

ABSTRACT
Human rhinovirus A89 (HRV-A89) and HRV-B14 bind to and are internalized by intercellular adhesion molecule 1 (ICAM-1); as demonstrated earlier, the RNA genome of HRV-B14 penetrates into the cytoplasm from endosomal compartments of the lysosomal pathway. Here, we show by immunofluorescence microscopy that HRV-A89 but not HRV-B14 colocalizes with transferrin in the endocytic recycling compartment (ERC). Applying drugs differentially interfering with endosomal recycling and with the pathway to lysosomes, we demonstrate that these two major-group HRVs productively uncoat in distinct endosomal compartments. Overexpression of constitutively active (Rab11-GTP) and dominant negative (Rab11-GDP) mutants revealed that uncoating of HRV-A89 depends on functional Rab11. Thus, two ICAM-1 binding HRVs are routed into distinct endosomal compartments for productive uncoating.
IMPORTANCE Based on similarity of their RNA genomic sequences, the more than 150 currently known common cold virus serotypes were classified as species A, B, and C. The majority of HRV-A viruses and all HRV-B viruses use ICAM-1 for cell attachment and entry. Our results highlight important differences of two ICAM-1 binding HRVs with respect to their intracellular trafficking and productive uncoating; they demonstrate that serotypes belonging to species A and B, but entering the cell via the same receptors, direct the endocytosis machinery to ferry them along distinct pathways toward different endocytic compartments for uncoating.
INTRODUCTION
Twelve of the genus A human rhinoviruses (HRV-As; the minor group) bind members of the low-density lipoprotein receptor (LDLR) family, whereas the remaining 90 A and B types (the major group) bind intercellular adhesion molecule-1 (ICAM-1) (1, 2); the HRV-C receptor was recently identified as CDHR3, a protein only marginally expressed in established tissue culture cells (3). The A and B types investigated so far are taken up by receptor-mediated endocytosis (4).
More than 40 years ago it was shown that several ligands, including low-density lipoproteins (LDL), once bound to their receptors, are internalized by clathrin-mediated endocytosis (5–7). Since then, internalization pathways and intracellular trafficking of many other ligands have been identified and characterized (8–10). LDL dissociate from LDL receptors (LDLRs) in mildly acidic (pH 6.5 to 6.0) early endosomes and are then transferred via late endosomes/multivesicular bodies (pH of ≤5.6) to lysosomes (pH of <5.0) (11), where degradation starts about 30 min after uptake (the lysosomal pathway) (Fig. 1) (12, 13). Depending on the cell type, transport from early endosomes to late endosomes may involve (multivesicular) endosomal carrier vesicles (ECV) (14, 15). In any case, late endosomes then mature until fusion with lysosomes takes place (16). Multivesicular late endosomes, and even more so lysosomes, are enriched in heavily glycosylated transmembrane proteins referred to as lysosome-associated membrane proteins (LAMPs). Consequently, LAMPs serve as markers for late endosomes and lysosomes (17).
FIG 1.

Endocytic pathways in HeLa cells and effect of inhibitors. After clathrin-mediated internalization of LDLR-bound LDL, the complex dissociates in the mildly acidic environment of early endosomes. Whereas LDL are transported via ECV and late endosomes to lysosomes for degradation, the LDLR recycles to the plasma membrane following the pathway also taken by transferrin receptor-bound apotransferrin. Recycling from early endosomes occurs by fast (short, red arrows) and slow (long arrows) routes. The slow route directs apotransferrin and various receptors to the ERC that has, in HeLa cells, a similarly low pH as ECV/late endosomes. Transport of ligands to lysosomes can be arrested in early endosomes by bafilomycin or EGA. In contrast, depolymerization of microtubules by nocodazole or inhibition of cytoplasmic dynein by ciliobrevin blocks transport to lysosomes as well as recycling via the slow route.
Another pathway is involved in recycling receptors, transporters, and other proteins back to the plasma membrane (Fig. 1). Transferrin and its receptor are prototypes for this route (18). After iron release from transferrin in early endosomes, receptor-bound apotransferrin is recycled via a fast route (half time, 2 min), as well as from the endocytic recycling compartment (ERC; also known as the perinuclear recycling compartment) via a slow route with a half time of 12 min (19). The pH in the ERC was found to vary in different cell types; e.g., in Chinese hamster ovary cells (CHO), it is about neutral (20) and thus high, as in early endosomes (19). On the other hand, in HepG2 cells (18) and HeLa cells (21), the pH is as acidic, as in late endosomes (pH of ≤5.6).
Since delivery to late endosomes and lysosomes (22, 23) as well as to the ERC (21, 24) depends on dynamic microtubules, transport to these compartments can be prevented by depolymerizing microtubules with nocodazole (Fig. 1). A similar effect, but via a distinct mechanism, is produced by the drug ciliobrevin A. Ciliobrevin A blocks the AAA+ ATPase motor cytoplasmic dynein and thereby the transport of minus-end directed, cargo-containing vesicles via microtubule gliding (22, 25, 26). Bafilomycin and 4-bromobenzaldehyde N-(2,6-dimethylphenyl) semicarbazone (EGA) arrest ligands directed into the lysosomal pathway in early endosomes without affecting the recycling pathways (Fig. 1). Bafilomycin inhibits the vacuolar proton-ATPase that is responsible for acidification of endocytic (and exocytic) compartments (27) and thus prevents the low-pH-dependent budding of ECV from early endosomes (28). In contrast, EGA does not affect the endosomal pH (29).
Rab proteins are small GTPases required for vesicle budding, uncoating, motility, tethering, and fusion at distinct stages of endocytic/exocytic transport (30). Rab proteins cycle between GTP- and GDP-bound forms. More than 60 different Rabs are localized to distinct endocytic compartments. Rab11 is a marker of the ERC, which is clustered around the microtubule-organizing center (MTOC) at the microtubule minus end (31). Furthermore, it is involved in transferrin transport to the ERC, in recycling to the plasma membrane, and in transport between the trans-Golgi network (TGN) and the ERC (32–34).
The minor-group prototype strain HRV-A2 enters HeLa cells preferentially via clathrin-mediated endocytosis (35) and uncoats in ECV/late endosomes, a process strictly dependent on acidic pH (36, 37). Unlike LDLR, ICAM-1 was found to facilitate uncoating in vitro and in acidic compartments in vivo to various degrees depending on the HRV type (38–40). At least for HRV-B14, RNA penetration takes place from endosomal compartments en route to lysosomes (41).
Recently, it was shown that HRV-A89, which also binds ICAM-1, is internalized into early endosomes in HeLa cells (42), but it was not detected in LAMP-2-positive compartments (B. Pfanzagl, personal communication). Here, we demonstrate that this major-group virus indeed does not uncoat in late endosomes but, rather, in the ERC, demonstrating that viruses using the same receptor might take different endocytic routes.
MATERIALS AND METHODS
Rat anti-HRV-B14 antibodies were obtained via three consecutive injections of HRV-B14 that had been heated for 10 min to 56°C to convert virions into empty capsids, according to established protocols in the Institut für Labortierkunde und Genetik, Himberg, Austria. We found that use of the empty particles as an immunogen resulted in the preferential generation of antibodies with low neutralization capacity but high affinity for subviral particles. On Western blots the antiserum stained VP1, VP2, and VP3 of HRV-B14. In immunofluorescence microscopy it detected native HRV-B14 as well as subviral particles, whereas the preimmune serum showed only background fluorescence (data not shown). Because of cross-reactivity with HRV-A89, the antiserum could not be used for simultaneous subcellular localization of HRV-B14 and HRV-A89.
