

ABSTRACT
Human cytomegalovirus (HCMV) is an enveloped double-stranded DNA virus that causes severe disease in newborns and immunocompromised patients. During infection, the host cell endosecretory system is remodeled to form the cytoplasmic virion assembly complex (cVAC). We and others previously identified the conserved, multifunctional HCMV virion tegument protein pUL103 as important for cVAC biogenesis and efficient secondary envelopment. To help define its mechanisms of action and predict additional functions, we used two complementary methods, coimmunoprecipitation (co-IP) and proximity biotinylation (BioID), to identify viral and cellular proteins that interact with pUL103. By using the two methods in parallel and applying stringent selection criteria, we identified potentially high-value interactions of pUL103 with 13 HCMV and 18 cellular proteins. Detection of the previously identified pUL103-pUL71 interaction, as well as verification of several interactions by reverse co-IP, supports the specificity of our screening process. As might be expected for a tegument protein, interactions were identified that suggest distinct roles for pUL103 across the arc of lytic infection, including interactions with proteins involved in cellular antiviral responses, nuclear activities, and biogenesis and transport of cytoplasmic vesicles. Further analysis of some of these interactions expands our understanding of the multifunctional repertoire of pUL103: we detected HCMV pUL103 in nuclei of infected cells and identified an ALIX-binding domain within the pUL103 sequence.
IMPORTANCE Human cytomegalovirus (HCMV) is able to reconfigure the host cell machinery to establish a virion production factory, the cytoplasmic virion assembly complex (cVAC). cVAC biogenesis and operation represent targets for development of novel HCMV antivirals. We previously showed that the HCMV tegument protein pUL103 is required for cVAC biogenesis. Using pUL103 as bait, we investigated viral and cellular protein-protein interactions to identify and understand the range of pUL103 functions. We found that pUL103 interacts with cellular antiviral defense systems and proteins involved in organelle biogenesis and transport of cytoplasmic vesicles and is present in infected cell nuclei. These results expand our understanding of the functional repertoire of pUL103 to include activities that extend from the earliest stages of infection through virion assembly and egress.
INTRODUCTION
Human cytomegalovirus (HCMV) induces a wide range of profound modifications of host cell biology, including changes in cell morphology, cell cycle, metabolism, intrinsic and innate immunity, and the endosecretory machinery. Alterations in the endosecretory machinery include remodeling of the Golgi and early endosomal compartments to form the cytoplasmic virion assembly compartment (cVAC), where the mature tegument is acquired, secondary envelopment occurs, and vesicles containing mature virions are transported to the plasma membrane for release (1–3).
cVAC biogenesis is contingent on the expression of specific HCMV microRNAs (miRNAs) (4) and late genes. We previously identified pUL48, pUL94, and pUL103 as important for cVAC biogenesis (5); when either pUL48 or pUL103 is deliberately degraded during infection, the cVAC fails to form properly. pUL103 is a virion tegument protein that is conserved across the Herpesviridae (herpes simplex virus [HSV] UL7 homolog, Pfam accession no. PF01677) (Table 1). pUL103 and its homologs are important for efficient production of infectious virions at low and high multiplicities of infection (MOI). Ahlqvist and Mocarski, as well as our lab, have identified roles for pUL103 in cell-to-cell spread, virion envelopment, and egress (5, 6). The mechanisms employed by HCMV pUL103 and its homologs remain elusive.
TABLE 1.
Properties of HCMV pUL103 and its alphaherpesvirus UL7 homologs
| Virus | Expression kinetics | Size (aa) | Properties |
Interactions | Reference(s) | |
|---|---|---|---|---|---|---|
| Deletion | Localization | |||||
| Herpes simplex virus 1 (HSV-1) | Delayed early or early-late | 296 | Essential or decrease in viral titers (up to 3 logs), 50% reduction in plaque size | Nuclear and juxtanuclear, present in mature virions | ANT2, pUL51 (homolog of HCMV pUL71) | 9, 10, 40 |
| Herpes simplex virus 2 (HSV-2) | Late | 296 | NTa | Nuclear and juxtanuclear, present in mature virions | A, B, and C capsids (weak) | 17 |
| Pseudorabies virus (PRV) | Delayed early or early-late | 266 | Decrease in viral titers (up to 0.5 log), 60% reduction in plaque size, inefficient secondary envelopment, and delay in neuroinvasion | NT | NT | 41, 42 |
| Bovine herpesvirus 1 (BHV-1) | Early | 300 | Reduced viral titers | Cytoplasmic, not detected in mature virions | NT | 43 |
| Human cytomegalovirus (HCMV) | Early-late | 250 | Decreased viral titers (up to 3.5 logs), 60% reduction in plaque size, delayed secondary envelopment and release of dense bodies and virions | Juxtanuclear, overlap with Golgi markers, present in mature virions | pUL103, pUL22A, pUL48N, pUL71 | 5–7 |
NT, not tested.
Identification of protein-protein interactions can provide insights into the activities of pUL103 during infection. Four HCMV proteins (pUL22, pUL48, pUL71, and pUL103) were previously identified as having interactions with pUL103: in a yeast two-hybrid analysis, interactions were detected between pUL103 and itself, pUL22A, and pUL48N (7), and an interaction between pUL103 and pUL71 was identified by coimmunoprecipitation (co-IP) and bimolecular fluorescence complementation (8). However, none of these interactions was confirmed in the context of HCMV infection. During HSV-1 infection, proper localization and incorporation into the virion of pUL7 (the HSV pUL103 homolog) is dependent on its interaction with pUL51 (homolog of HCMV pUL71) (9). In addition, HSV-1 pUL7 associates with the mitochondrial protein adenine nucleotide translocator (ANT2), an interaction of uncertain significance (10). Viral and cellular interaction partners of pUL103 have not been studied in HCMV-infected cells, leaving a void in understanding its biological roles and mechanisms of action.
To address this gap, we employed a set of complementary proteomics approaches, co-IP and proximity biotinylation (BioID system), to identify viral and cellular proteins that interact with pUL103. Co-IP and the BioID system are well suited for detection of interactions that are strong and direct. The BioID system also enables detection of weak, indirect, and transient interactions. The complementary differences in these methods provide internal controls that reduce the number of false positives; interactions that score highly in both approaches are more likely to be specific. Using these methods, we identified multiple novel interactions between pUL103 and other viral and cellular proteins. As might be expected for a virion tegument protein, pUL103 interacted with several cellular antiviral proteins, including IFI16. It also interacts with DNA-associated proteins, is present in nuclei, and binds to ALIX (a component of the ESCRT machinery) via a sequence similar to the “late” (L) domains by which retrovirus gag proteins bind to ALIX during envelopment.
MATERIALS AND METHODS
Cell culture and virus stocks.
Human foreskin fibroblasts (HFFs) below passage 20 were grown in Dulbecco modified Eagle's medium (DMEM) (HyClone-Thermo Fisher Scientific, Waltham, MA) supplemented with 10% fetal bovine serum (FBS), 2 mM GlutaMAX (Life Technologies, Grand Island, NY), and 1% minimal nonessential amino acids (HyClone-Thermo Fisher Scientific, Rockford, IL). Stocks of virus reconstituted from the pAD/Cre HCMV strain AD169 bacterial artificial chromosome (BAC) (kindly provided by Dong Yu) were propagated by infecting 80% confluent HFFs at an MOI of 0.01 and collecting supernatant every 4 days. Virions present in cell culture supernatants were purified by centrifugation at 2,700 rpm for 15 min to remove cell debris and then ultracentrifugation (70,000 × g for 1 h) through a 20% sorbitol cushion. Pelleted virions were resuspended and stored at −80°C in DMEM containing 10% FBS. Titers of virus stocks were determined by plaque assay on confluent monolayers.
Plasmids.
All cloning was performed in Escherichia coli DH5α. To construct the BioID-UL103 plasmid (p), full-length UL103 was amplified from pAD/Cre (primers listed in Table 2) and inserted into the pcDNA3.1 mycBioID (plasmid 35700; Addgene, Cambridge, MA) (11) using XhoI and AfIII. UL103-V5-His(v) described in the following section was used to generate UL103-V5-His(p) by inserting the V5-His-tagged UL103 gene into pcDNA3.1 between the KpnI and XbaI sites. Alanine substitutions and deletion mutants were created by overlap extension PCR, followed by incorporation into UL103-V5-His(p). All constructs were verified by DNA sequencing.
