeV. Lectin blot analysis of the SeV-infected cells indicated that neuraminidase activity of the HN protein had a specificity to α2,3-linked sialic acids. IAV infection, which indicated superior ability to remove α2,3-linked sialic acids, interfered with SeV superinfection. These results strengthen the characterization of SeV receptors. In this context, consideration of hPIV2 receptors is of interest. It has been shown that the minimal binding motif of hPIV2 contained α2,3-linked sialic acids (13). Our lectin blot analysis of the hPIV2-infected cells, which showed the specific decrease of α2,3-linked sialic acids, supported receptor specificity of hPIV2. hPIV1 also preferentially recognizes α2,3-linked sialic acids (29, 30). This result suggests that hPIVs utilize α2,3-linked sialic acids distributed in the upper respiratory tract (31).
It was of interest that SeV and hPIV2 infections tended to increase α2,6-linked sialic acids (Fig. 5B). The virus-infected cells produce a large amount of the HN and F glycoproteins, resulting in accumulation of sialoglycoproteins. However, if the neuraminidase activity of the HN proteins functions to release sialic acids, cell-associated sialic acids are decreased, as observed in Fig. 5A. Thus, the result suggests that the SeV and hPIV2 HN proteins have little or no ability to cleave α2,6-linked sialic acids. A recent report showed a possible role of sialylated antigens in induction of antigen-specific immune tolerance (32). The neuraminidase activity of SeV and hPIV2 may contribute to viral pathogenesis by a novel mechanism.
It turned out that lectin blot analysis was an easy method to characterize neuraminidase specificity. Our assay revealed that the neuraminidase activity of IAV had a specificity to both α2,3- and α2,6-linked sialic acids, with high affinity to α2,3 linkage. This result is consistent with Li et al. (33), who analyzed neuraminidase specificity using a microtiter plate-based high-throughput colorimetric assay method. Using the lectin blot assay, to our knowledge we have shown the specific activity of the SeV neuraminidase on α2,3-linked sialic acids for the first time. The lectin blot assay may accelerate analysis of neuraminidase specificity, which is limited compared to receptor specificity analysis.
ACKNOWLEDGMENTS
We thank Yoko Matsuzaki for kindly providing us with influenza A virus A/Puerto Rico/8/24 and A/WSN/33 and Takemasa Sakaguchi for rabbit serum and MAbs for SeV.
REFERENCES
- 1.Portner A, Kingsbury DW. 1971. Homologous interference by incomplete Sendai virus particles: changes in virus-specific ribonucleic acid synthesis. J Virol 8:388–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kingsbury DW, Portner A, Darlington RW. 1970. Properties of incomplete Sendai virions and subgenomic viral RNAs. Virology 42:857–871. doi: 10.1016/0042-6822(70)90335-1. [DOI] [PubMed] [Google Scholar]
- 3.Kingsbury DW, Portner A. 1970. On the genesis of incomplete Sendai virions. Virology 42:872–879. doi: 10.1016/0042-6822(70)90336-3. [DOI] [PubMed] [Google Scholar]
- 4.Shimazu Y, Takao SI, Irie T, Kiyotani K, Yoshida T, Sakaguchi T. 2008. Contribution of the leader sequence to homologous viral interference among Sendai virus strains. Virology 372:64–71. doi: 10.1016/j.virol.2007.10.026. [DOI] [PubMed] [Google Scholar]
- 5.Kimura Y, Norrby E, Nagata I, Ito Y, Shimokata K. 1976. Homologous interference induced by a temperature sensitive mutant derived from an HVJ (Sendai virus) carrier culture. J Gen Virol 33:333–343. doi: 10.1099/0022-1317-33-2-333. [DOI] [PubMed] [Google Scholar]
- 6.Lamb RA, Parks GD. 2013. Paramyxoviridae: the viruses and their replication, p 957–995. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 1 Lippincott Williams and Wilkins, Philadelphia, PA. [Google Scholar]
