

ABSTRACT
Congenital cytomegalovirus (CMV) infection is a leading cause of mental retardation and deafness in newborns. The guinea pig is the only small animal model for congenital CMV infection. A novel CMV vaccine was investigated as an intervention strategy against congenital guinea pig cytomegalovirus (GPCMV) infection. In this disabled infectious single-cycle (DISC) vaccine strategy, a GPCMV mutant virus was used that lacked the ability to express an essential capsid gene (the UL85 homolog GP85) except when grown on a complementing cell line. In vaccinated animals, the GP85 mutant virus (GP85 DISC) induced an antibody response to important glycoprotein complexes considered neutralizing target antigens (gB, gH/gL/gO, and gM/gN). The vaccine also generated a T cell response to the pp65 homolog (GP83), determined via a newly established guinea pig gamma interferon enzyme-linked immunosorbent spot assay. In a congenital infection protection study, GP85 DISC-vaccinated animals and a nonvaccinated control group were challenged during pregnancy with wild-type GPCMV (105 PFU). The pregnant animals carried the pups to term, and viral loads in target organs of pups were analyzed. Based on live pup births in the vaccinated and control groups (94.1% versus 63.6%), the vaccine was successful in reducing mortality (P = 0.0002). Additionally, pups from the vaccinated group had reduced CMV transmission, with 23.5% infected target organs versus 75.9% in the control group. Overall, these preliminary studies indicate that a DISC CMV vaccine strategy has the ability to induce an immune response similar to that of natural virus infection but has the increased safety of a non-replication-competent virus, which makes this approach attractive as a CMV vaccine strategy.
IMPORTANCE Congenital CMV infection is a leading cause of mental retardation and deafness in newborns. An effective vaccine against CMV remains an elusive goal despite over 50 years of CMV research. The guinea pig, with a placenta structure similar to that in humans, is the only small animal model for congenital CMV infection and recapitulates disease symptoms (e.g., deafness) in newborn pups. In this report, a novel vaccine strategy against congenital guinea pig cytomegalovirus (GPCMV) infection was developed, characterized, and tested for efficacy. This disabled infectious single-cycle (DISC) vaccine strategy induced a neutralizing antibody or a T cell response to important target antigens. In a congenital infection protection study, animals were protected against CMV in comparison to the nonvaccinated group (52% reduction of transmission). This novel vaccine was more effective than previously tested gB-based vaccines and most other strategies involving live virus vaccines. Overall, the DISC vaccine is a safe and promising approach against congenital CMV infection.
INTRODUCTION
Human cytomegalovirus (HCMV), a betaherpesvirus, has evolved very closely with its human host. Virus infection in a healthy host is normally asymptomatic but leads to a lifelong infection. In contrast, infection of an immunocompromised host (AIDS and transplant patients) or virus reactivation because of an impaired immune system can have severe consequences of morbidity and mortality, but established antiviral therapy can potentially reduce the impact of the disease in these patients (1). Another important aspect of cytomegalovirus disease is congenital infection, where the virus crosses the placenta and infects the fetus in utero. This occurs in approximately 2% of live births in the United States and can lead to serious symptomatic disease, including impaired vision, mental retardation, and sensorineural hearing loss (SNHL) in newborns (2–6). Established antiviral therapy cannot be used because of possible teratogenic and toxic side effects associated with transmission of the drugs to the fetus in utero (1). However, long-term (6-month) valganciclovir antiviral therapy is now recommended for infants with central nervous system (CNS) involvement to improve SNHL and development outcome (7). Importantly, the greatest risk of congenital infection is for mothers who acquire a primary infection during pregnancy; prior immunity can reduce the risk by up to 69% (8). Hence, the impact of a vaccine is potentially substantial, especially in the United States, European Union, and Japan, where up to 50% of women of child-bearing age are negative for HCMV (9–11) and therefore at a greater risk of primary infection during pregnancy.
Any proposed intervention for the prevention or treatment of HCMV infection should ideally be evaluated in a preclinical model. Unfortunately, HCMV is extremely species specific. Consequently, animal model pathogenicity, vaccine, and antiviral studies are carried out using animal-specific CMVs (12–16). The guinea pig is unique insofar as it is the only small animal model that allows the study of congenital CMV infection, unlike the mouse or rat model (17). Both human and guinea pig placentas are hemomonochorial, containing a homogenous layer of trophoblast cells separating maternal and fetal circulation (18–20). Additionally, as with human pregnancy, the guinea pig gestation period (approximately 65 days) can be divided into trimesters. Importantly, guinea pig CMV (GPCMV) congenital infection causes disease in the fetus and in newborn pups similar to that found in humans, including SNHL (21–23). Consequently, the guinea pig model is well suited for testing of intervention strategies aimed at preventing congenital CMV infection (1, 24, 25).
A major drawback in GPCMV research has largely been overcome by the recent sequencing of the viral genome and the development of infectious bacterial artificial chromosome (BAC) clones of GPCMV (15, 26–29). Manipulation of an infectious GPCMV BAC has allowed the preliminary study of some viral genes (1, 30–36). Analysis of the viral genome (15, 29) indicated that GPCMV encodes homologs to the HCMV glycoproteins (gB, gH, gL, gM, gN, and gO) in genes colinear with the HCMV genome (designated GP55, GP75, GP115, GP100, GP73, and GP74, respectively). In HCMV, these six glycoproteins (gB, gH, gL, gM, gN, and gO) are required for fibroblast cell entry, and they form the glycoprotein complexes gCI (gB), gCII (gM/gN), and gcIII (gH/gL/gO) on the viral membrane (37–39). In HCMV, these complexes are important neutralizing antibody targets and vaccine candidates (40–44). We recently demonstrated that GPCMV forms functionally similar glycoprotein complexes, and these complexes are essential for infection of fibroblast cells as well as serving as important target antigens (36). Additionally, GPCMV forms a homologous pentameric complex (gH/gL/UL128-131) that is necessary for epithelial tropism in HCMV (45) and GPCMV (103) as well as other cell types (46).
In both HCMV and GPCMV, the viral glycoprotein gB is the immunodominant neutralizing viral antigen (47–51). A recombinant HCMV gB has been investigated as a candidate subunit vaccine in phase 2 clinical trials, but this vaccine provides at best approximately 50% efficacy (41). This is despite high antibody titers which are effective in neutralizing virus on fibroblasts (41, 52). Importantly, separate studies of serum from gB-vaccinated individuals showed that it was less effective at neutralizing virus infection on endothelial and epithelial cells, in comparison to convalescent-phase sera from HCMV-infected individuals (53–55). This demonstrated the importance of other viral neutralizing target antigens for infection on these cell types. Consequently, other target antigens should be considered important in the development of a vaccine against congenital CMV infection. The importance of other target antigens can be demonstrated in the guinea pig model, where a gB vaccine, despite high antibody titers, fails to fully protect against congenital CMV infection (51, 56, 57). The gH/gL complex has been identified as a potentially important target antigen, including in a congenital GPCMV infection model. In this study, a novel antibody therapy strategy was found to be effective in reducing the incidence of congenital CMV infection (36, 58). Consequently, an effective immune response to both gB and gH/gL is likely an important factor for a successful vaccine against congenital CMV.
Although patients in the convalescent phase of HCMV infection have an antibody response to various important neutralizing antigens, they also have a heightened immune response to the major tegument protein pp65, which is considered the major T cell target antigen (59). The pp65 antigen has been explored as a T cell vaccine target in animal models and in clinical trials (60). GPCMV encodes a homolog to pp65 (30), and a pp65 homolog (GP83) vaccine strategy based on a defective alphavirus delivery approach resulted in partial protection against congenital GPCMV infection, indicating the potential importance of a T cell-mediated immune response against congenital infection (61). However, HCMV also encodes other T cell target antigens (immediate-early protein 1 [IE1, IE2, pp150, and gB) which are also likely important for the generation of a protective T cell immune response against HCMV and also most likely against congenital CMV infection (62). Importantly, GPCMV encodes homologs to these proteins. Undoubtedly, development of a successful vaccine strategy against congenital CMV may require an approach that induces an immune response to multiple target antigens, including both those that generate antibody responses and those that generate a T cell response and thus mimic a natural CMV infection, to induce long-term protection.
Potentially, one of the most effective vaccine strategies would be to use an attenuated virus, as it would mimic a natural infection. However, a live attenuated virus may have the potential to establish latency in the host and therefore could be considered a high-risk vaccine strategy. An alternative is the use of a non-replication-competent virus, which infects cells in a manner similar to a wild-type virus and expresses an array of viral antigens but is incapable of producing progeny virus because of knockout of a specific essential gene. The GPCMV vaccine strategy described in this study was based on a targeted knockout of a capsid gene which is essential for virus assembly but relatively unimportant as a vaccine target. Importantly, CMV capsid genes are highly conserved between HCMV and animal CMV. The process of capsid assembly in HCMV has been well studied and defined (63). The defective infectious single-cycle (DISC) vaccine strategy was based on a UL85 homolog (GP85) mutant that encodes the minor capsid protein, which dimerizes with itself and the minor capsid binding protein (UL46) to form a triplex as part of a fundamental building block for capsid assembly (63). The GP85 capsid gene was placed under the control of the Tet-Off Advanced transactivation system (Clontech) (64) in a GPCMV BAC, and mutant virus was propagated on a newly developed Tet-Off Advanced guinea pig fibroblast cell line. Animals vaccinated with the GP85 DISC GPCMV showed an immune response to important antibody target antigens (gB, gH/gL, and gM/gN). Additionally, DISC-vaccinated animals showed a T cell response to the pp65 homolog (GP83), which was evaluated in a novel gamma interferon (IFN-γ) enzyme-linked immunosorbent spot (ELISPOT) assay. Finally, in a congenital infection vaccine protection study, GP85 DISC-vaccinated animals demonstrated a highly protective immune response against congenital infection compared to a nonvaccinated control group. Overall, the GPCMV DISC vaccine strategy is a highly promising intervention strategy against congenital infection.
