

ABSTRACT
It is well established that glycosaminoglycans (GAGs) function as attachment factors for human metapneumovirus (HMPV), concentrating virions at the cell surface to promote interaction with other receptors for virus entry and infection. There is increasing evidence to suggest that multiple receptors may exhibit the capacity to promote infectious entry of HMPV into host cells; however, definitive identification of specific transmembrane receptors for HMPV attachment and entry is complicated by the widespread expression of cell surface GAGs. pgsA745 Chinese hamster ovary (CHO) cells are deficient in the expression of cell surface GAGs and resistant to HMPV infection. Here, we demonstrate that the expression of the Ca2+-dependent C-type lectin receptor (CLR) DC-SIGN (CD209L) or L-SIGN (CD209L) rendered pgsA745 cells permissive to HMPV infection. Unlike infection of parental CHO cells, HMPV infection of pgsA745 cells expressing DC-SIGN or L-SIGN was dynamin dependent and inhibited by mannan but not by pretreatment with bacterial heparinase. Parental CHO cells expressing DC-SIGN/L-SIGN also showed enhanced susceptibility to dynamin-dependent HMPV infection, confirming that CLRs can promote HMPV infection in the presence or absence of GAGs. Comparison of pgsA745 cells expressing wild-type and endocytosis-defective mutants of DC-SIGN/L-SIGN indicated that the endocytic function of CLRs was not essential but could contribute to HMPV infection of GAG-deficient cells. Together, these studies confirm a role for CLRs as attachment factors and entry receptors for HMPV infection. Moreover, they define an experimental system that can be exploited to identify transmembrane receptors and entry pathways where permissivity to HMPV infection can be rescued following the expression of a single cell surface receptor.
IMPORTANCE On the surface of CHO cells, glycosaminoglycans (GAGs) function as the major attachment factor for human metapneumoviruses (HMPV), promoting dynamin-independent infection. Consistent with this, GAG-deficient pgaA745 CHO cells are resistant to HMPV. However, expression of DC-SIGN or L-SIGN rendered pgsA745 cells permissive to dynamin-dependent infection by HMPV, although the endocytic function of DC-SIGN/L-SIGN was not essential for, but could contribute to, enhanced infection. These studies provide direct evidence implicating DC-SIGN/L-SIGN as an alternate attachment factor for HMPV attachment, promoting dynamin-dependent infection via other unknown receptors in the absence of GAGs. Moreover, we describe a unique experimental system for the assessment of putative attachment and entry receptors for HMPV.
INTRODUCTION
Human metapneumovirus (HMPV) can cause both upper and lower respiratory tract infections and is most commonly associated with disease in infants and young children but also in elderly and immunocompromised patients (reviewed in reference 1). HMPV is a member of the genus Metapneumovirus within the family Paramyxoviridae and shares structural, epidemiological, and clinical features with respiratory syncytial virus (RSV), a closely related paramyxovirus.
Airway epithelial cells are the predominant target of HMPV infection (2, 3); however, infection of airway macrophages may contribute to virus propagation during the early phase of HMPV infection (4). HMPV also infects dendritic cells (DCs), and this may play a role in immune evasion by interfering with the function of DCs, including their ability to activate CD4+ T cells (5–8). HMPV expresses 3 envelope glycoproteins, the putative attachment (G) protein, the F protein, and the small hydrophobic (SH) protein. For cellular infection to occur, HMPV must first attach to the cell surface and then fuse the viral and cellular membranes, a process that is driven by the F protein (reviewed in reference 9). To date, there is no evidence of a role for the SH protein in viral entry, and mutants lacking a functional SH protein replicate efficiently in vitro and in vivo (10, 11). Of interest, deletion mutants of HMPV that do not express the G protein also replicate efficiently in cell culture (11), suggesting that the F protein of HMPV can perform both attachment and fusion functions in the absence of the G protein. However, while HMPV lacking the G protein could infect African green monkeys, replication was attenuated compared to the wild-type virus, indicating that the G protein is required for full virulence in vivo (12). Thus, the G protein of HMPV might bind to cellular receptors expressed by only certain cell types, or it may mediate an entirely different function in the virus life cycle.
Recent studies suggest that HMPV can interact with multiple binding partners to facilitate virus attachment and subsequent entry into target cells. An integrin binding recognition sequence, Arg-Gly-Asp (RGD), has been identified in the F proteins of all known HMPV strains (13), and the HMPV F protein is capable of interacting with multiple RGD binding integrins (13–16). While not essential for virus attachment, interactions between the F protein and integrins are required to promote efficient HMPV entry and infection, at least for certain cell types (13, 14, 16). Of interest, Chang et al. reported that efficient HMPV infection of Vero and CHO-K1 cells depends on the expression of a proteinaceous receptor (17), which, in contrast to integrins, is sensitive to trypsin and proteinase K digestion (17, 18). Thus, HMPV entry and infection are likely to involve more than one cell surface receptor, and these receptors may be distinct for different cell types. Moreover, receptors utilized by HMPV to infect epithelial cells may differ from those involved in infectious entry into immune cells such as macrophages and DCs.
Glycosaminoglycans (GAGs) are long, unbranched polysaccharides that consist of repeating disaccharide subunits, of which heparan sulfate (HS) and, to a lesser extent, chondroitin sulfate (CS) can act as attachment factors for a range of human viruses (reviewed in reference 19). A number of studies have shown that HMPV infection can be inhibited in the presence of soluble GAGs by the enzymatic removal of GAGs from the cell surface or by the use of GAG-deficient cell lines (17, 20–22). For example, the G protein of certain lineages of HMPV can interact with GAGs (20, 21), and this has been proposed to modulate HMPV infection. However, Chang et al. reported that the initial interactions between HMPV and target cells occurred via the recognition of HS moieties on the cell surface by the F protein of HMPV (17). In the absence of HS, HMPV binding to and infection of CHO cells, CHO cell mutants, and Vero cells were drastically reduced, indicating that integrins (and/or other cell surface receptors) do not function efficiently to promote attachment and entry in the absence of cell surface GAGs (17). Thus, GAGs appear to be the first cell surface receptors engaged by HMPV. However, the specific receptors and mechanisms required to trigger subsequent fusion and cell entry are yet to be fully elucidated. HMPV entry may not be limited to fusion at the plasma membrane, as a growing body of evidence indicates that infectious entry can also occur following virus internalization via endocytosis and fusion with endosomal membranes (15, 23).
CHO pgsA745 cells represent a mutant of the CHO cell line lacking xylosyltransferase activity and are therefore deficient in the expression of cell surface GAGs (24). Moreover, multiple studies have demonstrated that GAG-deficient pgsA745 cells do not bind HMPV efficiently and are resistant to HMPV infection (17, 20, 21). Therefore, we hypothesized that putative receptors could be expressed in pgsA745 cells to assess their ability to function as attachment and/or entry receptors for HMPV in the absence of interactions with cell surface GAGs. C-type lectin receptors (CLRs) have been reported to bind to glycans expressed on the surface glycoproteins of a range of different viruses. In many studies, recognition by cell surface CLR has been associated with enhanced susceptibility to viral infection (reviewed in reference 25). Recently, Le Nouen et al. reported that macropinocytosis represented the predominant route for infectious entry of HMPV into monocyte-derived dendritic cells (MDDCs) and presented data implicating DC-SIGN-mediated endocytosis as an alternative pathway for HMPV entry and infection (26).
Here, we have characterized interactions between HMPV and CHO cells deficient in either GAGs (pgsA745) or sialic acid (SIA) (Lec2), confirming that pgsA745 cells did not bind HMPV efficiently and were resistant to HMPV infection. We then utilized pgsA745 cells to develop an experimental model to assess the ability of human DC-SIGN and L-SIGN to function as attachment and entry receptors for HMPV. Our studies demonstrate that stable transfected pgsA745 cell lines expressing either DC-SIGN or L-SIGN were able to bind HMPV in a Ca2+-dependent manner and showed enhanced susceptibility to dynamin-dependent HMPV infection. Moreover, DC-SIGN/L-SIGN could augment HMPV infection when expressed by CHO-K1 cells expressing cell surface GAGs.
MATERIALS AND METHODS
Cell lines.
CHO-Pro5 cells and the SIA-deficient mutant cell line Lec2 (27, 28), derived from CHO-Pro5 cells, were obtained from the American Type Culture Collection (ATCC), Manassas, VA, USA. CHO-K1 cells and the GAG-deficient mutant cell line pgsA745 (24), derived from CHO-K1 cells, were obtained from Mark Hulett, Department of Biochemistry, La Trobe University, Melbourne, Australia. All CHO cell lines were cultured in Dulbecco's modified Eagle's medium–nutrient mixture Ham F-12 medium (DMEM–F-12; Gibco BRL, New York, NY, USA) supplemented with 10% (vol/vol) fetal calf serum (FCS; JRH Biosciences, KS, USA), 4 mM l-glutamine, 100 IU penicillin, 10 μg of streptomycin/ml, nonessential amino acids (Gibco BRL, New York, NY, USA), and 50 μM β-mercaptoethanol. LLC-MK2 (ATCC CLL7.1) cells were maintained in Opti-MEM (Gibco BRL) supplemented with 5% FCS.
Viruses.
The CAN97-83 HMPV strain was propagated in LLC-MK2 cells at 32°C in the presence of 5 μg/ml tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK) trypsin (Worthington Biochemical, NJ, USA), based on methods reported previously (11). Virus was concentrated from cell-free supernatants by ultracentrifugation on a 30 to 60% (wt/vol) sucrose step gradient for 2 h at 26,000 rpm at 4°C by using an SW28 rotor (Beckman Coulter, CA, USA). The virus-containing band at the 30 to 60% interface was collected and diluted, and virus particles were pelleted by centrifugation for 90 min at 25,000 rpm by using an SW28 rotor (Beckman Coulter). The virus pellet was resuspended in serum-free Opti-MEM and frozen in aliquots at −80°C prior to use. Titers of infectious HMPV were determined by titration on LLC-MK2 monolayers, followed by immunofluorescence staining with a monoclonal antibody (MAb) against the HMPV N protein (MAB80121; Merck Millipore, MA), and titers are expressed as fluorescent-focus units (FFU) per milliliter. RSV strain A2 was propagated in HEp-2 cells by standard procedures (29). Titers of infectious RSV were determined by titration on CV-1 cells, followed by immunofluorescence staining with a MAb against the RSV fusion (F) protein (clone 133-1H; Millipore, MA, USA), and titers are expressed as FFU per milliliter. The influenza A virus (IAV) strain used in this study was BJx109 (H3N2), a high-yield reassortant of A/PR/8/34 (PR8) (H1N1) and A/Beijing/353/89 (H3N2) which expresses the H3N2 surface glycoproteins. IAV was grown in 10-day-old embryonated eggs by using standard procedures (30). Titers of infectious IAV were determined by a plaque assay on Madin-Darby canine kidney (MDCK) cells and are expressed as PFU per milliliter (30).
Generation of pgsA745 cells expressing DC-SIGN/L-SIGN or DC-SIGN/L-SIGN with deletions in the cytoplasmic tail.
