

ABSTRACT
Early HIV-1 infection is characterized by enhanced tryptophan catabolism, which contributes to immune suppression and disease progression. However, the mechanism by which kynurenine, a tryptophan-related metabolite, induces immune suppression remains poorly understood. Herein, we show that the increased production of kynurenine correlates with defective interleukin-2 (IL-2) signaling in memory CD4 T cells from HIV-infected subjects. Defective IL-2 signaling in these subjects, which drives reduced protection from Fas-mediated apoptosis, was also associated with memory CD4 T-cell loss. Treatment of memory CD4 T cells with the concentration of kynurenine found in plasma inhibited IL-2 signaling through the production of reactive oxygen species. We further show that IL-2 signaling in memory CD4 T cells is improved by the antioxidant N-acetylcysteine. Early initiation of antiretroviral therapy restored the IL-2 response in memory CD4 T cells by reducing reactive oxygen species and kynurenine production. The study findings provide a kynurenine-dependent mechanism through IL-2 signaling for reduced CD4 T-cell survival, which can be reversed by early treatment initiation in HIV-1 infection.
IMPORTANCE The persistence of functional memory CD4 T cells represents the basis for long-lasting immune protection in individuals after exposure to HIV-1. Unfortunately, primary HIV-1 infection results in the massive loss of these cells within weeks of infection, which is mainly driven by inflammation and massive infection by the virus. These new findings show that the enhanced production of kynurenine, a metabolite related to tryptophan catabolism, also impairs memory CD4 T-cell survival and interferes with IL-2 signaling early during HIV-1 infection.
INTRODUCTION
Progressive depletion of CD4 T cells by pyroptosis and activation-related apoptosis is the hallmark of HIV-1 infection (1–4). The loss of memory CD4 T cells and, in particular, central memory CD4 T cells (TCM cells), which are critical to ensure long-lasting immune protection, occurs from the onset of infection and independently predicts disease progression (5–8). Memory T-cell survival depends on signals provided by the gamma-chain-receptor cytokines, such as interleukin-2 (IL-2) and IL-7 (9–12). Previous data showed an impaired response to these cytokines in T cells during HIV-1 infection (13–16). However, our knowledge of the molecular mechanisms responsible for these immune defects is still incomplete.
CD4 T-cell depletion is also linked with persistent inflammation, which takes place in the gut and lymphoid tissues early after infection and in the bloodstream later after infection (17–19). This inflammation is clinically relevant, since it has not been fully abrogated by long-term antiretroviral therapy (ART) (20–23). Persistent inflammation during HIV-1 infection is associated with enhanced metabolic activity, which drives a Warburg-like effect on lipid, glucose, and amino acid pathways (22, 24–27). The catabolism of an essential amino acid like tryptophan into kynurenine (Kyn), which is mediated by the indoleamine 2,3-dioxygenase 1 (IDO-1) expressed in dendritic cells (DCs) and monocytes, is increased in HIV-1-infected subjects (22, 28–30). The enhanced tryptophan catabolism detected early during HIV-1 infection correlates with proinflammatory molecules like IL-6, soluble CD40 (sCD40), and gamma interferon (IFN-γ)-inducible protein 10 (IP-10) (22, 25, 31, 32) and is associated with CD4 T-cell decay (33–35). Many studies showed that the excessive tryptophan metabolism occurring during primary HIV-1 infection is associated with increased immune dysfunction. This state is characterized by the decrease of tryptophan availability and the increased production of the immunosuppressive compound Kyn. Enhanced tryptophan/kynurenine catabolism leads to the loss of Th17 and Th22 cells, which play key protective roles in the gut for microbial defense (36), the generation of regulatory T cells (25, 29, 32), and the inhibition of T-cell proliferation (37, 38).
In the present study, we investigated the impact of Kyn on memory CD4 T-cell survival in the early and chronic stages of HIV-1 infection. Our results identified a critical role of Kyn in the loss of memory CD4 T cells during HIV-1 infection by interfering with IL-2 signaling through the production of reactive oxygen species (ROS).
MATERIALS AND METHODS
Ethics statement.
Control subjects and HIV-1-infected subjects were participants in the Montreal, Quebec, Canada, primary HIV infection study that received approval from the McGill University Health Centre Ethical Review Board (ethics reference number SL-00.069). All subjects provided informed and written consent for participation in the study.
Study population.
Peripheral blood mononuclear cells (PBMCs) and plasma samples were collected from subjects in whom viral acquisition was estimated to have occurred less than 90 days earlier (primary HIV-1-infected [PHI] subjects), untreated ART-naive patients who had been infected with HIV-1 for a mean 4 years (chronically HIV-1-infected [CHI] subjects), and age-matched HIV-uninfected control subjects (HIVfree subjects) (n = 6 subjects in each group). All HIV-1-infected subjects included in the overall study were homogeneously selected, and clinical and virological data were similar for all HIV-1-infected subjects. Table 1 summarizes the clinical features of the 12 PHI and CHI subjects, including the viral load (VL) and CD4/CD8 ratio.
TABLE 1.
Clinical features of all HIV-1-infected subjects
| Characteristic | Result for study HIV-1-infected population |
|
|---|---|---|
| PHI patients | CHI patients | |
| Time of infection before the study (days) | ||
| Mean ± SD | 61.3 ± 18.4 | 647.1 ± 380.7 |
| Range | 31–90 | 167–1,278 |
| Age (yr) | ||
| Mean ± SD | 35.7 ± 8.1 | 37.2 ± 7.5 |
| Range | 22–48 | 21–47 |
| No. (%) of patients of male sex | 18 (90) | 12 (100) |
| CD4 count (no. of cells/μl) | ||
| Mean ± SD | 510 ± 238 | 489 ± 229 |
| Range | 240–1,120 | 260–1,128 |
| CD8 count (no. of cells/μl) | ||
| Mean ± SD | 1,094 ± 728 | 829 ± 319 |
| Range | 410–3,450 | 410–1,270 |
| CD4/CD8 ratio | ||
| Mean ± SD | 0.61 ± 0.41 | 0.66 ± 0.32 |
| Range | 0.13–1.40 | 0.32–1.30 |
| VL (log10 no. of copies/ml) | ||
| Mean ± SD | 5.11 ± 0.96 | 4.88 ± 0.65 |
| Range | 3.71–7.48 | 4.10–6.34 |
Reagents and antibodies.
RPMI 1640 medium, fetal bovine serum (FBS), antibiotics, and phosphate-buffered saline (PBS) were obtained from Wisent Inc. (Saint-Jean-Baptiste, QC, Canada). Anti-Fas CH11 antibody was from MBL International Corporation (Woburn, MA, USA). Recombinant cytokines (IL-2 and IL-7), hydrogen peroxide (H2O2) solution, phorbol myristate acetate, and ionomycin were provided from Sigma-Aldrich (Oakville, ON, Canada). We purchased all antibodies and reagents for flow cytometry from BD Biosciences (San Jose, CA, USA), except for the antibody to CD45RA-ECD, which was from Beckman Coulter (Montreal, QC, Canada). 7-Aminoactinomycin D (7-AAD) came from Invitrogen (Burlington, ON, Canada).
Quantitation of plasma markers.
The plasma levels of proinflammatory IL-6, soluble CD14 (sCD14), lipopolysaccharide (LPS)-binding protein (LBP), IP-10, and the tryptophan-related metabolite Kyn were measured by enzyme-linked immunosorbent assays (ELISAs) according to the manufacturers' instructions. Commercially available ELISA kits were purchased from R&D Systems (Minneapolis, MN, USA) for IL-6, IP-10, and sCD14; from Hycult Biotech for LBP; and Antibodies Online for Kyn.
Purification of CD4 T cells.
Total CD4 T cells were purified using an untouched CD4 isolation kit (EasySep human CD4 T cell enrichment kit; Stem Cell Technologies, Vancouver, BC, Canada), allowing more than 96% purification without any cell stimulation or apoptosis.
STAT5 phosphorylation assay.