Time course of HRV RNA release.
HeLa cells (ATCC CRL-1958) were grown on coverslips in minimal essential medium (MEM) containing 10% fetal calf serum (FCS), 1% l-glutamine (Glu), and 1% penicillin-streptomycin (P-S; diluted from 100× stock solutions) until 70% confluent. Cells were preincubated in MEM containing 30 mM MgCl2 and 1% Glu (MEM+) at 37°C for 30 min. HRV (∼300 50% tissue culture infective doses [TCID50]/cell for HRV-B14 and ∼100 TCID50/cell for HRV-A89) was allowed to attach at 4°C. After 1 h, the medium was changed to remove unbound virus, and cells were transferred to infection medium (MEM containing 2% FCS, 1% Glu, 30 mM MgCl2) at 37°C. At the times indicated in Fig. 2, they were washed with cold phosphate-buffered saline (PBS) plus 1 mM MgCl2 and 1 mM CaCl2 (PBS+) supplemented with 25 mM NH4Cl; infection medium containing 25 mM NH4Cl was added, and incubation was continued for 16 h. Cells were fixed with 4% paraformaldehyde (PFA) in PBS, quenched with 50 mM NH4Cl in PBS, permeabilized with 0.2% Triton X-100, and blocked with 10% goat serum in PBS. HRV-B14 proteins were detected by incubation with mouse monoclonal antibody (MAb) 17-IA (5.67 μg/ml) in 10% goat serum for 1 h at room temperature, followed by Alexa Fluor 488 goat anti-mouse IgG(H+L) at 1 μg/ml. HRV-A89 proteins were detected with P5, an IgG fraction affinity purified from an antiserum raised against recombinant HRV-A89 VP1 protein (43) at 1:1,000, followed by Alexa Fluor 488 goat anti-rabbit IgG(H+L) at 1 μg/ml (all fluorescent antibodies were from Life Technologies). Nuclei were labeled with 4′,6′-diamidino-2-phenylindole (DAPI; 1 μg/ml) for 10 min. Coverslips were mounted with 4 μl of Fluoromount-G. Images were recorded on a TissueFaxs automated microscope (TissueGnostics), and infected cells were determined using TissueQuest, version 3, software (44).
FIG 2.

HRV-A89 uncoats much more slowly than HRV-B14. (A) Time course of RNA release. Cells were incubated with HRV-A89 at 100 TCID50/cell and with HRV-B14 at 300 TCID50/cell at 4°C for 60 min; unbound virus was removed, and cells were transferred into infection medium. At the specified times, uncoating was halted via addition of NH4Cl, and incubation was continued for 16 h. Infected cells were then determined in a TissueFaxs. The number of infected cells at 120 min was arbitrarily set to 100% for each virus. Data shown are the means ± standard deviations from three independent experiments, each carried out in quadruplicate. (B) Time course of loss of infectivity. Viruses as specified were allowed to bind to the cells for 1 h at 4°C. Unbound virus was removed by washing three times with cold PBS. At time zero the cells were returned to 37°C in infection medium; they were frozen at the times indicated. Virus was released by freeze-thawing, debris was removed by centrifugation, and viral titer was determined in the supernatant as the TCID50 per milliliter. Values are expressed in percentages of the initial viral titer. The means ± standard deviations from two independent experiments each carried out in six replicates is shown. Note that no further decrease of infectivity was observed between 120 min and 180 min (data not shown).
Time course of HRV loss of infectivity.
HeLa cells were seeded in six-well plates in culture medium (MEM containing 10% FCS, 1% P-S, and 1% Glu) and grown until 90% confluent. Medium was changed to MEM+, and virus (100 TCID50/cell) was allowed to bind in a total volume of 1 ml for 1 h at 4°C. Six replicates were made for each time point. Unbound virus was removed by washing three times with cold PBS on ice. Infection medium was added to the wells at time zero (t = 0), and virus was allowed to internalize at 37°C for the times specified in Fig. 2 and then frozen at −80°C. Cells were disrupted by three freeze-thaw cycles, and the medium containing the released virus was collected in Eppendorf vials. Cell debris was pelleted in a table-top centrifuge, and 200 μl of the supernatant was added to 1.8 ml of infection medium. Further 2-fold dilutions were made in 96-deep-well plates (the initial virus dilution was 1:20, and a dilution series at 1:2 was performed in eight replicates). One hundred microliters of each dilution was added to 96-well plates seeded on the day before and grown until cells were about 40% confluent. The plates were incubated for 5 days at 34°C, and cells were stained with crystal violet. The TCID50 values were calculated using the Reed-Muench method (45). Values are expressed in percentages of the initial viral titer as determined at time zero.
Internalization and colocalization of HRV-A89 and HRV-B14 with Alexa Fluor 568-transferrin and LAMP-2.
Cells were grown on coverslips and cooled to 4°C, and HRV-A89 (100 TCID50/cell in MEM+) was bound for 60 min; unattached virus was removed, and cells were warmed to 37°C in MEM with 5 μg/ml Alexa Fluor 647-transferrin. HRV-B14 (100 TCID50/cell in MEM+) and 5 μg/ml Alexa Fluor 647-transferrin were cointernalized at 37°C. The somewhat different incubation conditions were necessary for obtaining similar signal strengths. Cells were cooled, and transferrin was removed from the plasma membrane by sequential washes with isotonic acetic acid at pH 3.5 and PBS+. Cells were fixed and processed for indirect immunofluorescence microscopy (see above). The following antibody combinations and dilutions were used: (i) for HRV-B14, rat antiserum (1:1,000) and Alexa Fluor 488 goat anti-rat IgG(H+L) at 1 μg/ml; (ii) for HRV-A89, rabbit P5 (1:1,000) and Alexa Fluor 488 goat anti-rabbit IgG(H+L) at 1 μg/ml; and (iii) for LAMP-2 (CD107b), purified mouse monoclonal antibody at 1:200 (H4B4; BD Pharmingen,) and Alexa Fluor 568 goat anti-mouse IgG(H+L) at 1 μg/ml. Confocal images were acquired with a PerkinElmer Ultraview ERS rapid confocal imager using Volocity, version 6.1, software. Z stacks were spaced by 0.4 μm. Colocalization statistics were calculated with ImageJ from single (perinuclear) z stacks, using 20 to 40 cells per sample from three to four fields of view. The Manders colocalization coefficient for the channel used for virus detection was taken as the percentage of virus colocalizing with the respective marker. Averages were calculated, and two-tailed t tests were performed using GraphPad Prism, version 6.
Internalization of HRV-A2 and colocalization with LAMP-1.
Cells were seeded on coverslips as described above, and HRV-A2 (100 TCID50/cell in MEM+) was internalized for 20 min at 37°C. Cells were cooled, unbound virus was removed, and cells were fixed and processed for indirect immunofluorescence microscopy (see above). The following antibodies and dilutions were used: (i) for HRV-A2, mouse monoclonal antibody 8F5 (46) at 1.45 μg/ml and Alexa Fluor 488 goat anti-mouse IgG(H+L) at 1 μg/ml; (ii) for LAMP-1, rabbit polyclonal antibody NB600-956 (1:200; Novus Biologicals) and Alexa Fluor 568 goat anti-rabbit IgG(H+L) at 1 μg/ml. Confocal images were acquired as described above.