TABLE 2.
Primers used for generation of recombinant viruses
| Plasmid or virus primer | Primer sequencea | Amplification target |
|---|---|---|
| Plasmid | ||
| BioID-UL103(p) | 5′-GAAGCGCAGAGAAGCTCGAGATGGAGGCCCTGATGATCCG-3′ | UL103 gene |
| 5′-ATCAGCGGTTTAAACTTAAGTCACTCTTCCTCTCCTCGTT-3′ | ||
| UL103-V5-His(p) | 5′-GTACCGAGCTCGGATCCACTATGGAGGCCCTGATGATCCG-3′ | UL103-V5-His gene |
| 5′-TAGACTCGAGCGGCCGCCACTCAATGGTGATGGTGATGAT-3′ | ||
| Virus | ||
| UL103-GalK/kan | 5′-GTTGCGTGTTTTTTTTTTTTCTATGATATGCGTGTCTAGTTCGCTTCTCAgctggagctccaccgcgggaagttc-3′ | GalK/Kan selection markers |
| 5′-TGCCCTCACCCCCCAAGCTGCCGCCGCGCTGGGAACGAGGAGAGGAAGAGatgggagtgcaggtggaaaccatc-3′ | ||
| UL103-BioID | 5′-CGTGTTGCGTGTTTTTTTTTTTTCTATGATATGCGTGTCTAGTTCGCTTCTCActtctctgcgcttctcagggag-3′ | mycBioID tag |
| 5′-TGCCCTCACCCCCCAAGCTGCCGCCGCGCTGGGAACGAGGAGAGGAAGAGgaacaaaaactcatctcagaagagg-3′ | ||
| UL103-V5-His | 5′-GTTGCGTGTTTTTTTTTTTTCTATGATATGCGTGTCTAGTTCGCTTCtcaatggtgatggtgatgatgaccggta-3′ | V5-His tag |
| 5′-TGCCCTCACCCCCCAAGCTGCCGCCGCGCTGGGAACGAGGAGAGGAAGAGggtaagcctatccctaaccctctcc-3′ |
The uppercase sequences in the virus primers correspond to the 50-bp segments of viral DNA needed for recombination into the viral genome. The lowercase sequences are regions needed to enable amplification of exogenous sequences (e.g., the V5 epitope) intended to be inserted into the BAC.
Construction of recombinant viruses.
Viruses were constructed by recombineering using the HCMV AD169 pAD/Cre bacterial artificial chromosome in SW105 cells (provided by the Court lab at the National Cancer Institute). The UL103-V5-His and UL103-BioID mutants were created by inserting a gene cassette at the extreme C terminus of UL103. The GalK/Kan cassette was PCR amplified from pYD-C630 (provided by Dong Yu) using 70-bp primers, 20 bp of which corresponded to flanking sequences in pYD-C630, with the remaining 50 bp homologous to sites upstream and downstream of the UL103 stop codon (Table 2). Induction of the lambda red recombinase in SW105 cells harboring pAD/Cre and the gene cassette led to homologous recombination followed by selection of transformed colonies on LB plates containing kanamycin. Single colonies were screened by BAC digestion and PCR.
To construct the scarless UL103-V5-His or UL103-BioID mutants, the GalK/kan cassette was replaced by homologous recombination with another cassette containing V5-His or BioID and the homologous ends described above (Table 2). SW105 cells carrying UL103 with the replacement cassette were negatively selected for on 2-deoxy-galactose (DOG) minimal plates. Galactose-negative colonies were streaked on MacConkey agar plates supplemented with chloramphenicol and galactose. Nonfermenting colonies were picked and screened by BAC digestion, PCR, and open reading frame sequence analysis.
Co-IP.
Cells (approximately 3.0 × 107) were either transfected with 30 μg of plasmid or inoculated at an MOI of 0.1. Transfected cells were harvested after 48 h, and infected cells were harvested 5 days postinfection (dpi) using radioimmunoprecipitation assay (RIPA) buffer (10 mM HEPES [pH 7.4], 1% sodium deoxycholate, 1× protease inhibitors [Roche, Indianapolis, IN], 150 mM NaCl, 1% Nonidet P-40, 0.1% SDS). Protein was extracted by passing the lysate through a 27-gauge needle 10 times, centrifugation at 13,000 rpm for 10 min, and collecting the supernatant. Antibodies were conjugated to the beads by mixing 20 μl of protein A/G Plus agarose (Santa Cruz Biotechnology, Dallas, TX) and 1 μg of antibody in 0.5 ml of phosphate-buffered saline (PBS) at 4°C overnight. Antibody-bead complexes were washed twice in PBS and then incubated with cell lysates overnight at 4°C in a tube rotator. Nonspecific interactions were stripped from the immunocomplex by washing three times with RIPA buffer. For mass spectrometry analysis, beads were spun at 2,000 × g and resuspended in 50 μl of 50 mM ammonium bicarbonate. For verification of interactions, immunocomplexes were washed with PBS, and the pellet was resuspended in 30 μl of 2× Laemmli sample buffer for immunoblot analysis.
BioID.
Transfected and infected cells were incubated with 50 μM biotin 24 h prior to harvesting. Cells lysed as described above were incubated at 4°C overnight with 500 μl of streptavidin conjugated to magnetic beads (New England BioLabs, Ipswich, MA). Beads were washed once in 1.5 ml of wash buffer 1 (2% SDS in H2O), once with wash buffer 2 (0.1% deoxycholate, 1% Triton X-100, 500 mM NaCl, 1 mM EDTA, and 50 mM HEPES [pH 7.5]), once with wash buffer 3 (250 mM LiCl, 0.5% NP-40, 0.5% deoxycholate, 1 mM EDTA, 10 mM Tris [pH 8.1]), and then twice with wash buffer 4 (50 mM Tris [pH 7.4], 50 mM NaCl). To evaluate sample integrity, 10% of the total was retained for immunoblots. The remaining beads were spun at 2,000 × g and resuspended in 50 μl of 50 mM ammonium bicarbonate for mass spectrometry.
Mass spectrometry.
Samples were digested overnight using sequencing-grade trypsin (Promega) in 25 mM ammonium bicarbonate and 10% acetonitrile. The resulting peptides were separated by reverse-phase chromatography (Acclaim PepMap100 C18 column; Thermo Scientific), followed by ionization with the Nanospray Flex ion source (Thermo Scientific), and introduced into an Orbitrap FusionTribrid mass spectrometer (Thermo Scientific). Primary data analysis was done using Proteome Discoverer 1.4 (Thermo), which incorporated the Mascot (Matrix Science) algorithm. Mascot searched the SwissProt_2014_06 and UniProt_Hum_Compl_All databases for viral and cellular proteins, respectively. Secondary analysis was performed with Scaffold 4.3.4 (Proteome Software) set to a minimum protein identification probability of ≥99% and 2 cellular or 3 viral unique peptides at a minimum peptide identification probability of ≥99%. Proteins identified by Scaffold were normalized by size using the normalized spectral abundance factor (NSAF) (12). Nonspecific cellular interactions were filtered out using the Contaminant Repository for Affinity Purification (CRAPome) (13).
Network association map of pUL103 interactions with cellular proteins.
The set of filtered, dual high-NSAF proteins that interact with pUL103 were analyzed using the STRING database to obtain a functional protein association network (14). The default settings were used for STRING network generation. The strengths of the STRING network relationships were visualized by assigning line weights to the compiled scores. The STRING network, line weight scores, and non-networked nodes were then imported into Cytoscape (15). The Gene Ontology (GO) plug-in GOlorize was used to color nodes with a biological process significance of P < 0.05 (16). Redundant terms and terms with only one protein associated were omitted.
Cell fractionation.
HFFs were collected and pelleted in ice cold PBS. Following pellet resuspension in 900 μl of mild lysis buffer (0.1% NP-40 in PBS), 300 μl was removed as whole-cell lysate. The remaining cell lysate was centrifuged for 10 s. Three hundred microliters of the supernatant was removed as the cytoplasmic fraction, while the pellet was washed in 1 ml of lysis buffer followed by another 10-s spin to pellet the nuclear fraction. The nuclear pellet was resuspended in 180 μl of 1× SDS-Laemmli buffer. Whole-cell lysates and nuclear fractions were sonicated prior to immunoblots.