Cells from PHI, CHI, and HIVfree subjects were stimulated with IL-2 (25 IU/ml) or IL-7 (0.3 ng/ml) for 15 min or were unstimulated. STAT5 phosphorylation was then measured by the BD Biosciences Phosflow protocol using anti-phospho-STAT (pSTAT5)-Alexa Fluor 647-specific antibody as previously described (39). The following multiparameter antibody cocktail was used: anti-CD3-phycoerythrin (PE), anti-CD4-V450, anti-CD45RA-ECD, anti-CD27-allophycocyanin (APC) H7, and anti-CCR7-PE Cy7 antibodies. The viability marker 7-AAD was always used to exclude dead cells from analysis. We collected approximately 20,000 gated events on a BD LSRII Fortessa flow cytometer (BD) and analyzed the data using DIVA and FlowJo software. Results shown are the expression levels of pSTAT5 within gated 7-AAD-negative CD3+ CD4+ CD45RA− memory CD4 T cells.
Fas-mediated apoptosis.
Similarly to the procedure described in our previous study (8), we first cultured cells from PHI, CHI, and HIVfree subjects with 1.25 μg/ml of anti-Fas CH11 antibody in the presence or absence of 25 IU/ml IL-2 for 24 h. We then detected apoptotic cells on gated CD3+ CD4+ CD45RA− memory CD4 T cells with annexin V-V450 labeling (BD Biosciences) using the following surface antibody cocktail: anti-CD3-PE, anti-CD4-APC H7, and anti-CD45RA-ECD antibodies. The results shown represent the percentage of cells protected from Fas-mediated apoptosis when cells were treated with IL-2. The percent inhibition of apoptosis by IL-2 was determined using the following formula: [(percentage of cells with Fas-mediated apoptosis for cells not treated with IL-2 − percentage of cells with Fas-mediated apoptosis for cells treated with IL-2)/percentage of cells with Fas-mediated apoptosis for cells not treated with IL-2] × 100.
Detection of intracellular ROS.
ROS measurement was assessed by flow cytometry using a CM-H2DCFDA (chloromethyl 2′,7′-dichlorodihydrofluorescein diacetate acetyl ester) probe (Life Technologies, Invitrogen) according to the manufacturer's protocol. Briefly, cells were incubated for 45 min with 10 μM CM-H2DCFDA in a cell incubator (37°C), then washed three times in 1× PBS, and, finally, subjected to flow cytometry analyses. Upon oxidation by ROS, the nonfluorescent probe CM-H2DCFDA is converted to the highly fluorescent 2′,7′-dichlorofluorescein (DCF), that is detectable at 525 nm (40).
Data analysis.
All statistical analyses comparing the groups of subjects (HIVfree, PHI, and CHI subjects) were performed using the nonparametric Mann-Whitney U test with the assumption of independent samples. This test, which uses the rank of the data rather than their raw values to calculate statistical significance, is an alternative to the t test when the assumption of normality cannot be tested in the case of a moderate sample size. A sample size of 6 subjects per group is sufficient to achieve a significant statistical power based on the observed changes. In contrast, we used the paired t test for analyzing the results of in vitro assays using noninfected memory CD4 T cells. Spearman's correlation test was used to identify the association among study clinical and immunological variables. We considered P values of less than 0.05 to be significant.
RESULTS
Loss of memory CD4 T cells during HIV-1 infection involves defective IL-2 signaling.
Early HIV-1 infection is characterized by the loss of CD4 T cells, including CD4 T cells from the memory compartment (5–7). To newly identify the underlying mechanisms associated with the depletion of memory CD4 T cells, we first measured their frequencies in primary HIV-1-infected (PHI), chronically HIV-1-infected (CHI), and HIVfree subjects. Table 1 summarizes the clinical features of the two groups of viremic subjects. The frequencies of viable CD3+ CD4+ CD45RA− memory CD4 T cells were significantly lower in PHI and CHI subjects than HIVfree donors (7.4% ± 4.3%, 9.7% ± 3.5%, and 14.7% ± 4.7%, respectively; P < 0.05; n = 6) (Fig. 1A). Since IL-2, IL-7, and IL-15 play critical roles in the long-term survival of memory T cells (9–12), we next investigated the response to these cytokines in memory CD4 T cells from all groups by measuring the phosphorylation of STAT5. Ex vivo, uninduced and IL-7- and IL-15-induced phospho-STAT5 (pSTAT5) expression levels in memory CD4 T cells were similar in all groups. In contrast, we found that IL-2-induced pSTAT5 levels were significantly reduced in PHI and CHI subjects compared with those in HIVfree donors (mean fluorescence intensities [MFI], 4,223.8 ± 2,060.1, 4,794 ± 2,178.9, and 9,033.7 ± 1,303.8, respectively; P < 0.01; n = 6) (Fig. 1B). Similarly, the frequency of pSTAT5-positive memory CD4 T cells upon IL-2 stimulation was lower in PHI and CHI subjects than HIVfree subjects (36.4% ± 16.5%, 44.9% ± 15.8%, and 61.1% ± 4.3%, respectively, for PHI, CHI, and HIVfree subjects) (data not shown). We found no significant difference in the levels of IL-2-induced pSTAT5 in naive CD45RA+ cells among the subjects (Fig. 1B). The reduced response to IL-2 in memory CD4 T cells during HIV-1 infection concerned key memory subsets, such as the CD27+ CCR7+ central memory CD4 T cells (TCM cells) and CD27+ CCR7− transitional memory CD4 T cells (TTM cells) (Fig. 1C) (41). The defective IL-2 response in memory CD4 T cells from HIV-1-infected subjects was not related to the decreased expression of the surface IL-2 receptor (CD25), since we found similar percentages of CD25-positive cells, including CD8 T cells and memory and naive CD4 T cells, in all subjects (Fig. 1D).
FIG 1.

Defective IL-2 signaling in memory CD4 T cells is associated with cell loss during HIV-1 infection. (A) Ex vivo frequency of memory CD4 T cells among PBMCs from PHI, CHI, and HIVfree subjects. (B) PBMCs from all groups were stimulated or not with cytokines for 15 min and then collected to assess pSTAT5 levels. (Inset) Representative dot plots for IL-2-induced pSTAT5 expression. (C) IL-2-induced pSTAT5 levels in viable CD3+ CD4+ CD45RA− CD27+ CCR7− TCM cells, CD27+ CCR7− TTM cells, and CD27− CCR7− TEM cells for all subject groups. (D) Percentage of CD25-positive cells. In panels A to D, data are for 6 subjects in each group. (E) Correlation between the level of IL-2-induced pSTAT5 and the frequency (i) (n = 18) or the absolute number (ii) (n = 12) of memory CD4 T cells. *, 0.05 > P > 0.01; **, 0.01 > P > 0.001.
We further determined whether the loss of memory CD4 T cells during HIV-1 infection is associated with defective IL-2 signaling. We confirmed correlations between the IL-2-induced pSTAT5 level and (i) the memory CD4 T-cell frequency determined in all study subjects or (ii) the absolute number of memory CD4 T cells by using cell counts, which were available only for HIV-1-infected subjects (P = 0.0127 [n = 18] and 0.0366 [n = 12], respectively) (Fig. 1E).
Finally, we assessed the protection from Fas-mediated apoptosis in memory CD4 T cells from PHI, CHI, and HIVfree subjects upon IL-2 stimulation. To this end, cells from all groups were first treated with anti-Fas CH11 antibody for 24 h in the presence or absence of IL-2 and then collected to assess apoptosis levels by annexin V staining. Memory CD4 T cells from PHI and CHI subjects displayed a higher sensitivity to Fas-mediated apoptosis and concomitantly showed increased expression of Fas receptor (CD95) compared to the response of memory CD4 T cells from HIVfree donors (Fig. 2A and B). IL-2 stimulation was effective at reducing Fas-mediated apoptosis in memory CD4 T cells in all subject groups (Fig. 2A). However, the levels of IL-2-associated protection were significantly lower in memory CD4 T cells from PHI and CHI subjects than in those from HIVfree donors (35.1% ± 12.8%, 44.8% ± 22.4%, and 80.2% ± 15.9%, respectively; P < 0.01; n = 6) (Fig. 2C). Interestingly, we found a significant correlation between the protection from Fas-mediated apoptosis with IL-2 treatment and the frequency of memory CD4 T cells for all study subjects (P = 0.0083; n = 18) (Fig. 2D).