Wild-type (WT) DC-SIGN and L-SIGN pcDNA3.1/V5-His-TOPO expression vectors were described previously (31). PCR-generated deletion (DEL) mutants of DC-SIGN or L-SIGN lacking the last 33 or 41 amino acids of the cytoplasmic tail, respectively, were also inserted into pcDNA3.1/V5-His-TOPO vectors as described previously (32). Nucleotide sequences of all DC-SIGN/L-SIGN constructs were confirmed by sequence analysis.
pgsA745 cells were transfected with pcDNA3.1/V5-His-TOPO expression vectors containing WT or DEL DC-SIGN/L-SIGN by using FuGene 6 transfection reagent (Roche Diagnostic, Switzerland) according to the manufacturer's instructions. As controls, CHO-K1 and pgsA745 cells were transfected with pcDNA3.1/V5-His-TOPO expressing cytoplasmic hen egg ovalbumin (OVA) lacking the sequence for cell surface trafficking, as described previously (31). Stable transfectants expressing wild-type (pgs-DC-SIGN-WT and pgs-L-SIGN-WT) or deleted (pgs-DC-SIGN-DEL and pgs-L-SIGN-DEL) forms of DC-SIGN/L-SIGN or cytoplasmic OVA (CHO-ctrl and pgs-ctrl) were selected in the presence of 1 mg/ml Geneticin (G418; Invitrogen). Transfected cells were screened for cell surface CLR expression by using a fluorescein-conjugated cross-reactive MAb (clone 120612; R&D Systems, Inc.) that detects both DC-SIGN and L-SIGN, and single cells with high cell surface CLR expression levels were isolated by using a FACSAria cell sorter (BD Biosciences) and expanded in culture for use in experiments.
Binding of lectins and viruses to cells.
To examine the binding of lectins and viruses to the cell surface, adherent cell lines were detached from tissue culture flasks by using 0.75 mM EDTA in phosphate-buffered saline (PBS) (0.05 M Na2HPO4·12H2O, 0.14 M NaCl [pH 7.4]) and washed and resuspended in binding buffer (0.05 M Tris-HCl in 0.15 M NaCl [pH 7.4] containing 10 mM CaCl2 and 1 mg/ml of bovine serum albumin [BSA]). All binding assays were performed at 4°C, and cells were washed in ice-cold binding buffer between incubations. In some experiments, CaCl2 was omitted from the binding buffer and replaced with 5 mM EDTA, as indicated.
Levels of cell surface SIA expression were determined by using the plant lectin Maackia amurensis agglutinin II (MAA; EY Laboratories, CA, USA), which binds specifically to α(2,3)-linked SIA. Cells were incubated with 5 μg/ml biotinylated MAA and washed, and bound MAA was detected with streptavidin conjugated to allophycocyanin (APC). Cell surface GAGs were detected by using mouse anti-human HS MAb 10E4 (US Biological, MA, USA), followed by fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse Ig (Millipore, MA). To examine HMPV binding, cells were incubated with 5 μg/ml of purified HMPV, and bound virus was detected by using a MAb specific for the F protein of HMPV (MAB80123; Merck Millipore, MA), followed by FITC-conjugated goat anti-mouse Ig. In some experiments, cell suspensions were pretreated for 30 min at 37°C with 200 mU/ml of broad-spectrum bacterial sialidase from Vibrio cholerae (type III; Sigma-Aldrich, MO, USA) or with 10 U/ml of bacterial heparinase from Flavobacterium heparinum (type I; Sigma-Aldrich, MO, USA) prior to use in binding assays. For all assays, cell surface binding of lectins, MAbs, and virus was then determined by using a FACSCalibur instrument (BD Biosciences, CA, USA).
Virus infection assays.
Cells were seeded into 8-well chamber slides (LabTek; Nunc, USA) and infected with IAV, HMPV, or RSV as described previously (25). Briefly, after culture overnight, slides with confluent cell monolayers were washed and incubated with virus in serum-free medium for 1 h at 37°C (to allow virus binding and entry). Unless otherwise stated, cells were infected with 107 PFU of IAV (multiplicity of infection [MOI] = 100), 3 × 105 FFU of HMPV (MOI = 3), and 2.5 × 105 FFU of RSV (MOI = 2.5) in 100 μl of serum-free medium. After removal of the virus inoculum, cells were washed and incubated for a further 6 to 8 h (for IAV) or 18 to 20 h (for HMPV/RSV) at 37°C in serum-free medium. Slides were washed with PBS and then fixed with 80% (vol/vol) acetone. IAV-infected cells were detected by immunofluorescence staining using MAb MP3.10g2.1C7 (WHO Collaborating Centre for Reference and Research on Influenza, Melbourne, Australia) specific for the nucleoprotein (NP) of type A influenza viruses, followed by FITC-conjugated goat anti-mouse Ig. HMPV- and RSV-infected cells were detected by using MAbs raised to the HMPV N protein (MAB80121; Merck Millipore, MA) and the RSV F protein (clone 133-1H; Millipore, MA, USA), respectively, followed by FITC-conjugated goat anti-mouse Ig. The percentage of infected cells was determined by costaining with 4′,6-diamidino-2-phenylindole (DAPI) and counting the total number of cells versus FITC-positive cells at a ×100 magnification. A minimum of four random fields were selected for counting, assessing at least 200 cells for each sample. In some experiments, cell monolayers were treated with 200 mU of bacterial sialidase or 10 U/ml of bacterial heparinase I for 30 min at 37°C and washed prior to virus infection. In other experiments, cells were pretreated with 10 mg/ml mannan (Sigma-Aldrich, MO, USA), 50 μM dynasore (Sigma-Aldrich, MO, USA), 20 mM NH4Cl, or 20 μM bafilomycin A1 (Baf-A1; Sigma-Aldrich, MO, USA) in serum-free medium for 30 min at 37°C. Cells were then infected with virus in the presence of the appropriate inhibitor for 1 h at 37°C, washed, and cultured for an additional 6 to 8 h (for IAV) or 18 to 20 h (for HMPV/RSV) in the presence of the inhibitor, prior to fixing and immunofluorescence staining.
Binding of recombinant human DC-SIGN/L-SIGN to HMPV by an enzyme-linked immunosorbent assay.
For enzyme-linked immunosorbent assays (ELISAs), 96-well polyvinyl microtiter plates were coated with 1 μg/ml of purified HMPV overnight and then blocked for 2 h at room temperature (RT) with BSA (10 mg/ml). Wells were washed with Tris-buffered saline (TBS) containing 0.05% (vol/vol) Tween 20 (TBST) and incubated at RT for 2 h with 1 μg/ml of recombinant human DC-SIGN or L-SIGN expressing a human Fc receptor tag (DC-SIGN/L-SIGN-Fc; R&D Systems, MN, USA) in TBS containing 5 mg/ml BSA and supplemented with either 20 mM CaCl2 or 10 mM EDTA. Wells were washed in TBST containing either 20 mM CaCl2 or 10 mM EDTA and then incubated with goat anti-human IgG conjugated to horseradish peroxidase (HRP; Millipore, MA, USA). Binding of DC-SIGN/L-SIGN-Fc to purified HMPV was detected by using 3,3′,5,5′-tetramethylbenzidine (TMB) substrate reagent (Perkin-Elmer, CT, USA), and the reaction was stopped with 1 M HCl.
Internalization of a DC-SIGN/L-SIGN-specific MAb by pgsA745 cells expressing WT or DEL forms of DC-SIGN/L-SIGN.
Cells seeded into duplicate 8-well chamber slides were cultured overnight, washed in serum-free medium, and incubated with MAb (clone 120612) (cross-reactive with DC-SIGN and L-SIGN) for 30 min at 4°C (to allow binding to the cell surface). Cells were then washed, and one chamber slide was moved to a temperature of 37°C for an additional 20 min (to allow MAb internalization and entry), while the duplicate chamber slide was held at 4°C. After this time, slides were washed with PBS and fixed in 80% (vol/vol) acetone in water. For binding and internalization of the MAb, detection of the MAb was enhanced by using Alexa Fluor 488 goat anti-mouse Ig (Life Technologies, Eugene, OR, USA). All slides were costained with DAPI to detect cell nuclei and examined by confocal microscopy. Images were acquired with a Zeiss LSM700 confocal microscope in conjunction with Zen2012 software.
Statistical analysis.
Graphing and statistical analysis of data were performed by using GraphPad Prism (GraphPad Software, San Diego, CA, USA). Unpaired Student's t test was used to compare two sets of data. For comparison of three or more sets of values, the data were analyzed by one-way analysis of variance (ANOVA) (nonparametric), followed by post hoc analysis using Tukey's multiple-comparison test. P values of ≤0.05 were considered significant.
RESULTS
Expression of SIA and GAGs by CHO-K1, pgsA745, CHO-Pro5, and Lec2 cells.
The Lec2 CHO cell line is a mutant of the CHO-Pro5 cell line that is deficient in terminal SIA due to a defect in the cytidine monophosphate (CMP)-SIA transporter (28, 33), whereas the pgsA745 CHO cell line is a mutant of the CHO-K1 cell line that lacks xylosyltransferase activity and therefore does not express cell surface GAGs (24). Initial studies were aimed at confirming the phenotype of parental and mutant CHO cell lines. As parental CHO lines (CHO-Pro5 and CHO-K1) express α(2,3)-linked but not α(2,6)-linked SIA, due to a deficiency in galactoside α2,6-sialyltransferase (34, 35), we used the plant lectin MAA to detect α(2,3)-linked SIA on the surface of CHO-Pro5, Lec2, CHO-K1, and pgsA745 cells. Representative histograms of MAA binding are shown in Fig. 1Ai, and geometric means representing fluorescence from triplicate samples are shown in Fig. 1Aii. CHO-K1 and pgsA745 cells bound the highest levels of MAA, with binding to CHO-Pro5 cells being significantly reduced compared to binding to CHO-K1 cells in multiple experiments. MAA binding to SIA-deficient Lec2 cells was negligible and markedly reduced compared to binding to all other cell lines (Fig. 1A). As expected, desialylation of cells with bacterial sialidase led to major and significant reductions in cell surface SIA levels and therefore in MAA binding to CHO-K1, pgsA745, and CHO-Pro5 cells (Fig. 1A). Desialylation of Lec2 cells was also associated with a modest, but not significant, reduction in MAA binding (Fig. 1A), consistent with previous studies reporting that SIA levels are drastically reduced, but not absent, compared to those in CHO-Pro5 cells (28, 36).
FIG 1.