MATERIALS AND METHODS
Cells, viruses, and oligonucleotides.
GPCMV (strain 22122, ATCC VR682) and first-generation GPCMV BAC-derived viruses (26, 27) were propagated on guinea pig fibroblast lung cells (GPL; ATCC CCL 158) or Tet-Off GPL cells (see below) in F-12 medium supplemented with 10% fetal calf serum (FCS; Life Technologies), 10,000 IU of penicillin/liter, 10 mg of streptomycin/liter (Life Technologies), and 7.5% NaHCO3 (Life Technologies) at 37°C, 5% CO2. Virus titrations were carried out on six-well plates. Plaques were stained with 10% Giemsa stain or visualized by fluorescence microscopy. High-titer virus stocks were generated as previously described (36). Pathogenic wild-type GPCMV used in congenital CMV challenge studies was serially maintained as salivary gland stocks from infected guinea pigs. All oligonucleotides were synthesized by Sigma-Genosys (The Woodlands, TX) and are listed in Table 1.
TABLE 1.
Oligonucleotides
| Oligonucleotide | Sequence |
|---|---|
| FEcGP85FLK | 5′GATAGAGAATTCGTTAAACTCAGCCTTTGAAGATACAAGTAGCTGATTTGTTGTC |
| RBmGP85FLK | 5′CAGACATGGATCCGACATGGAGACCACGACGTTCTGCACCTTCG |
| FBmGP86FLK | 5′TAGAGTCGGATCCGAGATCAGAGCGAGTTAAATAAAATACTGTCCTGGAG |
| RHdGP86FLK | 5′GAGATAGCAAGCTTGTTTAAACTACGACGACGAGGCCGCGCGCCGCATGCTCTATAACCAC |
| FBgXhBmKm | 5′AGATAGAAGATCTCTCGAGGATCCGATTTATTCAACAAAGCCACG |
| RBgEcVSlKm | 5′AGCATGAAGATCTGATATCGTCGACGCCAGTGTTACAACCAATTAACC |
| FSV40AEcSl | 5′ATAGATAGAATTCGTCGACTTAATTAATCTCTAGAGGATCATAATCAGCCATACCACATTTGTAG |
| RSV40AEcSl | 5′AGATAGAGAATTCGTCGACGATATCGCAGTGAAAAAAATGCTTTATTTGTGAAATTTG |
| FTREBm | 5′ATAGATAGGATCCTCGAGTTTACTCCCTATCAGTGATAG |
| RTREBm | 5′AAGATAGGATCCAGGCGATCTGACGGTTCACTAAACGAGC |
Tet-Off Advanced GPL cell line.
In order to generate a Tet-Off Advanced GPL cell line, GPL cells in 6-well plates were transfected with pTet-Off Advanced plasmid (4 μg/well; Clontech Laboratories), and cells were maintained under neomycin, G418 (Life Technologies), and antibiotic (400 to 600 μg/ml) selection in complete F-12 medium as described above. Selection medium was changed every third day. Individual colonies of cells were identified and clonally isolated by cloning rings and seeding into separate T-25 flasks and expanded under G418 selection (400 μg/ml) as separate cell lines. Thirty individual cell lines were generated, and frozen low-pass stocks were maintained in liquid nitrogen following a standard protocol. G418-resistant GPL cell lines were screened for expression of the tTA2 transactivator (Clontech Laboratories) based on the ability of the cell line to enable expression of a luciferase reporter gene placed under pTREtight promoter control in a recombinant expression plasmid, pTREtightLUC (Clontech Laboratories). Results of a transactivation/expression assay for luciferase expression were analyzed by bioluminescence imaging of the plasmid-transfected wells. GPL or presumptive GPL Tet-Off (tTA-positive) cells in separate 6-well dishes were transfected with pTRELuc plasmid (2 μg/well; Clontech Laboratories) following a standard transfection protocol (36). At 24 h postexpression, the medium in the wells was replaced with fresh F-12 complete medium (without G418). D-luciferin substrate (100 μg/ml; Promega) was also added to the transfected wells and incubated at 37°C. At 15 min postincubation, the plates were imaged (IVIS 50; Xenogen) (35) for 5 min. Control GPL cells in 6-well plates and transfected with pTREtightLuc were also imaged. As a positive control for bioluminescence imaging, separate GPL cells in six-well dishes were transfected with a second luciferase expression plasmid, pcDNA3-Luc (A. McGregor, unpublished data). This plasmid harbors the luciferase reporter cassette under HCMV IE promoter control on the backbone of a pcDNA3.0 plasmid (Life Technologies). The pcDNA3-Luc construct provided the maximal level of luciferase expression/bioluminescence on both GPL and Tet-Off GPL cell lines and was not dependent upon tTa2 transactivation for expression. On pTREtightLuc-transfected cells, bioluminescent signal was only detected on cells expressing tTA2 (Tet-Off Advanced transactivator).
Cloning of GPCMV genes and generation of GP85 locus shuttle vectors.
The GPCMV sequence was based on the complete 22122 viral genome sequence (GenBank accession AB592928.1). Our methods for generation of individual shuttle vectors for specific gene knockout and construction of transient-expression vectors are described in more detail below. For generation of GP85 5′ untranslated region (UTR) mutants, an initial construct was generated with flanking sequences (GP85 and GP86 coding sequences) for recombination to enable deletion of the original GP85 5′ UTR sequence and substitution of specific sequences. The targeted deletion (Fig. 1) removed all of the intergenic noncoding sequences between the GP85 and GP86 open reading frame (ORF) except for 3 bases prior to the GP85 ATG start and 4 bases after the stop codon of GP86 (deletion GPCMV coordinates [bases] 135907 to 136179). The left GP85-flanking sequence was PCR amplified with primers FEcGP85FLK and RBmGP85FLK, and the 0.9-kb sequence (GPCMV coordinates 134992 to 135906) was cloned as an EcoRI/BamHI fragment into pUC19 to generate pGP85FLK. The right GP86-flanking sequence was PCR amplified with primers FBmGP86FLK and RHdGP86FLK, and the 0.5-kb sequence (GPCMV coordinates 136180 to 136681) was cloned as a BamHI/HindIII fragment into pUC19 to generate pGP86FLK. Both constructs were sequenced to verify the GPCMV sequence (data not shown). Next, the GP86FLK cassette was isolated from pGP86FLK as a BamHI/HindIII fragment and cloned into pGP85FLK cut with BamHI/HindIII to generate a construct carrying both left- and right-flanking recombination arms separated by a BamHI site. The modified construct was designated pGP85/GP86. Next, a kanamycin (Km) cassette from pACYC177 (NEB) (36) was PCR amplified with primers FBgXhBmKm and RBgEcVSlKm. The 1.1-kb PCR product was isolated by agarose gel electrophoresis, digested with BglII, and cloned into pGP85/GP86 cut with BamHI. The correct orientation of the construct carried the Km cassette running in the opposite direction to the GP85 and GP86 genes and was designated pGP8586Km+ (data not shown). Next, the simian virus 40 (SV40) poly(A) sequence from peGFP-C1 (Clontech Laboratories) was PCR amplified with the primers FSV40AEcSl and RSV40AEcSl. The 330-bp fragment had novel EcoRI and SalI sites introduced into both the 5′ and 3′ ends and was cloned initially into pNEB193 (NEB) cut with EcoRI to generate pNEBSV40polyA. The SV40 poly(A) portion was next isolated as a SalI fragment from pNEBSV40polyA and cloned into pGP8586Km+ cut with SalI, which introduced the SV40 poly(A) sequence downstream of the GP86 stop codon in a plasmid designated pGP8586SV40AKm. Next, the TREtight promoter was PCR amplified as a BamHI fragment from pTREtight (Clontech Laboratories) and cloned initially into pNEB193 (NEB) to generate pNEBTRE, and the promoter sequence was verified (data not shown). The TREtight promoter as a BamHI fragment was then isolated and cloned into pGP8586SV40AKm cut with BamHI, and its orientation was determined by XhoI digestion. The correct construct placed the pTREtight promoter directly upstream of the GP85 coding sequence. This construct, which was used to generate the DISC virus strain, was designated pGP8586TRESV40AKm. The cloning strategy for the shuttle vectors is summarized in Fig. 2. In order to further demonstrate the essential nature of the GP85 protein, an additional GP85-knockout shuttle vector was generated synthetically (via DNA2.0), which deleted the majority of the GP85 coding sequence of pSYDGP85 (codons 58 to 241 deleted). This shuttle vector was further modified by the insertion of a Km BamHI cassette (36) to enable selection of the GPCMV GP85 knockout mutant BAC. The GP85 deletion knockout vector was designated pSYDGP85Km.
FIG 1.