Studies with chemical inhibitors.
HeLa cells grown on coverslips were preincubated in serum-free MEM+ with dimethyl sulfoxide (DMSO; solvent control), 20 μM nocodazole (Sigma-Aldrich), 20 μM EGA (ChemBridge), 30 μM ciliobrevin A (Tocris) (all in DMSO, which was kept at a final concentration 0.1%), or 25 mM NH4Cl. HRV-A2, HRV-B14, or HRV-A89 (all at 100 TCID50/cell) was then internalized at 37°C for 60 min in the presence or absence of the same drugs. The medium containing drug and nonattached virus was removed, and cells were washed with cold PBS+ containing 25 mM NH4Cl for 15 min on ice, transferred to warm infection medium containing 25 mM NH4Cl, and further incubated for 16 h at 37°C to allow viral replication. Cells were fixed and processed for indirect immunofluorescence microscopy using the antibodies and protocol described above. For HRV-A2, mouse monoclonal antibody 8F5 (1.45 μg/ml) was used (see above). In the experiment with nocodazole, MAb J2 at 1:3,000 (0.66 μg/ml; English and Scientific Consulting, Bt. Szirák, Hungary) was applied to detect double-stranded RNA (dsRNA) stemming from replicating virus (47). Numbers of infected cells were determined using TissueQuest software (see above) and normalized to the level in the control (i.e., the percentage of cells infected in the absence of drug was set to 100%). Statistics were calculated via ordinary one-way analysis of variance (ANOVA) with Dunnett's test as a post hoc multiple comparison procedure. Every inhibitor was compared to the control. Analyses were run in GraphPad Prism, version 6. The data on the effect of nocodazole on HRV-A2 were obtained under similar conditions previously (37) and were added for the sake of completeness.
Studies with Rab11 mutants.
Cells were seeded in 24-well plates on coverslips, grown until 70% confluent, and transfected with plasmids encoding enhanced green fluorescent protein (EGFP)-tagged constitutively active (Q70L) or dominant negative (S25N) Rab11 mutants (32, 48). Opti-MEM (250 μl) was mixed with 1 μg of plasmid DNA, and then 2 μl of peqFECT was added; the tubes were shortly vortexed and incubated for 25 min at room temperature, and the mixture was then added to the well. On the following day, the cells were preincubated with MEM+, and virus was allowed to internalize for 60 min at 37°C at 100 TCID50/cell in 200 μl of MEM+. Unbound virus was washed away with cold PBS+ containing 25 mM NH4Cl. Warm infection medium (containing 25 mM NH4Cl) was added, the plates were incubated for 6.5 h at 37°C, and the cells were processed for indirect immunofluorescence microscopy using the following antibodies: (i) for HRV-A2, mouse 8F5 (1.45 μg/ml) and Alexa Fluor 555 goat anti-mouse IgG(H+L) at 1 μg/ml; (ii) for HRV-B14, mouse 17-IA (5.67 μg/ml) and Alexa Fluor 555 goat anti-mouse IgG(H+L) at 1 μg/ml; and (iii) for HRV-A89, rabbit P5 (1:1,000) and Alexa Fluor 568 goat anti-rabbit IgG(H+L) at 1 μg/ml. Nuclei were stained using DAPI (1 μg/ml) for 10 min, and coverslips were mounted with 5 μl of Mowiol on glass slides. Images were recorded on a TissueFaxs automated microscope; the numbers of infected and EGFP-expressing cells were determined using TissueQuest software. In short, cells doubly positive for the EGFP-tagged plasmid and the red-labeled secondary antibody recognizing infected cells were related to the number of EGFP-expressing cells. Averages were calculated, and two-tailed t tests were performed using GraphPad Prism, version 6.
RESULTS
HRV-A89 and HRV-B14 differ with respect to time course of internalization, RNA release, and generation of subviral particles.
Upon internalization, HRVs undergo structural changes resulting in (noninfectious) A and B subviral particles. Pfanzagl and collaborators noticed that internalization, as well as production of these particles from HRV-A89, was rather slow and inefficient; even 3 h after synchronized internalization into HeLa cells, residual infectious virus was detected (42). In contrast, the minor-group virus HRV-A2 is taken up efficiently (almost 100% within 25 min), with about 80% of the input virus being converted into noninfectious particles already by 10 min (49). HRV-B14 exhibited intermediate efficiency of uptake (30% within 60 min) and of inactivation (15% at 10 min). For confirmation under our internalization conditions, we investigated the time course of cell-mediated RNA release and concomitant inactivation of HRV-A89 and HRV-B14 in parallel experiments. Virus was bound to HeLa cells at 4°C, and the cells were transferred to 37°C to initiate internalization, capsid modification, and RNA release. At the times indicated in Fig. 2, NH4Cl was added to block further uncoating, and incubation was continued to allow for viral replication. Previously, we had shown that the number of viral particles productively uncoated and the number of cells producing viral antigen are strongly correlated (44). This applies also for the generation of dsRNA produced as a consequence of viral replication and detected with MAb J2 (47). We thus determined the number of infected cells at 16 h postinfection (p.i.), using TissueFaxs and TissueQuest software as described previously (44), via immunofluorescence detection of de novo-synthesized viral proteins of the respective virus by using suitable antibodies. As depicted in Fig. 2A, uncoating of HRV-B14 was comparatively quick; when NH4Cl was added at 5 min p.i., about 42% of the cells were already infected relative to the percentage at 120 min p.i., which was arbitrarily set to 100% infection for both virus types. Uncoating of HRV-A89 was found to be much slower; on blocking uncoating at the same time (5 min p.i.), only about 10% of cells were recorded as infected. Infection by HRV-A2 is prevented by monensin, bafilomycin-A1, and NH4Cl (36, 50) (see Fig. 4B), whereas infection by HRV-B14 (40) and HRV-A89 (51) was seen to be strongly reduced but not prevented when NH4Cl or bafilomycin was present throughout the experiment (Fig. 2A; see also Fig. 4B). Similar kinetics were found when uncoating was measured as inactivation of the input virus. Cells were infected, nonattached virus was removed, and the decline of infectivity with time was determined from cell extracts and measured as the number of infectious units. Accordingly, measurements of viral infectivity following entry showed that the HRV-B14 titer decreased quickly: at 5 min p.i. infectivity was about 60% of the input, whereas, at the same time, HRV-A89 infectivity decreased only marginally (Fig. 2B). This fundamental difference in the rate of uncoating was maintained over 2 h, whereupon the infectivity did not further decrease, suggesting that a large proportion (∼20%) of cell-associated HRV-B14 was not undergoing uncoating. This was even more evident for HRV-A89, with ∼50% of the input infectivity remaining at 120 min. The slow inactivation of HRV-A89 is in agreement with the observation that only about 30% of bound virus is internalized within this time period (data not shown).
FIG 4.