Immunoblots.
Protein was quantified by bicinchoninic acid (BCA) assay (Pierce Biotechnology, Rockford, IL). For cell lysates, 50 μg of protein was solubilized in 2× SDS-Laemmli buffer followed by SDS-PAGE. Separated proteins were transferred to nitrocellulose membranes (Whatman, Florham Park, NJ), probed with primary antibodies, and then reacted with horseradish peroxidase (HRP)-conjugated goat anti-mouse/rabbit IgG secondary antibodies (Thermo Scientific, Rockford, IL). Reactions were detected with the Supersignal West Pico chemiluminescent substrate (Bio-Rad, Hercules, CA) on autoradiography film (GE Healthcare, Pittsburgh, PA). Antibodies are listed in Table 3.
TABLE 3.
Antibodies used in IFA and immunoblot assays
| Antibody target | Host/isotype, clone | Source, catalog no. |
|---|---|---|
| Epitope tag | ||
| V5 (14 aa) | Mouse monoclonal/IgG2a | Life Technologies, R960-25 |
| His (6 aa) | Rabbit polyclonal | Rockland 600-401-382 |
| Cellular | ||
| GAPDH (36 kDa) | Mouse monoclonal/IgG1, clone GA1R | Thermo Scientific, MA5-15738 |
| GM130 (130 kDa) | Mouse monoclonal/IgG1(κ), clone 35/GM130 | BD Biosciences, 610822 |
| EEA1 (180 kDa) | Rabbit polyclonal | Abcam, ab2900 |
| IFI16 (85–95 kDa) | Mouse monoclonal/IgG1, clone 1G7 | Santa Cruz, sc-8023 |
| ALIX (96 kDa) | Rabbit polyclonal | Abcam, ab76608 |
| HCMV | ||
| IE1 (72 kDa) and IE2 (86 kDa) | Mouse monoclonal/IgG1(κ), clone CH160 | Virusys, P1215 |
| IE2 (86 kDa) | Mouse monoclonal, clone 8B1.2 | Chemicon (Millipore), MAB810 |
| pUL44 (CMV ICP36) (46 kDa) | Mouse monoclonal/IgG1(κ), clone 10D8 | Virusys, CA006-100 |
| UL34-R4 | Rabbit polyclonal | Bonita Biegalke (Ohio University) |
IFA.
For the immunofluorescence assays (IFA), HFFs were seeded at 80% confluence 1 day prior to infection on 0.2% gelatin-coated 8-well glass chamber slides (Thermo Fisher Scientific, Waltham, MA). The following day, cells were infected at an MOI of 0.1 and then fixed in paraformaldehyde and stained at 120 h postinfection (hpi) as previously described (5). Primary antibodies are listed in Table 3. Fluor-tagged secondary antibodies (Alexa Fluor 488-conjugated goat anti-rabbit IgG and Alexa Fluor 568-conjugated goat anti-mouse IgG) were from Life Technologies (Grand Island, NY). Mounting was done with Vectashield containing DAPI (4′,6-diamidino-2-phenylindole) (Vector Laboratories, Inc., Burlingame, CA). Imaging was done on a Nikon E800 fluorescence microscope and a Leica TCS SP5 laser scanning confocal microscope.
RESULTS
Experimental design.
Affinity purification followed by mass spectrometry can be used to identify proteins pulled down by a bait protein. The proteins identified can be true interacting partners, but false positives are common, arising due to reasons that can include nonspecific interactions with the beads, antibody, protein tag, and sticky or highly abundant proteins. To increase the likelihood of identifying biologically meaningful interactions, we limited confounding by false positives through the use of two independent methods of affinity purification: coimmunoprecipitation (co-IP) and proximity-labeled biotinylation (BioID). Co-IP detects proteins that strongly interact with the bait protein, while the BioID system uses BirA*, a promiscuous biotin ligase that biotinylates proteins within ∼10 nm of the tagged protein (11). Each method uses different beads, antibodies, tags, and binding/elution conditions. The differences in these complementary methods provide internal controls that reduce the number of false positives, while proteins highly enriched in both approaches are more likely to be specific.
Our experiments involved plasmids (p) and recombinant viruses (v) with tags fused to pUL103. Plasmids included one construct that expresses the BirA* protein alone, one with BirA* fused to the pUL103 N terminus [BioID-pUL103(p)], and one in which the V5-His tag was added to the C terminus of pUL103 [UL103-V5-His(p)]. Two recombinant viruses were also used, in which the tags were attached to the C terminus of pUL103 [UL103-BioID(v) and UL103-V5-His(v)]. For infection experiments, we inoculated cells at an MOI of 0.1 with either UL103-V5-His(v) or UL103-BioID(v) and harvested the cells 5 days postinfection. Use of low-MOI infections enabled analysis of cells at multiple stages of infection in a single specimen, giving us a broad survey of the viral and cellular proteins that interact with pUL103.
Collectively, these constructs and conditions enable identification of interactions of pUL103 with (i) HCMV proteins in infected cells, (ii) cellular proteins in the absence of other viral proteins, and (iii) cellular proteins in infected cells. Comparison of the latter two categories enabled identification of interactions with cellular proteins that are enhanced during infection.
Effect of tags on virus replication, expression, and localization of pUL103.
Reagents are not available to examine the localization of native (untagged) pUL103. To determine whether the affinity tags have different effects on the localization or activity of pUL103, we conducted immunofluorescence and virus replication plaque assays. In cells transfected with C-terminally [UL103-V5-His(p)] or N-terminally [BioID-UL103(p)]-tagged pUL103, staining was abundant in perinuclear regions that correspond to the endoplasmic reticulum (ER) and Golgi structures (Fig. 1A), while BioID(p) alone was dispersed diffusely throughout the cell cytoplasm. During infection, UL103-V5-His(v) and UL103-BioID(v) localized to regions corresponding to the cVAC, consistent with previous reports (Fig. 1A) (5, 6). In multistep growth experiments, UL103-BioID(v) produced titers similar to those of the parent, while UL103-V5-His(v) replication was delayed, taking 6 days to reach parental levels (Fig. 1B). Our previous results using an FKBP tag also showed a lag in extracellular virus production, suggesting that some tags at the C terminus of pUL103 perturb virion production at earlier times after infection (5). It is likely that use of tagged proteins had some effect on the set of pUL103-interacting proteins we detected.
FIG 1.

Effect of tagged pUL103 on localization and viral growth. (A) HFFs were either transfected (p) for 48 h or infected (v) at an MOI of 0.2 for 120 h and then analyzed by fluorescence microscopy. pUL103 was visualized using a V5 antibody against the V5-His epitope or a myc antibody targeting the BioID tag. (B) Replication of UL103-V5-His and UL103-BioID recombinant viruses was evaluated by multistep growth curves (MOI of 0.1). Determination of virus titers was performed in triplicate.
Viral proteins identified by affinity purification mass spectrometry.
Mass spectrometry returned 5,566 total viral spectral hits, which correspond to 49 HCMV proteins using co-IP and 47 proteins with BioID (outlined in Fig. 2A). Altogether, 53 viral proteins were detected, indicating high overlap between the two methods. Larger proteins tend to generate higher spectral counts than smaller proteins, so we used the normalized spectral abundance factor (NSAF) to normalize the results based on protein size. To identify interactions more likely to be significant, we generated NSAF scatter plots of the UL103-BioID versus the UL103-V5-His results (Fig. 2B). The highest NSAF values were for the highly abundant tegument protein, pp65 (NSAF of 21.6 for UL103-BioID and 23.4 for UL103-V5-His), but pp65 was also pulled down in untagged AD169 controls, arguing against a specific interaction between pp65 and pUL103 (data not shown). The second highest NSAF value for UL103-V5-His was the pUL103 NSAF of 13.25; this demonstrated sufficient immunoprecipitation of the pUL103-V5-His protein with the V5 antibody, but does not prove pUL103 self-interaction. The tegument hub protein, pUL25, also scored highly in both approaches, but lack of a pUL25-specific antibody precludes immediate verification. Other proteins with a dual NSAF of >0.5 were pUL34, pUL44, pUL45, pUL47, pUL50, pUL56, pUL71, pUL84, pUL86, pUL112/113, and IE2. Identification of pUL71 by our pulldowns extends the result obtained in transfected cells by Fischer (8), providing evidence that pUL103 interacts with pUL71 during infection (8). In addition, Roller and Fetters showed that the homologs of pUL103 and pUL71 (HSV pUL7 and pUL51) form a complex in HSV1-infected cells (9). A manually curated network map of the interactions of pUL103 with other HCMV proteins is shown in Fig. 2D.