FIG 2.

Memory CD4 T cells from HIV-1-infected subjects displayed reduced protection from apoptosis upon IL-2 stimulation. (A) Fas-mediated apoptosis of memory CD4 T cells for PHI, CHI, and HIVfree subjects in the presence or absence of IL-2 treatment. This was calculated using the following formula: percent apoptosis of cells treated with anti-Fas antibody − percent apoptosis of anti-Fas-untreated cells. (B) Ex vivo percentage of CD95-positive memory CD4 T cells. (C) Levels of apoptosis protection when cells were treated with IL-2. In panels A to C, data are for 6 subjects in each group. (D) Correlation between the level of apoptosis protection with IL-2 and the ex vivo frequency of memory CD4 T cells from all subjects (n = 18). *, 0.05 > P > 0.01; **, 0.01 > P > 0.001.
Overall, these data demonstrate that memory CD4 T cells, which are depleted during HIV-1 infection, showed a reduced capacity to resist Fas-mediated apoptosis in the presence of IL-2.
Defective IL-2 signaling in memory CD4 T cells is associated with the Kyn level during HIV-1 infection.
As early HIV-1 infection is characterized by persistent inflammation and enhanced tryptophan catabolism (22, 24–27, 42), we first evaluated the levels of several proinflammatory molecules and Kyn in plasma from the PHI, CHI, and HIVfree subjects. Consistent with earlier reports (22, 25, 43), we found higher levels of IL-6, sCD14, IP-10, and Kyn in PHI and CHI subjects than HIVfree donors (Fig. 3A to D). However, we did not observe any significant differences for LPS-binding protein (LBP) (Fig. 3E). More importantly, we found significant correlations between IL-2-induced pSTAT5 levels in memory CD4 T cells from all subjects and the CD4/CD8 ratio or the IL-6, sCD14, IP-10, or Kyn level (P = 0.0159, 0.023, 0.0267, 0.0149, and 0.0028, respectively; n = 18) (Fig. 3F).
FIG 3.

Reduced IL-2 signaling correlates with inflammation and Kyn levels during HIV-1 infection. (A to E) Levels of proinflammatory soluble factors and Kyn in plasma samples from all subject groups (n = 6). (F) Correlations between IL-2-induced pSTAT5 levels and the CD4/CD8 ratio (i), the viral load (VL) (ii), the concentrations of proinflammatory molecules (iii to v), or the Kyn concentration (vi) for all subjects (n = 18). Tryp, tryptophan. *, 0.05 > P > 0.01; **, 0.01 > P > 0.001; ns, not significant.
Collectively, these results indicate that HIV-1 disease progression, inflammation, and, in particular, the level of Kyn production were associated with reduced IL-2 signaling in memory CD4 T cells from infected subjects.
Treatment of CD4 T cells with Kyn inhibits IL-2 signaling through ROS.
From the plasma markers studied (Fig. 3), our observations indicated that the enhanced production of Kyn was the best correlate of reduced levels of IL-2-induced pSTAT5 expression during HIV-1 infection. To determine whether Kyn directly interferes with IL-2 signaling in memory CD4 T cells, purified CD4 T cells from HIVfree donors were treated in vitro with different concentrations of recombinant Kyn (0 to 50 μM) before being stimulated with IL-2. Our data showed that Kyn treatment led to a dose-dependent inhibition of IL-2-induced pSTAT5 levels in memory CD4 T cells, starting with a 5 μM concentration (which reduced the pSTAT5 levels 23% ± 8.9% compared to the level in untreated cells) (Fig. 4A). Of note, no apoptosis of memory CD4 T cells was induced after 48 h following Kyn treatments (with 2.5 to 50 μM concentrations) (data not shown). Since oxidative stress has been reported to be responsible for the apoptosis of natural killer cells mediated by concentrations of Kyn (44), we hypothesized that defective IL-2 signaling in memory CD4 T cells treated with Kyn could implicate ROS. To test this hypothesis, we first investigated whether 5 μM Kyn treatment resulted in ROS production by memory CD4 T cells. Our results confirmed that memory CD4 T cells cultured with 5 μM Kyn for 24 h displayed higher levels of expression of intracellular ROS than untreated cells (MFI, 1,798.4 ± 376.9 and 1,277.2 ± 214.5, respectively; P = 0.0385; n = 5) (Fig. 4B). We also preincubated CD4 T cells from uninfected subjects with increasing concentrations of H2O2 (0.1 to 100 μM), a powerful oxidant, prior to the stimulation with IL-2. Treatment with H2O2 (starting with a 1 μM concentration) led to both higher levels of ROS expression by memory CD4 T cells (Fig. 4B) and a decrease in the level of IL-2-induced pSTAT5 (Fig. 4C), confirming the contribution of the oxidative stress pathway.
FIG 4.

Kyn treatment inhibits the protection from Fas-mediated apoptosis provided by IL-2 and involves ROS production. (A) (Bottom) IL-2-induced pSTAT5 level on memory CD4 T cells in response to various Kyn concentrations. The percent inhibition of IL-2 signaling is indicated for each Kyn concentration. (Top) Representative histograms. (B) ROS expression by memory CD4 T cells in the presence or absence of 5 μM Kyn. We also stimulated CD4 T cells with increasing concentrations of H2O2. (C) CD4 T cells were preincubated with H2O2 for 1 h, stimulated with IL-2 for 15 min, and then collected to determine pSTAT5 levels in memory cells. (D) After 24 h of Kyn and/or NAC treatment, CD4 T cells were stimulated with IL-2 and pSTAT5 levels in memory cells were assessed. (E) Protection from Fas-mediated apoptosis by IL-2 in memory CD4 T cells in the presence or absence of Kyn and/or NAC. In panels A to E, data are for 5 subjects in each group. *, 0.05 > P > 0.01; **, 0.01 > P > 0.001; ***, P < 0.001.
To further demonstrate that Kyn-mediated defective IL-2 signaling in memory CD4 T cells involved ROS production, cells treated with Kyn were stimulated or not with the antioxidant N-acetylcysteine (NAC). Briefly, CD4 T cells were cultured with 5 μM Kyn for 24 h in the presence or absence of 5 mM NAC and then stimulated with IL-2 for 15 min. Cells were collected to determine pSTAT5 levels in the memory CD4 T-cell subset. Our results showed that the induction of pSTAT5 mediated by IL-2 was not affected by NAC treatment alone (Fig. 4D). We confirmed that treatment of cells with 5 μM Kyn for 24 h led to a significant reduction of IL-2-induced pSTAT5 levels. Defective IL-2 signaling in memory CD4 T cells treated with Kyn was partially ROS dependent, since the addition of NAC led to the significant but incomplete restoration of pSTAT5 levels (Fig. 4D). Kyn treatment of CD4 T cells also resulted in reduced protection of memory cells from Fas-mediated apoptosis by IL-2 (P = 0.0206 compared to the results for Kyn-untreated cells) (Fig. 4E). Similarly, the addition of NAC when cells were treated with Kyn led to significant improvements of memory CD4 T-cell survival (Fig. 4E).
Altogether, these data indicate that the oxidative stress generated by elevated levels of Kyn production during HIV-1 infection significantly interferes with IL-2 signaling in memory CD4 T cells.
ROS inhibits IL-2 signaling in memory CD4 T cells during HIV-1 infection.
We next investigated the involvement of oxidative stress in the reduced response to IL-2 in memory CD4 T cells from HIV-1-infected subjects. We first measured ex vivo the expression of intracellular ROS in memory CD4 T cells from PHI, CHI, and HIVfree subjects. We showed that the levels of ROS expression measured in memory CD4 T cells from PHI and CHI subjects were significantly increased compared to those measured in memory CD4 T cells from HIVfree subjects (MFI, 2,225.2 ± 568.9, 2,344.5 ± 469, and 1,267.2 ± 237.7, respectively; P = 0.0022; n = 6) (Fig. 5A). All memory cell subsets, including TCM cells, TTM cells, and CD27− CCR7− effector memory CD4 T cells (TEM cells), from HIV-1-infected subjects displayed increased levels of ROS expression compared to the memory cell subsets from HIVfree donors (Fig. 5B). Correlations between the levels of ROS expression detected in memory CD4 T cells and the plasma Kyn level (Fig. 5C) or the level of IL-2-induced pSTAT5 expression (Fig. 5D) were observed for all subjects (P = 0.0022 and P < 0.0001, respectively; n = 18).