Attachment to and infection of CHO-K1, pgsA745, CHO-Pro5, and Lec2 cells by HMPV. (A) Binding of biotinylated MAA to SIA on CHO-K1 (K1), pgsA745 (pgs), CHO-Pro5 (Pro5), and Lec2 cells was determined by flow cytometry. Black histograms represent cells incubated in medium alone, white histograms represent cells incubated in medium supplemented with 100 mU/ml of bacterial sialidase, and gray histograms represent cells stained with FITC-conjugated rabbit anti-mouse IgM/IgG alone [No MAA (−)]. (i) Representative histograms of triplicate samples. (ii) MAA binding represented as the geometric mean fluorescence intensity (±1 standard error of the means) of histograms for triplicate samples. * indicates that binding to sialidase-treated cells is significantly reduced compared to binding to mock-treated cells, and # indicates that binding to CHO-Pro5 mock cells is significantly reduced compared to binding to CHO-K1 mock cells (P < 0.05, as determined by one-way ANOVA followed by Tukey's post hoc test). ns, not significant. (B) Anti-HS MAb binding to GAGs on different CHO cell lines. Cells were incubated in medium alone (black histograms) or medium supplemented with 10 U/ml of bacterial heparinase I (white histograms) and washed, and binding of anti-HS MAb was determined. Gray histograms represent cells stained with FITC-conjugated rabbit anti-mouse IgM/IgG alone [No HS mAb (−)]. (i) Representative histograms for triplicate samples. (ii) Anti-HS MAb binding represented as the geometric mean fluorescence intensity (±1 standard error of the mean) of histograms for triplicate samples. * indicates that anti-HS MAb binding to heparinase-treated cells is significantly different from binding to mock-treated cells (P < 0.05, as determined by one-way ANOVA followed by Tukey's post hoc test). (C) Binding of HMPV to CHO cell lines. Cells were incubated in serum-free medium alone (black histograms) or pretreated with either 100 mU/ml of bacterial sialidase (white histograms) or 10 U/ml of bacterial heparinase I (light gray histograms) and then washed, and virus binding was determined. Dark gray histograms represent cells stained with primary and secondary antibodies only (no virus). (i) Representative histograms of HMPV binding for triplicate samples. (ii) HMPV binding represented as the geometric mean fluorescence intensity (±1 standard error of the mean) of histograms for triplicate samples. * indicates that HMPV binding to heparinase-treated cells is significantly different from binding to mock- and sialidase-treated cells (P < 0.05, as determined by one-way ANOVA followed by Tukey's post hoc test). (D) Infection of CHO cell lines by HMPV. Cell monolayers were incubated with 3 × 105 FFU (MOI of 3 FFU/cell) of HMPV for 1 h at 37°C, washed, incubated for a further 1 h and 17 h, and then fixed and stained for the expression of newly synthesized viral N and DAPI (to stain cell nuclei for total cell counts). (i) Representative images of HMPV-infected CHO-K1 and pgsA745 cells at 2 and 18 h were taken by confocal microscopy. (ii) The percentage of HMPV-infected cells was determined by counting the total number of DAPI+ cells versus FITC+ cells at a ×100 magnification at 18 h postinfection. Data show mean percentages of infection (±1 standard error of the mean) from a minimum of four independent fields per chamber and are representative of results from three independent experiments. * indicates results that are significantly different compared to mock- and sialidase-treated cells for HMPV.
To assess cell surface expression of GAGs, cell lines were incubated with a mouse MAb specific for HS (10E4 epitope), followed by FITC-conjugated rabbit anti-mouse IgM/IgG. Representative histograms are shown in Fig. 1Bi, and the geometric means for triplicate samples are shown in Fig. 1Bii. As expected, HS was expressed at high levels by CHO-K1, CHO-Pro5, and Lec2 cells but not by pgsA745 cells. As expected, pretreatment of CHO-K1, CHO-Pro5, and Lec2 cells with bacterial heparinase I resulted in major reductions in cell surface expression of HS but did not alter expression by pgsA745 cells, confirming the severe deficiency in the cell surface expression of GAGs in these cells (24).
Ability of HMPV to bind to and infect CHO-K1, pgsA745, CHO-Pro5, and Lec2 cells.
Cell surface GAGs have been implicated as the major attachment factor for HMPV (17, 20–22). To confirm these findings and to determine the role of cell surface SIA in HMPV attachment and entry, we examined the ability of HMPV to bind to parental and mutant CHO cell lines. HMPV bound efficiently to the surface of CHO-K1, CHO-Pro5, and Lec2 cells, and binding was abrogated by pretreatment of cells with bacterial heparinase but not sialidase (Fig. 1Ci). Statistical analyses confirmed that for each of these cell lines, heparinase treatment reduced HMPV attachment significantly, whereas sialidase treatment did not (Fig. 1Cii). HMPV did not bind efficiently to GAG-deficient psgA745 cells, and pretreatment with bacterial sialidase or heparinase did not significantly affect binding (Fig. 1Ci and ii). In additional experiments, we confirmed that pretreatment of CHO-K1 cells with the same concentration of sialidase reduced the binding of purified IAV by >90% (data not shown). Thus, the attachment of HMPV to CHO cell lines used in these studies occurs primarily via the recognition of cell surface GAGs.
Next, we determined the susceptibility of CHO cell lines to infection by HMPV. In these studies, cell monolayers were inoculated with HMPV (3 × 105 FFU) for 1 h at 37°C. After washing, one set of monolayers was incubated for a further 1 h and then fixed and stained for the expression of viral NP. Duplicate monolayers fixed at 18 h postinfection were also stained for viral NP and examined by immunofluorescence microscopy. Representative images of CHO-K1 and pgsA745 cells infected with HMPV (Fig. 1Di) are shown. Negligible HMPV staining was evident at 2 h postinfection (Fig. 1Di, top), whereas HMPV NP localized to punctate cytoplasmic bodies in CHO-K1 cells at 18 h (Fig. 1Di, bottom). In contrast, few pgsA745 cells showed expression of HMPV NP at 18 h postinfection. These studies confirmed that susceptibility to HMPV infection, as assessed by immunofluorescence at 18 h, corresponds to the detection of newly synthesized viral proteins rather than detection of residual virus inoculum.
To determine the percentage of HMPV-infected cells at 18 h postinfection, DAPI-positive (DAPI+) and fluorescent NP+ cells were counted. As shown in Fig. 1Dii, CHO-K1, CHO-Pro5, and Lec2 cells were all highly susceptible to infection by HMPV, whereas pgsA745 cells were largely resistant. Consistent with HMPV binding data (Fig. 1C), pretreatment of CHO-K1, CHO-Pro5, and Lec2 cells with heparinase, but not with sialidase, abrogated susceptibility to HMPV infection (Fig. 1Dii). Note that pretreatment of CHO-K1 cells with an equivalent concentration of sialidase reduced IAV infection from 80% to <5% when assessed by immunofluorescence at 8 to 10 h postinfection (data not shown). Confirming the findings reported previously by Chang et al. (17), we report that attachment to and infection of CHO cell lines by HMPV strain CAN97-83 are dependent on cell surface GAGs. However, it is currently unclear if GAG-dependent binding and infection are broadly applicable to different HMPV strains, particularly given that binding to GAGs can be acquired as a result of adaptation to growth in tissue culture (reviewed in reference 37). For the purposes of our study, we focus on the expression of putative attachment and entry receptors for HMPV in pgsA745 cells, removing the complicating factor of GAG-dependent interactions that might contribute to infection.
Expression of DC-SIGN or L-SIGN by GAG-deficient pgsA745 CHO cells is associated with Ca2+-dependent binding of HMPV.
A previous study implicated DC-SIGN-mediated recognition of HMPV in entry into DCs (26), although direct binding interactions were not investigated. We first examined the ability of recombinant human DC-SIGN or L-SIGN to bind to HMPV by an ELISA. In these studies, wells coated with purified HMPV were incubated with 1 μg/ml of recombinant DC-SIGN-Fc or L-SIGN-Fc in the presence of 10 mM Ca2+ or 5 mM EDTA, and bound CLRs were detected as described in Materials and Methods. As shown in Fig. 2A, both DC-SIGN-Fc and L-SIGN-Fc bound to HMPV in the presence of Ca2+ but not EDTA, confirming the Ca2+-dependent recognition of HMPV by both CLRs.
FIG 2.

Expression of DC-SIGN or L-SIGN on the surface of GAG-deficient pgsA745 CHO cells is associated with Ca2+-dependent binding of HMPV. (A) ELISA plates coated with purified HMPV were incubated with 1 μg/ml of recombinant DC-SIGN-Fc or L-SIGN-Fc in the presence of 10 mM Ca2+ or 5 mM EDTA. After washing, levels of DC-SIGN/L-SIGN binding were determined as described in Materials and Methods. Data represent the mean optical densities (OD) from triplicate samples (±1 standard error of the mean) and are representative of results from two independent experiments. * indicates that HMPV binding was significantly reduced in the presence of EDTA (P < 0.0001, as determined by unpaired two-tailed Student's t test). (B) Cell surface expression of DC-SIGN or L-SIGN on pgsA745 cells (pgs-DC/L-SIGN) or pgsA745 cells expressing an irrelevant intracellular protein (pgs-ctrl cells) was determined by flow cytometry using a cross-reactive MAb that detects both DC-SIGN and L-SIGN. (C) Binding of HMPV to the surface of CHO-K1-ctrl (K1-ctrl), pgsA745-ctrl (pgs-ctrl), pgs-DC-SIGN, and pgs-L-SIGN cells was determined by flow cytometry in the presence of 10 mM CaCl2 (Ca2+) or 5 mM EDTA. A sample that received no HMPV but was stained with all relevant antibodies was included for comparison (no virus). Representative histograms for triplicate samples are shown. (D) Geometric mean HMPV binding to cells in the presence of 10 mM CaCl2 or 5 mM EDTA (±1 standard error of the mean) from histograms for triplicate samples. Data are representative of results from three independent experiments. * indicates that HMPV binding to pgs-DC-SIGN/L-SIGN cells was significantly reduced with EDTA compared to Ca2+ (P < 0.0001, as determined by two-tailed unpaired Student's t test).
To examine the potential of these CLRs to act as attachment and/or entry receptors for HMPV, we transfected pgsA745 cells with human DC-SIGN or L-SIGN and selected clones showing stable cell surface expression of each receptor. Flow cytometric analyses confirmed the cell surface expression of either DC-SIGN (Fig. 2B, left) or L-SIGN (Fig. 2B, right) on pgsA745-DC-SIGN or pgsA745-L-SIGN cells, respectively, compared to control cells expressing intracellular OVA (pgsA745-ctrl). Stable transfectants expressed higher cell surface levels of L-SIGN than of DC-SIGN, as detected by using a MAb that is cross-reactive with DC-SIGN and L-SIGN.
DC-SIGN and L-SIGN contain C-type CRDs that bind to mannose-rich oligosaccharides in a Ca2+-dependent manner. Therefore, we assessed the ability DC-SIGN and L-SIGN expressed by GAG-deficient pgsA745 cells to bind to HMPV in the presence of Ca2+ or EDTA. Representative histograms of HMPV binding are shown in Fig. 2C, and the mean fluorescence from triplicate samples is shown in Fig. 2D. HMPV bound to K1-ctrl cells, but not to GAG-deficient pgs-ctrl cells, in the presence of Ca2+ or EDTA, consistent with Ca2+-independent recognition of GAGs on the surface of K1-ctrl cells. In contrast, the expression of either DC-SIGN or L-SIGN by pgsA745 cells was associated with enhanced HMPV binding in the presence of Ca2+, and this was abolished in the presence of EDTA (Fig. 2C and D). Thus, DC-SIGN/L-SIGN expressed on the surface of GAG-deficient pgsA745 cells could mediate C-type lectin-mediated recognition of HMPV.
DC-SIGN and L-SIGN facilitate GAG-independent infection of cells by HMPV but not by RSV.
Next, we investigated the ability of DC-SIGN or L-SIGN to rescue the permissivity of pgsA745 cells to infection by HMPV. In these studies, an HMPV inoculum was added to cells for 1 h at 37°C and removed by washing, and cells were then cultured for an additional 17 h. Infected cells were fixed, stained for the HMPV N protein, and examined by immunofluorescence. Consistent with data shown in Fig. 1D, K1-ctrl cells were highly susceptible to HMPV infection, whereas pgs-ctrl cells were largely resistant (Fig. 3A). Moreover, the expression of either DC-SIGN or L-SIGN by pgsA745 cells was associated with a significant enhancement in susceptibility to HMPV infection (Fig. 3A). These findings indicate that the poor ability of HMPV to infect pgsA745 cells does not represent a defect in the intrinsic ability of pgsA745 cells to support HMPV infection but rather that pgsA745 cells lack appropriate cell surface receptors for virus attachment and/or entry. Note that pgs-L-SIGN cells were more susceptible than pgs-DC-SIGN cells to HMPV infection, consistent with higher cell surface expression levels of CLRs on pgs-L-SIGN cells (Fig. 2B). As an alternative approach, flow cytometry confirmed that the expression of either DC-SIGN or L-SIGN by pgsA745 cells was associated with an enhanced susceptibility to infection (Fig. 3B). K1-ctrl, pgs-DC-SIGN, and pgs-L-SIGN cells showed a marked enhancement of the expression of the HMPV N protein at between 2 and 18 h postinfection, confirming that detection at 18 h represents the synthesis of new viral N and therefore true infection by HMPV.