Nucleotide sequence of the GP85/GP86 locus. The sense genome strand sequence is shown. Both GP85 and GP86 are encoded on the complementary strand. The sequence used for flanking arms in shuttle vector (Fig. 2) recombination are highlighted. The GP85 flanking sequence includes GPCMV bases 134992 to 135906; GP86 flanking sequence includes bases 136180 to 136681; the deleted intergenic sequence includes bases 135907 to 136179.
FIG 2.

Cloning strategy for generation of a shuttle vector encoding GP85 under Tet-Off (TRE-tight) promoter control. (Steps 1 and 2) Parts of the GP85 (5′) and GP86 (3′) coding sequences were separately cloned via PCR into pUC19 to generate pGP85FLK and pGP86FLK. (Step 3) The GP86 coding sequence was then cloned into pGP85FLK as a BamHI/HindIII fragment to generate pGP85GP86, which lacked the intergenic sequence between GP85 and GP86. (Steps 4 to 6) A BglII Km cassette was introduced to generate pGP8586Km+ (steps 4 and 5), which was digested with SalI to enable cloning of a SV40 poly(A) cassette to generate pGP8586SV40AKm (step 6). (Step 7) The TREtight promoter from pTREtight (Clontech) was PCR cloned as a BamHI fragment immediately upstream of the GP85 coding sequence to generate pGP8586TRESV40AKm. Shuttle vectors (steps 5, 6, and 7) linearized with PmeI were used to modify the GP85/GP86 intergenic locus in the GPCMV BAC via homologous recombination in separate reactions to introduce the modified sequence, as described in Materials and Methods, to generate the DISC GPCMV BAC and other BAC mutants.
Generation of mutant GPCMV BACmids and analysis of GPCMV BAC mutants.
An inducible ET recombination system (Gene Bridges) was introduced into DH10B bacterial cells containing a first-generation GPCMV BAC plasmid (26, 27), and mutagenesis of the GPCMV BAC was performed using linearized shuttle vectors encoding a kanamycin marker as previously described (32). Isolated mutant GPCMV BAC colonies were characterized by separate EcoRI and HindIII restriction digestions of BAC DNA to verify the accuracy of the predicted genome configuration after mutation (26, 27). Insertion of the Km drug resistance cassette into the viral genome introduced a novel HindIII restriction enzyme site at the site of mutation to enable verification of locus modification. Specific gene modifications were confirmed by comparative PCR analysis between wild-type and mutant GPCMV BACs using common flanking primers for each gene (Table 1). PCRs were carried out under conditions described by McGregor et al. (32), except the extension time at 72°C was modified based on the size of each gene (based on 30 s of extension per 500 bases). The gene knockout for mutants was further verified by sequencing of the cloned PCR product.
Generation of mutant GPCMV.
For generation of recombinant viruses, large-scale GPCMV BAC DNA was purified from E. coli strain DH10B by using a maxiplasmid kit (Qiagen). BAC DNA was transfected onto GPL or GPL Tet-Off cells in six-well dishes by using Lipofectamine 2000 (Invitrogen) as previously described (33). GPCMV BAC transfections were carried out with two independent clones for each gene knockout/modification. Transfections were followed for at least 4 weeks for the production of viral plaques. Green fluorescent protein (GFP)-positive viral plaques were detected via microscopy (33). Noninfectious mutants produced only single GFP-positive cells that did not progress to viral plaques. GPCMV mutant BAC transfections were carried out multiple times (minimum of 6 times) for each clone. Rescue of lethal knockout mutants was by cotransfection of the mutant GPCMV BAC DNA with a 1.7-kb PCR product of the wild-type GP85/GP86 locus generated from wild-type GPCMV BAC DNA with primers FEcGP85FLK and RHdGP86FLK (Table 1).
Ethics.
Guinea pig (Hartley) animal studies were carried out under IACUC guidelines (Texas A&M University or University of Minnesota). All study procedures were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and published by the National Research Council (65). Animals were observed daily by trained animal care staff, and animals that required care were referred to the attending veterinarian for immediate care or euthanasia. Terminal euthanasia was carried out by lethal CO2 overdose followed by cervical dislocation in accordance with IACUC protocols and NIH guidelines. Animals purchased from Charles River Laboratories were verified seronegative for GPCMV by toe nail clip bleed and anti-GPCMV ELISA analysis of serum samples as previously described (36). Animal studies were carried out to determine the following: (i) the immune (antibody) response to the GPCMV DISC vaccine; (ii) the T cell immune response to the GPCMV pp65 homolog protein; (iii) the level of protection against congenital GPCMV infection provided by the DISC vaccine. Initial DISC vaccine regimen studies were carried as described below in Results. Congenital GPCMV protection studies were carried out (described below). Additionally, animals hyperimmune to wild-type GPCMV were evaluated for the antibody response to GPCMV and specific glycoprotein complexes via ELISA, and neutralizing antibody titers were also determined. Guinea pigs (n = 5) were made hyperimmune to GPCMV by three injections of wild-type GPCMV via the subcutaneous route (105 PFU per injection). Each injection was separated by an interval of 4 weeks. Animals were evaluated after the first and last injections for anti-GPCMV titer. Animals were euthanized at approximately 4 weeks after the last injection, and sera were pooled for evaluation of antibody titers (anti-GPCMV, anti-gB, anti gH/gL, and anti-gM/gN). Additionally, splenocytes from individual animals were used to evaluate the T cell response to GP83 via gamma interferon ELISPOT assay as described above.
Congenital GPCMV vaccine protection studies.
Seronegative female guinea pigs were randomly assigned to two different groups. Group 1 (DISM; n = 14) was vaccinated subcutaneously with the GPCMV DISC vaccine (1 × 103 PFU) and boosted with a second inoculation 1 month later with a repeat dose (1 × 103 PFU). Animals were confirmed seroconverted for GPCMV and paired with seronegative males for mating. Dams were confirmed pregnant by palpitation at approximately day 20 to 25 of gestation. A second control group of nonvaccinated seronegative females (NOD; group 2; n = 15) was also paired for mating. At late second trimester/early third trimester, pregnant animals in both groups were challenged with the salivary gland stock of wild-type GPCMV (105 PFU) via subcutaneous inoculation, and animals were allowed to go to term. The viral loads in target organs (liver, lung, spleen, brain) of live and stillborn pups were evaluated by real-time PCR.
Real-time PCR.
Tissues were collected from euthanized guinea pigs to determine viral loads. For pups from congenital infection studies, tissues (lung, liver, spleen, brain) were collected within 3 days postbirth. Pup-specific placenta was collected and preserved for DNA extraction when applicable. For tissue DNA extraction, FastPrep 24 (MP Biomedical) was used to homogenize tissues as a 20% (wt/vol) homogenate in lysing matrix D (MP Biomedicals). DNA was extracted using the QIAxtractor apparatus (Qiagen) according to the manufacturer's tissue protocol instructions. Viral load was determined by real-time PCR on a LightCycler 480 system (Roche Applied Science). Primers and hydrolysis probe were designed using the LightCycler Probe Design2 program to amplify a product from the GPCMV GP44 gene: forward primer, 5′TCTCCACGGTGAAAGAGTTGT; reverse primer, 5′GTGCTGTCGGACCACGATA; hydrolysis probe, 5′–6-carboxyfluorescein–TCTTGCTCTGCAGGTGGACGA–black hole quencher 1. The PCR mixture contained LightCycler probes master mix (Roche Life Sciences), 0.4 μM primers and 0.1 μM probe, 0.4 U uracil N-glycosylase (UNG) in a 25-μl total reaction volume, including 10 μl of DNA per reaction mixture. Standard controls and no-template controls (NTC) were run with each assay for quantification. LightCycler 480 amplification parameters were as follows: UNG step for 10 min at 40°C, followed by activation at 95°C for 10 min, then 45 cycles of denaturation at 95°C for 15 s, annealing at 56°C for 15 s, and elongation at 72°C for 10 s. Data were collected by “single” acquisition during the extension step. Data were analyzed with the LightCycler data analysis software (version 1.5.1; Roche). A standard curve was generated using serial dilutions of GPCMV GP44 plasmid (33) at known concentrations for quantification and determination of assay sensitivity. The sensitivity of the assay was determined to be 5 copies/reaction mixture. Viral loads are expressed as the copy number per milligram of tissue. Results were calculated as the mean values of triplicate PCR runs per sample.
ELISAs, GPCMV neutralization assays, and Western analysis.
An anti-GPCMV ELISA and specific glycoprotein complex ELISAs (anti-gB, anti-gH/gL, and anti-gM/gN) were carried out as previously described (36). MaxiSorp ELISA plates (Nunc) were coated with 0.25 μg of either Ag+ or Ag− preparations diluted in carbonate coating buffer overnight at 4°C, washed in phosphate-buffered saline (PBS) with Tween 20, and then blocked with 2% nonfat dry milk. Test sera were diluted in blocking buffer in doubling dilutions from 1:80 to 1:5,120, incubated for 2 h at 37°C, and then reacted with anti-guinea pig IgG–horseradish peroxidase (HRP)-conjugated antibody (Sigma) diluted (1:1,000) in blocking buffer for an additional 1 h at 37°C before reacting with tetramethylbenzidine membrane peroxidase substrate (KPL). The net optical density at 450 nm (OD450) was attained by subtracting the OD of Ag− preparations from the OD of Ag+ preparations. ELISA reactivity was considered positive if the net OD was greater than or equal to 0.2 for GPCMV-negative serum. GPCMV neutralization assays were performed on GPL fibroblast cells with a GFP-tagged GPCMV (vAM403) (26), and neutralization assays were performed as previously described (36). The final neutralizing antibody titer was the highest dilution producing a 50% or greater reduction in plaques compared to the virus-only control. Western blot assays were carried out as previously described (36) using wild-type virus-infected cell lysate or sucrose-purified virions plus control mock-infected GPL cell lysate. Sera from vaccinated animals from treatment regimen 1 (R1; 1:2,000 dilution) were used in conjunction with anti-guinea pig IgG–HRP conjugate (1:500 dilution; Sigma).