FIG 5.

Reduced IL-2 signaling in memory CD4 T cells during HIV-1 infection involves ROS production. (A) (Bottom) ROS levels in ex vivo memory CD4 T cells from PHI, CHI, and HIVfree subjects (n = 6 subjects in each group). (Top) Representative histograms, including histograms for H2O2 as a positive control. (B) ROS level in gated TCM, TTM, and TEM cell subsets for all subjects (n = 6 subjects in each group). (C and D) Correlation between ROS and plasma Kyn level (C) or IL-2-induced pSTAT5 levels in memory CD4 T cells (D) (n = 18). (E) PBMCs from PHI, CHI, and HIVfree subjects were first incubated or not incubated with NAC for 24 h and stimulated with IL-2 for 15 min, and then pSTAT5 levels in memory CD4 T cells were assessed (n = 6). *, 0.05 > P > 0.01; **, 0.01 > P > 0.001.
Finally, PBMCs from PHI, CHI, and HIVfree subjects were cultured for 24 h with NAC, stimulated with IL-2 for 15 min, and then collected to assess the level of IL-2-induced pSTAT5 in memory CD4 T cells. Pretreatment of memory CD4 T cells from HIV-1-infected subjects with NAC resulted in a significant improvement in the level of pSTAT5 expression (P = 0.0079 and 0.0317 for PHI and CHI subjects, respectively; n = 6), but the levels did not reach those for HIVfree subjects (Fig. 5E).
Together, these results showed that ROS are involved with the inability of memory CD4 T cells to optimally respond to IL-2 during HIV-1 infection.
ART improves IL-2 signaling in memory CD4 T cells by reducing ROS and Kyn levels.
In addition to improving the life expectancy of treated patients by controlling HIV-1 replication, ART inhibits the immunosuppressive tryptophan catabolism (22, 34, 45, 46). To test whether ART could restore the ability of memory CD4 T cells from PHI subjects to respond to IL-2, we collected plasma and PBMCs from HIV-1-infected subjects before treatment (pre-ART) and after 1 year of effective treatment (on ART). Figure 6A summarizes the clinical features of HIV-1-infected subjects pre-ART and on ART. First, we showed by analyzing plasma concentrations that ART was effective in reducing Kyn production in HIV-1-treated subjects (2.0 ± 0.5 μM and 1.3 ± 0.4 μM, respectively, for the pre- and on-ART conditions; P = 0.0317; n = 5) (Fig. 6B). Although the Kyn concentration appeared to be lower in plasma from HIVfree donors (0.61 μM ± 0.03), only a trend between Kyn levels from subjects on ART and uninfected subjects was noted (P = 0.1905; n = 5). We next determined the levels of ROS expression by memory CD4 T cells from HIVfree donors and subjects pre-ART and on ART. Whereas memory CD4 T cells from subjects pre-ART displayed significantly increased ROS levels compared to those of memory CD4 T cells from subjects on ART and HIVfree subjects (MFI, 2,128 ± 981.9, 933.8 ± 156.2, and 993.4 ± 147.1, respectively; P = 0.0079; n = 5), we found a complete suppression of oxidative stress in ART recipients (Fig. 6C). PBMCs from HIVfree subjects and subjects pre-ART and on ART were also stimulated for 15 min with IL-2 to determine pSTAT5 levels in memory CD4 T cells. Memory CD4 T cells from subjects pre-ART displayed lower pSTAT5 levels than memory CD4 T cells from subjects on ART and HIVfree subjects (P = 0.0079 for both comparisons; n = 5) (Fig. 6D). Once again, we found no difference between pSTAT5 levels in memory CD4 T cells from ART recipients and those from uninfected donors (P = 0.5476; n = 5). Importantly, we observed correlations between the plasma concentrations of Kyn and (i) ROS or (ii) IL-2-induced pSTAT5 levels with all study subjects (P = 0.002 and 0.014, respectively; n = 15) (Fig. 6E).
FIG 6.

ART restores proper IL-2 signaling in memory CD4 T cells by reducing ROS and Kyn levels. (A) Clinical features of 5 HIV-1-infected patients in each subject group at the pre- and on-ART longitudinal time points. d, number of days; undetectable#, the VL was <40 copies/ml. (B to D) Measurement of Kyn (B), ROS (C), and IL-2-induced pSTAT5 (D) levels for all subject groups. In panels A to D, data are for 5 subjects in each group. (E) Correlations between Kyn and ROS (i) or IL-2-induced pSTAT5 (ii) levels for subjects pre-ART, subjects on ART, and HIV-1free subjects (n = 15). *, 0.05 > P > 0.01; **, 0.01 > P > 0.001.
Collectively, these results provide evidence that 1 year of ART restored the response to IL-2 and reduced the levels of both Kyn production and oxidative stress in memory CD4 T cells from treated HIV-1-infected patients (Fig. 7).
FIG 7.
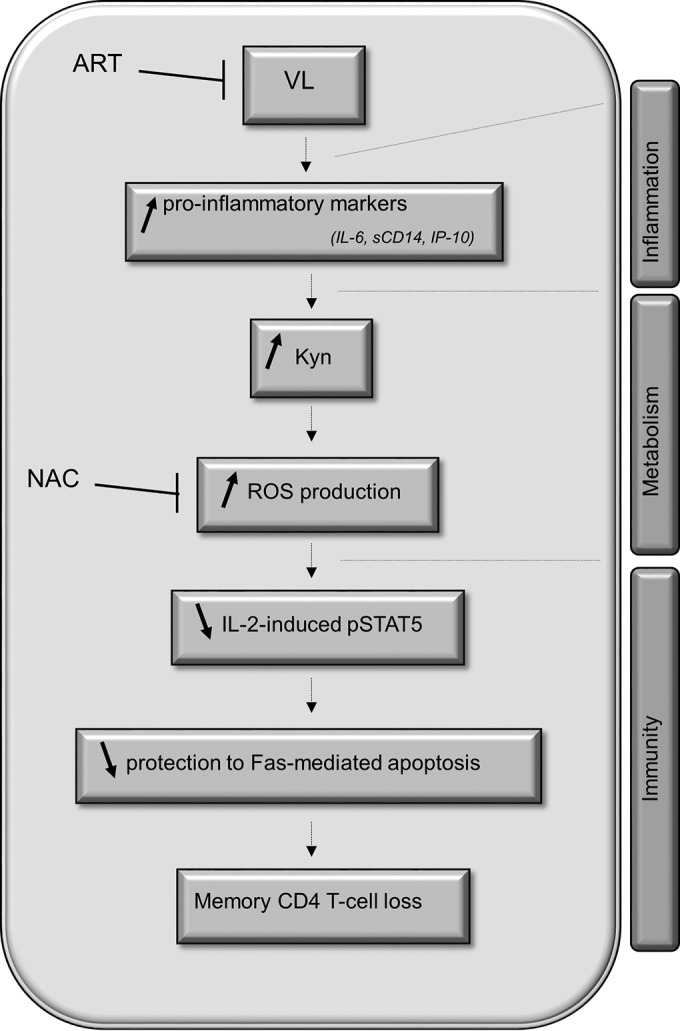
Interplay between inflammation, increased kynurenine levels, and reduced IL-2 signaling in memory CD4 T cells during HIV-1 infection.
DISCUSSION
Herein, we revealed a newly discovered metabolism-related mechanism responsible for inhibition of the IL-2 response and the subsequent loss of memory CD4 T cells during HIV-1 infection. Although we found no significant difference in the IL-7 and IL-15 responses in memory CD4 T cells from all subject groups, our results also showed that following cytokine stimulation, CD27− CCR7− TEM cells from HIVfree donors displayed lower pSTAT5 levels than those from PHI and CHI subjects, as previously reported (14) (P = 0.055 and 0.026, respectively, for IL-7 and P = 0.0087 and 0.0043, respectively, for IL-15).