Uncoating and infection of HRV-A89 are inhibited by ciliobrevin A and nocodazole but not by EGA. (A) Experimental setup. Cells were preincubated with and without (control) the respective drug for 30 min and challenged with the respective HRV for 1 h. Unabsorbed virus was washed away at 4°C with buffer containing NH4Cl to halt uncoating, and cells were further incubated for 16 h without drug but in the presence of NH4Cl. As a negative control, NH4Cl was kept present throughout the experiment. Percent infected cells was determined in a TissueFaxs and related to the number of cells infected when NH4Cl was added at t = 120 min (control arbitrarily set to 100%). (B) Nocodazole and ciliobrevin A inhibit HRV-A89 but not HRV-B14 or HRV-A2. EGA inhibits HRV-A2 and HRV-B14 but not HRV-A89. Note the following: (i) that the lack of inhibition of HRV-A2 by nocodazole was deduced from previous experiments in Bayer et al. (37), and the bar was just added for completeness; ii) that both major-group viruses are not completely inhibited by NH4Cl. Averages and standard deviations were calculated with GraphPad Prism, version 6, from 3 to 14 independent experiments carried out in quadruplicate, and the significance was determined by ordinary one-way ANOVA with Dunnett's test as a post hoc multiple comparison procedure by comparing every inhibitor to the positive control. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001; ns, not significant. (C and D) Virus (at 100 TCID50/cell) was internalized at 37°C in the absence and presence of nocodazole for 20 min (HRV-A2) or 60 min (HRV-B14). Cells were then cooled, washed, and processed for detection of HRV-A2 (shown in green) and LAMP-1 (shown in red) or HRV-B14 (shown in green) and LAMP-2 (shown in red), as described in Materials and Methods. (E) HRV-A89 at 100 TCID50/cell was bound to HeLa cells for 60 min at 4°C. Unbound virus was removed, and cells were warmed to 37°C for 60 min in the presence of 5 μg/ml Alexa Fluor 647-transferrin. Subsequently, cells were cooled, transferrin at the plasma membrane was removed, and HRV-A89 (shown in green) was detected by indirect immunofluorescence microscopy (see Materials and Methods). To facilitate detection of colocalization between HRV-A89 and transferrin, transferrin is shown in red. (F) Schematic of HRV localization in the absence and presence of nocodazole. Confocal images are shown in panels C to E. Arrows point to colocalization of virus with LAMP (C, D, and F) or with transferrin (E and F) in large perinuclear endosomes in the absence of nocodazole. In the presence of nocodazole, arrowheads indicate colocalization of the respective virus with LAMPs (C and D) or transferrin (E) in peripheral vesicles. Previous publications (21, 37) have shown that in the presence of nocodazole HRV-A2 and most likely HRV-B14 are arrested in ECV where uncoating can occur, whereas transferrin and thus HRV-A89 are not transported to the ERC, preventing virus uncoating (F).
Subcellular localization of HRV-A89 and HRV-B14.
We then asked whether the observed differences in the uncoating kinetics of HRV-A89 and -B14 are related to differences in the endocytic pathways; to this end we assessed eventual colocalization of internalized virus with either late (LAMP-2-positive) or transferrin-recycling endosomes by indirect immunofluorescence microscopy. For these experiments we chose the 60-min time point for virus internalization based on results of the experiment shown in Fig. 2A. HRV-A89 was bound to HeLa cells at 4°C, unbound virus was washed away, and cells were incubated in medium containing Alexa Fluor 647-transferrin for 60 min. Since transferrin recycles with a half time of about 15 min, it was kept present during internalization of the virus (21). To increase the low signal recorded when HRV-B14 was bound at 4°C, virus and Alexa Fluor 647-transferrin were internalized for 60 min at 37°C. Cells were cooled, and transferrin remaining at the plasma membrane was removed. Cells were then fixed, quenched, and permeabilized, and viral proteins were detected with IgG P5 (HRV-A89) or rat anti-HRV-B14 antiserum, followed by the respective Alexa Fluor-labeled secondary antibody. To allow detection of LAMP-2 (CD107b), mouse monoclonal antibody H4B4 and the respective Alexa Fluor-labeled secondary antibody were applied. Remarkably, confocal images revealed colocalization of HRV-A89 with transferrin in the ERC, whereas colocalization with LAMP-2 was only marginal (Fig. 3A, upper panel). This contrasts with HRV-B14, which does not colocalize with transferrin to any significant extent but does colocalize with LAMP-2 (Fig. 3A, lower panel). The colocalization of each virus with the respective endocytic marker was quantitated in single confocal layers using ImageJ (Fig. 3B). In agreement with the data shown in Fig. 3A, HRV-A89 exhibited significant colocalization with transferrin (78%), and HRV-B14 was colocalized with LAMP-2 (80%), demonstrating that the viruses are indeed transferred into distinct endocytic compartments.
FIG 3.
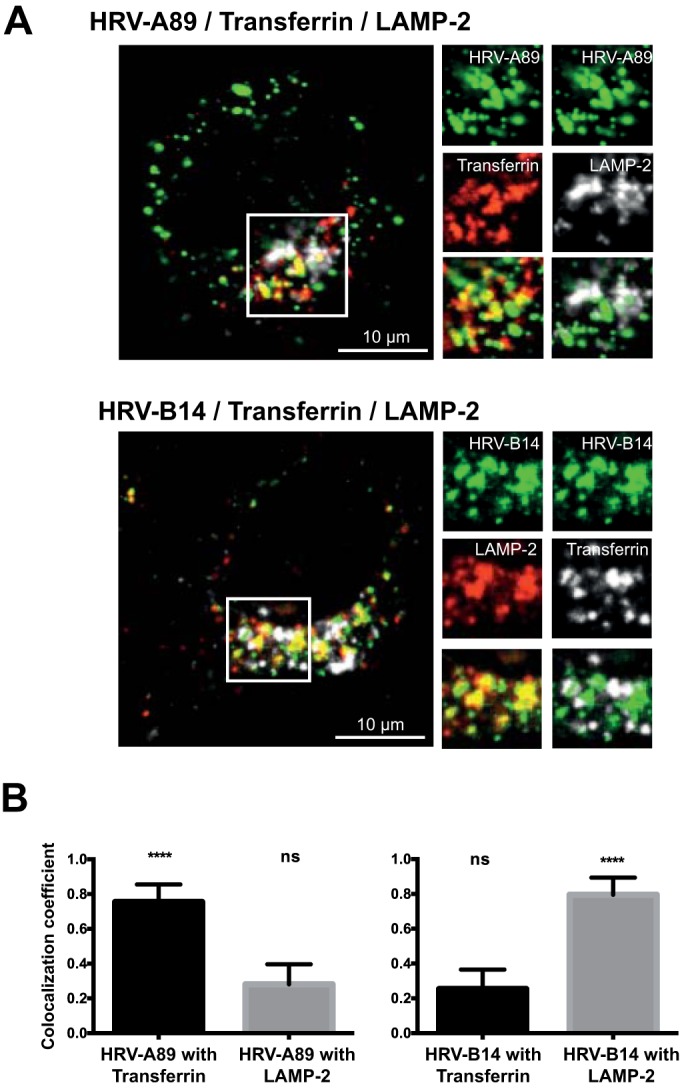
HRV-A89 is routed to the endocytic recycling compartment, and HRV-B14 is routed to late endosomes. (A) HRV-A89 was bound (at 100 TCID50/cell) to HeLa cells at 4°C; cells were transferred into medium containing Alexa Fluor 647-transferrin and incubated for 60 min at 37°C. HRV-B14 (at 100 TCID50/cell) and Alexa Fluor 647-transferrin were internalized for 60 min at 37°C to obtain a stronger signal. Cells were cooled, transferrin remaining at the plasma membrane was removed, and cells were fixed with PFA, quenched, and permeabilized. HRV-A89 was detected with rabbit antiserum P5, and HRV-B14 was detected with rat anti-HRV-B14 antiserum followed by the respective Alexa Fluor 488-labeled secondary antibodies. LAMP-2 was detected with mouse anti-LAMP-2 antibody H4B4 and Alexa Fluor 568 goat anti mouse IgG. Images are confocal. To facilitate detection of colocalization between HRV-A89 and transferrin, transferrin is shown in red. (B) Colocalization was determined by analyzing single confocal layers of 20 cells with ImageJ. Means ± standard deviations as determined with GraphPad Prism, version 6, are shown. ****, P ≤ 0.0001; ns, not significant.