Guinea pig IFN-γ ELISPOT assay.
Anti-guinea pig IFN-γ monoclonal antibodies used in the assay were based on previously characterized monoclonal antibodies against guinea pig IFN-γ (66, 67). Hybridoma cell lines for the production of the monoclonal antibodies were a generous gift from H. Schäfer, Robert Koch-Institute, Germany. Large-scale antibody production and purification were carried out by Genscript. IFN-γ ELISPOT assays were performed in polyvinylidene difluoride membrane 96-well plates. Membranes were presoaked with 70% ethanol, then washed with 1× PBS to thoroughly remove the alcohol prior to coating with 0.5 μg guinea pig IFN-γ capture antibody (V-E4), and incubated overnight at 4°C. Membranes were washed with 1× PBS and then blocked with RPMI plus 10% FCS for 2 h at room temperature. Splenocytes were prepared as previously described (68). Briefly, isolated spleens harvested from euthanized animals were immediately homogenized in PBS, homogenates were put through a 70-μm cell strainer, and red blood cells were lysed. Splenocytes were washed with 1× PBS three times before being resuspended in RPMI medium containing 10% FBS and antibiotic/antimycotic. Cells were counted via trypan blue exclusion, and 1 × 105 cells were used per well. Blocked membranes were washed once with PBS before splenocytes were added. GPCMV GP83 peptide pools (see below) were added to each well of cells at a final concentration of 5 μg/ml. Concanavalin A (ConA; 10 μg/ml) was used as a positive control, and other controls included cell-only controls, a dimethyl sulfoxide (DMSO) control (peptide background), GFP (nonspecific peptide control), and medium-only control. Plates were covered with foil and then incubated at 37°C in a 5% CO2 cell culture incubator for 18 h. Membranes were washed with wash buffer (1× PBS plus 0.1% Tween 20) 5 times before a detection antibody, biotinylated N-G3, was added and mixtures were incubated at room temperature for 2 h. Detection antibody was diluted to 1 μg/ml with diluent (1× PBS, 1% bovine serum albumin, 0.05% Tween 20), and the mixture was filtered through a 0.2-μm filter before use. Membranes were washed with wash buffer 3 times before 100 μl of streptavidin-alkaline phosphatase (R&D Systems), diluted 1:3,200 in diluent, was added and the mixture was incubated for 1.5 h at room temperature. A 100-μl volume of 5-bromo-4-chloro-3-indolyl-phosphate/nitroblue tetrazolium (Life Technologies) was added to membranes, which were then washed 3 times in wash buffer before incubation for 30 min at room temperature, protected from light. Membranes were washed 2 times in distilled water, inverted onto blots, and dried before counting the spots on an ImmunoSpot S6 analyzer (CTL). Final counts were calculated based on the number of spot-forming cells (SFC) per 106 cells, after subtraction of the number of background spots (cells only, without any stimulation).
A total of 140 PEPscreen 9-amino-acid peptides overlapping by 5 amino acids were generated (Sigma-Aldrich, The Woodlands, TX) to create the GPCMV GP83 peptide library, expanding the full-length gene. Nine-amino-acid peptides were used to target CD8+ T cell activation (69). Each peptide was reconstituted to 10 mg/ml with DMSO. Peptide pools were generated using a configuration matrix previously described (69, 70). The GP83 matrix consisted of 24 peptide pools in a 12-by-12 grid, with each pool containing 12 peptides. Pools IX, X, XI, and XII contained 11 peptides, and pool XXIV had 8 peptides. Additional DMSO was added to keep the concentration the same in all pools. All peptide pools were diluted to a 10-μg/ml working stock in RPMI for stimulation. The matrix was designed for each peptide to be included in exactly 2 pools, keeping the number of pools at a minimum. The intersection of positive pools corresponded to the stimulating peptides.
Statistical analysis.
Mean antibody titers (ELISA and neutralizing antibody tests) in the vaccine study and hyperimmune GPCMV-infected animals were compared with the Mann-Whitney test. In congenital infection studies, pup outcome and transmission rates were compared by using Fisher's exact test. GPCMV viral loads in specific target organs of pups were compared with the Student t test or/and Mann-Whitney test, depending on sample size. All comparisons were two-tailed tests.
RESULTS
Generation of a GPCMV DISC virus strain.
A lethal gene knockout was engineered into the GPCMV genome to render the virus incapable of productive infection unless the mutant virus was grown on a complementing cell line. This mutant virus was the basis of the GPCMV vaccine strategy and is referred to as a DISC (defective infectious single-cycle) virus vaccine. This terminology was originally developed for a herpes simplex virus (HSV) vaccine strategy, which was based on a glycoprotein (gH) knockout virus (71). The GPCMV DISC vaccine strategy was based on a targeted knockout of a capsid gene, as the gH glycoprotein was considered potentially important for glycoprotein complex formation and consequently an important target antigen(s). In contrast, the viral capsid genes are essential for virus assembly but in themselves are relatively unimportant vaccine targets. Additionally, a capsid mutant was considered unlikely to interfere with expression of other viral proteins or the assembly of viral glycoprotein complexes, since capsid assembly occurs independently in the nucleus. Importantly, CMV capsid genes are highly conserved between HCMV and animal CMV, and the process of capsid assembly in HCMV is well studied and defined (63). Additionally, capsid assembly is conserved between alphaherpesviruses (e.g., HSV) and HCMV (72). GPCMV encodes homologs of the HCMV capsid genes, and earlier electron microscopy studies demonstrated a similar icosahedral capsid assembly process in the nucleus as well as a similar virus maturation process (15, 73). The GPCMV DISC vaccine strategy was based on a UL85 homolog (GP85) mutant that was predicted to encode the minor capsid protein (GP85). In HCMV, UL85 dimerizes with itself and the minor capsid binding protein (UL46) to form a triplex as part of a fundamental building block for capsid assembly (63). GPCMV encodes a UL46 homolog (GP46) which enables triplex formation with GP85. A BLAST alignment of the predicted GP85 and UL85 nucleotide sequences demonstrated a high level of sequence conservation between the open reading frames (56% identity) (Fig. 3). Transient plasmid expression of the GP85 protein in GPL cells demonstrated that the protein was located in both the nucleus and cytoplasm (data not shown). A targeted deletion knockout of the GP85 gene (codons 58 to 241) in a GPCMV BAC resulted in lethal knockout of the virus, which demonstrated the essential nature of GP85 (data not shown).
FIG 3.

BLAST alignment of the predicted minor capsid protein (mCP) nucleotide sequence from GPCMV (GP85) and HCMV Towne strain (UL85). (i) Colinear location of GP85 and UL85 genes in GPCMV and HCMV, respectively. (ii) BLAST search of the GP85 protein identified it as a member of the herpes virus V23 (capsid protein) superfamily. (iii) BLAST (NCBI BLASTp) alignment of GP85 and UL85 proteins. Score, 354 bits (908); E, = 2e−120; method used, compositional matrix adjust; identities, 172/305 (56%); positives, 226/305 (74%); gaps, 4/305 (1%).
The first stage in the generation of a DISC virus was the development of a complementing cell line for virus growth. In the case of the GPCMV GP85 DISC strain, the classical approach, which entails use of a cell line that expresses the missing gene in trans, was replaced by another strategy that employed the Tet-Off Advanced system, where cells express the transactivator (64). Control of GP85 gene expression in a recombinant virus was achieved by placing the gene under the control of the Tet-Off transactivating protein tTA2 (64). Potentially, this approach has the advantage of placing multiple viral genes under the control of the same Tet-Off system/Tet-Off transactivating protein and therefore bypasses the requirement for a cell line expressing the essential target gene(s) in trans. Establishment of the Tet-Off Advanced system required the development of a GPL cell line that expressed the Tet-Off transactivator tTA2 (Clontech Laboratories). (The procedure for generation of the GPL Tet-Off cell lines is described in Materials and Methods.) Overall, 30 potential Tet-Off Advanced cell lines were isolated and screened for the tTA2 transactivator by transfection of a luciferase reporter plasmid (Clontech Laboratories) under TREtight (Tet-Off) promoter control (pTREtightLUC). Expression of luciferase was assayed by bioluminescence imaging of transfected plates at 24 h posttransfection. Four cell lines (A4, A20, A21, and D7) were identified which continued to exhibit consistent levels of tTA2 expression, determined in a luciferase reporter gene transactivation bioluminescence assay. Figure 4 shows the results of the transient-expression assays used to screen candidate cell lines. Plate bioluminescence imaging was employed to ensure that the assay demonstrated activity across the complete intact monolayer. Control plasmid transfections (pTREtightLUC and pTet-Off-Advanced) were performed on GPL cells to provide an idea of the minimal acceptable level of activity of reporter gene transactivation. Additionally, pCDNALuc (luciferase under the HCMV major immediate-early [MIE] protein control) was used to provide a guide for efficient direct expression of the reporter gene (Fig. 1). Cell lines A20 and A21 (Fig. 1) were subsequently used for the propagation of the GP85 DISC mutant virus strain, as they consistently exhibited the highest levels of reporter gene transactivation.