FIG 2.

Experimental scheme and graphical representation of enriched HCMV proteins. (A) HFFs were infected for 120 h at an MOI of 0.1 with UL103-V5-His(v) or UL103-BioID(v), and immunocomplexes were pulled down with antibodies to V5 or biotin, respectively. Mass spectrometry spectral counts were normalized based on size using the normalized spectral abundance factor (NSAF) formula. (B) Scatter plot of viral protein NSAFs from coimmunoprecipitation and proximity-labeled biotinylation experiments. The dotted line separates the viral proteins with an NSAF of >0.5 (colored) from those less enriched (gray). The pp65 protein was excluded from the scatter plot due to nonspecific interaction with the V5 antibody. The asterisk (*) indicates a known interaction. (C) Bar graph of all the viral proteins identified at increasing NSAF thresholds. The lines indicate proteins shared by both methods. (D) Network of dual high-scoring interactions of pUL103 with other HCMV proteins. Previously identified interactions are indicated with solid lines, and novel interactions identified here are indicated with dashed lines. Line weights are uniform, and no statistical analysis was applied.
Verification of interactions with viral proteins.
We detected several novel interactions of pUL103 with other HCMV proteins (Fig. 2D). Four of the highest-scoring viral proteins are known to associate with DNA (pUL34, pUL44, pUL84, and IE2). This was somewhat surprising, since we and others have detected pUL103 immunofluorescence staining predominantly in the cytoplasm of infected cells (5, 6) (Fig. 1A). However, the HSV1 and HSV2 homologs of pUL103 (HSV pUL7) have been detected in nuclei (10, 17). Although there appears to be some nuclear staining for pUL103-V5 in transfected cells shown in Fig. 1A, robust analysis of nuclear localization of pUL103 is not available from this experiment, because the images were not obtained by confocal microscopy. To assess the nuclear presence of pUL103, we performed three-dimensional confocal microscopy on HCMV-infected cells. Although much less intense than the cytoplasmic fluorescence, in central slices of the z-series (approximately equidistant from apical and basal surfaces), weak but unambiguous nuclear staining for pUL103 was present (Fig. 3A). We also performed cellular fractionation followed by immunoblotting and found that pUL103 is present in nuclear fractions that are not contaminated with the cytoplasmic proteins glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and BiP, the latter being resident in the endoplasmic reticulum, which is contiguous with the nuclear membrane (Fig. 3B). The absence of BiP in the nuclear fraction strengthens the case for it being relatively free of cytoplasmic contamination. We conclude that a portion of pUL103 is indeed present in infected cell nuclei.
FIG 3.
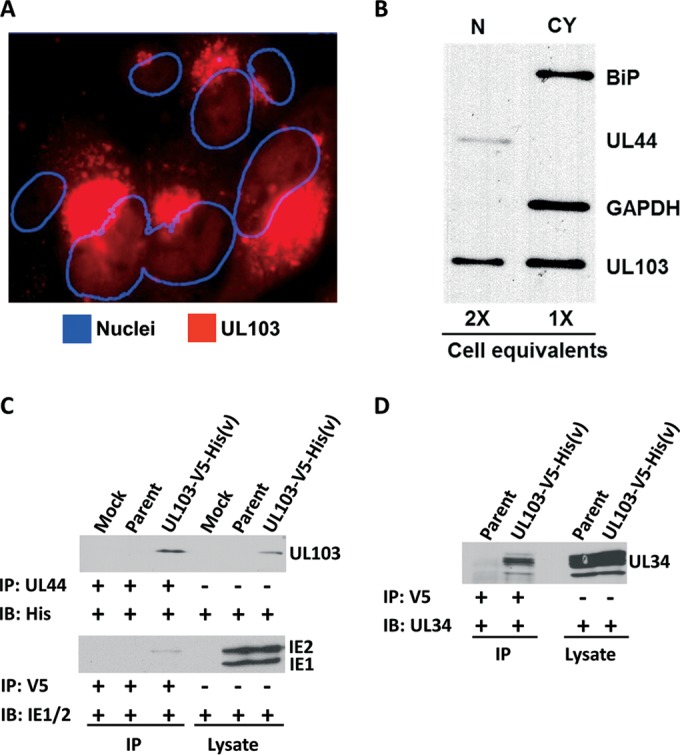
Localization of pUL103 in infected cells and coimmunoprecipitation verification of viral proteins. (A and B) HFFs were infected with UL103-V5-His(v) for 96 h at an MOI of 0.1 and probed with anti-V5 antibody. (A) The pUL103-V5-His-tagged protein was analyzed by confocal microscopy. The center slice of a z-stack is shown with the nuclei outlined using ImageJ. (B) Cellular fractionation of infected cells using antibodies against BiP, UL44, and GAPDH as markers for endoplasmic reticulum, nucleus (N), and cytoplasm (CY), respectively. (C and D) Pulldowns were performed with anti-UL44 or anti-V5 on HFFs infected for 120 h at an MOI of 0.1. The immunoprecipitating antibody (IP) and the probing antibody (IB) are listed to the left of each blot. Immunoblots were probed with antibodies against His-tagged UL103, IE1/2, and UL34.
We employed immunoprecipitation and immunoblotting to verify interactions of pUL103 with three of the DNA-associated viral proteins (IE2, UL34, and UL44). IE2 and pUL34 were verified by pulldowns with a V5 antibody followed by immunoblotting. Each blot showed a band in the UL103-V5-His(v) IP lane but not in the parent or control lanes (Fig. 3C and D). pUL44 is approximately 46 kDa and is obscured by the 50-kDa heavy-chain antibody band. Because of this, pUL103-V5-His was immunoprecipitated with a pUL44 antibody and then probed with anti-His. There was a clear band in UL103-V5-His(v) (Fig. 3C, lane 3) that comigrates with pUL103 in the total cell lysates (Fig. 3C, lane 6). These results provide evidence for the specificity obtained by merging results from two independent affinity-based pulldown approaches and reveal previously undetected interactions of pUL103 with proteins predominately located in infected cell nuclei.
Interactions of pUL103 with cellular proteins.
We used the dual affinity purification approach to identify pUL103 interaction partners in transfected and in HCMV-infected cells. Mass spectrometry detected a total of 2,052 cellular proteins pulled down by both methods. To eliminate proteins likely to represent nonspecific background, we carried forward only proteins that had a UL103-BioID(v) NSAF value higher than the NSAF of the BioID(p) control and a CRAPome score lower than 100. We then generated two forms of NSAF-based scatter plots: one that includes all of the cellular protein interactions that passed the filtering described above (Fig. 4A) and another comparing NSAF fold change of infected cells versus uninfected (transfected) cells to detect interactions that are enhanced during infection (Fig. 4C). To further increase the likelihood of identifying pUL103 interaction partners of biological significance, we employed a dual high cutoff of >2 for both the V5 and for the BioID proteomic analyses (Fig. 4A and C) to generate the list of proteins employed in subsequent analyses. A total of 18 cellular proteins met these criteria (Table 4).
FIG 4.

Dual NSAF analyses of interactions between pUL103 and cellular proteins. (A) After filtering out nonspecific interactions, the cellular NSAF scores of UL103-V5-His(v) and UL103-BioID(v) were plotted against each other. The functions of the most enriched interactions (NSAF of >3) are shown. (C) NSAF fold change selects for cellular interactions enhanced during infection. Fold changes greater than 2 highlight multiple antiviral proteins. (B and D) Charts showing all the cellular proteins at increasing cutoff values. The horizontal lines across each pair of bars indicate the number of proteins in common between the two pulldown methods. (E) Network of dual high-scoring interactions of pUL103 with cellular proteins. Node colors depict biological process GO categories, and the line width of connected nodes represents the STRING confidence score.
TABLE 4.