FIG 3.

DC-SIGN and L-SIGN facilitate GAG-independent infection of cells by HMPV but not by RSV. (A and B) Monolayers of K1-ctrl, pgs-ctrl, pgs-DC-SIGN, and pgs-L-SIGN cells were incubated with 3 × 105 FFU (MOI of 3 FFU/cell) of HMPV for 60 min at 37°C and then washed and incubated for a further 17 h. (A) Cells were fixed and stained by immunofluorescence for the expression of newly synthesized viral N as described in Materials and Methods. Data represent the mean percentages of HMPV-infected cells (±1 standard error of the mean) from a minimum of four independent fields per chamber and are representative of results of three independent experiments. * indicates values that are significantly increased compared to values for pgs-ctrl cells (P < 0.05, as determined by one-way ANOVA followed by Tukey's post hoc test). (B) Cells were fixed, permeabilized, stained for expression of the HMPV N protein, and then examined by flow cytometry. Histograms show the expression of the HMPV N protein 2 h and 18 h after exposure to HMPV. The percentages of HMPV-infected cells at 18 h postinfection are indicated. (C) Monolayers were infected with 3 × 105 FFU (MOI of 3 FFU/cell) of RSV as described above and assessed by immunofluorescence at 18 h postinfection. Data represent the mean percentages of RSV-infected cells (±1 standard error of the mean) from a minimum of four independent fields per chamber and are representative of results from two independent experiments. ns, not significantly different compared to pgs-ctrl cells (as determined by one-way ANOVA followed by Tukey's post hoc test). (D and E) Monolayers of K1-ctrl, pgs-DC-SIGN, or pgs-L-SIGN cells were incubated at 37°C for 30 min in serum-free medium with 10 mg/ml mannan (+ mannan) or without mannan (virus only) prior to the addition of HMPV (D) or pretreated with 10 U/ml bacterial heparinase at 37°C for 30 min and washed prior to infection with HMPV (E). Note that K1-ctrl cells were inoculated with either a high dose (MOI = 3 FFU/cell) or a low dose (MOI = 0.3 FFU/cell) of HMPV (i), whereas all other cells were inoculated with only a high dose (ii). Cells were incubated for 1 h at 37°C after virus addition, washed, and then cultured for an additional 17 h in the presence (+ mannan) or absence (virus only) of 10 mg/ml mannan. After this time, cells were fixed and stained by immunofluorescence for the presence of newly synthesized viral N. Data represent the mean percentages of infected cells (±1 standard error of the mean) and are pooled from two independent experiments. * indicates that values for mannan-treated cells are significantly reduced compared to those for untreated cells for pgs-DC-SIGN (P < 0.0001) and pgs-L-SIGN (P < 0.0001) cells, as determined by using unpaired two-tailed Student's t test.
GAGs have been implicated as attachment factors that are essential for the initiation of infection by RSV (38–40), a paramyxovirus closely related to HMPV. Johnson et al. reported that DC-SIGN and L-SIGN bind to the G protein of RSV; however, stable K562, Raji, or NIH 3T3 cell lines expressing either DC-SIGN or L-SIGN did not show enhanced susceptibility to RSV infection (41). We therefore examined the susceptibility GAG-deficient pgsA745 cells expressing DC-SIGN/L-SIGN to infection by RSV. As expected, K1-ctrl cells were susceptible to RSV, whereas GAG-deficient pgs-ctrl cells were resistant (Fig. 3C), confirming the importance of GAGs in mediating RSV infection of CHO cells. Moreover, the expression of cell surface CLRs by pgs-DC-SIGN or pgs-L-SIGN cells did not result in enhanced susceptibility to RSV infection relative to pgs-ctrl cells (Fig. 3C). Together, these data indicate that the expression of DC-SIGN/L-SIGN by GAG-deficient pgsA745 cells can promote infectious entry of HMPV but not RSV.
To determine if HMPV infection of pgsA745 cells expressing DC-SIGN/L-SIGN occurred independently of residual GAGs and involved lectin-mediated recognition of virus, cells were either (i) treated with bacterial heparinase prior to infection or (ii) incubated with mannan, a multivalent inhibitor of DC-SIGN and L-SIGN, prior to and during infection with HMPV. For infections of K1-ctrl cells, we included treatments at the standard inoculum dose (high [MOI = 3 FFU/cell]), resulting in ∼80% infection, as well as a lower inoculum dose (low [MOI = 0.3 FFU/cell]), resulting in ∼25% infection. We rationalized that lower levels of HMPV infection of K1-ctrl cells would be more sensitive to the effects of various inhibitors and treatments used in infection assays. For all other cell lines, the standard inoculum dose was used. At both high and low inoculum doses, HMPV infected K1-ctrl cells, and treatment with bacterial heparinase reduced HMPV infection to <5% (Fig. 3Di). However, pretreatment with heparinase had only negligible effects on HMPV infection of pgs-DC-SIGN or pgs-L-SIGN cells, confirming the GAG-independent infection of these cell lines (Fig. 3Dii). In the presence of mannan, HMPV infection of K1-ctrl cells was largely unaffected (Fig. 3Ei), whereas mannan blocked HMPV infection of both pgsA745-DC-SIGN and pgsA745-L-SIGN cells to significant levels (Fig. 3Eii). Thus, infectious entry of HMPV into CHO-K1 cells is blocked by pretreatment with heparinase but is unaffected in the presence of mannan. In contrast, HMPV infection of pgsA745 cells expressing DC-SIGN/L-SIGN is unaffected by pretreatment with heparinase but is blocked in the presence of mannan.
DC-SIGN/L-SIGN-mediated infection of GAG-deficient pgsA745 cells by HMPV occurs via a dynamin-dependent, pH-independent route.
Following attachment to the cell surface, a number of entry pathways have been implicated in HMPV infection, including direct fusion, macropinocytosis, and endocytosis (reviewed in reference 9). Dynasore is a small-molecule inhibitor of the GTPase dynamin 2, which is essential for endocytic vesicle formation, as it pinches the endosome from the plasma membrane to form early endosomes (42). First, we confirmed that 50 μM dynasore was sufficient to inhibit IAV infection of K1-ctrl cells, as this virus is known to infect CHO cells via dynamin-dependent endocytosis (Fig. 4Aiii). Next, we examined the effects of dynasore on HMPV infection of K1-ctrl cells, compared to pgsA745 cells expressing DC-SIGN or L-SIGN. The addition of dynasore (50 μM) had no effect on HMPV infection of K1-ctrl cells, even when a lower inoculum dose of virus was used, which led to reduced infection levels of K1-ctrl cells (Fig. 4Ai). However, HMPV infection of pgsA745-DC-SIGN and pgsA745-L-SIGN cells was completely blocked in the presence of dynasore (Fig. 4Aii), indicating that CLR-mediated infection of pgsA745 cells expressing DC-SIGN or L-SIGN occurred via dynamin-dependent endocytosis. Of interest, the low levels of HMPV infection in pgs-ctrl cells were also significantly blocked in the presence of dynasore, suggesting that dynamin-dependent endocytosis of HMPV might occur at low levels in this GAG-deficient cell line.
FIG 4.

DC-SIGN/L-SIGN-mediated infection of GAG-deficient pgsA745 cells by HMPV occurs via a dynamin-dependent, pH-independent route. Monolayers of K1-ctrl, pgs-ctrl, pgs-DC-SIGN, or pgs-L-SIGN cells were treated with 50 μM dynasore (+ dynasore) or mock treated (virus only) (A), with 20 mM NH4Cl (+ NH4Cl) or mock treated (virus only) (B), or with 20 μM bafilomycin (+ bafilomycin) or mock treated (virus only) (C) for 30 min at 37°C and then infected with HMPV. Note that K1-ctrl cells were inoculated with either a high dose (MOI = 3 FFU/cell) or a low dose (MOI = 0.3 FFU/cell) of HMPV (i), whereas all other cells were inoculated with only a high dose (ii). Additional monolayers were inoculated with a standard dose of IAV (iii). After incubation with virus, cells were washed, cultured in serum-free medium alone (virus only) or supplemented with either 50 μM dynasore (+ dynasore) or 20 mM NH4Cl (+ NH4Cl) for an additional 7 h (IAV) or 17 h (HMPV), and then fixed and stained by immunofluorescence for the expression of viral NP/N. Data represent the mean percentages of infected cells (±1 standard error of the mean) from a minimum of four independent fields per chamber and are pooled from results of two independent experiments. In panel A, * indicates that HMPV infection is significantly different from infection of untreated cells (virus only) for pgs-ctrl (P = 0.0100), pgs-DC-SIGN (P < 0.0001), and pgs-L-SIGN (P < 0.0001) cells, and # indicates that IAV infection is significantly different from infection of untreated cells (virus only) for K1-ctrl cells (P < 0.0001) (as determined by two-tailed unpaired Student's t test). In panels B and C, # indicates that IAV infection is significantly different from infection of untreated cells (virus only) for K1-ctrl cells (P < 0.0001) (as determined by two-tailed unpaired Student's t test).
IAV enters and infects host cells in a pH-dependent manner, and endosomal acidification is required to induce an irreversible conformational change in viral hemagglutinin (HA), activating its membrane fusion activity and facilitating the release of the viral capsid into the cytoplasm (reviewed in reference 43). Consistent with this, incubation of K1-ctrl cells with 20 mM NH4Cl, a weak base that inhibits endosomal acidification (44), completely inhibited susceptibility to IAV infection (Fig. 4Biii). While HMPV infection of cells is generally considered to occur via pH-independent pathways, some studies have indicated that fusion and/or infectious entry of certain strains can be enhanced under acidic conditions (23, 45, 46). More recently, Mas et al. reported that there was no correlation between the effects of inhibitors of endosomal acidification on infectivity and the sensitivity of the F proteins of different strains to low pH in a syncytium formation assay (47). In our studies, while 20 mM NH4Cl completely inhibited IAV infection of K1-ctrl cells, it had no significant effects on HMPV infection of K1-ctrl cells (Fig. 4Bi) or pgsA745 cells expressing either DC-SIGN or L-SIGN (Fig. 4Bii). Similar results were obtained by using 20 μM bafilomycin A1, an inhibitor of the vacuolar-type H+-ATPase, which also prevents endosomal acidification (48), which potently inhibited IAV infection of K1-ctrl cells (Fig. 4Ciii) but did not alter the susceptibility of K1-ctrl (Fig. 4Ci), pgs-DC-SIGN, or pgs-L-SIGN cells to HMPV infection (Fig. 4Cii). Note that the concentrations of dynasore, NH4Cl, and bafilomycin A1 used did not result in the detachment of adherent cells (as determined by counting the total number of propidium iodide-positive [PI+] cells after 18 h of incubation compared to mock-treated controls), confirming that these concentrations were not toxic (data not shown).