FIG 4.

Characterization of GPL Tet-Off cell lines via luciferase reporter gene transactivation. GPL cells (A to D) or candidate GP Tet-Off cells (E to H) were transfected with a luciferase reporter plasmid under TREtight promoter control (pTREtightLUC). Wells A and C were additionally transfected with the Tet-Off transactivator (tTA2) expression plasmid. Well B was transfected with a luciferase reporter gene plasmid under HCMV MIE promoter control (pcDNA3LUC). At 24 h posttransfection, luciferase substrate (D-luciferin) was added, and plates were imaged for 5 min to evaluate bioluminescence (IVIS 50; Xenogen). Shown are black and white images of six-well plates, with superimposed photon emission intensities. Vertical color bar ranges indicate the highest (red) to lowest (purple) levels of bioluminescence (in photons per second per square centimeter per steradian) for imaged samples. Tet-Off cell line assays included duplicate wells for A20 (E and G) and A21 (F and H) cells.
In order to generate the GP85 DISC GPCMV mutant, the first-generation GPCMV BAC was modified at the GP85/GP86 intergenic locus. The UTR upstream of the GP85 ORF was deleted (Fig. 1) and replaced by partial or complete sequences to generate a GP85 DISC mutant as described in Materials and Methods, using specific GP85/GP86 locus shuttle vectors (Fig. 2). Three specific GP85 GPCMV BAC mutants were generated (Fig. 4). The first mutant replaced the GP85 UTR sequence (272 bp; coordinates 135907 to 136179) with a Km cassette (designated GP85/GP86Km GPCMV BAC). The second mutant replaced the UTR with a Km cassette and SV40 poly(A) sequence downstream of the GP86 coding sequence (designated GP85/GP86Km/poly(A) GPCMV BAC). The third mutant replaced the GP85 UTR with a Km cassette and SV40 poly(A) sequence and also introduced a TREtight promoter directly upstream of the GP85 ORF. This third GPCMV BAC mutant was designated TREGP85 DISC GPCMV BAC. Verification of the GP85 locus mutations was confirmed by HindIII restriction profile analysis of wild-type and mutant GPCMV BACs. Insertion of a Km cassette introduced a novel HindIII site into the GP85/GP86 locus and disrupted the HindIII A fragment (approximately 44 kb; GPCMV coordinates 102379 to 146446), which was modified to produce two novel fragments in the TREGP85 DISC GPCMV BAC profile of approximately 34.5 kb and 10.8 kb after inclusion of the extra cassette sequence and deletion sequence. Other modifications also produced a similar pattern of the modified HindIII A fragment (Fig. 5). Specific modification to the GP85/GP86 locus was also verified by PCR analysis of the modified locus based on primers used to create the shuttle vector (left-arm coordinates 134992 to 135906, right-arm coordinates 136180 to 136681). The GP85/GP86 locus and deleted intergenic sequence are shown in Fig. 1. Figure 5, panel iv, shows the 1.7-kb wild-type locus (GPCMV coordinates 134992 to 136681) and the 2.5-kb PCR product of the locus with the GP85 5′ UTR deletion and insertion of a 1.1-kb Km cassette. PCR of the TREGP85 GPCMV BAC GP85/GP86 locus demonstrated a modified locus of 3.1 kb as a result of the insertion of the Km/SV40 poly(A)/pTREtight cassette and deletion of the original GP85 5′ UTR. Note that the GP85/GP86/Km/poly(A) GPCMV BAC was correctly modified based on its restriction profile and PCR analyses (data not shown, to limit redundancy).
FIG 5.

Modification of the GP85 5′ UTR in a GPCMV BAC and generation of a DISC GP85 strain BAC. (i) Schematic of the GPCMV genome, with location of the BAC insertion and HindIII sites indicated. The GP85/GP86 locus is encoded in the HindIII A fragment. (ii) Modifications to the 5′ UTR in the GP85/GP86 locus in the wild type (wt; 1) and mutants GP85/GP86KmR BAC (2, top), GP85/GP86KmRpoly(A) BAC (2, bottom), and TRE GP85 (DISC) BAC (3). Also shown are the approximate sizes of the wild-type and mutant loci, based on the external primer used to generate the original GP85/GP86 shuttle vector. (iii) EcoRI restriction profile analysis of wild-type and mutant GPCMV BACs described in panel ii, including wild-type GPCMV BAC (1), GP85/GP86KmR BAC (2), and TRE GP85 (DISC) BAC (3). The original HindIII A genomic fragment is indicated in blue, and the modified fragment indicated by red dots. (iv) PCR amplification of the GP85/GP86 locus in wild-type and mutant GPCMV BACs: wild type (1), GP85/GP86 KmR (2), and TRE GP85 DISC (3). The DNA ladder (measurements in kilobases) was obtained from Invitrogen.
All three mutant GPCMV BAC clones were transfected onto both GPL and GPL Tet-Off cell lines as described in Materials and Methods. Separate transfections of the three GP85 GPCMV BAC mutants failed to generate infectious virus on GPL cells, and individually transfected cells remained GFP positive, but viral plaques and virus spread across the monolayer failed to occur (Fig. 6). The GP85 mutants could be returned to wild type by cotransfection of the BAC DNA with rescue with the PCR product of the wild-type GP85/GP86 locus (Fig. 5). Separate transfections of all three GP85 mutant GPCMV BACs onto the GPL Tet-Off cells resulted in viable virus production for the TREGP85 GPCMV BAC, which produced GFP-positive virus that spread across the monolayer. In contrast, cells in the other GP85 mutant-transfected wells remained single GFP-positive cells (data not shown). Monolayers were monitored daily for 3 to 4 weeks with frequent medium changes. Transfection of each mutant was carried out multiple times (>40) and additional mutants were also tested (data not shown), but the same result was obtained each time. We concluded that the tTA2 transactivator in the Tet-Off cell line specifically enabled expression of the GP85 gene when under TREtight promoter control in the TREGP85 GPCMV BAC-derived virus. Virus stocks of the GP85 DISC virus strain were generated on Tet-Off GPL cells, and the growth kinetics of the mutant virus on the complementing Tet-Off GPL cell line were evaluated. The results in Fig. 7 demonstrate that the DISC virus grew with attenuated growth kinetics on the Tet-Off cell line compared to wild-type virus, but the normal growth kinetics of the wild-type virus demonstrated that expression of the Tet-Off transactivator did not interfere with GPCMV growth. Potentially, the expression kinetics of the GP85 protein (with the gene under Tet-Off control) compared to the that for the other triplex components of the UL46 protein (with the gene under GPCMV promoter control) impaired the stoichiometry of protein expression to enable optimal triplex formation. However, an in-depth evaluation of protein expression would require the development of custom antibodies to GP85 and GP46 for future Western blot assays. The GP85 DISC virus was able to successfully infect normal GPL cells as verified by GFP reporter gene expression in infected GPL cells, but it did not produce any titratable progeny virus during a 7-day infection of GPL cells (data not shown). We concluded that the GP85 DISC strain was restricted to the Tet-Off GPL cells for production of progeny virus but retained the ability to infect noncomplementing cells. The GP85 DISC strain expressed an array of viral antigens on nonsupporting GPL cells which could be detected by Western blot analysis using hyperimmune sera from convalescent guinea pigs (data not shown). The immune response to GPCMV was also evaluated by Western blotting. Figure 8 shows a typical result for Western blot assays of sera from R1 DISC-vaccinated animals. Evaluation was carried out in Western blot assays of sucrose-purified virus as well as GPCMV virus-infected GPL cell lysates (Fig. 8). These results demonstrated that antibodies were generated against both structural and nonstructural proteins.
FIG 6.
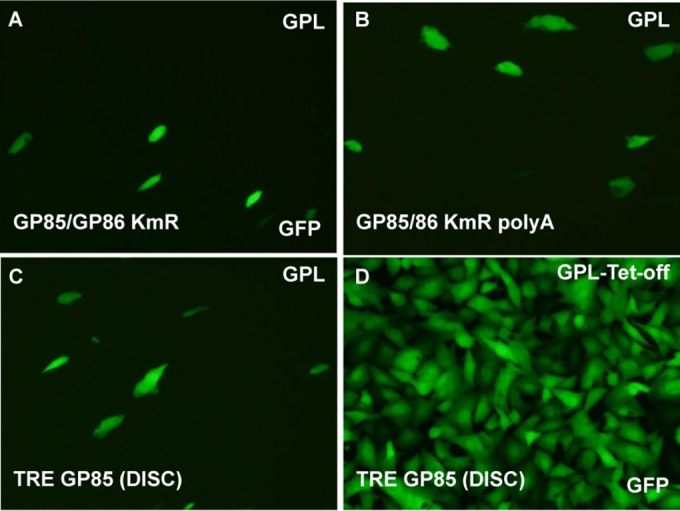
Regeneration of a DISC GP85 GPCMV from GP85 BAC mutants requires a TRE promoter and a cell line expressing a Tet-Off transactivator (tTA2). (A to C) GP85 mutant GPCMV BACs were individually transfected into GPL cells: GP85/GP86 KmR (A), GP85/86 KmR poly(A) (B), and TRE GP85 DISC (C). (D) Mutants were also transfected onto GPL Tet-Off cells. The TRE GP85 DISC is shown. Individual transfected cells expressed GFP encoded by the BAC. Virus spread was detected by GFP spread across the cell monolayer. Images were taken at 16 days posttransfection.