Properties of the cellular proteins with dual high NSAF or NSAF-fold changea
| Gene product ID | Property | Infected |
Transfected, NSAF for BioID(p) | CRAPome score | |||
|---|---|---|---|---|---|---|---|
| NSAF |
NSAF FCb |
||||||
| V5 | BioID | V5 | BioID | ||||
| TUBB3 | Microtubule assembly | 19.1 | 13.5 | 1.1 | 1.1 | 10.4 | NAc |
| LMNA | Structural protein of inner nuclear membrane | 18.5 | 2.2 | 6.7 | 2.2 | 0.0 | 92 |
| HSPB1 | Stress resistance and actin organization | 14.6 | 1.7 | 3.3 | 2.2 | 0.8 | 71 |
| SERPINH1 (HSP70) | Collagen biosynthesis and folding | 10.6 | 4.8 | 3.1 | 6.1 | 0.8 | 78 |
| STAT1 | Antiviral transcription activator | 6.2 | 4.6 | 3.9 | 8.9 | 0.5 | 23 |
| ISG15 | Ubiquitin-like protein with antiviral activity | 6.0 | 3.1 | 6.0 | 3.1 | 0.0 | 5 |
| DYNLL1 | Links dynein to cargo and adapter proteins | 5.4 | 2.4 | 1.8 | 64 | ||
| IFI16 | Viral DNA sensor that triggers innate immune response | 4.9 | 21.5 | 4.9 | 21.5 | 0.0 | 9 |
| MYO1C | Intracellular vesicle transport; nuclear isoform functions in transcription initiation | 4.7 | 9.3 | 3.2 | 12.6 | 0.7 | 66 |
| IFIT1 | RNA-binding protein that inhibits viral replication and translation initiation | 4.2 | 3.1 | 4.2 | 3.1 | 0.0 | 4 |
| COPA | Mediates budding from Golgi membranes | 3.9 | 2.5 | 0.8 | 2.3 | 1.1 | 86 |
| SEC61A1 | Associated with ER ribosomes for assembly of membrane and secretory proteins | 3.9 | 3.5 | 0.8 | 2.5 | 1.4 | 28 |
| PDCD6IP (ALIX) | Associated with ESCRT machinery sorting of protein cargo into MVBs | 3.6 | 5.9 | 0.7 | 3.3 | 1.8 | 55 |
| COPG1 | Mediates budding from Golgi membranes | 3.3 | 3.0 | 1.0 | 1.8 | 1.6 | 78 |
| COPB1 | Mediates budding from Golgi membranes | 3.0 | 3.6 | 0.6 | 1.9 | 1.9 | 92 |
| SURF4 | Structural maintenance of Golgi compartment and endoplasmic reticulum-Golgi intermediate compartment | 2.3 | 2.4 | 1.7 | 2.4 | 0.0 | 35 |
| CAND1 | Exchange factor involved in neddylation and deneddylation | 2.3 | 4.0 | 0.5 | 1.0 | 4.1 | 88 |
| MYD88 | Adapter protein in interleukin-1 and Toll-like receptor signaling pathways | 1.2 | 1.4 | 8.2 | 5.3 | 0.3 | 0 |
A high NASF represents an NASF of >2.
FC, fold change.
NA, not applicable.
Several proteins involved in innate immune responses were among the highly enriched proteins (IFI16, IFIT1, ISG15, MYD88, and STAT1). In cells transfected with pUL103 expression vectors, all of these antiviral proteins either had low NSAF scores or were not detected. In GO analyses, the associated biological processes had corrected P values of <0.01 (Table 5 and Fig. 4E). Although it is possible that some of these interactions might be attributed to elevated protein expression in infected cells, mRNA levels of IFI16 decreased by 71% and 62% in fibroblasts infected for 96 h with HCMV strains Towne and AD169 (18); a similar decrease in abundance of the protein is evident in immunoblots of infected cell lysates (Fig. 5A). Importantly, the IFI16 interaction was verified by forward and reverse co-IP. These results are consistent with some aspect of infection leading to enhanced interaction of pUL103 with IFI16 in infected versus uninfected transfected cells and suggest a novel role for pUL103 in the regulation of innate immune signaling during infection (Fig. 5A).
TABLE 5.
Enriched biological process GO terms
| GO ID | Term | P value | Corrected P value | Frequency (%) |
Genes | |
|---|---|---|---|---|---|---|
| Cluster | Total | |||||
| Golgi transport and localization | ||||||
| 48200 | Golgi transport vesicle coating | 1.11E−07 | 1.73E−05 | 3/15 (20.0) | 10/14,305 (0.0) | COPA, COPB1, COPG1 |
| 48205 | COPI coating of Golgi vesicle | 1.11E−07 | 1.73E−05 | 3/15 (20.0) | 10/14,305 (0.0) | COPA, COPB1, COPG1 |
| 48194 | Golgi vesicle budding | 1.53E−07 | 1.73E−05 | 3/15 (20.0) | 11/14,305 (0.0) | COPA, COPB1, COPG1 |
| 48199 | Vesicle targeting to, from, or within Golgi compartment | 2.04E−07 | 1.73E−05 | 3/15 (20.0) | 12/14,305 (0.0) | COPA, COPB1, COPG1 |
| 6901 | Vesicle coating | 6.29E−07 | 4.28E−05 | 3/15 (20.0) | 17/14,305 (0.1) | COPA, COPB1, COPG1 |
| 6900 | Membrane budding | 8.95E−07 | 5.07E−05 | 3/15 (20.0) | 19/14,305 (0.1) | COPA COPB1 COPG1 |
| 6890 | Retrograde vesicle-mediated transport, Golgi compartment to ER | 1.23E−06 | 5.96E−05 | 3/15 (20.0) | 21/14,305 (0.1) | COPA, COPB1, COPG1 |
| 6903 | Vesicle targeting | 1.42E−06 | 6.03E−05 | 3/15 (20.0) | 22/14,305 (0.1) | COPA, COPB1, COPG1 |
| 51650 | Establishment of vesicle localization | 4.99E−06 | 1.89E−04 | 3/15 (20.0) | 33/14,305 (0.2) | COPA, COPB1, COPG1 |
| 51648 | Vesicle localization | 7.70E−06 | 2.62E−04 | 3/15 (20.0) | 38/14,305 (0.2) | COPA, COPB1, COPG1 |
| 16050 | Vesicle organization | 2.37E−05 | 7.32E−04 | 3/15 (20.0) | 55/14,305 (0.3) | COPA, COPB1, COPG1 |
| 51656 | Establishment of organelle localization | 5.11E−05 | 1.45E−03 | 3/15 (20.0) | 71/14,305 (0.4) | COPA, COPB1, COPG1 |
| 51640 | Organelle localization | 1.26E−04 | 2.85E−03 | 3/15 (20.0) | 96/14,305 (0.6) | COPA, COPB1, COPG1 |
| 48193 | Golgi vesicle transport | 3.22E−04 | 6.09E−03 | 3/15 (20.0) | 132/14,305 (0.9) | COPA, COPB1, COPG1 |
| Protein localization | ||||||
| 15031 | Protein transport | 7.08E−05 | 1.85E−03 | 6/15 (40.0) | 756/14,305 (5.2) | SEC61A1, COPA, PDCD6IP, MYO1C, COPB1, COPG1 |
| 45184 | Establishment of protein localization | 7.68E−05 | 1.87E−03 | 6/15 (40.0) | 767/14,305 (5.3) | SEC61A1, COPA, PDCD6IP, MYO1C, COPB1, COPG1 |
| 8104 | Protein localization | 2.12E−04 | 4.50E−03 | 6/15 (40.0) | 921/14,305 (6.4) | SEC61A1, COPA, PDCD6IP, MYO1C, COPB1, COPG1 |
| 46907 | Intracellular transport | 4.35E−04 | 7.30E−03 | 5/15 (33.3) | 665/14,305 (4.6) | SEC61A1, COPA, MYO1C, COPB1, COPG1 |
| 6886 | Intracellular protein transport | 4.51E−04 | 7.30E−03 | 4/15 (26.6) | 364/14,305 (2.5) | SEC61A1, COPA, COPB1, COPG1 |
| 33036 | Macromolecule localization | 5.88E−04 | 9.08E−03 | 6/15 (40.0) | 1111/14,305 (7.7) | SEC61A1, COPA, PDCD6IP, MYO1C, COPB1, COPG1 |
| Innate immunity | ||||||
| 44419 | Interspecies interaction between organisms | 3.00E−04 | 6.00E−03 | 4/15 (26.6) | 327/14,305 (2.2) | PDCD6IP, STAT1, COPB1, ISG15 |
| 9615 | Response to virus | 3.99E−04 | 7.14E−03 | 3/15 (20.0) | 142/14,305 (0.9) | IFI16, STAT1, ISG15 |
FIG 5.