Collectively, the ex vivo and in vitro data demonstrated that the enhanced production of Kyn was one of the molecular mechanisms associated with memory CD4 T-cell loss and interference with IL-2 signaling (Fig. 7). Tryptophan catabolism into Kyn is driven by the IDO-1 enzyme expressed by monocyte/macrophages and DCs. Several soluble factors associated with immune activation, including IFN-γ, tumor necrosis factor alpha, IL-1β, sCD40, IL-32, and Toll-like receptor ligands, are known to induce IDO-1 activity in these cells (42). Strain-dependent HIV-1 infection and Tat expression also lead to increased IDO-1 activity in innate immune cells (47). Interestingly, the in vitro assays using Kyn treatment indicated that a low 5 μM concentration was sufficient to reduce the ability of noninfected memory CD4 T cells to respond to IL-2 (Fig. 4A). Similar concentrations were previously reported in plasma from viremic subjects (22, 29).
The fact that NAC partially improved memory CD4 T-cell survival from Fas-mediated apoptosis demonstrates that ROS production is one of the mechanisms contributing to defective cell survival during HIV-1 infection. Similarly, previous observations showed that oxidative stress is responsible for the induction of apoptosis in several cell types, including natural killer cells, enterocytes, podocytes, and astrocytes, during HIV-1 infection (44, 48–50). Recent studies, such as those conducted by Kalinowska et al. (14) and Trautmann et al. (16), also showed that decreased IL-7-induced pSTAT5 levels in CD8 T cells during HIV-1 infection were associated with oxidative stress.
Kyn-induced defective IL-2 signaling in memory CD4 T cells during HIV-1 infection not only was mediated by elevated oxidative stress but also involved other ROS-independent mechanisms. Although in the present study the results of the in vitro assays using Kyn treatment implicated oxidative stress, the addition of NAC only partially restored IL-2 signaling and cell survival compared to the levels of IL-2 signaling and survival found for cells cultured in the absence of Kyn (Fig. 4D and E). These observations were further supported by similar results with memory CD4 T cells from PHI and CHI subjects and those from HIVfree donors (Fig. 5E). Finally, our data also showed that 1 μM H2O2 treatment resulted in similar levels of ROS production following IL-2 treatment and Kyn treatment (Fig. 4B) but led to lower levels of reduction of pSTAT5 levels following IL-2 treatment than Kyn treatment (Fig. 4C and D). Collectively, such results indicate underlying mechanisms which are Kyn dependent but ROS independent. A previous study demonstrated that the activation of the aryl hydrocarbon receptor, which is triggered by several agonists, including Kyn (51), interferes with the activation of STAT5 by IL-2 through direct binding with the STAT protein (52). Therefore, we cannot exclude the possibility of the involvement of the aryl hydrocarbon receptor in reducing IL-2 signaling in memory CD4 T cells as an additional mechanism which does not necessarily involve oxidative stress.
Our data also indicate that the loss of memory CD4 T cells and interference with IL-2 signaling during HIV-1 infection depend on other Kyn-independent mechanisms. In agreement with findings reported in the literature, we demonstrated that the diminished STAT5 phosphorylation found during the early phase of HIV-1 infection correlated not only with enhanced ROS and Kyn levels but also with enhanced levels of several inflammation markers, such as IL-6, sCD14, and IP-10 (Fig. 3A to C) (22, 25, 29). Therefore, we cannot exclude the possibility that other mechanisms, which could involve the release of intermediate molecules such as IFN type I and other proinflammatory markers by monocyte/macrophages and DCs, are at play in our observations. This is particularly true considering that previous reports have indicated that the IFN-α produced by plasmacytoid DCs interferes with the ability of CD4 T cells to respond to IL-2 and survive (53, 54). Novel discoveries on the interplay between inflammation and viral persistence shed new light on the key role of the inflammasome, especially for CD4 T-cell loss, during HIV-1 infection (1, 24, 55). The inflammasome is a key signaling platform that detects ROS, pathogen-associated molecular patterns, and metabolic perturbations (56–58). Inflammasome activation accounts for the majority of CD4 T-cell death by pyroptosis in HIV-1-infected lymphoid tissues (1, 2, 59). In fact, the results of studies with macaques indicate that inflammasome activation represents one of the earliest events which follow mucosal simian immunodeficiency virus infection (60). Interestingly, Sagulenko et al. recently demonstrated that the inflammasome activates both apoptotic and pyroptotic cell death, whose balance depended on the amounts of stimuli (61). Therefore, since the results implicate both inflammation and ROS in the loss of memory CD4 T cells in HIV-1-infected patients, inflammasome activation could represent an additional mechanism at play in our study.
Gut damage and the subsequent microbial translocation from tissue to the periphery have also been recognized to be major contributors to HIV-1-related inflammation (21, 62). Results from a study by Gaardbo et al. showed that microbe-related effects in the gut are associated with CD4 T-cell loss during HIV-1 infection (35). The dysbiosis that occurs in the gut of HIV-1-infected subjects persisting on ART describes the imbalance of bacterial diversity that is illustrated by increased proinflammatory pathogenic strains (63, 64). Importantly, the enhanced tryptophan metabolism related to the composition of the gut microbiota has been linked to the disease progression contributed by enhanced Kyn production in the gut (65). Conversely, the administration of probiotic bacteria in HIV-1-infected subjects transiently increases the CD4 T-cell count and reduces gut inflammation (66, 67). Therefore, because of the accumulation of Kyn, the gut microbial imbalance could interfere with memory CD4 T-cell survival.
Finally, our data reinforce the notion that the early initiation of ART could limit metabolic and CD4 T-cell immune dysfunction. Despite relatively low HIV-1-specific CD8 T-cell responses, recent observations demonstrate a beneficial effect of early initiation of ART in preserving mucosal CD4 T cells, possibly limiting the release of microbial products associated with immune activation (68–70). It is important to note that the complete reestablishment of IDO-1 activity occurs only when ART is initiated early, in contrast to the partial normalization that occurs when ART is initiated during the chronic phase of infection (34, 45, 46). Accordingly, we also found a drastic suppression of the Kyn-dependent inhibition of the IL-2 response in memory CD4 T cells after 1 year of ART when ART was initiated during the first months of HIV-1 infection (initiation at 124.6 ± 106.4 days postinfection) (Fig. 6). However, in ART recipients, Kyn levels were not fully normalized, while the oxidative stress and IL-2-induced pSTAT5 levels were similar to those seen in HIVfree subjects (Fig. 6B to D). These data strongly suggest that the reduced memory CD4 T-cell survival occurring during HIV-1 infection depends on multiple parameters, including Kyn-independent mechanisms.
In conclusion, our study provides another piece of evidence indicating that the enhanced metabolic activity occurring at the onset of primary infection plays a key role in interfering with the host molecular signaling involved in the survival of memory CD4 T cells. This information should be considered in the development of therapeutic strategies.
ACKNOWLEDGMENTS
We are grateful to all subjects participating in this study, their physicians, and attending staff members from the Réseau SIDA/Maladies Infectieuses of the Fonds de la Recherche Québec-Santé (FRQ-S; Montreal, QC, Canada). We thank Danica Albert, Angie Massicotte, Natacha Cotta-Grand, and Mario Legault for technical and administrative assistance. We also thank Yu She and Zhong He for generating several sets of flow cytometry data, including pSTAT5 measures, for all groups of study subjects.