FIG 7.
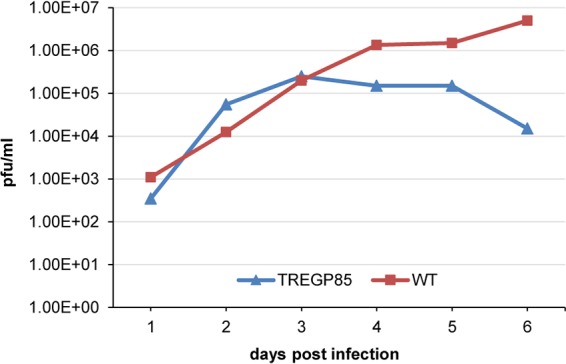
Growth kinetics of DISC GP85 GPCMV versus wild-type GPCMV on GPL Tet-Off cells. GPL Tet-Off cells were infected at a multiplicity of infection of 1 PFU/cell with each respective virus in separate wells of six-well dishes. Samples were taken on different days postinfection and titrated in duplicate as previously described (33). Results are plotted as the virus titer versus day postinfection.
FIG 8.
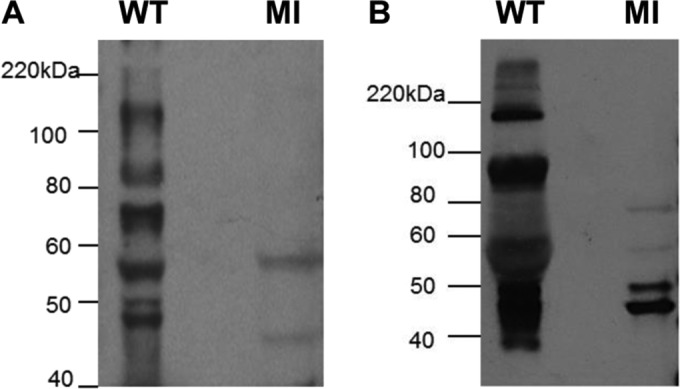
Western blots of wild-type GPCMV. GPCMV (purified virus particles or total cell lysate) were separated by SDS-PAGE and immunoblotted with anti-GPCMV sera (1:2,000 dilution) from DISC-vaccinated animals and with anti-guinea pig IgG–HRP (1:500; Sigma) as described in Materials and Methods. (A) Sucrose-purified GPCMV. (B) GPCMV-infected GPL cell lysate. MI, control lanes with uninfected GPL cells. Protein sizes are indicated in kilodaltons (Bio-Rad ladder).
Antibody immune response against viral glycoprotein complexes: vaccine regimen 1 (two-shot vaccination) versus vaccine regimen 2 (three-shot vaccination).
Two vaccination regimens were investigated to evaluate the antibody response to the GPCMV GP85 DISC vaccine (Fig. 9). In the first vaccine study, R1, GPCMV-negative female guinea pigs (n = 6) were vaccinated subcutaneously with 103 PFU DISC virus and subsequently boosted 4 weeks post-initial infection with an equivalent dose. Sera collected 4 weeks postbooster (8 weeks post-initial infection) were tested for GPCMV seroconversion and the level of neutralizing antibodies. All animals seroconverted and showed relatively high anti-GPCMV ELISA titers (1:1,280 to 1:5,120; mean, 1:2,987), but this was lower than the result with pooled hyperimmune animal sera convalescent for wild-type GPCMV infection (mean titer, 1:5,000). The immune responses to specific glycoproteins (gB, gH/gL, and gM/gN) were also evaluated for DISC-vaccinated animals by using newly developed ELISAs (36). ELISA titers of the DISC-vaccinated animals for anti-gB were not as robust as those for wild-type GPCMV hyperimmune sera (1:640 versus 1:1,600; P < 0.05). Similarly, the glycoprotein complex gH/gL titer was lower (1:216 versus 1:960; P < 0.05) (Fig. 10A). However, the immune response of DISC-vaccinated animals to the gM/gN complex was slightly stronger than, but not statistically significant, the response to hyperimmune sera (1:704 versus 1:480). The neutralizing antibody titer of DISC-vaccinated animals on GPL cells was lower, 1:960 (P < 0.05) than seen with a 1:4,200 dilution of hyperimmune sera (Fig. 10B).
FIG 9.

Overview of DISC GP85 vaccination schedules and the preconception vaccine study. (a and b) In the initial characterization of the immune response to the DISC vaccine, two vaccine regimens were employed: R1 (a) and R2 (b). DISC virus inoculations are indicated with the arrow along with a virus symbol. Animals were bled for immune response measurements at the indicated weeks post-initial vaccination. Animals were euthanized at the end of the vaccination schedule (and after confirmation of seroconversion) to determine the T cell response via an ELISPOT assay as described in Materials and Methods. (c) Preconception vaccine study (group 1) following vaccine regimen R1. During the late second trimester of pregnancy, animals were challenged with wild-type GPCMV (105 PFU) and then allowed to go to term. (d) Control nonvaccinated animals (group 2) from the congenital vaccine study.
FIG 10.
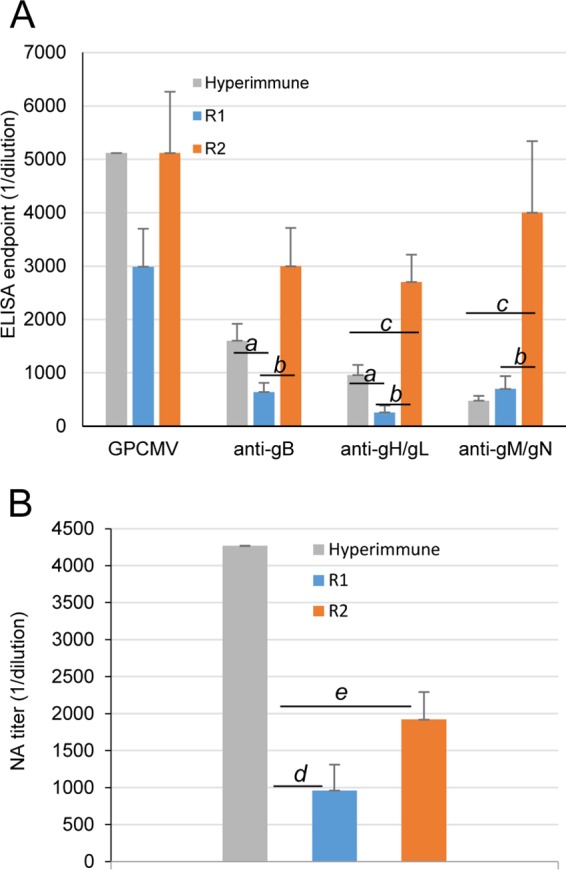
Antibody immune responses (determined in ELISAs) to GPCMV and specific viral glycoprotein complexes (gB, gH/gL, and gM/gN) in DISC GP85-vaccinated guinea pigs. (A) Immune responses of animals vaccinated with DISC GP85 under the R1 or R2 regimen were compared to animals hyperimmunized with wild-type GPCMV. Results shown are means for each group with standard deviations represented by error bars. Statistically significant differences (P < 0.05; Mann-Whitney test) are shown by lowercase italic letters for comparisons as follows: a, hyperimmune versus R1; b, R1 versus R2; c, hyperimmune versus R2. (B) Neutralizing antibody titers on fibroblast cells from hyperimmune sera, compared to those from the R1 and R2 regimens. Statistically significant differences (P < 0.05; Mann-Whitney test) are indicated by lowercase italic letters for comparisons as follows: d, hyperimmune versus R1; e, hyperimmune versus R2.
In the second vaccine study, regimen 2, GPCMV-negative female guinea pigs (n = 5) were vaccinated subcutaneously with 103 PFU DISC virus and subsequently received equivalent booster inoculations of 103 DISC virus at 4 and 8 weeks post-initial inoculation (Fig. 9). Sera collected 4 weeks post-second boost (12 weeks post-initial vaccination) were tested for anti-GPCMV titers and the specific immune response to glycoprotein complexes, as described for R1 samples. The additional DISC vaccination in R2 animals appeared to enhance the immune response to GPCMV. The anti-GPCMV ELISA titers ranged from 1:2,560 to 1:10,240, with a mean titer of 1:5,120, resembling titers for hyperimmune GPCMV animals (Fig. 10A). Specific glycoprotein complex ELISA results demonstrated an increase in the immune response to both gB (1:3,000 titer, versus hyperimmune titer of 1:1,600) and gH/gL (1:2,700 titer versus 1:960 in hyperimmune group; P < 0.05). The antibody titer against gM/gN also increased (1:4,000 versus hyperimmune titer of 1:480; P < 0.05). Although the R2 booster strategy increased the immune response to all specific complexes tested, the impact on the gM/gN immune response was relatively unusual, as titer levels were above those of hyperimmune animals. The reason for this dramatic change in the gM/gN immune response is undetermined at this time and is worthy of further investigation. It should be noted that although our vaccine studies compared the immune response with that in hyperimmune animals, the actual immune response to the GPCMV glycoprotein complexes as well as the anti-GPCMV titers are more varied and lower in naturally infected animals, based on results obtained from GPCMV-positive animals purchased from various animal vendors (data not shown).