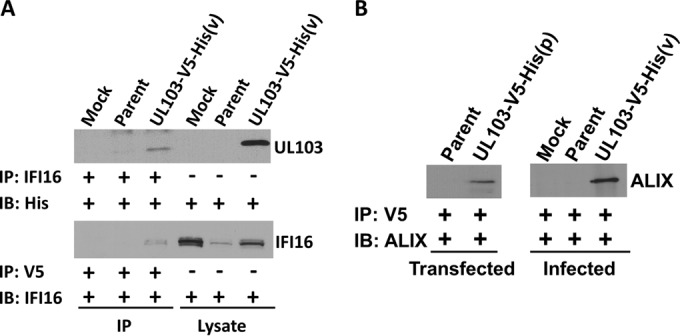
Coimmunoprecipitation verification of IFI16 and ALIX interactions. (A) HFFs were infected for 120 h at an MOI of 0.1, and affinity purification was conducted on the left three lanes, while the right three lanes contain protein from cell lysates. The immunoprecipitating antibody (IP) and the probing antibody (IB) are listed to the left of each blot. pUL103 was purified with IFI16 in reciprocal pulldown assays. (B) Affinity purification of pUL103 using a V5 antibody verified the interaction with ALIX in transfected and infected cells.
Another category of proteins identified in these analyses includes proteins involved in formation, transport, and targeting of intracellular vesicles (COPA, COPB1, COPG1, MYO1C, PDCD6IP, and SEC61A1) (Table 5 and Fig. 4E). P values for the associations with these biological processes were highly significant, ranging from 6.09 × 10−3 to 1.73 × 10−5. These interactions may be important during cVAC biogenesis and subsequently during secondary envelopment and egress.
Pulldowns in transfected and infected cells confirmed the binding of pUL103 to the ESCRT-associated protein, ALIX (Fig. 5B). Interestingly, amino acid motif analysis of pUL103 revealed two potential “late” or “L” domains that bind ALIX at the C terminus (Fig. 6A). Late domains are well-characterized motifs present in retrovirus gag proteins that are sites of interaction with ALIX important for virion budding at the plasma membrane (reviewed in reference 19). These results led us to further explore the pUL103-ALIX interaction.
FIG 6.

pUL103 amino acid sequence schematic and mutational analysis of late domains. (A) Map of the HCMV pUL103 gene depicting a herpesvirus conserved region (GFX8EDX12R), along with HCMV-specific helicase and late domains. (B and C) Coimmunoprecipitations of pUL103 using a V5 antibody in transfected cells. (B) Deletion or alanine substitutions within the first or second ALIX binding motif reduced the quantity of ALIX pulled down with pUL103. (C) The quantity of ALIX immunoprecipitated per condition was measured by densitometry and divided by normalized pUL103 (the ratio of GAPDH to pUL103 in each lane).
Mutational analysis of ALIX-binding motifs in uninfected cells.
We identified two closely spaced potential ALIX-binding late motifs in pUL103 (Fig. 6A). The first is a sequence, G215WPVGLGLL, that conforms to both the type 1 (YPXnL, where X is any residue and n can range from 1 to 3) and type 3 (ΦYXnΦXnL, where Φ is any hydrophobic residue) ALIX-binding late domain motifs (19). The chemical and structural similarities between tryptophan and tyrosine permit them to be interchanged, as seen in simian immunodeficiency virus Gag p6, in which the YPXL motif is replaced by WPXL (20). The second sequence, Y230PNL, corresponds to the type 1 late domain motif, YPXnL. In experiments in which levels of pUL103 were normalized relative to GAPDH, replacement of multiple residues with alanines within either of the putative pUL103 late domains or deletion of the G215WPVGLGLL motif led to at least 50% reduction in ALIX pulled down in uninfected cells (Fig. 6B and C). Single alanine substitutions in the YPNL motif had little discernible effect on pUL103-ALIX interaction (data not shown). These results confirm the specificity of the interactions seen in the proteomics analysis and are consistent with pUL103 containing two closely spaced motifs involved in its interaction with ALIX.
DISCUSSION
HCMV pUL103 is a conserved herpesvirus virion tegument protein that is involved in several processes, including cell-to-cell spread, cVAC biogenesis, and virion envelopment and egress. Several pUL103 interaction partners were previously identified in transfection and yeast two-hybrid systems (7, 8), but it is important to study the protein in the context of infection because in the absence of infection, interactions among viral proteins, as well as interactions of other viral proteins with cellular proteins (e.g., antiviral proteins), do not occur. To address these gaps and to provide novel insights into its molecular functions and biological roles, we employed recombinant viruses expressing forms of pUL103 that enabled use of independent affinity purification methods (co-IP and BioID) followed by mass spectrometry to identify its interacting partners. By restricting our analysis to interactions that scored highly in both of the complementary methods, we increased the likelihood of identifying specific, high-value biologically relevant interactions worthy of further study, while limiting the number of false positives and incidental interactions. The set of viral and cellular interaction partners we identified suggests that pUL103, and possibly its homologs in other herpesviruses, interacts with nuclear proteins that bind DNA and play roles in the regulation of antiviral responses and in the formation, transport, and targeting of intracellular vesicles. The latter activities may be important during cVAC biogenesis and subsequently during secondary envelopment and egress.
Interactions of pUL103 with other HCMV proteins.
The pUL103-interacting viral proteins that met our screening criteria belong to several distinct categories, including tegument proteins, DNA-binding proteins, and capsid and nuclear egress proteins (Fig. 2D). pUL103 and pUL71 were previously shown to interact outside the context of infection in cotransfection and bimolecular fluorescence complementation experiments (8), and interactions between the HSV homologs (pUL7 and UL51) of these proteins were detected in infected cells (9). The high score of this interaction provides validation for our dual pulldown approach.
Of the interactions that scored highly in both the V5 and BioID experiments, follow-up pulldown experiments were performed with antibodies against pUL34, pUL44, and IE2. In each instance, the interactions were detected only in cells infected with the UL103-V5-His virus and not in mock-infected cells or cells infected with untagged parental virus. These results provide further evidence that the interactions characterized by high NSAF scores in both the V5- and BioID-based proteomic analyses are sufficiently specific to justify follow-up studies.
The highest pair of NSAF scores was for the interaction between pUL103 and the HCMV tegument protein pUL25. pUL25 is expressed at late times postinfection, strongly localizes to the cVAC, and is incorporated into the three major types of virus particles: mature virions, noninfectious enveloped particles, and dense bodies (21, 22). The reported interactions of pUL25 with several other tegument proteins suggest it serves as an organizing hub for incorporating viral proteins into enveloped particles (7). Further analysis of the pUL103-pUL25 interaction is warranted.
We did not detect the previously described interactions between pUL103 and HCMV pUL22A and pUL48N, which were identified in a yeast two-hybrid system but not verified by co-IP in transfected cells (7). In yeast cells, pUL103 may lack posttranslational modification(s) or have alternative folding that exposes or creates artificial binding sites. It is also possible that those interactions would have been inhibited by either or both of the affinity tags we used.
Nuclear pUL103.
Identification of five viral DNA-associated proteins with dual NSAF values above 0.5 (Fig. 2B) and co-IP verification of three of these (pUL34, pUL44, and IE2) suggest a nuclear role for pUL103. This was unexpected given that pUL103 had appeared to be strictly cytoplasmic by IFA in HCMV-infected cells. Only after substantially increased exposure times were we able to detect a faint but discernible pUL103 signal in nuclei (Fig. 3A). Immunoblots of fractionated cells confirmed the presence of pUL103 in nuclear fractions that were not contaminated with the endoplasmic reticulum marker BiP. HSV-1 and -2 homologs (pUL7) of pUL103 have been detected in infected cell nuclei by IFA and transmission immunoelectron microscopy (9, 17). The pUL103 homologs of all members of the Herpesviridae contain a conserved domain that is homologous with a segment of DNA topoisomerase III of Schizosaccharomyces pombe; this protein releases supercoil tension that is introduced during DNA replication and transcription (17, 23). The presence of this conserved domain, in conjunction with its observed nuclear localization and interaction with HCMV DNA-associated proteins, suggests a heretofore undefined role for pUL103 in nuclear events.