- 7.Karron RA, Collins PL. 2013. Parainfluenza viruses, p 996–1023. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 1 Lippincott Williams and Wilkins, Philadelphia, PA. [Google Scholar]
- 8.Horga MA, Gusella GL, Greengard O, Poltoratskaia N, Porotto M, Moscona A. 2000. Mechanism of interference mediated by human parainfluenza virus type 3 infection. J Virol 74:11792–11799. doi: 10.1128/JVI.74.24.11792-11799.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morrison TG, McGinnes LW. 1989. Avian cells expressing the Newcastle disease virus hemagglutinin-neuraminidase protein are resistant to Newcastle disease virus infection. Virology 171:10–17. doi: 10.1016/0042-6822(89)90505-9. [DOI] [PubMed] [Google Scholar]
- 10.Markwell MA, Paulson JC. 1980. Sendai virus utilizes specific sialyloligosaccharides as host cell receptor determinants. Proc Natl Acad Sci U S A 77:5693–5697. doi: 10.1073/pnas.77.10.5693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Markwell MA, Svennerholm L, Paulson JC. 1981. Specific gangliosides function as host cell receptors for Sendai virus. Proc Natl Acad Sci U S A 78:5406–5410. doi: 10.1073/pnas.78.9.5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suzuki Y, Suzuki T, Matsunaga M, Matsumoto M. 1985. Gangliosides as paramyxovirus receptor. Structural requirement of sialo-oligosaccharides in receptors for hemagglutinating virus of Japan (Sendai virus) and Newcastle disease virus. J Biochem 97:1189–1199. [DOI] [PubMed] [Google Scholar]
- 13.Tappert MM, Smith DF, Air GM. 2011. Fixation of oligosaccharides to a surface may increase the susceptibility to human parainfluenza virus 1, 2, or 3 hemagglutinin-neuraminidase. J Virol 85:12146–12159. doi: 10.1128/JVI.05537-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suzuki Y, Ito T, Suzuki T, Holland RE Jr, Chambers TM, Kiso M, Ishida H, Kawaoka Y. 2000. Sialic acid species as a determinant of the host range of influenza A viruses. J Virol 74:11825–11831. doi: 10.1128/JVI.74.24.11825-11831.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tozawa H, Komatsu H, Ohkata K, Nakajima T, Watanabe M, Tanaka Y, Arifuku M. 1986. Neutralizing activity of the antibodies against two kinds of envelope glycoproteins of Sendai virus. Arch Virol 91:145–161. doi: 10.1007/BF01316735. [DOI] [PubMed] [Google Scholar]
- 16.Nishio M, Tsurudome M, Ito M, Kawano M, Komada H, Ito Y. 2003. Characterization of Sendai virus persistently infected L929 cells and Sendai virus pi strain: recombinant Sendai viruses having Mpi protein show lower cytotoxicity and are incapable of establishing persistent infection. Virology 314:110–124. doi: 10.1016/S0042-6822(03)00404-5. [DOI] [PubMed] [Google Scholar]
- 17.Bontron S, Ucla C, Mach B, Steimle V, Bontron V, Ucla C, Mach B. 1997. Efficient repression of endogenous major histocompatibility complex class II expression through dominant negative CIITA mutants isolated by a functional selection strategy. Mol Cell Biol 17:4249–4258. doi: 10.1128/MCB.17.8.4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buchholz UJ, Finke S, Conzelmann KK. 1999. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J Virol 73:251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moss B, Elroy-Stein O, Mizukami T, Alexander WA, Fuerst TR. 1990. Product review. New mammalian expression vectors. Nature 348:91–92. [DOI] [PubMed] [Google Scholar]
- 20.Reed L, Muench H. 1938. A simple method of estimating fifty per cent endpoints. Am J Epidemiol 27:493–497. [Google Scholar]