Inhibitors blocking different steps in the entry pathway have different impacts on infection by different HRV serotypes.
To verify whether the transport of HRV-A89 to the ERC and of HRV-B14 to late endosomes leads to productive uncoating, cells were treated with various drugs that differently interfere with the lysosomal and the recycling routes. To assess productive uncoating (i.e., RNA release resulting in infection), we used the protocol outlined in Fig. 4A. Cells were incubated for 30 min at 37°C either without or with NH4Cl as a control for nearly complete inhibition of uncoating and the following drugs: EGA (29), ciliobrevin A (25, 26), and nocodazole (21, 52). Figure 1 schematically depicts the endosomal compartments where transport is arrested in the presence of these drugs in HeLa cells. Because no data on the effect of either EGA or ciliobrevin A on HRV-A2 infection were available, this serotype was included in the experiment. Following preincubation with the individual drugs, virus was internalized at 37°C for 60 min; medium containing drug and nonattached virus was removed, cells were washed with cold PBS+ containing NH4Cl, and incubation was continued for 16 h at 37°C in the presence of NH4Cl to halt further uncoating and allow replication excluding possible secondary effects of the drugs.
EGA reduced HRV-A2 infection to 16%, presumably because the virus was retained in early endosomes that are not sufficiently acidic to trigger uncoating (27, 29). EGA also reduced infection by HRV-B14 (to about 28%), whereas the effect on HRV-A89 was only minor (Fig. 4B). HRV-A2 and HRV-B14 were virtually unaffected by ciliobrevin A, whereas HRV-A89 infection was strongly reduced. This is in agreement with cytoplasmic dynein being involved in sorting and recycling of transferrin and in maturation of epidermal growth factor (EGF)-containing vesicles into degradative compartments (31, 53). By the same token, HRV-A89 infection was equally reduced by nocodazole, whereas HRV-A2, HRV-B14 (Fig. 4B), HRV-B3, and HRV-A16 were almost unaffected under similar conditions (37, 39). We then studied the influence of nocodazole on subcellular localization of internalized virus. In general, as expected, the presence of the drug leads to a relocalization of the labeled vesicles to a more peripheral position. In the absence of the drug, HRV-A2 and HRV-B14 localize to perinuclear LAMP-positive late endosomes and lysosomes, respectively, whereas in its presence they accumulate in peripheral vesicles (Fig. 4C and D). Furthermore, the colocalization of HRV-A2 or HRV-B14 with LAMP seen under control conditions was reduced in the presence of the drug. This is in agreement with the lower expression of LAMPs on ECV than in late endosomes and lysosomes (22). For HRV-A2, subcellular fractionation had shown previously that nocodazole led to accumulation of the virus in ECV (37). Nevertheless, since ECV establish a low pH similar to that of late endosomes (pH of ≤5.5), uncoating takes place (Fig. 4F). On the other hand, nocodazole (21) and ciliobrevin (data not shown) block transport of transferrin and of HRV-A89 (Fig. 4E) to the ERC, which has a pH of ≤5.6 in HeLa cells (21). Thus, in the presence of the drug, HRV-A89 remains in peripheral early and/or fast-recycling endosomes where productive uncoating cannot take place (Fig. 4F). Taken together, these data strongly suggest that HRV-A2 and HRV-B14 productively uncoat in compartments of the lysosomal pathway, whereas HRV-A89 uncoats in the ERC.
Rab11-GTP is required for HRV-A89 infection.
Rab11 is involved in transferrin transport to the ERC and in recycling to the plasma membrane (32–34). We thus investigated the effect of the constitutively active (Rab11-Q70L) and dominant negative (Rab11-S25N) mutants on infection by the three viral serotypes. Cells were transfected with the Rab11 mutant fused to EGFP to allow detection of expression at about 24 h posttransfection. Cells were infected with an HRV, and at 6 h p.i. de novo-produced viral proteins were detected with suitable antibodies. The percentage of Rab11-expressing cells producing viral proteins was determined by TissueFaxs as described above. As depicted in Fig. 5, expression of the dominant negative mutant Rab11-S25N reduced the number of cells actively synthesizing HRV-A2 and HRV-B14 by 5% and 2% compared to the level with the constitutively active form Rab11-Q70L. However, infection by HRV-A89 was virtually abolished. Overexpression of the constitutively active GTP form of Rab11 has been shown to result in transferrin accumulation in the ERC as (slow) recycling from the ERC was blocked (32). In contrast, overexpression of the dominant negative mutant Rab11-S25N prevents transfer from sorting endosomes to the ERC as well as recycling to the plasma membrane via the fast pathway (32). The subcellular localization of transferrin is similar to that of the Rab11 proteins: Rab11-S25N exhibits a peripheral localization in small vesicles, whereas Rab11-Q70L is found in large, perinuclear compartments (32). This is in agreement with the results shown here on HRV-A89 infection. Furthermore, cointernalization of HRV-A89 and Alexa Fluor 647-transferrin resulted in accumulation of both ligands in the ERC in the presence of Rab11-Q70L (Fig. 5C). Since the ERC has a low-pH environment (pH of ≤5.6), this allows RNA uncoating, while overexpression of Rab11-S25N inhibits transfer to the ERC (Fig. 5C) and thus infection (Fig. 5B). For HRV-A2 we have shown earlier that a fraction of internalized virus that is converted to altered subviral particles is recycled into the medium. This is indicative for either recycling from the ECV or passage through the ERC (36). Thus, the minor effect of Rab11-S25N on HRV-A2 infection may be due to inhibition of recycling and transport to the ERC.
FIG 5.