Interactions of pUL103 with cellular proteins.
To identify interactions of pUL103 that are likely to be biologically significant, we applied the stringent screening criteria outlined in Fig. 2A, with the understanding that we may have excluded some true interactions. For instance, HSV pUL7 can pull down the mitochondrial protein ANT2 (10). We found a dual high-scoring interaction between ANT2 and pUL103 in transfected and in infected cells, but because ANT2 has a CRAPome score of 223, it did not pass our screening filter. Thus, even though the interaction might be biologically meaningful, we did not pursue it here.
Most of the proteins that passed our screening criteria are involved in innate antiviral responses and vesicle formation/trafficking (Fig. 4 and 5 and Tables 4 and 5). A cellular protein from each category was verified in reciprocal pulldown experiments, providing evidence that these represent true interactions.
A novel role for pUL103 in innate immunity?
A possible role for pUL103 in regulation of innate immunity is difficult to evaluate in uninfected cells; with the exception of STAT1, none of the antiviral interaction partners was detected in our transfection experiments. In infected cells, the presence of pathogen-associated molecular patterns (PAMPs) activates the antiviral response. Relocalization, increased expression, or phosphorylation can lead to interactions of innate immune sensors and effectors with viral partners. We identified five antiviral proteins that displayed dual high NSAF scores and verified the interaction with IFI16 by reciprocal co-IP. The number of antiviral proteins detected and their functional interrelatedness demonstrated by STRING analysis (Fig. 4E) suggest that pUL103 is likely to modulate innate immune responses.
Our proteomic experiments involved low-MOI infections (MOI of 0.1). At the time of harvest 5 dpi, the cultures included nonsynchronously infected cells spanning the earliest and late stages of infection. Virion tegument proteins are introduced into the host cell immediately upon fusion of the viral and host cell membranes. The release of tegument proteins pp65 and pp71 into host cells is critical for subverting innate immune responses and helping to establish an environment conducive to productive infection (24, 25). As a virion-associated tegument protein (26), pUL103 may have analogous or complementary antiviral activities. In addition, recent work by Xie and colleagues showed that HCMV makes use of interferon-induced transmembrane proteins (IFITMs) for cVAC assembly and virion production (27). Thus, virion-associated, as well as newly synthesized pUL103 could interact with antiviral proteins to facilitate cVAC biogenesis.
Potential roles for pUL103 in biogenesis and trafficking of intracellular vesicles.
Identification of several proteins (e.g., COPB1, MYO1C, and ALIX) involved in formation and trafficking of vesicles suggests mechanistic connections to the known role of pUL103 in virion envelopment and egress. COPB1 and its relatives mediate Golgi fragmentation during the M phase of the cell cycle and retrograde transport from the Golgi compartment to the ER (28, 29). MYO1C is involved in trafficking GLUT4 to the plasma membrane and participates in lipid raft organization (30–32). These cellular proteins have functions that encompass activities of the sort expected to be involved in cVAC biogenesis and trafficking of virions to the cytoplasmic membrane, a subject for future study.
ALIX is involved in envelopment of several viruses. Proteins that bind to ALIX typically interact via ALIX-specific late domains that are important for virion production in many RNA and DNA viruses (33–37). Late domains in retrovirus gag proteins can bind the V domain of ALIX to facilitate viral budding at the cell membrane (reviewed in references 19, 36, and 37). For DNA viruses, vaccinia viruses use an ALIX-binding domain for extracellular virion production, and Epstein-Barr virus, a gammaherpesvirus, exploits ALIX for vesicle formation and nuclear egress of capsids (33, 35). The use of ALIX and ESCRT machinery is thus a common theme for production of enveloped virions.
By sequence analysis, we identified two adjacent ALIX-binding motifs in pUL103: one that conforms to canonical type 1 and type 3 motifs and one that matches the type 1 motif (Fig. 6A). We evaluated mutations in these motifs in transfection pulldown experiments and found that changes in the type 1 motif had little effect by themselves, while deletion of the type 1/3 motif or replacement of five critical residues with alanines led to a substantial reduction in the amount of ALIX pulled down by pUL103-V5-His.
Demonstration of an interaction between ALIX and pUL103 suggests involvement of ALIX in HCMV envelopment and egress. However, studies involving small interfering RNA (siRNA) knockdown and dominant-negative versions of ALIX had no effect on HCMV virion production (38, 39). This raises the possibility that HCMV employs redundant ESCRT-related pathways during the late stages of virion biogenesis. Recombinant viruses with ALIX-binding motif mutations are under construction to elucidate the role of the pUL103-ALIX interaction during infection.
In summary, the collection of viral and cellular proteins identified here led to novel insights into the biological functions of pUL103 and, by extension, its respective homologs. This provides a robust foundation for future studies of this conserved multifunctional herpesvirus protein.
ACKNOWLEDGMENTS
We thank Kyle Roux from the Wandless lab for the BioID plasmid, Bonita Biegalke (Ohio University) for anti-pUL34, Dong Yu (Washington University) for the HCMV(AD169) BAC, the Court lab at the National Cancer Institute for bacterial strains used for generation of BAC mutants, and the Wayne State University and Karmanos Cancer Center Proteomics Core, which is supported by NIH grants P30 ES020957, P30 CA022453, and S10 OD010700. We also thank Ashley Anderson for contributions to the text and the other members of the Pellett laboratory for advice and encouragement.
Funding Statement
The funding agencies had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Alwine JC. 2012. The human cytomegalovirus assembly compartment: a masterpiece of viral manipulation of cellular processes that facilitates assembly and egress. PLoS Pathog 8:e1002878. doi: 10.1371/journal.ppat.1002878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Das S, Vasanji A, Pellett PE. 2007. Three-dimensional structure of the human cytomegalovirus cytoplasmic virion assembly complex includes a reoriented secretory apparatus. J Virol 81:11861–11869. doi: 10.1128/JVI.01077-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tandon R, Mocarski ES. 2012. Viral and host control of cytomegalovirus maturation. Trends Microbiol 20:392–401. doi: 10.1016/j.tim.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hook LM, Grey F, Grabski R, Tirabassi R, Doyle T, Hancock M, Landais I, Jeng S, McWeeney S, Britt W, Nelson JA. 2014. Cytomegalovirus miRNAs target secretory pathway genes to facilitate formation of the virion assembly compartment and reduce cytokine secretion. Cell Host Microbe 15:363–373. doi: 10.1016/j.chom.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Das S, Ortiz DA, Gurczynski SJ, Khan F, Pellett PE. 2014. Identification of human cytomegalovirus genes important for biogenesis of the cytoplasmic virion assembly complex. J Virol 88:9086–9099. doi: 10.1128/JVI.01141-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahlqvist J, Mocarski E. 2011. Cytomegalovirus UL103 controls virion and dense body egress. J Virol 85:5125–5135. doi: 10.1128/JVI.01682-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.To A, Bai Y, Shen A, Gong H, Umamoto S, Lu S, Liu F. 2011. Yeast two hybrid analyses reveal novel binary interactions between human cytomegalovirus-encoded virion proteins. PLoS One 6:e17796. doi: 10.1371/journal.pone.0017796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fischer D. 2012. Dissecting functional motifs of the human cytomegalovirus tegument protein pUL71. Ph.D. thesis. University of Ulm, Ulm, Germany. [Google Scholar]