DC-SIGN/L-SIGN expression augments HMPV infection of CHO cells expressing cell surface GAGs.
GAG-deficient pgsA745 cells provide a unique experimental system to study interactions between HMPV and putative attachment/entry receptors, in the absence of additional interactions with cell surface GAGs. However, under physiological conditions, CLRs such as DC-SIGN and L-SIGN will be expressed on the surface of cells that also express GAGs. Therefore, it is important to determine if DC-SIGN/L-SIGN augments or inhibits HMPV infection by cells expressing cell surface GAGs. To examine this, we generated lines of parental CHO-K1 cells expressing either DC-SIGN or L-SIGN (Fig. 5A). Next, we infected K1-ctrl, K1-DC-SIGN, and K1-L-SIGN cells with a low inoculum dose and determined the percentage of HMPV-infected cells at 18 h postinfection in the presence or absence of dynasore. As shown in Fig. 5B, expression of DC-SIGN or L-SIGN was associated with a significant enhancement of the percentage of HMPV-infected cells. Of interest, the enhanced levels of HMPV infection were reduced to background levels equivalent to those of CHO-K1-ctrl cells in the presence of dynasore. Thus, in CHO-K1 cells expressing DC-SIGN/L-SIGN, both dynamin-independent (i.e., the typical pathway of HMPV infection of CHO-K1 cells) and dynamin-dependent (i.e., an additional pathway of HMPV infection introduced following the expression of CLRs) pathways of HMPV infection occur simultaneously.
FIG 5.
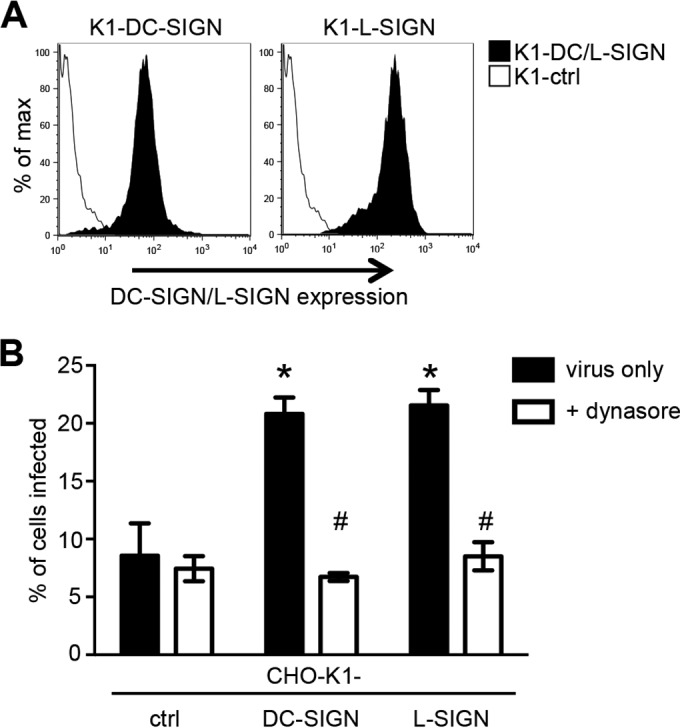
DC-SIGN/L-SIGN expression augments HMPV infection of CHO cells expressing cell surface GAGs. (A) Cell surface expression of DC-SIGN or L-SIGN on CHO-K1 cells (K1-DC/L-SIGN) or CHO-K1 cells expressing an irrelevant intracellular protein (K1-ctrl cells) was determined by flow cytometry using a cross-reactive MAb that detects both DC-SIGN and L-SIGN. (B) Monolayers of K1-ctrl, K1-DC-SIGN, or K1-L-SIGN cells were treated with 50 μM dynasore (+ dynasore) or mock treated (virus only) for 30 min at 37°C. Cells were then infected with 3.7 × 104 FFU (MOI of 0.37 FFU/cell) of HMPV for 1 h at 37°C, washed, and cultured in serum-free medium alone (virus only) or supplemented with either 50 μM dynasore or 20 mM NH4Cl (+ NH4Cl) for an additional 17 h. At this time, cells were fixed and stained by immunofluorescence for the expression of viral N. Data represent the mean percentages of infected cells (±1 standard error of the mean) from a minimum of four independent fields per chamber and are representative of results from three independent experiments. In panel B, * indicates that CHO-K1-DC/L-SIGN cells are significantly enhanced compared to CHO-K1-ctrl cells (P < 0.01, as determined by one-way ANOVA followed by Tukey's post hoc test), and # indicates that for K1-DC-SIGN and K1-L-SIGN cells, mock treatment is significantly different from dynasore treatment for each cell line (P < 0.01, as determined by one-way ANOVA followed by Tukey's post hoc test).
Susceptibility of pgsA745 cells expressing endocytosis-defective mutants of DC-SIGN/L-SIGN to HMPV infection.
In previous studies, endocytosis-defective mutants of different CLRs were generated via mutation or deletion of the intracellular domain (32, 49, 50). Therefore, we generated pgsA745 cell lines expressing DC-SIGN or L-SIGN with the entire N-terminal intracellular domain of DC-SIGN (33 amino acids) or L-SIGN (41 amino acids) deleted (DEL). Flow cytometry was used to confirm cell surface expression of DC-SIGN-DEL or L-SIGN-DEL on pgsA745 cell lines (Fig. 6A). Note that clones of DC-SIGN/L-SIGN-DEL mutants expressed higher levels of CLR than clones expressing the relevant WT receptor.
FIG 6.

pgsA745 cells expressing endocytosis-defective mutants of DC-SIGN/L-SIGN remain susceptible to HPMV infection. (A) Flow cytometry was used to determine cell surface expression of DC-SIGN (left) or L-SIGN (right) on pgsA745 cells expressing WT or DEL forms of DC-SIGN/L-SIGN. pgsA745-ctrl cells were included to confirm the specificity of binding. (B) Endocytic capacity of pgsA745 cells expressing different forms of DC-SIGN/L-SIGN following cross-linking with antibody. Cell monolayers expressing WT or DEL forms of DC-SIGN/L-SIGN were incubated with an anti-DC-SIGN/L-SIGN MAb for 1 h at 4°C, washed, and incubated for a further 20 min at either 4°C (left) or 37°C (right). Cells were then fixed, stained with Alexa Fluor 488 anti-mouse Ig (green) (to detect anti-DC-SIGN/L-SIGN MAb) and DAPI (blue) (to stain the nucleus), and examined by confocal microscopy. (C) Binding of HMPV to pgsA745 cells expressing WT or DEL forms of DC-SIGN/L-SIGN was determined by flow cytometry. Representative histograms of HMPV binding to pgsA745 cells expressing WT or DEL forms of DC-SIGN/L-SIGN in the presence of 20 mM Ca2+ are shown. pgsA745-ctrl cells were included to confirm the specificity of binding. (D) Cells were incubated with 3 × 105 FFU (MOI of 3 FFU/cell) of HMPV for 1 h, washed, cultured for an additional 17 h, and then fixed and stained for the expression of the HMPV N protein. Data show the mean percentages of infection (±1 standard deviation). * indicates that values are significantly different from those for pgsA745-ctrl cells (P < 0.01, as determined by one-way ANOVA followed by Tukey's post hoc test). No significant differences were observed between pgsA745 cells expressing WT or DEL DC-SIGN and those expressing WT or DEL L-SIGN. (E) To examine endocytic entry during the first 1 h, cells were incubated with 3 × 105 FFU (MOI of 3 FFU/cell) of HMPV for 1 h and washed, and 50 μM dynasore was added during subsequent culture to prevent further endocytosis [+ Dyn (60 min)]. As controls, either cells were pretreated with 50 μM dynasore for 30 min at 37°C and infected with HMPV for 60 min, and dynasore was included in all washing and culture steps [+ Dyn (pretreated)], or cells were infected and incubated with medium alone (untreated). After 18 h, cells were fixed, stained for the expression of the HMPV N protein, and examined by fluorescence microscopy. Data show the mean percentages of infection (±1 standard deviation). * indicates that values for infected cells treated with dynasore are significantly different from those for cells pretreated with dynasore (P < 0.01, as determined by one-way ANOVA followed by Tukey's post hoc test). (F) To gain insight regarding the rate of endocytic entry of HMPV, cells were incubated with 3 × 105 FFU (MOI of 3 FFU/cell) of HMPV for either 60, 30, or 15 min and then washed, and 50 μM dynasore was added to prevent further endocytosis. At 18 h postinfection, cells were fixed, stained, and examined by fluorescence microscopy. * indicates that infection of DEL cells is significantly reduced compared to that of the appropriate WT controls (P < 0.01, as determined by two-tailed unpaired Student's t test).
To confirm that the deletion of the N-terminal intracellular domain abrogated the endocytic capacity of DC-SIGN/L-SIGN, cell monolayers were incubated with an anti-DC-SIGN/L-SIGN MAb at 4°C to facilitate binding to cell surface CLRs and then moved to a temperature of 37°C to promote internalization. Following incubation at 4°C, MAb bound to the surface of pgsA745 cells expressing WT and DEL forms of DC-SIGN (Fig. 6B, left). However, when moved to a temperature of 37°C, the MAb was internalized into intracellular compartments in cells expressing WT DC-SIGN/L-SIGN but remained at the surface of cells expressing DEL DC-SIGN/L-SIGN, consistent with major defects in the endocytic capacity (Fig. 6B, right).
Next, we compared the abilities of pgsA745-DC-SIGN-WT and pgsA745-DC-SIGN-DEL cells to bind to HMPV in the presence or absence of Ca2+. Representative histograms confirmed that cell lines expressing WT or DEL DC-SIGN/L-SIGN recognized HMPV in a Ca2+-dependent manner (Fig. 6C), consistent with CLR-mediated recognition of virus. Analysis of the geometric mean fluorescence intensities from triplicate samples indicated no significant differences in the abilities of cells expressing WT and DEL DC-SIGN/L-SIGN to bind HMPV, and all lines showed significantly enhanced binding in Ca2+ compared to pgs-ctrl cells (data not shown).
Finally, we compared the abilities of HMPV to infect pgsA745 cells expressing WT or endocytosis-defective DEL forms of DC-SIGN/L-SIGN. We first conducted a standard infection assay, with the addition of the virus inoculum for 1 h, washing, and incubation for an additional 17 h before cells were fixed and stained by immunofluorescence. Surprisingly, we observed that cells expressing DEL mutants of DC-SIGN/L-SIGN were equally as susceptible to HMPV infection as cells expressing the appropriate WT receptor (Fig. 6D), and similar results were obtained when lower doses of the virus inoculum (representing MOIs of 1.5, 0.75, and 0.25 FFU/cell) were used (data not shown). These findings imply that following capture by either WT or DEL DC-SIGN/L-SIGN, the virus is passed to other (unknown) receptors on pgsA745 cells to promote virus entry.
Next, we modified our standard infection assay such that the virus inoculum was added for 1 h at 37°C and then washed prior to the addition of 50 μM dynasore to block subsequent endocytic virus entry during the 17-h culture period (Fig. 6E). As a control, cells were pretreated with 50 μM dynasore, and this was retained throughout infection and culture. The addition of dynasore after 1 h resulted in a significant reduction of HMPV infection of cells expressing WT DC-SIGN/L-SIGN; however, this reduction was more profound in cells expressing DEL DC-SIGN/L-SIGN (Fig. 6E). These studies suggest that cells expressing WT DC-SIGN/L-SIGN promote infectious entry of HMPV more efficiently during the first 60 min of infection than cells expressing DEL DC-SIGN/L-SIGN.