Overall, the DISC vaccine strategy in vaccinated animals was more successful with a two-booster approach (R2) rather than the single-booster approach (R1). In the R2 study, the anti-GPCMV titers increased and, importantly, the antibody responses for the specific glycoprotein complexes were also increased compared to those in R1. However, despite improved values in the R2 study with titers equivalent to or better than those for the hyperimmune sera, the neutralizing titers for animals in the R2 study were below those for hyperimmune sera (1:2,000 versus 1:4,200; P < 0.05) (Fig. 10B) and only slightly higher than in the R1 study. Potentially, a missing component of the immune response in DISC-vaccinated animals compared to wild-type virus-infected animals was an immune response to the homologous pentameric complex (58), as the DISC vaccine was based on a lab-adapted virus that lacked a full-length UL128-131 homologous locus (15, 26). The immune response to the homologous pentameric complex is under investigation in our laboratory via the use of attenuated GPCMV mutants as well as a second-generation GPCMV DISC vaccine.
T cell immune response to the pp65 homolog protein.
The T cell response to the DISC virus vaccine was evaluated in a newly developed guinea pig-specific IFN-γ ELISPOT assay as described in Materials and Methods. Pools of 9-mer peptides designed to target the CD8+ T cell response were used to pulse isolated splenocytes from animals in the R1 and R2 study groups as well as wild-type GPCMV-infected animals. The peptide pool matrix was designed to optimally utilize the largest number of peptides in the most efficient and cost-effective manner (69, 70). The GP83 protein has a predicted length of 565 amino acids, which generate 140 overlapping 9-mer peptides. This enabled 24 pools of peptides in a 12-by-12 square matrix, with each peptide appearing in two pools. The intersection of positive pools identified the stimulating peptides, narrowing the potential candidate peptides that could be tested individually. The concentration of peptide pools used was determined by testing different concentrations based on HCMV IFN-γ ELISPOT results (74, 75). For the GP83 peptide pools, a final concentration of 5 μg/ml gave the highest stimulation of IFN-γ-producing cells. ConA (positive control) produced an average of 5,840 SFC per 106 stimulated cells (± 235 [standard deviation]). Unstimulated cells (634 ± 5.8 SFC/106 cells) and DMSO-stimulated cells (548 ± 16 SFC/106 cells) produced similar results, which were used to establish the baseline background level. Each animal's response to GP83 peptide stimulation was slightly different, but at least 50% of the animals responded to peptide pools V, VIII, X, and XX (data not shown). An increase in IFN-γ-secreting cells in response to peptide pools X and XX occurred in all animals. Spot-forming cells from group R1 animals responding to pool X (2,621 ± 508 SFC/106 cells) or pool XX (2,227 ± 590 SFC/106 cells) were similar to those seen in wild-type GPCMV-infected cells stimulated with pool X (2,900 ± 468 SFC/106 cells) and pool XX (3,220 ± 1,362 SFC/106 cells). Furthermore, there were no significant differences in responses to pool X (3,220 ± 1,114 SFC/106 cells) or pool XX (2,918 ± 990 SFC/106 cells) in group R2 (Fig. 11), despite a higher humoral response to GPCMV antigens. Certain GP83 peptide pools (pools II and IV) failed to routinely stimulate the splenocytes above background levels in infected animals (Fig. 11).
FIG 11.
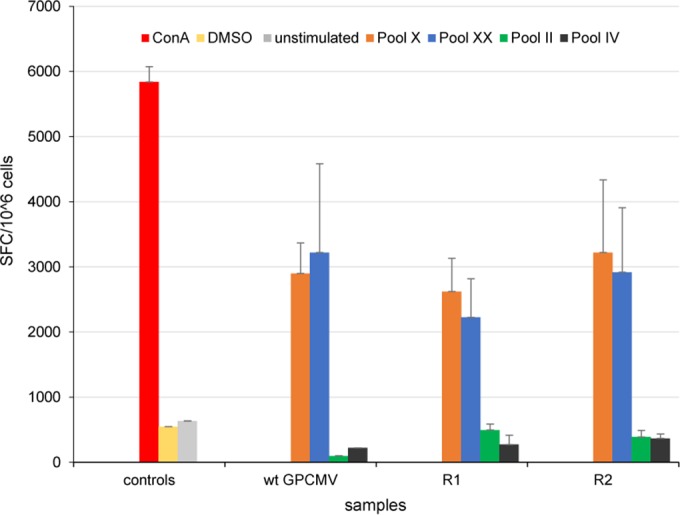
Evaluation of animal T cell response to the GPCMV vaccine. Gamma interferon ELISPOT assay results are shown for splenocyte responses to GPCMV GP83 peptide pools in R1 or R2 vaccinated and wild-type (wt) GPCMV-infected animals. The assays were carried out as described in Materials and Methods, with splenocytes isolated from DISC GP85-vaccinated animals (vaccine regimens R1 and R2) or wild-type GPCMV-infected animals. Overlapping peptides (9-mers) spanning the complete GP83 protein sequence were assigned to 24 peptide pools (I to XXIV). Results are shown for selected GP83 peptide pools: II, IV, X, and XX. ConA was used as a positive control, and negative controls included unstimulated and DMSO-treated cells. Final counts were calculated based on SFC per 106 cells after background spots (cells only, without any stimulation) were subtracted. GP83 peptide pools X and XX produced highly stimulated cells, whereas pools II and IV resulted in poorly stimulated cells.
Overall, the DISC vaccine was confirmed to induce a T cell response against the pp65 homologous protein, which is considered a major T cell target with HCMV. Encouragingly, the DISC vaccine T cell response was similar to that in animals challenged with wild-type live virus. An analysis of the immune response in naturally infected animals remains to be investigated to determine if there is a difference in response in vaccinated or hyperimmune animals. The stimulation of splenocytes from DISC-vaccinated animals by GP83 peptide pools allowed the identification of a hot spot region within the GP83 protein, which will enable a narrower range of peptides to be used in future studies and enable identification of the most reactive T cell epitope. However, the use of outbred Harley guinea pigs in future studies may have to be changed to the use of inbred strain 2 animals to limit animal-to-animal variation in the GP83 T cell response.
Congenital GPCMV protection study (2-shot vaccination, regimen 1).
Previous sections described how the DISC vaccine strategy induced effective antibody and T cell responses against GPCMV in vaccinated animals. Consequently, the DISC virus vaccine strategy was evaluated for the ability to protect against congenital GPCMV infection under vaccine regimen 1 (Fig. 9). Initially, 29 female guinea pigs seronegative for GPCMV were randomly assigned into two groups: group 1, vaccinated (DISM; n = 14); and group 2, control unvaccinated (NOD; n = 15). Group 1 animals were vaccinated with 103 PFU subcutaneously with the GP85 DISC virus, and 1 month postvaccination the animals received an equivalent booster dose of the DISC strain. Evaluation of their serum status revealed that the animals had a similar range of mixed anti-GPCMV ELISA titers, which ranged from 1:160 to 1:2,560, and neutralizing antibody titers as the animals from the R1 study (data not shown). Once animals were confirmed to have an immune response to the DISC vaccine, they were paired with seronegative male guinea pigs for mating. Control group 2 (nonvaccinated animals) were also paired for mating at this time with seronegative males. Dams were confirmed pregnant by palpation (at 20 to 25 days of gestation). At approximately late second trimester, dams were challenged subcutaneously with 105 PFU of wild-type salivary gland stock virus, and animals were allowed to go to term. The viral loads in target organs (liver, lung, spleen, and brain) of live or stillborn pups were evaluated by real-time PCR. Table 2 summarizes the pregnancy outcomes in the two animal groups. Overall, more dams carried pregnancy to term in the vaccinated group (100% versus 69.2%), which also had a higher proportion of live pups (94.1% versus 63.6%). Additionally, the DISM group had a greater overall number of live pups (48 versus 28). Although there were 3 dead pups in the vaccinated animal group, all of these pups were determined to be negative for GPCMV and death was attributed to a complication of pregnancy. In contrast, all 16 stillborn pups in the control nonvaccinated group were positive for GPCMV, with 80% of tissue analyzed (liver, lung, spleen, or brain) positive for GPCMV. Additionally, in the nonvaccinated group, two litters were reabsorbed by the dam. Based on the outcome (live versus stillborn pups) between groups, the DISC vaccine was considered effective in protecting against congenital CMV mortality (P = 0.0002, based on live births of 94.1% versus 63.6%). In this study, the CMV mortality rate in the control group was 36.4% based on dead pups for term litters. This excluded 2 litters reabsorbed by the dams in the control group. In previously published congenital GPCMV infection studies, with a challenge virus dose of 105 PFU the mortality rate ranged from 34 to 81% (51, 61, 76–80). The results of the present study fall within this range. However, in previous studies the higher mortality rates included animals that aborted within 2 weeks of virus challenge. This scenario did not occur in our studies, but reabsorbed litters were excluded from the present study, which could have increased the reported mortality rate.
TABLE 2.
Congenital infection outcomes for live versus dead pupsa
| Treatment group (n) | Litter outcomes |
Outcomes for total pups (no. [%]) |
|||||
|---|---|---|---|---|---|---|---|
| Total | Live only | Dead only | Mixed | Reabsorbed | Live borna | Stillborn | |
| DISM (14) | 13 | 11 | 0 | 2 | 0 | 48* (94.1) | 3 (5.9) |
| NOD controls (15) | 13 | 8 | 4 | 1 | 2 | 28* (63.6) | 16 (36.4) |
DISM, DISC-vaccinated mothers. *, P = 0.0002 compared to controls (Fisher's exact test).