- 9.Roller RJ, Fetters R. 2015. The herpes simplex virus 1 UL51 protein interacts with the UL7 protein and plays a role in its recruitment into the virion. J Virol 89:3112–3122. doi: 10.1128/JVI.02799-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanaka M, Sata T, Kawaguchi Y. 2008. The product of the herpes simplex virus 1 UL7 gene interacts with a mitochondrial protein, adenine nucleotide translocator 2. Virol J 5:125. doi: 10.1186/1743-422X-5-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roux KJ, Kim DI, Raida M, Burke B. 2012. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol 196:801–810. doi: 10.1083/jcb.201112098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zybailov B, Mosley AL, Sardiu ME, Coleman MK, Florens L, Washburn MP. 2006. Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J Proteome Res 5:2339–2347. doi: 10.1021/pr060161n. [DOI] [PubMed] [Google Scholar]
- 13.Mellacheruvu D, Wright Z, Couzens AL, Lambert JP, St-Denis NA, Li T, Miteva YV, Hauri S, Sardiu ME, Low TY, Halim VA, Bagshaw RD, Hubner NC, Al-Hakim A, Bouchard A, Faubert D, Fermin D, Dunham WH, Goudreault M, Lin ZY, Badillo BG, Pawson T, Durocher D, Coulombe B, Aebersold R, Superti-Furga G, Colinge J, Heck AJ, Choi H, Gstaiger M, Mohammed S, Cristea IM, Bennett KL, Washburn MP, Raught B, Ewing RM, Gingras AC, Nesvizhskii AI. 2013. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat Methods 10:730–736. doi: 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovic M, Bork P, von Mering C. 2009. STRING 8—a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res 37:D412–D416. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia O, Saveanu C, Cline M, Fromont-Racine M, Jacquier A, Schwikowski B, Aittokallio T. 2007. GOlorize: a Cytoscape plug-in for network visualization with Gene Ontology-based layout and coloring. Bioinformatics 23:394–396. doi: 10.1093/bioinformatics/btl605. [DOI] [PubMed] [Google Scholar]
- 17.Nozawa N, Daikoku T, Yamauchi Y, Takakuwa H, Goshima F, Yoshikawa T, Nishiyama Y. 2002. Identification and characterization of the UL7 gene product of herpes simplex virus type 2. Virus Genes 24:257–266. doi: 10.1023/A:1015332716927. [DOI] [PubMed] [Google Scholar]
- 18.Gurczynski SJ, Das S, Pellett PE. 2014. Deletion of the human cytomegalovirus US17 gene increases the ratio of genomes per infectious unit and alters regulation of immune and endoplasmic reticulum stress response genes at early and late times after infection. J Virol 88:2168–2182. doi: 10.1128/JVI.02704-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Votteler J, Sundquist WI. 2013. Virus budding and the ESCRT pathway. Cell Host Microbe 14:232–241. doi: 10.1016/j.chom.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liegeois F, Lafay B, Formenty P, Locatelli S, Courgnaud V, Delaporte E, Peeters M. 2009. Full-length genome characterization of a novel simian immunodeficiency virus lineage (SIVolc) from olive colobus (Procolobus verus) and new SIVwrcPbb strains from western red colobus (Piliocolobus badius badius) from the Tai Forest in Ivory Coast. J Virol 83:428–439. doi: 10.1128/JVI.01725-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Battista MC, Bergamini G, Boccuni MC, Campanini F, Ripalti A, Landini MP. 1999. Expression and characterization of a novel structural protein of human cytomegalovirus, pUL25. J Virol 73:3800–3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baldick CJ Jr, Shenk T. 1996. Proteins associated with purified human cytomegalovirus particles. J Virol 70:6097–6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Win TZ, Goodwin A, Hickson ID, Norbury CJ, Wang SW. 2004. Requirement for Schizosaccharomyces pombe Top3 in the maintenance of chromosome integrity. J Cell Sci 117:4769–4778. doi: 10.1242/jcs.01351. [DOI] [PubMed] [Google Scholar]
- 24.Saffert RT, Kalejta RF. 2006. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J Virol 80:3863–3871. doi: 10.1128/JVI.80.8.3863-3871.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Browne EP, Shenk T. 2003. Human cytomegalovirus UL83-coded pp65 virion protein inhibits antiviral gene expression in infected cells. Proc Natl Acad Sci U S A 100:11439–11444. doi: 10.1073/pnas.1534570100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Varnum SM, Streblow DN, Monroe ME, Smith P, Auberry KJ, Pasa-Tolic L, Wang D, Camp DG, Rodland K, Wiley S, Britt W, Shenk T, Smith RD, Nelson JA. 2004. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J Virol 78:10960–10966. doi: 10.1128/JVI.78.20.10960-10966.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xie M, Xuan B, Shan J, Pan D, Sun Y, Shan Z, Zhang J, Yu D, Li B, Qian Z. 2015. Human cytomegalovirus exploits interferon-induced transmembrane proteins to facilitate morphogenesis of the virion assembly compartment. J Virol 89:3049–3061. doi: 10.1128/JVI.03416-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nickel W, Brugger B, Wieland FT. 2002. Vesicular transport: the core machinery of COPI recruitment and budding. J Cell Sci 115:3235–3240. [DOI] [PubMed] [Google Scholar]
- 29.Misteli T. 1996. Molecular mechanisms in the disassembly and reassembly of the mammalian Golgi apparatus during M-phase. FEBS Lett 389:66–69. doi: 10.1016/0014-5793(96)00518-2. [DOI] [PubMed] [Google Scholar]
- 30.Brandstaetter H, Kendrick-Jones J, Buss F. 2012. Molecular roles of Myo1c function in lipid raft exocytosis. Commun Integr Biol 5:508–510. doi: 10.4161/cib.21201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brandstaetter H, Kishi-Itakura C, Tumbarello DA, Manstein DJ, Buss F. 2014. Loss of functional MYO1C/myosin 1c, a motor protein involved in lipid raft trafficking, disrupts autophagosome-lysosome fusion. Autophagy 10:2310–2323. doi: 10.4161/15548627.2014.984272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen XW, Leto D, Chiang SH, Wang Q, Saltiel AR. 2007. Activation of RalA is required for insulin-stimulated Glut4 trafficking to the plasma membrane via the exocyst and the motor protein Myo1c. Dev Cell 13:391–404. doi: 10.1016/j.devcel.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 33.Honeychurch KM, Yang G, Jordan R, Hruby DE. 2007. The vaccinia virus F13L YPPL motif is required for efficient release of extracellular enveloped virus. J Virol 81:7310–7315. doi: 10.1128/JVI.00034-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bardens A, Doring T, Stieler J, Prange R. 2011. Alix regulates egress of hepatitis B virus naked capsid particles in an ESCRT-independent manner. Cell Microbiol 13:602–619. doi: 10.1111/j.1462-5822.2010.01557.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee CP, Liu PT, Kung HN, Su MT, Chua HH, Chang YH, Chang CW, Tsai CH, Liu FT, Chen MR. 2012. The ESCRT machinery is recruited by the viral BFRF1 protein to the nucleus-associated membrane for the maturation of Epstein-Barr Virus. PLoS Pathog 8:e1002904. doi: 10.1371/journal.ppat.1002904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhai Q, Landesman MB, Robinson H, Sundquist WI, Hill CP. 2011. Identification and structural characterization of the ALIX-binding late domains of simian immunodeficiency virus SIVmac239 and SIVagmTan-1. J Virol 85:632–637. doi: 10.1128/JVI.01683-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Freed EO. 2002. Viral late domains. J Virol 76:4679–4687. doi: 10.1128/JVI.76.10.4679-4687.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fraile-Ramos A, Pelchen-Matthews A, Risco C, Rejas MT, Emery VC, Hassan-Walker AF, Esteban M, Marsh M. 2007. The ESCRT machinery is not required for human cytomegalovirus envelopment. Cell Microbiol 9:2955–2967. doi: 10.1111/j.1462-5822.2007.01024.x. [DOI] [PubMed] [Google Scholar]
- 39.Tandon R, AuCoin DP, Mocarski ES. 2009. Human cytomegalovirus exploits ESCRT machinery in the process of virion maturation. J Virol 83:10797–10807. doi: 10.1128/JVI.01093-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patel AH, MacLean JB. 1995. The product of the UL6 gene of herpes simplex virus type 1 is associated with virus capsids. Virology 206:465–478. doi: 10.1016/S0042-6822(95)80062-X. [DOI] [PubMed] [Google Scholar]
- 41.Fuchs W, Granzow H, Klopfleisch R, Klupp BG, Rosenkranz D, Mettenleiter TC. 2005. The UL7 gene of pseudorabies virus encodes a nonessential structural protein which is involved in virion formation and egress. J Virol 79:11291–11299. doi: 10.1128/JVI.79.17.11291-11299.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dijkstra JM, Fuchs W, Mettenleiter TC, Klupp BG. 1997. Identification and transcriptional analysis of pseudorabies virus UL6 to UL12 genes. Arch Virol 142:17–35. doi: 10.1007/s007050050056. [DOI] [PubMed] [Google Scholar]
- 43.Schmitt J, Keil GM. 1996. Identification and characterization of the bovine herpesvirus 1 UL7 gene and gene product which are not essential for virus replication in cell culture. J Virol 70:1091–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]