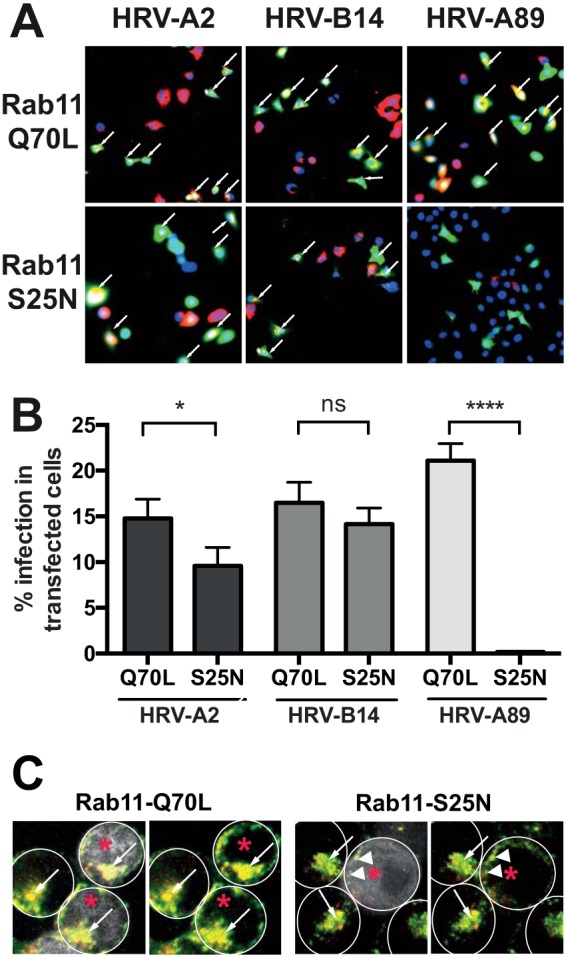
Rab11 GTPase is required for infection with HRV-A89 but not for infection with HRV-A2 and HRV-B14. Cells were seeded in 24-well plates on coverslips, grown until 70% confluent, and transfected with EGFP-tagged plasmids for the expression of constitutively active (Q70L) or dominant negative (S25N) Rab11 mutants as indicated. (A and B) Twenty-four hours later, the respective virus (at 100 TCID50/cell) was allowed to internalize for 60 min; unbound virus was washed away with cold PBS+ containing 25 mM NH4Cl for 15 min, and replication was allowed to proceed for 6.5 h at 37°C in infection medium containing NH4Cl. Cells were fixed, quenched, permeabilized, and blocked, and de novo-synthesized viral protein was detected with the respective antibodies, followed by suitable fluorescent secondary antibodies. Images were viewed and recorded on a TissueFaxs, and cells expressing EGFP and/or viral proteins were determined by using the TissueQuest software. The numbers of transfected and infected cells (A, white arrows) are shown in panel B as the means ± standard deviations from one typical experiment carried out in triplicate. The significance was calculated as described in the legend of Fig. 3. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001; ns, not significant. (C) HRV-A89 at 100 TCID50/cell and Alexa Fluor 647-transferrin (5 μg/ml MEM+) were internalized for 60 min at 37°C. After removal of transferrin from the plasma membrane, HRV-A89 was detected by indirect immunofluorescence (see Materials and Methods). Colocalization of HRV-A89 (red) and transferrin (green) in cells expressing the respective Rab11-EGFP by confocal fluorescence microscopy. Cells expressing Rab-EGF are marked with red asterisks; on the respective left panels they are also shown in white. Individual cells are encircled with white lines. In control and Rab11-Q70L-expressing cells, virus and transferrin accumulate in the ERC (white arrows), whereas in Rab11-S25N-expressing cells, virus and transferrin accumulate in peripheral vesicles (white arrowheads).
Recently, the ERC has been shown to play a major role not only in pathogen entry and infection but also in the innate (beta interferon) immune response to viruses (54). Although a number of viruses traffic through the ERC during entry (55–57), their infection was not inhibited by Rab11-S25N, which is indicative of the ERC not being involved in penetration into the cytoplasm. Nevertheless, some viruses (e.g., dengue virus) productively uncoat in the ERC, whereas others (e.g., influenza A virus) depend on the ERC for replication and assembly (58).
DISCUSSION
We have shown that, despite binding the same receptor, HRV-A89 and HRV-B14 exploit different endocytic pathways for infection. What differences might account for this behavior? In vitro, these viruses become inactivated (i.e., converted into subviral particles) at pHs of ≤4.8 and ≤5.4, respectively (data not shown). HRV-A89 infection is inhibited by recombinant soluble ICAM-1 (sICAM-1) with a 50% inhibitory concentration (IC50) of 70 nM, whereas HRV-B14 needs 3,272 nM for the same effect (59). These differences might also explain the failure of removing surface-bound HRV-A89 via washing with acidic buffer (data not shown). Presumably, the high affinity of ICAM-1 for HRV-A89 directs this virus into the recycling pathway, whereas HRV-B14 may dissociate from ICAM-1 and is thus routed to lysosomes. It is generally assumed that uncoating of the major-group viruses depends on the catalytic action of ICAM-1 and temperatures above 26°C (38, 39). This holds true for HRV-B3, HRV-B14, and HRV-A16 in vitro (38) and for HRV-B14 in vivo (41). However, HRV-A89 can uncoat at 20°C in vivo (Ganjian et al., unpublished data). Thus, either the temperature dependence of ICAM-1-induced uncoating is distinct for various serotypes, or ICAM-1 is not required for uncoating of all major-group HRVs. Notwithstanding that sICAM-1 might compete with the cellular receptor as well as inactivate the virus via promoting uncoating (60), it is conceivable that, in the cell, the concomitant actions of ICAM-1 and of the acidic pH differentially trigger the transient and reversible externalization of the amphiphilic N-terminal sequences of the capsid protein VP1 (61). This might result in membrane interactions of the respective viral particle taking place at different stations on the endocytic route directing it to different destinations. Further experiments along these lines will clarify whether the acid lability or the affinity for ICAM-1 or both are indeed responsible for the different behavior of the two viral serotypes.
ACKNOWLEDGMENTS
We are grateful to Rudolf Valenta for the generous gift of IgG P5, Tom Smith for MAb 17-IA, Marino Zerial for the Rab11 mutants, Christin Zietz for performing the experiment shown in Fig. 5C, and Irene Goesler for virus preparations and titer determinations.
Funding Statement
Funding was provided by Austrian Science Fund (FWF) projects P27444-B13 and P23308-B13) to Dieter Blaas.