To explore this further, we incubated the virus inoculum with cells for 60, 30, and 15 min prior to washing and the addition of dynasore to block subsequent endocytic entry and infection. Compared to cells expressing WT DC-SIGN/L-SIGN, the addition of dynasore at 60, 30, or 15 min resulted in significant reductions in HMPV infection of cells expressing DEL DC-SIGN/L-SIGN (Fig. 6F). Overall, HMPV infection of cells expressing WT CLRs was progressively reduced by the shorter incubation periods prior to the addition of dynasore; however, infection of cells expressing DEL DC-SIGN/L-SIGN was largely undetectable if dynasore was added after 30 or 15 min of incubation with the virus inoculum. These experiments confirm that DC-SIGN and L-SIGN can act as bona fide entry receptors for HMPV, internalizing virus rapidly to promote infection. Thus, using endocytosis-defective mutants, different incubation times following HMPV inoculation, and dynasore to block endocytic entry, we demonstrate that HMPV can enter pgsA745 cells by more than one endocytic route following the recognition of HMPV by cell surface DC-SIGN/L-SIGN. In addition to rapid CLR-mediated entry (i.e., increased infection of cells expressing WT CLRs with shorter incubation times), CLRs act as attachment factors to promote endocytic entry by other (unknown) receptors on pgsA745 cells (seen by delayed infection kinetics in cells expressing DEL mutants).
DISCUSSION
To date, evidence suggests that the attachment and entry of HMPV into host cells may be a multistep process, involving sequential recognition by attachment factors (i.e., GAGs) (17, 20, 21) to localize virus on the cell surface and promote subsequent interactions with integrins (13–16) and/or other proteinaceous receptors (17). Moreover, it is possible that these interactions might also occur within endosomal compartments to promote the infectious entry of HMPV (15, 23). It should be noted that the specific HMPV receptor(s) utilized for attachment and infectious entry may also be cell type specific (e.g., between airway epithelial cells and immune cells in the lung) and could differ between in vitro and in vivo settings and between different species. As GAG-deficient pgsA745 cells did not bind HMPV (Fig. 1C) (17) and were resistant to HMPV infection (Fig. 1D) (17), this study defines an experimental system in which GAG-independent interactions between HMPV and putative cell surface receptors can be investigated. Moreover, as the infectious entry of HMPV is rescued by the expression of a single putative receptor in GAG-deficient cells, these approaches allow characterization of specific receptor-mediated HMPV entry pathways and may be exploited in future studies to determine specific domains and residues within cell surface receptors that are necessary for the infectious entry of HMPV.
Recombinant DC-SIGN/L-SIGN (Fig. 2A) and DC-SIGN/L-SIGN expressed on the surface of GAG-deficient pgsA745 cells (Fig. 2C and D) recognized HMPV in a Ca2+-dependent manner. Moreover, the expression of DC-SIGN or L-SIGN restored the capacity of GAG-deficient pgsA745 cells to support HMPV infection (Fig. 3A). A recent study by Le Nouen et al. highlighted the relevance of DC-SIGN in enhancing the infectious entry of HMPV into primary human immune cells, with HMPV infection of MDDCs being partially blocked in the presence of mannan or an anti-DC-SIGN MAb (26). Of interest, this pathway was dependent on the presence of HMPV SH and/or G glycoproteins, as infection by a deletion mutant lacking G and SH was not blocked by these treatments, suggesting that glycans expressed by HMPV G/SH, but not the F protein, were the primary ligands for DC-SIGN. In future studies, it will be of interest to assess appropriate HMPV deletion mutants in our experimental system to confirm which viral glycoproteins are recognized by DC-SIGN/L-SIGN to promote infectious HMPV entry. Moreover, variation in the location and number of potential sites of N-linked glycosylation on HMPV glycoproteins between virus strains is likely to modulate the efficiency of CLR-mediated recognition, as reported previously for other viruses such as IAV (reviewed in reference 51). As the dynamin-independent HMPV entry pathways observed by using CHO-K1 cells do not operate efficiently in GAG-deficient pgsA745 cells, CLR-mediated enhancement of HMPV infection can be studied more effectively in this model.
For many years, paramyxovirus fusion was thought to occur at the plasma membrane in a pH-independent manner; however, growing evidence suggests that a number of paramyxoviruses may enter and infect cells, at least in part, via endocytic pathways. For example, several studies have implicated endocytic pathways in the infectious entry of RSV into human cell lines (52–54). Schowalter et al. reported that HMPV infection of Vero cells was sensitive to treatment with inhibitors of dynamin or clathrin-mediated endocytosis (23), and recent studies confirmed that HMPV particles were internalized into human bronchial epithelial cells via clathrin-mediated endocytosis in a dynamin-dependent manner (15). Using GAG-deficient cells that are resistant to HMPV infection, we demonstrate that the expression of DC-SIGN/L-SIGN restored permissivity to dynamin-dependent infection, implicating endocytosis as the sole route of infectious entry under these conditions. While infection of parental CHO-K1 cells by HMPV occurred via dynamin-independent pathways, the expression of DC-SIGN/L-SIGN opened up an alternative dynamin-dependent pathway of HMPV entry, resulting in enhanced levels of infection (Fig. 4). These findings confirm that the receptors expressed on the surface of different cell types are likely to modulate HMPV entry pathways, and multiple entry pathways may operate simultaneously. Consistent with this, the uptake of HMPV by MDDCs was reported to occur primarily by macropinocytosis, although DC-SIGN-mediated endocytosis represented an alternative entry pathway (26). This concept is not limited to HMPV infection. For example, IAV can enter cells via multiple pathways, including clathrin-mediated endocytosis, caveolin-mediated endocytosis, and macropinocytosis (reviewed in reference 55), and the particular pathway utilized may be influenced by structural features of the virion (e.g., spherical or filamentous), cell type, experimental culture conditions, and/or differential expression of cell surface receptors.
The HMPV F protein contains a conserved RGD motif that plays a critical role during HMPV infection of human cells by interacting with RGD-binding integrins at the cell surface to promote cell-cell fusion and infectious entry (13–16). In addition to virus binding, RGD-binding integrins are required for a subsequent entry step that occurs after fusion but before virus transcription (14), with recent studies implicating their importance for vesicle trafficking during HMPV entry (15). Previous reports demonstrated that CHO-K1 cells express high levels of β1 integrin (17) but may also express additional RGD-binding integrins such as αvβ5, αvβ6, and/or αvβ8 that might also bind HMPV F as well as other (unknown) proteinaceous receptors (17). By using a combination of anti-αv, anti-α5, and anti-β1 MAbs to block all available RGD integrins on human BEAS-2B cells, Cox et al. demonstrated a 90% reduction of HMPV infectivity (14). However, our experimental system was dependent on the use of pgsA745 cells, a GAG-deficient mutant of the CHO cell line, and we were unable to identify appropriate MAbs to recognize and block hamster homologues of all RGD integrins. Given that GAG-deficient cells expressing endocytosis-defective mutants of DC-SIGN/L-SIGN remained susceptible to HMPV infection using our standard experimental approaches (Fig. 6D), our data suggest that these CLRs can act as attachment factors and concentrate the virus at the cell surface to promote dynamin-dependent entry via alternative receptors, possibly RGD binding integrins. HMPV F has been reported to bind RGD binding integrins in the absence of G, and HMPV expressing a mutated RGD motif (RAE) in its F protein was profoundly impaired in its ability to replicate in cell culture (14). Moreover, HMPV deletion mutants lacking G and SH did not appear to utilize DC-SIGN-mediated endocytosis to infect MDDCs (26). Therefore, it would be of interest to compare HMPV mutants lacking SH/G or with mutations within the RGD motif of the F protein to gain insights regarding the viral determinants that contribute to CLR-mediated enhancement of HMPV infection in GAG-deficient cells.
Schowalter et al. reported that syncytium formation promoted by the HMPV F protein in transfected cells required the exposure of cells to short pulses of low pH (46), although subsequent studies demonstrated that this was a rare strain-dependent phenomenon (45). More recently, Mas et al. implicated four variable residues as the main determinants of pH-dependent syncytium formation in HMPV-infected cultures and reported no correlation between the sensitivity of the F protein to low pH in a syncytium formation assay and the requirement for a low-pH step to promote virus entry into cells (47). In our studies, we have focused only on the infectious entry of HMPV into cells. We report that the dynamin-dependent entry of CAN97-83 into pgsA745 cells expressing DC-SIGN/L-SIGN, or into CHO-ctrl cells, was unaffected in the presence of inhibitors of endosomal acidification (NH4Cl and Baf-A1), consistent with pH-independent entry. In contrast, both NH4Cl and Baf-A1 treatments completely blocked pH-dependent infection of cells by IAV (Fig. 4B/C). It should be noted that Schowalter et al. reported that NH4Cl or Baf-A1 treatment of Vero/A549 cells resulted in marked reductions in HMPV infection by strain CAN97-83 (23), perhaps reflecting differences in cell types and/or culture conditions or, in particular, virus stocks. In our hands, CAN97-83 infection of Vero cells was unaffected in the presence of NH4Cl or Baf-A1 at concentrations that completely blocked IAV infection of these cells (data not shown).
DC-SIGN and L-SIGN have been implicated in enhancing cellular infection by a range of different viruses, although in many cases, it is not clear whether enhanced infection results from CLR-mediated endocytosis or if CLR capture promotes subsequent interactions with other entry receptors (reviewed in reference 25). However, studies have reported that cells expressing either WT or endocytosis-defective (DEL) mutants of DC-SIGN/L-SIGN showed enhanced infection by dengue virus (50) or Ebola virus (56), highlighting their ability to cooperate with other cellular receptors to enhance infection. In contrast, while WT DC-SIGN/L-SIGN enhanced cellular infection by IAV (32) and phleboviruses (57), endocytosis-defective mutants did not. Here, we demonstrate that the expression of DC-SIGN/L-SIGN by GAG-deficient cells rescued permissivity to HMPV infection. Moreover, pgsA745 cells expressing endocytosis-defective mutants of DC-SIGN/L-SIGN remained susceptible to HMPV infection in our standard infection assay (inoculum at 37°C for 60 min, wash, and culture for 18 to 20 h), indicating that they can act as attachment factors to promote dynamin-dependent HMPV entry via other (unknown) receptors (Fig. 6D). However, modifying our infection assays by (i) adding dynasore immediately after 60 min of incubation with the HMPV inoculum (Fig. 6E) or (ii) reducing incubation times with the HMPV inoculum to 30 or 15 min prior to addition of dynasore (Fig. 6F) provided evidence that CLR-mediated endocytosis can also contribute to HMPV infection. Thus, in the context of the expression of DC-SIGN/L-SIGN by GAG-deficient pgsA745 cells, we provide evidence that CLRs can act as attachment factors and/or as bona fide entry receptors for HMPV.