Funding Statement
This work was supported in part by the Fonds de la Recherche Québec-Santé (FRQ-S): Réseau SIDA/Maladies infectieuses and Thérapie cellulaire, the Canadian Institutes of Health Research (CIHR) (grants MOP 103230 and CTN 257), and the Canadian HIV Cure Enterprise (CanCURE) (Team Grant HIG-133050). CanCURE is a partnership of the CIHR, the Canadian Foundation for AIDS Research (CANFAR), and the International AIDS Society (IAS). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, Hunt PW, Hatano H, Sowinski S, Munoz-Arias I, Greene WC. 2014. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 505:509–514. doi: 10.1038/nature12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galloway NL, Doitsh G, Monroe KM, Yang Z, Munoz-Arias I, Levy DN, Greene WC. 2015. Cell-to-cell transmission of HIV-1 is required to trigger pyroptotic death of lymphoid-tissue-derived CD4 T cells. Cell Rep 12:1555–1563. doi: 10.1016/j.celrep.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Joshi A, Sedano M, Beauchamp B, Punke EB, Mulla ZD, Meza A, Alozie OK, Mukherjee D, Garg H. 2016. HIV-1 Env glycoprotein phenotype along with immune activation determines CD4 T cell loss in HIV patients. J Immunol 196:1768–1779. doi: 10.4049/jimmunol.1501588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Massanella M, Curriu M, Carrillo J, Gomez E, Puig J, Navarro J, Dalmau J, Martinez-Picado J, Crespo M, Cabrera C, Negredo E, Clotet B, Blanco J. 2013. Assessing main death pathways in T lymphocytes from HIV infected individuals. Cytometry A 83:648–658. doi: 10.1002/cyto.a.22299. [DOI] [PubMed] [Google Scholar]
- 5.Douek DC, Brenchley JM, Betts MR, Ambrozak DR, Hill BJ, Okamoto Y, Casazza JP, Kuruppu J, Kunstman K, Wolinsky S, Grossman Z, Dybul M, Oxenius A, Price DA, Connors M, Koup RA. 2002. HIV preferentially infects HIV-specific CD4+ T cells. Nature 417:95–98. doi: 10.1038/417095a. [DOI] [PubMed] [Google Scholar]
- 6.Sun Y, Permar SR, Buzby AP, Letvin NL. 2007. Memory CD4+ T-lymphocyte loss and dysfunction during primary simian immunodeficiency virus infection. J Virol 81:8009–8015. doi: 10.1128/JVI.00482-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yates A, Stark J, Klein N, Antia R, Callard R. 2007. Understanding the slow depletion of memory CD4+ T cells in HIV infection. PLoS Med 4:e177. doi: 10.1371/journal.pmed.0040177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Grevenynghe J, Procopio FA, He Z, Chomont N, Riou C, Zhang Y, Gimmig S, Boucher G, Wilkinson P, Shi Y, Yassine-Diab B, Said EA, Trautmann L, El Far M, Balderas RS, Boulassel MR, Routy JP, Haddad EK, Sekaly RP. 2008. Transcription factor FOXO3a controls the persistence of memory CD4(+) T cells during HIV infection. Nat Med 14:266–274. doi: 10.1038/nm1728. [DOI] [PubMed] [Google Scholar]
- 9.Jaleco S, Swainson L, Dardalhon V, Burjanadze M, Kinet S, Taylor N. 2003. Homeostasis of naive and memory CD4+ T cells: IL-2 and IL-7 differentially regulate the balance between proliferation and Fas-mediated apoptosis. J Immunol 171:61–68. doi: 10.4049/jimmunol.171.1.61. [DOI] [PubMed] [Google Scholar]
- 10.Leone A, Picker LJ, Sodora DL. 2009. IL-2, IL-7 and IL-15 as immuno-modulators during SIV/HIV vaccination and treatment. Curr HIV Res 7:83–90. doi: 10.2174/157016209787048519. [DOI] [PubMed] [Google Scholar]
- 11.Riou C, Yassine-Diab B, Van Grevenynghe J, Somogyi R, Greller LD, Gagnon D, Gimmig S, Wilkinson P, Shi Y, Cameron MJ, Campos-Gonzalez R, Balderas RS, Kelvin D, Sekaly RP, Haddad EK. 2007. Convergence of TCR and cytokine signaling leads to FOXO3a phosphorylation and drives the survival of CD4+ central memory T cells. J Exp Med 204:79–91. doi: 10.1084/jem.20061681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schluns KS, Lefrancois L. 2003. Cytokine control of memory T-cell development and survival. Nat Rev Immunol 3:269–279. doi: 10.1038/nri1052. [DOI] [PubMed] [Google Scholar]
- 13.Bazdar DA, Kalinowska M, Sieg SF. 2009. Interleukin-7 receptor signaling is deficient in CD4+ T cells from HIV-infected persons and is inversely associated with aging. J Infect Dis 199:1019–1028. doi: 10.1086/597210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalinowska M, Bazdar DA, Lederman MM, Funderburg N, Sieg SF. 2013. Decreased IL-7 responsiveness is related to oxidative stress in HIV disease. PLoS One 8:e58764. doi: 10.1371/journal.pone.0058764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kryworuchko M, Pasquier V, Keller H, David D, Goujard C, Gilquin J, Viard JP, Joussemet M, Delfraissy JF, Theze J. 2004. Defective interleukin-2-dependent STAT5 signalling in CD8 T lymphocytes from HIV-positive patients: restoration by antiretroviral therapy. AIDS 18:421–426. doi: 10.1097/00002030-200402200-00007. [DOI] [PubMed] [Google Scholar]
- 16.Trautmann L, Mbitikon-Kobo FM, Goulet JP, Peretz Y, Shi Y, Van Grevenynghe J, Procopio FA, Boulassel MR, Routy JP, Chomont N, Haddad EK, Sekaly RP. 2012. Profound metabolic, functional, and cytolytic differences characterize HIV-specific CD8 T cells in primary and chronic HIV infection. Blood 120:3466–3477. doi: 10.1182/blood-2012-04-422550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Biancotto A, Grivel JC, Iglehart SJ, Vanpouille C, Lisco A, Sieg SF, Debernardo R, Garate K, Rodriguez B, Margolis LB, Lederman MM. 2007. Abnormal activation and cytokine spectra in lymph nodes of people chronically infected with HIV-1. Blood 109:4272–4279. doi: 10.1182/blood-2006-11-055764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Breen EC. 2002. Pro- and anti-inflammatory cytokines in human immunodeficiency virus infection and acquired immunodeficiency syndrome. Pharmacol Ther 95:295–304. doi: 10.1016/S0163-7258(02)00263-2. [DOI] [PubMed] [Google Scholar]
- 19.Hamlyn E, Fidler S, Stohr W, Cooper DA, Tambussi G, Schechter M, Miro JM, McClure M, Weber J, Babiker A, Porter K. 2014. Interleukin-6 and D-dimer levels at seroconversion as predictors of HIV-1 disease progression. AIDS 28:869–874. doi: 10.1097/QAD.0000000000000155. [DOI] [PubMed] [Google Scholar]
- 20.Ananworanich J, Schuetz A, Vandergeeten C, Sereti I, de Souza M, Rerknimitr R, Dewar R, Marovich M, van Griensven F, Sekaly R, Pinyakorn S, Phanuphak N, Trichavaroj R, Rutvisuttinunt W, Chomchey N, Paris R, Peel S, Valcour V, Maldarelli F, Chomont N, Michael N, Phanuphak P, Kim JH. 2012. Impact of multi-targeted antiretroviral treatment on gut T cell depletion and HIV reservoir seeding during acute HIV infection. PLoS One 7:e33948. doi: 10.1371/journal.pone.0033948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, Blazar BR, Rodriguez B, Teixeira-Johnson L, Landay A, Martin JN, Hecht FM, Picker LJ, Lederman MM, Deeks SG, Douek DC. 2006. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med 12:1365–1371. [DOI] [PubMed] [Google Scholar]
- 22.Jenabian MA, El-Far M, Vyboh K, Kema I, Costiniuk CT, Thomas R, Baril JG, LeBlanc R, Kanagaratham C, Radzioch D, Allam O, Ahmad A, Lebouche B, Tremblay C, Ancuta P, Routy JP. 2015. Immunosuppressive tryptophan catabolism and gut mucosal dysfunction following early HIV infection. J Infect Dis 212:355–366. doi: 10.1093/infdis/jiv037. [DOI] [PubMed] [Google Scholar]