REFERENCES
- 1.Uncapher CR, Dewitt CM, Colonno RJ. 1991. The major and minor group receptor families contain all but one human rhinovirus serotype. Virology 180:814–817. doi: 10.1016/0042-6822(91)90098-V. [DOI] [PubMed] [Google Scholar]
- 2.Vlasak M, Roivainen M, Reithmayer M, Goesler I, Laine P, Snyers L, Hovi T, Blaas D. 2005. The minor receptor group of human rhinovirus (HRV) includes HRV23 and HRV25, but the presence of a lysine in the VP1 HI loop is not sufficient for receptor binding. J Virol 79:7389–7395. doi: 10.1128/JVI.79.12.7389-7395.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bochkov YA, Watters K, Ashraf S, Griggs TF, Devries MK, Jackson DJ, Palmenberg AC, Gern JE. 2015. Cadherin-related family member 3, a childhood asthma susceptibility gene product, mediates rhinovirus C binding and replication. Proc Natl Acad Sci U S A 112:5485–5490. doi: 10.1073/pnas.1421178112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fuchs R, Blaas D. 2012. Productive entry pathways of human rhinoviruses. Adv Virol 2012:826301. doi: 10.1155/2012/826301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldstein JL, Brown MS. 1974. Binding and degradation of low density lipoproteins by cultured human fibroblasts. Comparison of cells from a normal subject and from a patient with homozygous familial hypercholesterolemia. J Biol Chem 249:5153–5162. [PubMed] [Google Scholar]
- 6.Pearse BM. 1976. Clathrin: a unique protein associated with intracellular transfer of membrane by coated vesicles. Proc Natl Acad Sci U S A 73:1255–1259. doi: 10.1073/pnas.73.4.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anderson RG, Vasile E, Mello RJ, Brown MS, Goldstein JL. 1978. Immunocytochemical visualization of coated pits and vesicles in human fibroblasts: relation to low density lipoprotein receptor distribution. Cell 15:919–933. doi: 10.1016/0092-8674(78)90276-3. [DOI] [PubMed] [Google Scholar]
- 8.Elkin SR, Lakoduk AM, Schmid SL. 2016. Endocytic pathways and endosomal trafficking: a primer. Wien Med Wochenschr 166:196–204. doi: 10.1007/s10354-016-0432-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mellman I. 1996. Endocytosis and molecular sorting. Annu Rev Cell Dev Biol 12:575–625. doi: 10.1146/annurev.cellbio.12.1.575. [DOI] [PubMed] [Google Scholar]
- 10.Mercer J, Schelhaas M, Helenius A. 2010. Virus entry by endocytosis. Annu Rev Biochem 79:803–833. doi: 10.1146/annurev-biochem-060208-104626. [DOI] [PubMed] [Google Scholar]
- 11.Mindell JA. 2012. Lysosomal acidification mechanisms. Annu Rev Physiol 74:69–86. doi: 10.1146/annurev-physiol-012110-142317. [DOI] [PubMed] [Google Scholar]
- 12.Goldstein JL, Brown MS, Anderson RG, Russell DW, Schneider WJ. 1985. Receptor-mediated endocytosis: concepts emerging from the LDL receptor system. Annu Rev Cell Biol 1:1–39. doi: 10.1146/annurev.cb.01.110185.000245. [DOI] [PubMed] [Google Scholar]
- 13.Davis CG, Goldstein JL, Sudhof TC, Anderson RG, Russell DW, Brown MS. 1987. Acid-dependent ligand dissociation and recycling of LDL receptor mediated by growth factor homology region. Nature 326:760–765. doi: 10.1038/326760a0. [DOI] [PubMed] [Google Scholar]
- 14.Gu F, Gruenberg J. 1999. Biogenesis of transport intermediates in the endocytic pathway. FEBS Lett 452:61–66. doi: 10.1016/S0014-5793(99)00561-X. [DOI] [PubMed] [Google Scholar]
- 15.Gruenberg J, Stenmark H. 2004. The biogenesis of multivesicular endosomes. Nat Rev Mol Cell Biol 5:317–323. doi: 10.1038/nrm1360. [DOI] [PubMed] [Google Scholar]
- 16.Huotari J, Helenius A. 2011. Endosome maturation. EMBO J 30:3481–3500. doi: 10.1038/emboj.2011.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saftig P, Schroder B, Blanz J. 2010. Lysosomal membrane proteins: life between acid and neutral conditions. Biochem Soc Trans 38:1420–1423. doi: 10.1042/BST0381420. [DOI] [PubMed] [Google Scholar]
- 18.Dautry-Varsat A, Ciechanover A, Lodish HF. 1983. pH and the recycling of transferrin during receptor-mediated endocytosis. Proc Natl Acad Sci U S A 80:2258–2262. doi: 10.1073/pnas.80.8.2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maxfield FR, McGraw TE. 2004. Endocytic recycling. Nat Rev Mol Cell Biol 5:121–132. doi: 10.1038/nrm1315. [DOI] [PubMed] [Google Scholar]
- 20.van der Sluijs P, Hull M, Webster P, Male P, Goud B, Mellman I. 1992. The small GTP-binding protein Rab4 controls an early sorting event on the endocytic pathway. Cell 70:729–740. doi: 10.1016/0092-8674(92)90307-X. [DOI] [PubMed] [Google Scholar]
- 21.Baravalle G, Schober D, Huber M, Bayer N, Murphy RF, Fuchs R. 2005. Transferrin recycling and dextran transport to lysosomes is differentially affected by bafilomycin, nocodazole, and low temperature. Cell Tissue Res 320:99–113. doi: 10.1007/s00441-004-1060-x. [DOI] [PubMed] [Google Scholar]
- 22.Aniento F, Emans N, Griffiths G, Gruenberg J. 1993. Cytoplasmic dynein-dependent vesicular transport from early to late endosomes. J Cell Biol 123:1373–1387. doi: 10.1083/jcb.123.6.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kornilova ES. 2014. Receptor-mediated endocytosis and cytoskeleton. Biochemistry 79:865–878. doi: 10.1134/S0006297914090041. [DOI] [PubMed] [Google Scholar]
- 24.Daro E, van der Sluijs P, Galli T, Mellman I. 1996. Rab4 and cellubrevin define different early endosome populations on the pathway of transferrin receptor recycling. Proc Natl Acad Sci U S A 93:9559–9564. doi: 10.1073/pnas.93.18.9559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Firestone AJ, Weinger JS, Maldonado M, Barlan K, Langston LD, O'Donnell M, Gelfand VI, Kapoor TM, Chen JK. 2012. Small-molecule inhibitors of the AAA+ ATPase motor cytoplasmic dynein. Nature 484:125–129. doi: 10.1038/nature10936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roossien DH, Miller KE, Gallo G. 2015. Ciliobrevins as tools for studying dynein motor function. Front Cell Neurosci 9:252. doi: 10.3389/fncel.2015.00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mellman I, Fuchs R, Helenius A. 1986. Acidification of the endocytic and exocytic pathways. Annu Rev Biochem 55:663–700. doi: 10.1146/annurev.bi.55.070186.003311. [DOI] [PubMed] [Google Scholar]
- 28.Aniento F, Gu F, Parton RG, Gruenberg J. 1996. An endosomal beta COP is involved in the pH-dependent formation of transport vesicles destined for late endosomes. J Cell Biol 133:29–41. doi: 10.1083/jcb.133.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gillespie EJ, Ho CL, Balaji K, Clemens DL, Deng G, Wang YE, Elsaesser HJ, Tamilselvam B, Gargi A, Dixon SD, France B, Chamberlain BT, Blanke SR, Cheng G, de la Torre JC, Brooks DG, Jung ME, Colicelli J, Damoiseaux R, Bradley KA. 2013. Selective inhibitor of endosomal trafficking pathways exploited by multiple toxins and viruses. Proc Natl Acad Sci U S A 110:E4904–4912. doi: 10.1073/pnas.1302334110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stenmark H. 2009. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol 10:513–525. doi: 10.1038/nrm2728. [DOI] [PubMed] [Google Scholar]
- 31.Horgan CP, Hanscom SR, Jolly RS, Futter CE, McCaffrey MW. 2010. Rab11-FIP3 links the Rab11 GTPase and cytoplasmic dynein to mediate transport to the endosomal-recycling compartment. J Cell Sci 123:181–191. doi: 10.1242/jcs.052670. [DOI] [PubMed] [Google Scholar]