Expression of putative receptors in pgsA745 cells removed the confounding factor of multiple low-affinity interactions between HMPV and cell surface GAGs. While others have reported the critical presence of cell surface GAGs for efficient HMPV infection of Vero cells (17), we found that low-level HMPV infection of GAG-deficient pgsA745 cells was also inhibited in the presence of dynasore (Fig. 4A), suggesting that an alternative pathway of GAG-independent HMPV infection might also operate albeit inefficiently. Thus, HMPV entry can occur independently of GAGs (i.e., pgsA745 cells), although the efficiency of infection was enhanced if virions were concentrated at the cell surface by low-affinity interactions with GAGs (i.e., CHO cells) or by recognition via DC-SIGN/L-SIGN (i.e., pgsA745-DC/L-SIGN). Of interest, attachment of HMPV to GAGs (expressed by CHO-K1 cells) or to DC-SIGN/L-SIGN (expressed by pgsA745 transfectants) promoted dynamin-independent and dynamin-dependent HMPV infection, respectively. These findings suggest that interactions between HMPV and different attachment factors (i.e., GAGs on CHO cells and CLR on pgsA745-DC/L-SIGN cells, respectively) might modify subsequent pathways of virus entry, possibly by modulating interactions with particular entry receptors or coreceptors. However, while our studies using CHO/pgsA745 cells define an experimental system to examine putative HMPV receptors in the presence or absence of GAGs, the relevance of these receptors and pathways to other cellular targets of HMPV infection is unclear. For example, GAGs have been implicated in the attachment of CAN97-83 to Vero cells (17), yet infection of Vero cells by CAN97-83 was largely inhibited by dynasore (23), which is in contrast to our findings that GAG-dependent infection of CHO-K1 cells by CAN97-83 occurred via dynamin-independent pathways (Fig. 4A). Recently, dynamin-dependent HMPV infection of BEAS-2B airway epithelial cells was also reported (15), confirming the importance of endocytosis for HMPV entry in other epithelial cell types. Thus, while we report dynamin-dependent infection of GAG-deficient pgsA745 cells expressing DC-SIGN/L-SIGN, it is likely that other endocytic receptors facilitate HMPV infection of a range of different cell types, including airway epithelial cells. The specific identity of these receptors and the role of GAGs in modulating infectious entry into cell types more relevant to HMPV infection (including airway epithelial cells and macrophages/DCs) remain to be determined.
Recognition of viruses by DC-SIGN/L-SIGN can result in a range of outcomes, including modulation of host immunity (e.g., DC maturation and migration) and virus uptake for infection and/or virus dissemination (reviewed in references 58 and 59). Currently, relatively little is known regarding the consequences of the recognition of paramyxoviruses by CLRs. DC-SIGN has been reported to act as an attachment factor for measles virus, thereby enhancing CD46/CD150-mediated infection of DCs in cis (60). In addition, measles virus suppresses RIG-I and Mda5 by activating DC-SIGN signaling events, resulting in reduced type I interferon (IFN) responses and enhanced virus replication in DCs (61). Given that RSV is the human pneumovirus most closely related to HMPV, it is of interest that DC-SIGN/L-SIGN expression by pgsA745 cells did not promote RSV infection (Fig. 3C). Consistent with this, previous studies by Johnson et al. reported that DC-SIGN/L-SIGN binds the G glycoprotein of RSV; however, the expression of these CLRs by RSV-resistant K562 and Raji cells, or by semipermissive NIH 3T3 cells, did not result in increased susceptibility to infection (41). Instead, interactions between the RSV G glycoprotein and DC-SIGN/L-SIGN were implicated in intracellular signaling and phosphorylation of extracellular signal-regulated kinase 1 (ERK1) and ERK2, which were proposed to limit DC maturation and cytokine/chemokine production (41). While DC-SIGN has been implicated as an alternative pathway for HMPV entry into MDDCs, the functional consequences of this interaction are not yet fully understood (26). Moreover, as DC-SIGN is also expressed on alveolar macrophages (62) and L-SIGN is expressed on endothelial cells, including those in the lungs and lymph nodes, as well as lung alveolar epithelial cells (63, 64), it will be of interest to assess the contribution of these and other CLRs in modulating the responses of different airway cell types to HMPV.
ACKNOWLEDGMENT
The Melbourne WHO Collaborating Centre for Reference and Research on Influenza is supported by the Australian Government Department of Health.
REFERENCES
- 1.Kroll JL, Weinberg A. 2011. Human metapneumovirus. Semin Respir Crit Care Med 32:447–453. doi: 10.1055/s-0031-1283284. [DOI] [PubMed] [Google Scholar]
- 2.Kuiken T, van den Hoogen BG, van Riel DA, Laman JD, van Amerongen G, Sprong L, Fouchier RA, Osterhaus AD. 2004. Experimental human metapneumovirus infection of cynomolgus macaques (Macaca fascicularis) results in virus replication in ciliated epithelial cells and pneumocytes with associated lesions throughout the respiratory tract. Am J Pathol 164:1893–1900. doi: 10.1016/S0002-9440(10)63750-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wyde PR, Chetty SN, Jewell AM, Schoonover SL, Piedra PA. 2005. Development of a cotton rat-human metapneumovirus (hMPV) model for identifying and evaluating potential hMPV antivirals and vaccines. Antiviral Res 66:57–66. doi: 10.1016/j.antiviral.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 4.Kolli D, Gupta MR, Sbrana E, Velayutham TS, Chao H, Casola A, Garofalo RP. 2014. Alveolar macrophages contribute to the pathogenesis of human metapneumovirus infection while protecting against respiratory syncytial virus infection. Am J Respir Cell Mol Biol 51:502–515. doi: 10.1165/rcmb.2013-0414OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guerrero-Plata A, Casola A, Suarez G, Yu X, Spetch L, Peeples ME, Garofalo RP. 2006. Differential response of dendritic cells to human metapneumovirus and respiratory syncytial virus. Am J Respir Cell Mol Biol 34:320–329. doi: 10.1165/rcmb.2005-0287OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kolli D, Bao X, Liu T, Hong C, Wang T, Garofalo RP, Casola A. 2011. Human metapneumovirus glycoprotein G inhibits TLR4-dependent signaling in monocyte-derived dendritic cells. J Immunol 187:47–54. doi: 10.4049/jimmunol.1002589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Le Nouen C, Munir S, Losq S, Winter CC, McCarty T, Stephany DA, Holmes KL, Bukreyev A, Rabin RL, Collins PL, Buchholz UJ. 2009. Infection and maturation of monocyte-derived human dendritic cells by human respiratory syncytial virus, human metapneumovirus, and human parainfluenza virus type 3. Virology 385:169–182. doi: 10.1016/j.virol.2008.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan MC, Battini L, Tuyama AC, Macip S, Melendi GA, Horga MA, Gusella GL. 2007. Characterization of human metapneumovirus infection of myeloid dendritic cells. Virology 357:1–9. doi: 10.1016/j.virol.2006.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cox RG, Williams JV. 2013. Breaking in: human metapneumovirus fusion and entry. Viruses 5:192–210. doi: 10.3390/v5010192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Biacchesi S, Murphy BR, Collins PL, Buchholz UJ. 2007. Frequent frameshift and point mutations in the SH gene of human metapneumovirus passaged in vitro. J Virol 81:6057–6067. doi: 10.1128/JVI.00128-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biacchesi S, Skiadopoulos MH, Yang L, Lamirande EW, Tran KC, Murphy BR, Collins PL, Buchholz UJ. 2004. Recombinant human metapneumovirus lacking the small hydrophobic SH and/or attachment G glycoprotein: deletion of G yields a promising vaccine candidate. J Virol 78:12877–12887. doi: 10.1128/JVI.78.23.12877-12887.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biacchesi S, Pham QN, Skiadopoulos MH, Murphy BR, Collins PL, Buchholz UJ. 2005. Infection of nonhuman primates with recombinant human metapneumovirus lacking the SH, G, or M2-2 protein categorizes each as a nonessential accessory protein and identifies vaccine candidates. J Virol 79:12608–12613. doi: 10.1128/JVI.79.19.12608-12613.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cseke G, Maginnis MS, Cox RG, Tollefson SJ, Podsiad AB, Wright DW, Dermody TS, Williams JV. 2009. Integrin alphavbeta1 promotes infection by human metapneumovirus. Proc Natl Acad Sci U S A 106:1566–1571. doi: 10.1073/pnas.0801433106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cox RG, Livesay SB, Johnson M, Ohi MD, Williams JV. 2012. The human metapneumovirus fusion protein mediates entry via an interaction with RGD-binding integrins. J Virol 86:12148–12160. doi: 10.1128/JVI.01133-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cox RG, Mainou BA, Johnson M, Hastings AK, Schuster JE, Dermody TS, Williams JV. 2015. Human metapneumovirus is capable of entering cells by fusion with endosomal membranes. PLoS Pathog 11:e1005303. doi: 10.1371/journal.ppat.1005303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei Y, Zhang Y, Cai H, Mirza AM, Iorio RM, Peeples ME, Niewiesk S, Li J. 2014. Roles of the putative integrin-binding motif of the human metapneumovirus fusion (F) protein in cell-cell fusion, viral infectivity, and pathogenesis. J Virol 88:4338–4352. doi: 10.1128/JVI.03491-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang A, Masante C, Buchholz UJ, Dutch RE. 2012. Human metapneumovirus (HMPV) binding and infection are mediated by interactions between the HMPV fusion protein and heparan sulfate. J Virol 86:3230–3243. doi: 10.1128/JVI.06706-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Henning W, Bohn W, Nebe B, Knopp A, Rychly J, Strauss M. 1994. Local increase of beta 1-integrin expression in cocultures of immortalized hepatocytes and sinusoidal endothelial cells. Eur J Cell Biol 65:189–199. [PubMed] [Google Scholar]
- 19.Jolly CL, Sattentau QJ. 2013. Attachment factors. Adv Exp Med Biol 790:1–23. doi: 10.1007/978-1-4614-7651-1_1. [DOI] [PubMed] [Google Scholar]
- 20.Adamson P, Thammawat S, Muchondo G, Sadlon T, Gordon D. 2012. Diversity in glycosaminoglycan binding amongst hMPV G protein lineages. Viruses 4:3785–3803. doi: 10.3390/v4123785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thammawat S, Sadlon TA, Hallsworth PG, Gordon DL. 2008. Role of cellular glycosaminoglycans and charged regions of viral G protein in human metapneumovirus infection. J Virol 82:11767–11774. doi: 10.1128/JVI.01208-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wyde PR, Moylett EH, Chetty SN, Jewell A, Bowlin TL, Piedra PA. 2004. Comparison of the inhibition of human metapneumovirus and respiratory syncytial virus by NMSO3 in tissue culture assays. Antiviral Res 63:51–59. doi: 10.1016/j.antiviral.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 23.Schowalter RM, Chang A, Robach JG, Buchholz UJ, Dutch RE. 2009. Low-pH triggering of human metapneumovirus fusion: essential residues and importance in entry. J Virol 83:1511–1522. doi: 10.1128/JVI.01381-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Esko JD, Stewart TE, Taylor WH. 1985. Animal cell mutants defective in glycosaminoglycan biosynthesis. Proc Natl Acad Sci U S A 82:3197–3201. doi: 10.1073/pnas.82.10.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Londrigan SL, Tate MD, Brooks AG, Reading PC. 2012. Cell-surface receptors on macrophages and dendritic cells for attachment and entry of influenza virus. J Leukoc Biol 92:97–106. doi: 10.1189/jlb.1011492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Le Nouen C, Hillyer P, Brock LG, Winter CC, Rabin RL, Collins PL, Buchholz UJ. 2014. Human metapneumovirus SH and G glycoproteins inhibit macropinocytosis-mediated entry into human dendritic cells and reduce CD4+ T cell activation. J Virol 88:6453–6469. doi: 10.1128/JVI.03261-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eckhardt M, Gotza B, Gerardy-Schahn R. 1998. Mutants of the CMP-sialic acid transporter causing the Lec2 phenotype. J Biol Chem 273:20189–20195. doi: 10.1074/jbc.273.32.20189. [DOI] [PubMed] [Google Scholar]