- 21.Goto H, Kawaoka Y. 1998. A novel mechanism for the acquisition of virulence by a human influenza A virus. Proc Natl Acad Sci U S A 95:10224–10228. doi: 10.1073/pnas.95.17.10224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baumann CA, Neubert WJ. 2010. Neuraminidase-deficient Sendai virus HN mutants provide protection from homologous superinfection. Arch Virol 155:217–227. doi: 10.1007/s00705-009-0567-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.von Magnus P. 1952. Propagation of the PR8 strain of influenza A virus in chick embryos. IV. Studies on the factors involved in the formation of incomplete virus upon serial passage of undiluted virus. Acta Pathol Microbiol Scand 30:311–335. [PubMed] [Google Scholar]
- 24.Roux L, Holland JJ. 1979. Role of defective interfering particles of Sendai virus in persistent infections. Virology 93:91–103. doi: 10.1016/0042-6822(79)90278-2. [DOI] [PubMed] [Google Scholar]
- 25.Roux L, Waldvogel FA. 1981. Establishment of Sendai virus persistent infection: biochemical analysis of the early phase of a standard plus defective interfering virus infection of BHK cells. Virology 112:400–410. doi: 10.1016/0042-6822(81)90287-7. [DOI] [PubMed] [Google Scholar]
- 26.Garcin D, De Melo M, Roux L, Kolakofsky D, Curran J. 1994. Presence of a truncated form of the Sendai virus P protein in a long-term persistent infection: implications for the maintenance of the persistent state. Virology 201:19–25. doi: 10.1006/viro.1994.1261. [DOI] [PubMed] [Google Scholar]
- 27.Eguchi A, Kondoh T, Kosaka H, Suzuki T, Momota H, Masago A, Yoshida T, Taira H, Ishii-Watabe A, Okabe J, Hu J, Miura N, Ueda S, Suzuki Y, Taki T, Hayakawa T, Nakanishi M. 2000. Identification and characterization of cell lines with a defect in a post-adsorption stage of Sendai virus-mediated membrane fusion. J Biol Chem 275:17549–17555. doi: 10.1074/jbc.M910004199. [DOI] [PubMed] [Google Scholar]
- 28.Yoshida T, Nagai Y, Maeno K, Iinuma M, Hamaguchi M, Matsumoto T, Nagayoshi S, Hoshino M. 1979. Studies on the role of M protein in virus assembly using a ts mutant of HVJ (Sendai virus). Virology 92:139–154. doi: 10.1016/0042-6822(79)90220-4. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki T, Portner A, Scroggs RA, Uchikawa M, Koyama N, Matsuo K, Suzuki Y, Takimoto T. 2001. Receptor specificities of human respiroviruses. J Virol 75:4604–4613. doi: 10.1128/JVI.75.10.4604-4613.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fukushima K, Takahashi T, Ito S, Takaguchi M, Takano M, Kurebayashi Y, Oishi K, Minami A, Kato T, Park EY, Nishimura H, Takimoto T, Suzuki T. 2014. Terminal sialic acid linkages determine different cell infectivities of human parainfluenza virus type 1 and type 3. Virology 464–465:424–431. doi: 10.1016/j.virol.2014.07.033. [DOI] [PubMed] [Google Scholar]
- 31.Nicholls JM, Bourne AJ, Chen H, Guan Y, Peiris JSM. 2007. Sialic acid receptor detection in the human respiratory tract: evidence for widespread distribution of potential binding sites for human and avian influenza viruses. Respir Res 8:73. doi: 10.1186/1465-9921-8-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perdicchio M, Ilarregui JM, Verstege MI, Cornelissen LA, Schetters ST, Engels S, Ambrosini M, Kalay H, Veninga H, den Haan JM, van Berkel LA, Samsom JN, Crocker PR, Sparwasser T, Berod L, Garcia-Vallejo JJ, van Kooyk Y, Unger WW. 2016. Sialic acid-modified antigens impose tolerance via inhibition of T-cell proliferation and de novo induction of regulatory T cells. Proc Natl Acad Sci U S A 113:3329–3334. doi: 10.1073/pnas.1507706113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Y, Cao H, Dao N, Luo Z, Yu H, Chen Y, Xing Z, Baumgarth N, Cardona C, Chen X. 2011. High-throughput neuraminidase substrate specificity study of human and avian influenza A viruses. Virology 415:12–19. doi: 10.1016/j.virol.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]