- 32.Ren M, Xu G, Zeng J, De Lemos-Chiarandini C, Adesnik M, Sabatini DD. 1998. Hydrolysis of GTP on rab11 is required for the direct delivery of transferrin from the pericentriolar recycling compartment to the cell surface but not from sorting endosomes. Proc Natl Acad Sci U S A 95:6187–6192. doi: 10.1073/pnas.95.11.6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ullrich O, Reinsch S, Urbe S, Zerial M, Parton RG. 1996. Rab11 regulates recycling through the pericentriolar recycling endosome. J Cell Biol 135:913–924. doi: 10.1083/jcb.135.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kelly EE, Horgan CP, McCaffrey MW. 2012. Rab11 proteins in health and disease. Biochem Soc Trans 40:1360–1367. doi: 10.1042/BST20120157. [DOI] [PubMed] [Google Scholar]
- 35.Snyers L, Zwickl H, Blaas D. 2003. Human rhinovirus type 2 is internalized by clathrin-mediated endocytosis. J Virol 77:5360–5369. doi: 10.1128/JVI.77.9.5360-5369.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prchla E, Kuechler E, Blaas D, Fuchs R. 1994. Uncoating of human rhinovirus serotype 2 from late endosomes. J Virol 68:3713–3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bayer N, Schober D, Prchla E, Murphy RF, Blaas D, Fuchs R. 1998. Effect of bafilomycin A1 and nocodazole on endocytic transport in HeLa cells: implications for viral uncoating and infection. J Virol 72:9645–9655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoover-Litty H, Greve JM. 1993. Formation of rhinovirus-soluble ICAM-1 complexes and conformational changes in the virion. J Virol 67:390–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nurani G, Lindqvist B, Casasnovas JM. 2003. Receptor priming of major group human rhinoviruses for uncoating and entry at mild low-pH environments. J Virol 77:11985–11991. doi: 10.1128/JVI.77.22.11985-11991.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bayer N, Prchla E, Schwab M, Blaas D, Fuchs R. 1999. Human rhinovirus HRV14 uncoats from early endosomes in the presence of bafilomycin. FEBS Lett 463:175–178. doi: 10.1016/S0014-5793(99)01610-5. [DOI] [PubMed] [Google Scholar]
- 41.Schober D, Kronenberger P, Prchla E, Blaas D, Fuchs R. 1998. Major and minor receptor group human rhinoviruses penetrate from endosomes by different mechanisms. J Virol 72:1354–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pfanzagl B, Andergassen D, Edlmayr J, Niespodziana K, Valenta R, Blaas D. 2014. Entry of human rhinovirus 89 via ICAM-1 into HeLa epithelial cells is inhibited by actin skeleton disruption and by bafilomycin. Arch Virol 159:125–140. doi: 10.1007/s00705-013-1797-1. [DOI] [PubMed] [Google Scholar]
- 43.Edlmayr J, Niespodziana K, Popow-Kraupp T, Krzyzanek V, Focke-Tejkl M, Blaas D, Grote M, Valenta R. 2011. Antibodies induced with recombinant VP1 from human rhinovirus exhibit cross-neutralisation. Eur Respir J 37:44–52. doi: 10.1183/09031936.00149109. [DOI] [PubMed] [Google Scholar]
- 44.Khan AG, Pickl-Herk A, Gajdzik L, Marlovits TC, Fuchs R, Blaas D. 2011. Entry of a heparan sulphate-binding HRV8 variant strictly depends on dynamin but not on clathrin, caveolin, and flotillin. Virology 412:55–67. doi: 10.1016/j.virol.2010.12.042. [DOI] [PubMed] [Google Scholar]
- 45.Blake K, O'Connell S. 1993. Virus culture, p 81–122. In Harper DR. (ed), Virology labfax. Blackwell Scientific Publications, London United Kingdom. [Google Scholar]
- 46.Skern T, Neubauer C, Frasel L, Grundler P, Sommergruber W, Zorn M, Kuechler E, Blaas D. 1987. A neutralizing epitope on human rhinovirus type 2 includes amino acid residues between 153 and 164 of virus capsid protein VP2. J Gen Virol 68:315–323. doi: 10.1099/0022-1317-68-2-315. [DOI] [PubMed] [Google Scholar]
- 47.Jurgeit A, Moese S, Roulin P, Dorsch A, Lotzerich M, Lee WM, Greber UF. 2010. An RNA replication-center assay for high content image-based quantifications of human rhinovirus and coxsackievirus infections. Virol J 7:264. doi: 10.1186/1743-422X-7-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sonnichsen B, De Renzis S, Nielsen E, Rietdorf J, Zerial M. 2000. Distinct membrane domains on endosomes in the recycling pathway visualized by multicolor imaging of Rab4, Rab5, and Rab11. J Cell Biol 149:901–914. doi: 10.1083/jcb.149.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lonberg-Holm K, Korant BD. 1972. Early interaction of rhinoviruses with host cells. J Virol 9:29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Neubauer C, Frasel L, Kuechler E, Blaas D. 1987. Mechanism of entry of human rhinovirus 2 into HeLa cells. Virology 158:255–258. doi: 10.1016/0042-6822(87)90264-9. [DOI] [PubMed] [Google Scholar]
- 51.Reischl A, Reithmayer M, Winsauer G, Moser R, Gosler I, Blaas D. 2001. Viral evolution toward change in receptor usage: adaptation of a major group human rhinovirus to grow in ICAM-1-negative cells. J Virol 75:9312–9319. doi: 10.1128/JVI.75.19.9312-9319.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gruenberg J, Griffiths G, Howell K. 1989. Characterization of the early endosome and putative endocytic carrier vesicles in vivo and with an assay of vesicle fusion in vitro. J Cell Biol 108:1301–1316. doi: 10.1083/jcb.108.4.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Driskell OJ, Mironov A, Allan VJ, Woodman PG. 2007. Dynein is required for receptor sorting and the morphogenesis of early endosomes. Nat Cell Biol 9:113–120. doi: 10.1038/ncb1525. [DOI] [PubMed] [Google Scholar]
- 54.Guichard A, Nizet V, Bier E. 2014. RAB11-mediated trafficking in host-pathogen interactions. Nat Rev Microbiol 12:624–634. doi: 10.1038/nrmicro3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harbison CE, Lyi SM, Weichert WS, Parrish CR. 2009. Early steps in cell infection by parvoviruses: host-specific differences in cell receptor binding but similar endosomal trafficking. J Virol 83:10504–10514. doi: 10.1128/JVI.00295-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weir DL, Laing ED, Smith IL, Wang LF, Broder CC. 2014. Host cell virus entry mediated by Australian bat lyssavirus G envelope glycoprotein occurs through a clathrin-mediated endocytic pathway that requires actin and Rab5. Virol J 11:40. doi: 10.1186/1743-422X-11-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zila V, Difato F, Klimova L, Huerfano S, Forstova J. 2014. Involvement of microtubular network and its motors in productive endocytic trafficking of mouse polyomavirus. PLoS One 9:e96922. doi: 10.1371/journal.pone.0096922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vale-Costa S, Amorim MJ. 2016. Recycling endosomes and viral infection. Viruses 8:64. doi: 10.3390/v8030064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ohlin A, Hoover-Litty H, Sanderson G, Paessens A, Johnston SL, Holgate ST, Huguenel E, Greve JM. 1994. Spectrum of activity of soluble intercellular adhesion molecule-1 against rhinovirus reference strains and field isolates. Antimicrob Agents Chemother 38:1413–1415. doi: 10.1128/AAC.38.6.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Crump CE, Arruda E, Hayden FG. 1993. In vitro inhibitory activity of soluble ICAM-1 for the numbered serotypes of human rhinovirus. Antivir Chem Chemother 4:323–327. doi: 10.1177/095632029300400603. [DOI] [Google Scholar]
- 61.Lin J, Lee LY, Roivainen M, Filman DJ, Hogle JM, Belnap DM. 2012. Structure of the Fab-labeled “breathing” state of native poliovirus. J Virol 86:5959–5962. doi: 10.1128/JVI.05990-11. [DOI] [PMC free article] [PubMed] [Google Scholar]