- 28.Stanley P, Sudo T, Carver JP. 1980. Differential involvement of cell surface sialic acid residues in wheat germ agglutinin binding to parental and wheat germ agglutinin-resistant Chinese hamster ovary cells. J Cell Biol 85:60–69. doi: 10.1083/jcb.85.1.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghildyal R, Hartley C, Varrasso A, Meanger J, Voelker DR, Anders EM, Mills J. 1999. Surfactant protein A binds to the fusion glycoprotein of respiratory syncytial virus and neutralizes virion infectivity. J Infect Dis 180:2009–2013. doi: 10.1086/315134. [DOI] [PubMed] [Google Scholar]
- 30.Anders EM, Hartley CA, Jackson DC. 1990. Bovine and mouse serum beta inhibitors of influenza A viruses are mannose-binding lectins. Proc Natl Acad Sci U S A 87:4485–4489. doi: 10.1073/pnas.87.12.4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Londrigan SL, Turville SG, Tate MD, Deng YM, Brooks AG, Reading PC. 2011. N-linked glycosylation facilitates sialic acid-independent attachment and entry of influenza A viruses into cells expressing DC-SIGN or L-SIGN. J Virol 85:2990–3000. doi: 10.1128/JVI.01705-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gillespie L, Roosendahl P, Ng WC, Brooks AG, Reading PC, Londrigan SL. 2016. Endocytic function is critical for influenza A virus infection via DC-SIGN and L-SIGN. Sci Rep 6:19428. doi: 10.1038/srep19428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.North SJ, Huang HH, Sundaram S, Jang-Lee J, Etienne AT, Trollope A, Chalabi S, Dell A, Stanley P, Haslam SM. 2010. Glycomics profiling of Chinese hamster ovary cell glycosylation mutants reveals N-glycans of a novel size and complexity. J Biol Chem 285:5759–5775. doi: 10.1074/jbc.M109.068353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Conradt HS, Egge H, Peter-Katalinic J, Reiser W, Siklosi T, Schaper K. 1987. Structure of the carbohydrate moiety of human interferon-beta secreted by a recombinant Chinese hamster ovary cell line. J Biol Chem 262:14600–14605. [PubMed] [Google Scholar]
- 35.Lee EU, Roth J, Paulson JC. 1989. Alteration of terminal glycosylation sequences on N-linked oligosaccharides of Chinese hamster ovary cells by expression of beta-galactoside alpha 2,6-sialyltransferase. J Biol Chem 264:13848–13855. [PubMed] [Google Scholar]
- 36.Lim SF, Lee MM, Zhang P, Song Z. 2008. The Golgi CMP-sialic acid transporter: a new CHO mutant provides functional insights. Glycobiology 18:851–860. doi: 10.1093/glycob/cwn080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu W, Li J, Liang G. 2011. How does cellular heparan sulfate function in viral pathogenicity? Biomed Environ Sci 24:81–87. doi: 10.3967/0895-3988.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 38.Hallak LK, Collins PL, Knudson W, Peeples ME. 2000. Iduronic acid-containing glycosaminoglycans on target cells are required for efficient respiratory syncytial virus infection. Virology 271:264–275. doi: 10.1006/viro.2000.0293. [DOI] [PubMed] [Google Scholar]
- 39.Kwilas S, Liesman RM, Zhang L, Walsh E, Pickles RJ, Peeples ME. 2009. Respiratory syncytial virus grown in Vero cells contains a truncated attachment protein that alters its infectivity and dependence on glycosaminoglycans. J Virol 83:10710–10718. doi: 10.1128/JVI.00986-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martinez I, Melero JA. 2000. Binding of human respiratory syncytial virus to cells: implication of sulfated cell surface proteoglycans. J Gen Virol 81:2715–2722. doi: 10.1099/0022-1317-81-11-2715. [DOI] [PubMed] [Google Scholar]
- 41.Johnson TR, McLellan JS, Graham BS. 2012. Respiratory syncytial virus glycoprotein G interacts with DC-SIGN and L-SIGN to activate ERK1 and ERK2. J Virol 86:1339–1347. doi: 10.1128/JVI.06096-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Doherty GJ, McMahon HT. 2009. Mechanisms of endocytosis. Annu Rev Biochem 78:857–902. doi: 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]
- 43.Russell CJ. 2014. Acid-induced membrane fusion by the hemagglutinin protein and its role in influenza virus biology. Curr Top Microbiol Immunol 385:93–116. doi: 10.1007/82_2014_393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ohkuma S, Poole B. 1978. Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc Natl Acad Sci U S A 75:3327–3331. doi: 10.1073/pnas.75.7.3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herfst S, Mas V, Ver LS, Wierda RJ, Osterhaus AD, Fouchier RA, Melero JA. 2008. Low-pH-induced membrane fusion mediated by human metapneumovirus F protein is a rare, strain-dependent phenomenon. J Virol 82:8891–8895. doi: 10.1128/JVI.00472-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schowalter RM, Smith SE, Dutch RE. 2006. Characterization of human metapneumovirus F protein-promoted membrane fusion: critical roles for proteolytic processing and low pH. J Virol 80:10931–10941. doi: 10.1128/JVI.01287-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mas V, Herfst S, Osterhaus AD, Fouchier RA, Melero JA. 2011. Residues of the human metapneumovirus fusion (F) protein critical for its strain-related fusion phenotype: implications for the virus replication cycle. J Virol 85:12650–12661. doi: 10.1128/JVI.05485-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perez L, Carrasco L. 1994. Involvement of the vacuolar H(+)-ATPase in animal virus entry. J Gen Virol 75(Part 10):2595–2606. [DOI] [PubMed] [Google Scholar]
- 49.Engering A, Geijtenbeek TB, van Vliet SJ, Wijers M, van Liempt E, Demaurex N, Lanzavecchia A, Fransen J, Figdor CG, Piguet V, van Kooyk Y. 2002. The dendritic cell-specific adhesion receptor DC-SIGN internalizes antigen for presentation to T cells. J Immunol 168:2118–2126. doi: 10.4049/jimmunol.168.5.2118. [DOI] [PubMed] [Google Scholar]
- 50.Lozach PY, Burleigh L, Staropoli I, Navarro-Sanchez E, Harriague J, Virelizier JL, Rey FA, Despres P, Arenzana-Seisdedos F, Amara A. 2005. Dendritic cell-specific intercellular adhesion molecule 3-grabbing non-integrin (DC-SIGN)-mediated enhancement of dengue virus infection is independent of DC-SIGN internalization signals. J Biol Chem 280:23698–23708. doi: 10.1074/jbc.M504337200. [DOI] [PubMed] [Google Scholar]
- 51.Tate MD, Job ER, Deng YM, Gunalan V, Maurer-Stroh S, Reading PC. 2014. Playing hide and seek: how glycosylation of the influenza virus hemagglutinin can modulate the immune response to infection. Viruses 6:1294–1316. doi: 10.3390/v6031294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kolokoltsov AA, Deniger D, Fleming EH, Roberts NJ Jr, Karpilow JM, Davey RA. 2007. Small interfering RNA profiling reveals key role of clathrin-mediated endocytosis and early endosome formation for infection by respiratory syncytial virus. J Virol 81:7786–7800. doi: 10.1128/JVI.02780-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krzyzaniak MA, Zumstein MT, Gerez JA, Picotti P, Helenius A. 2013. Host cell entry of respiratory syncytial virus involves macropinocytosis followed by proteolytic activation of the F protein. PLoS Pathog 9:e1003309. doi: 10.1371/journal.ppat.1003309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.San-Juan-Vergara H, Sampayo-Escobar V, Reyes N, Cha B, Pacheco-Lugo L, Wong T, Peeples ME, Collins PL, Castano ME, Mohapatra SS. 2012. Cholesterol-rich microdomains as docking platforms for respiratory syncytial virus in normal human bronchial epithelial cells. J Virol 86:1832–1843. doi: 10.1128/JVI.06274-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lakadamyali M, Rust MJ, Zhuang X. 2004. Endocytosis of influenza viruses. Microbes Infect 6:929–936. doi: 10.1016/j.micinf.2004.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marzi A, Moller P, Hanna SL, Harrer T, Eisemann J, Steinkasserer A, Becker S, Baribaud F, Pohlmann S. 2007. Analysis of the interaction of Ebola virus glycoprotein with DC-SIGN (dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin) and its homologue DC-SIGNR. J Infect Dis 196(Suppl 2):S237–S246. doi: 10.1086/520607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lozach PY, Kuhbacher A, Meier R, Mancini R, Bitto D, Bouloy M, Helenius A. 2011. DC-SIGN as a receptor for phleboviruses. Cell Host Microbe 10:75–88. doi: 10.1016/j.chom.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 58.Khoo US, Chan KY, Chan VS, Lin CL. 2008. DC-SIGN and L-SIGN: the SIGNs for infection. J Mol Med 86:861–874. doi: 10.1007/s00109-008-0350-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang F, Ren S, Zuo Y. 2014. DC-SIGN, DC-SIGNR and LSECtin: C-type lectins for infection. Int Rev Immunol 33:54–66. doi: 10.3109/08830185.2013.834897. [DOI] [PubMed] [Google Scholar]
- 60.de Witte L, Abt M, Schneider-Schaulies S, van Kooyk Y, Geijtenbeek TB. 2006. Measles virus targets DC-SIGN to enhance dendritic cell infection. J Virol 80:3477–3486. doi: 10.1128/JVI.80.7.3477-3486.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mesman AW, Zijlstra-Willems EM, Kaptein TM, de Swart RL, Davis ME, Ludlow M, Duprex WP, Gack MU, Gringhuis SI, Geijtenbeek TB. 2014. Measles virus suppresses RIG-I-like receptor activation in dendritic cells via DC-SIGN-mediated inhibition of PP1 phosphatases. Cell Host Microbe 16:31–42. doi: 10.1016/j.chom.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Soilleux EJ, Morris LS, Leslie G, Chehimi J, Luo Q, Levroney E, Trowsdale J, Montaner LJ, Doms RW, Weissman D, Coleman N, Lee B. 2002. Constitutive and induced expression of DC-SIGN on dendritic cell and macrophage subpopulations in situ and in vitro. J Leukoc Biol 71:445–457. [PubMed] [Google Scholar]
- 63.Engering A, van Vliet SJ, Hebeda K, Jackson DG, Prevo R, Singh SK, Geijtenbeek TB, van Krieken H, van Kooyk Y. 2004. Dynamic populations of dendritic cell-specific ICAM-3 grabbing nonintegrin-positive immature dendritic cells and liver/lymph node-specific ICAM-3 grabbing nonintegrin-positive endothelial cells in the outer zones of the paracortex of human lymph nodes. Am J Pathol 164:1587–1595. doi: 10.1016/S0002-9440(10)63717-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jeffers SA, Tusell SM, Gillim-Ross L, Hemmila EM, Achenbach JE, Babcock GJ, Thomas WD Jr, Thackray LB, Young MD, Mason RJ, Ambrosino DM, Wentworth DE, Demartini JC, Holmes KV. 2004. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc Natl Acad Sci U S A 101:15748–15753. doi: 10.1073/pnas.0403812101. [DOI] [PMC free article] [PubMed] [Google Scholar]