- 23.Vinikoor MJ, Cope A, Gay CL, Ferrari G, McGee KS, Kuruc JD, Lennox JL, Margolis DM, Hicks CB, Eron JJ. 2013. Antiretroviral therapy initiated during acute HIV infection fails to prevent persistent T-cell activation. J Acquir Immune Defic Syndr 62:505–508. doi: 10.1097/QAI.0b013e318285cd33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aounallah M, Dagenais-Lussier X, El-Far M, Mehraj V, Jenabian MA, Routy JP, van Grevenynghe J. 2016. Current topics in HIV pathogenesis. Part 2. Inflammation drives a Warburg-like effect on the metabolism of HIV-infected subjects. Cytokine Growth Factor Rev 28:1–10. doi: 10.1016/j.cytogfr.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 25.Favre D, Mold J, Hunt PW, Kanwar B, Loke P, Seu L, Barbour JD, Lowe MM, Jayawardene A, Aweeka F, Huang Y, Douek DC, Brenchley JM, Martin JN, Hecht FM, Deeks SG, McCune JM. 2010. Tryptophan catabolism by indoleamine 2,3-dioxygenase 1 alters the balance of TH17 to regulatory T cells in HIV disease. Sci Transl Med 2:32ra36. doi: 10.1126/scitranslmed.3000632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pedersen KK, Pedersen M, Troseid M, Gaardbo JC, Lund TT, Thomsen C, Gerstoft J, Kvale D, Nielsen SD. 2013. Microbial translocation in HIV infection is associated with dyslipidemia, insulin resistance, and risk of myocardial infarction. J Acquir Immune Defic Syndr 64:425–433. doi: 10.1097/QAI.0b013e31829f919d. [DOI] [PubMed] [Google Scholar]
- 27.Zidar DA, Juchnowski S, Ferrari B, Clagett B, Pilch-Cooper HA, Rose S, Rodriguez B, McComsey GA, Sieg SF, Mehta NN, Lederman MM, Funderburg NT. 2015. Oxidized LDL levels are increased in HIV infection and may drive monocyte activation. J Acquir Immune Defic Syndr 69:154–160. doi: 10.1097/QAI.0000000000000566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huengsberg M, Winer JB, Gompels M, Round R, Ross J, Shahmanesh M. 1998. Serum kynurenine-to-tryptophan ratio increases with progressive disease in HIV-infected patients. Clin Chem 44:858–862. [PubMed] [Google Scholar]
- 29.Jenabian MA, Patel M, Kema I, Kanagaratham C, Radzioch D, Thebault P, Lapointe R, Tremblay C, Gilmore N, Ancuta P, Routy JP. 2013. Distinct tryptophan catabolism and Th17/Treg balance in HIV progressors and elite controllers. PLoS One 8:e78146. doi: 10.1371/journal.pone.0078146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maneglier B, Malleret B, Guillemin GJ, Spreux-Varoquaux O, Devillier P, Rogez-Kreuz C, Porcheray F, Therond P, Dormont D, Clayette P. 2009. Modulation of indoleamine-2,3-dioxygenase expression and activity by HIV-1 in human macrophages. Fundam Clin Pharmacol 23:573–581. doi: 10.1111/j.1472-8206.2009.00703.x. [DOI] [PubMed] [Google Scholar]
- 31.Fuchs D, Moller AA, Reibnegger G, Werner ER, Werner-Felmayer G, Dierich MP, Wachter H. 1991. Increased endogenous interferon-gamma and neopterin correlate with increased degradation of tryptophan in human immunodeficiency virus type 1 infection. Immunol Lett 28:207–211. doi: 10.1016/0165-2478(91)90005-U. [DOI] [PubMed] [Google Scholar]
- 32.Jenabian MA, Patel M, Kema I, Vyboh K, Kanagaratham C, Radzioch D, Thebault P, Lapointe R, Gilmore N, Ancuta P, Tremblay C, Routy JP. 2014. Soluble CD40-ligand (sCD40L, sCD154) plays an immunosuppressive role via regulatory T cell expansion in HIV infection. Clin Exp Immunol 178:102–111. doi: 10.1111/cei.12396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bipath P, Levay PF, Viljoen M. 2015. The kynurenine pathway activities in a sub-Saharan HIV/AIDS population. BMC Infect Dis 15:346. doi: 10.1186/s12879-015-1087-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen J, Shao J, Cai R, Shen Y, Zhang R, Liu L, Qi T, Lu H. 2014. Anti-retroviral therapy decreases but does not normalize indoleamine 2,3-dioxygenase activity in HIV-infected patients. PLoS One 9:e100446. doi: 10.1371/journal.pone.0100446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gaardbo JC, Troseid M, Stiksrud B, Midttun O, Ueland PM, Ullum H, Nielsen SD. 11 July 2015. Increased tryptophan catabolism is associated with increased frequency of CD161+ Tc17/MAIT cells, and lower CD4+ T cell count in HIV-1 infected patients on cART after two years of follow-up. J Acquir Immune Defic Syndr. doi: 10.1097/QAI.0000000000000758. [DOI] [PubMed] [Google Scholar]
- 36.Page EE, Greathead L, Metcalf R, Clark SA, Hart M, Fuchs D, Pantelidis P, Gotch F, Pozniak A, Nelson M, Boasso A, Gazzard B, Kelleher P. 2014. Loss of Th22 cells is associated with increased immune activation and IDO-1 activity in HIV-1 infection. J Acquir Immune Defic Syndr 67:227–235. doi: 10.1097/QAI.0000000000000294. [DOI] [PubMed] [Google Scholar]
- 37.Boasso A, Herbeuval JP, Hardy AW, Anderson SA, Dolan MJ, Fuchs D, Shearer GM. 2007. HIV inhibits CD4+ T-cell proliferation by inducing indoleamine 2,3-dioxygenase in plasmacytoid dendritic cells. Blood 109:3351–3359. doi: 10.1182/blood-2006-07-034785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Planes R, Bahraoui E. 2013. HIV-1 Tat protein induces the production of IDO in human monocyte derived-dendritic cells through a direct mechanism: effect on T cells proliferation. PLoS One 8:e74551. doi: 10.1371/journal.pone.0074551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Grevenynghe J, Cubas RA, Noto A, DaFonseca S, He Z, Peretz Y, Filali-Mouhim A, Dupuy FP, Procopio FA, Chomont N, Balderas RS, Said EA, Boulassel MR, Tremblay CL, Routy JP, Sekaly RP, Haddad EK. 2011. Loss of memory B cells during chronic HIV infection is driven by Foxo3a- and TRAIL-mediated apoptosis. J Clin Invest 121:3877–3888. doi: 10.1172/JCI59211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eruslanov E, Kusmartsev S. 2010. Identification of ROS using oxidized DCFDA and flow-cytometry. Methods Mol Biol 594:57–72. doi: 10.1007/978-1-60761-411-1_4. [DOI] [PubMed] [Google Scholar]
- 41.Fritsch RD, Shen X, Sims GP, Hathcock KS, Hodes RJ, Lipsky PE. 2005. Stepwise differentiation of CD4 memory T cells defined by expression of CCR7 and CD27. J Immunol 175:6489–6497. doi: 10.4049/jimmunol.175.10.6489. [DOI] [PubMed] [Google Scholar]
- 42.Dagenais-Lussier X, Mouna A, Routy JP, Tremblay C, Sekaly RP, El-Far M, van Grevenynghe J. 2015. Current topics in HIV-1 pathogenesis: the emergence of deregulated immuno-metabolism in HIV-infected subjects. Cytokine Growth Factor Rev 26:603–613. doi: 10.1016/j.cytogfr.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 43.Decrion AZ, Dichamp I, Varin A, Herbein G. 2005. HIV and inflammation. Curr HIV Res 3:243–259. doi: 10.2174/1570162054368057. [DOI] [PubMed] [Google Scholar]
- 44.Song H, Park H, Kim YS, Kim KD, Lee HK, Cho DH, Yang JW, Hur DY. 2011. l-Kynurenine-induced apoptosis in human NK cells is mediated by reactive oxygen species. Int Immunopharmacol 11:932–938. doi: 10.1016/j.intimp.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 45.Neurauter G, Zangerle R, Widner B, Quirchmair G, Sarcletti M, Fuchs D. 2003. Effective antiretroviral therapy reduces degradation of tryptophan in patients with HIV-1 infection. Adv Exp Med Biol 527:317–323. doi: 10.1007/978-1-4615-0135-0_35. [DOI] [PubMed] [Google Scholar]
- 46.Zangerle R, Widner B, Quirchmair G, Neurauter G, Sarcletti M, Fuchs D. 2002. Effective antiretroviral therapy reduces degradation of tryptophan in patients with HIV-1 infection. Clin Immunol 104:242–247. doi: 10.1006/clim.2002.5231. [DOI] [PubMed] [Google Scholar]
- 47.Samikkannu T, Saiyed ZM, Rao KV, Babu DK, Rodriguez JW, Papuashvili MN, Nair MP. 2009. Differential regulation of indoleamine-2,3-dioxygenase (IDO) by HIV type 1 clade B and C Tat protein. AIDS Res Hum Retroviruses 25:329–335. doi: 10.1089/aid.2008.0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Buccigrossi V, Laudiero G, Nicastro E, Miele E, Esposito F, Guarino A. 2011. The HIV-1 transactivator factor (Tat) induces enterocyte apoptosis through a redox-mediated mechanism. PLoS One 6:e29436. doi: 10.1371/journal.pone.0029436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pollicita M, Muscoli C, Sgura A, Biasin A, Granato T, Masuelli L, Mollace V, Tanzarella C, Del Duca C, Rodino P, Perno CF, Aquaro S. 2009. Apoptosis and telomeres shortening related to HIV-1 induced oxidative stress in an astrocytoma cell line. BMC Neurosci 10:51. doi: 10.1186/1471-2202-10-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reshi ML, Su YC, Hong JR. 2014. RNA viruses: ROS-mediated cell death. Int J Cell Biol 2014:467452. doi: 10.1155/2014/467452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. 2010. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol 185:3190–3198. doi: 10.4049/jimmunol.0903670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kimura A, Naka T, Nohara K, Fujii-Kuriyama Y, Kishimoto T. 2008. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc Natl Acad Sci U S A 105:9721–9726. doi: 10.1073/pnas.0804231105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Herbeuval JP, Hardy AW, Boasso A, Anderson SA, Dolan MJ, Dy M, Shearer GM. 2005. Regulation of TNF-related apoptosis-inducing ligand on primary CD4+ T cells by HIV-1: role of type I IFN-producing plasmacytoid dendritic cells. Proc Natl Acad Sci U S A 102:13974–13979. doi: 10.1073/pnas.0505251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nguyen TP, Bazdar DA, Mudd JC, Lederman MM, Harding CV, Hardy GA, Sieg SF. 2015. Interferon-alpha inhibits CD4 T cell responses to interleukin-7 and interleukin-2 and selectively interferes with Akt signaling. J Leukoc Biol 97:1139–1146. doi: 10.1189/jlb.4A0714-345RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guo H, Gao J, Taxman DJ, Ting JP, Su L. 2014. HIV-1 infection induces interleukin-1beta production via TLR8 protein-dependent and NLRP3 inflammasome mechanisms in human monocytes. J Biol Chem 289:21716–21726. doi: 10.1074/jbc.M114.566620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lamkanfi M, Dixit VM. 2009. The inflammasomes. PLoS Pathog 5:e1000510. doi: 10.1371/journal.ppat.1000510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martinon F. 2012. Dangerous liaisons: mitochondrial DNA meets the NLRP3 inflammasome. Immunity 36:313–315. doi: 10.1016/j.immuni.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 58.McGettrick AF, O'Neill LA. 2013. How metabolism generates signals during innate immunity and inflammation. J Biol Chem 288:22893–22898. doi: 10.1074/jbc.R113.486464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Monroe KM, Yang Z, Johnson JR, Geng X, Doitsh G, Krogan NJ, Greene WC. 2014. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science 343:428–432. doi: 10.1126/science.1243640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barouch DH, Ghneim K, Bosche WJ, Li Y, Berkemeier B, Hull M, Bhattacharyya S, Cameron M, Liu J, Smith K, Borducchi E, Cabral C, Peter L, Brinkman A, Shetty M, Li H, Gittens C, Baker C, Wagner W, Lewis MG, Colantonio A, Kang HJ, Li W, Lifson JD, Piatak M Jr, Sekaly RP. 2016. Rapid inflammasome activation following mucosal SIV infection of rhesus monkeys. Cell 165:656–667. doi: 10.1016/j.cell.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sagulenko V, Thygesen SJ, Sester DP, Idris A, Cridland JA, Vajjhala PR, Roberts TL, Schroder K, Vince JE, Hill JM, Silke J, Stacey KJ. 2013. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ 20:1149–1160. doi: 10.1038/cdd.2013.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Somsouk M, Estes JD, Deleage C, Dunham RM, Albright R, Inadomi JM, Martin JN, Deeks SG, McCune JM, Hunt PW. 2015. Gut epithelial barrier and systemic inflammation during chronic HIV infection. AIDS 29:43–51. doi: 10.1097/QAD.0000000000000511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mutlu EA, Keshavarzian A, Losurdo J, Swanson G, Siewe B, Forsyth C, French A, Demarais P, Sun Y, Koenig L, Cox S, Engen P, Chakradeo P, Abbasi R, Gorenz A, Burns C, Landay A. 2014. A compositional look at the human gastrointestinal microbiome and immune activation parameters in HIV infected subjects. PLoS Pathog 10:e1003829. doi: 10.1371/journal.ppat.1003829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nowak P, Troseid M, Avershina E, Barqasho B, Neogi U, Holm K, Hov JR, Noyan K, Vesterbacka J, Svard J, Rudi K, Sonnerborg A. 2015. Gut microbiota diversity predicts immune status in HIV-1 infection. AIDS 29:2409–2418. doi: 10.1097/QAD.0000000000000869. [DOI] [PubMed] [Google Scholar]
- 65.Vujkovic-Cvijin I, Dunham RM, Iwai S, Maher MC, Albright RG, Broadhurst MJ, Hernandez RD, Lederman MM, Huang Y, Somsouk M, Deeks SG, Hunt PW, Lynch SV, McCune JM. 2013. Dysbiosis of the gut microbiota is associated with HIV disease progression and tryptophan catabolism. Sci Transl Med 5:193ra191. doi: 10.1126/scitranslmed.3006438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cunningham-Rundles S, Ahrne S, Johann-Liang R, Abuav R, Dunn-Navarra AM, Grassey C, Bengmark S, Cervia JS. 2011. Effect of probiotic bacteria on microbial host defense, growth, and immune function in human immunodeficiency virus type-1 infection. Nutrients 3:1042–1070. doi: 10.3390/nu3121042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Monachese M, Cunningham-Rundles S, Diaz MA, Guerrant R, Hummelen R, Kemperman R, Kerac M, Kort R, Merenstein D, Panigrahi P, Ramakrishna B, Safdar N, Shane A, Trois L, Reid G. 2011. Probiotics and prebiotics to combat enteric infections and HIV in the developing world: a consensus report. Gut Microbes 2:198–207. doi: 10.4161/gmic.2.3.16106. [DOI] [PubMed] [Google Scholar]
- 68.Krebs SJ, Ananworanich J. 2015. Immune activation during acute HIV infection and the impact of early antiretroviral therapy. Curr Opin HIV AIDS 11:163–172. doi: 10.1097/COH.0000000000000228. [DOI] [PubMed] [Google Scholar]
- 69.Macatangay BJ, Rinaldo CR. 2015. Preserving HIV-specific T cell responses: does timing of antiretroviral therapy help? Curr Opin HIV AIDS 10:55–60. doi: 10.1097/COH.0000000000000124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schuetz A, Deleage C, Sereti I, Rerknimitr R, Phanuphak N, Phuang-Ngern Y, Estes JD, Sandler NG, Sukhumvittaya S, Marovich M, Jongrakthaitae S, Akapirat S, Fletscher JL, Kroon E, Dewar R, Trichavaroj R, Chomchey N, Douek DC, O'Connell RJ, Ngauy V, Robb ML, Phanuphak P, Michael NL, Excler JL, Kim JH, de Souza MS, Ananworanich J, RV254/SEARCH 010 and RV304/SEARCH 013 Study Groups. 2014. Initiation of ART during early acute HIV infection preserves mucosal Th17 function and reverses HIV-related immune activation. PLoS Pathog 10:e1004543. doi: 10.1371/journal.ppat.1004543. [DOI] [PMC free article] [PubMed] [Google Scholar]