Based on pup tissue positive for GPCMV, a transmission rate of 75.9% for the control group was reduced to 23.5% in the vaccine group (P = 0.0001). The transmission rate in the control group was roughly similar to the congenital GPCMV transmission rate observed for control nonvaccinated groups in other published studies that used challenge virus at a dose of 105 PFU. In previous studies, the GPCMV transmission rate ranged from 50 to 85%, with the majority in the range of 65 to 85% (average rate, 74.4%) (51, 76–80). In one study with a virus challenge dose of 106 PFU, the transmission rate was 70% (81), which indicated that a higher dose of challenge virus does not necessarily result in a higher transmission rate. Analysis of viral loads in pup tissues from both groups demonstrated that the tissues from the control nonvaccinated group had a higher frequency of GPCMV-positive tissue (Table 3). A 52.4% reduction in GPCMV transmission in the vaccinated group (23.5% transmission) compared to the control group (75.9% transmission) indicated that the DISC vaccine strategy was more effective than levels reported for studies with live GPCMV vaccine strains (78, 80) or gB-based vaccine strategies (76, 77, 79, 82). A comparison of the outcome of our study versus those of previous GPCMV vaccine studies is covered in more detail in the Discussion.
TABLE 3.
Impact of DISC vaccine on GPCMV transmission, based on frequencies of GPCMV-positive pups in each specific tissue group
| Vaccine group (n) | No. (%) of pups positive for GPCMV in specific tissue [P value]a |
No. (%) of CMV+ pupsb [P value] | |||
|---|---|---|---|---|---|
| Lung | Liver | Spleen | Brain | ||
| DISM (51) | 7 (13.7) [0.0001] | 2 (3.9) [0.0013] | 2 (3.9) [0.0001] | 5 (9.8) [0.0006] | 12 (23.5)[0.0001] |
| NOD controls (29) | 17 (58.62) | 9 (31.03) | 19 (65.52) | 13 (44.83) | 22 (75.9) |
P values are based on comparisons using Fisher's exact test of the number of animals CMV+ for the specific organ in the vaccinated group versus the NOD control group.
Number (percentage) of pups in which GPCMV was detected in at least one organ.
The majority of pup tissues in the vaccinated group were negative for virus, but a lower number of pups were positive. However, while the viral loads in target tissues of these individual positive pups were lower than those found in comparable tissues of the nonvaccinated group, the differences were not statistically significant except for the brain (Table 4). An important benchmark for the vaccine strategy was the prevention of CMV infection in pup brains. Consequently, although the vaccine strategy was successful in reducing congenital infection, the approach failed to completely prevent viral infection of pup brains, with 5/51 positive for a low level of virus. In the nonvaccinated group, 13/29 pups had brain infection, and the viral loads were higher than in the DISM group (Table 4) (P = 0.05).
TABLE 4.
Congenital infection outcome, based on viral load in target tissues of pups
| Treatment group | Viral load in target tissue (mean ± SD) [P value]a |
|||
|---|---|---|---|---|
| Lung | Liver | Spleen | Brain | |
| DISM | 2.5 ± 2 | 2.1 ± 1.7 | 2.2 ± 1.5 | 2.9 ± 2.6 |
| NOD | 2.6 ± 2 | 2.6 ± 2.1 | 3.2 ± 2.9 | 3.3 ± 2.7 |
| P value | [NSb,c] | [NSb] | [NSb] | [0.05c] |
Viral loads are expressed as log10 genome copies per milligram of tissue. P values are shown within brackets; NS, not significant (P > 0.05).
P value derived via Student's t test.
P value derived via Mann-Whitney test.
The congenital transmission rate from dams with an anti-GPCMV titer of 1:640 or less was 32.4%, compared to 5.9% (P = 0.042) for dams with an anti-GPCMV titer of 1:1,280 or greater (Table 5). Interestingly, a comparative analysis of individual animal anti-GPCMV antibody titers with congenital CMV transmission demonstrated that a high anti-GPCMV ELISA titer was not necessarily a predictor for protection against congenital infection. However, the incidence of congenital CMV infection in the high-titer group was significantly lower. If anti-GPCMV ELISA titers were grouped into low (1:160), medium (1:320 to 1:640), and high (1:1,280 to 1:2,560) categories, then congenital infection occurred across all groups, as follows: low titer (2 pups); medium titer (9 pups); high titer (1 pup) (Table 6). Potentially, an additional booster vaccination could have induced a stronger immune response, as indicated with DISC vaccine R2 studies. However, the rationale for testing efficacy with the R1 vaccination strategy was to match previous recombinant vaccine studies that used live attenuated GPCMV strains (78, 80, 81, 83). The outcome in comparison to previous GPCMV vaccine studies is covered in more detail in the Discussion. Overall, the DISC vaccine strategy was effective in reducing congenital infection compared to the response in the control group. Additionally, the DISC vaccine was more effective than described in previously published studies for most of the attenuated live GPCMV vaccines.
TABLE 5.
Congenital CMV infection outcomes in relation to anti-GPCMV titers
| Anti-GPCMV titer group | No. of live pups/no. of dead pups | No. of CMV+ pups/total pupsa (%) |
|---|---|---|
| Low to mid-range (1:160–1:640) | 33/1 | 11/34* (32.4%) |
| High (1:1280–1:2,560) | 15/2 | 1/17* (5.9%) |
The “total pups” includes live pups and dead pups. *, P = 0.042 (Fisher's exact test).
TABLE 6.
Individual congenital CMV infection outcomes in relation to anti-GPCMV titers
| DISC-vaccinated mother no. | Anti-GPCMV titer (titer group)a | Pups (no. live/no. dead) | No. of CMV+ pups |
|---|---|---|---|
| DISM11 | 1:160 (L) | 3/0 | 2 |
| DISM12 | 1:160 (L) | 5/0 | 0 |
| DISM6 | 1:320 (M) | 1/0 | 0 |
| DISM14 | 1:320 (M) | 4/0 | 4 |
| DISM1 | 1:640 (M) | 3/0 | 0 |
| DISM3 | 1:640 (M) | 5/0 | 0 |
| DISM5 | 1:640 (M) | 4/0 | 2 |
| DISM7 | 1:640 (M) | 4/0 | 1 |
| DISM10 | 1:640 (M) | 4/1 | 2 |
| DISM2 | 1:1,280 (H) | 3/0 | 0 |
| DISM9 | 1:1,280 (H) | 4/0 | 0 |
| DISM13 | 1:1,280 (H) | 3/2 | 1 |
| DISM8 | 1:2,560 (H) | 5/0 | 0 |
Anti-GPCMV titer ranges were grouped as follows: L, low (≤1:160); M, mid-range (1:320–1:640); H, high (≥1:1,280).
DISCUSSION
Vaccines strategies against congenital CMV that have gone forward to human clinical trials have mainly focused on a single target antigen (e.g., gB glycoprotein) and neutralizing antibodies. However, these strategies have at best provided 50% efficacy (41). Potentially, a vaccine against congenital CMV has to induce both an antibody to key neutralizing glycoprotein complexes that are essential for virus entry and additionally target important T cell target antigens to provide a comprehensive protective immune response against CMV. It is unlikely that a subunit vaccine or a recombinant vector delivery system (e.g., modified vaccinia virus Ankara [MVA]) could deliver such a complicated array of antigens and successfully evoke an immune response equivalent to that in CMV convalescent patients. An attenuated live recombinant CMV vaccine strategy is potentially an effective approach, since it mimics a natural infection and in theory induces an immune response equivalent to convalescent CMV immunity. However, attenuation covers a wide spectrum of viral mutants, and potentially some or all attenuated live vaccine strains have a safety risk associated with them, especially if the virus is able to establish latency in the host and contribute to disease later in life. A potentially safer CMV vaccine strategy entails the use of a non-replication-competent virus. This approach was investigated for guinea pig CMV infection, and the term DISC virus was adopted for the type of virus vaccine studied. The term DISC vaccine (defective infectious single-cycle vaccine) describes accurately an important feature of our vaccine strategy. The recombinant virus lacked the ability to express an essential gene (GP85, encoding the small capsid protein), and as such the virus could infect cells but lacked the ability to make progeny virus because of a defect in capsid assembly. Consequently, the DISC virus stock could only be propagated on a complementing cell line. The term DISC virus was originally used to describe an HSV-1 vaccine strategy, where the gH glycoprotein had been knocked out and the gH protein was supplied in trans in a complementing cell line (71). The DISC GPCMV approach deviated from this original strategy for HSV, as the gH glycoprotein in GPCMV was considered highly important for the generation of target antigen complexes (36). A capsid mutant was deemed more suitable, as capsid proteins are essential for the generation of progeny virus but in themselves are relatively unimportant target antigens for CMV. However, a rhesus CMV (RhCMV) gH knockout mutant virus was generated (84) and is under investigation for use as a CMV vaccine in rhesus macaques. In an additional further development for the DISC vaccine strategy, the essential capsid gene was not expressed by a complementing cell line in trans. Instead, the Tet-Off Advanced system was introduced into guinea pig fibroblast cells to enable generation of a complementing cell line. This also required modifying the GP85 gene in GPCMV to place gene expression under the control of the Tet-Off transactivator (tTA2) to enable GP85 DISC virus mutant growth on the Tet-Off cell line. This approach potentially enabled the development of next-generation DISC virus strains with additional mutations (e.g., pp71 expression) under Tet-Off promoter control with a single controlling complementing cell line. In murine CMV (MCMV), a spread-deficient virus vaccine strategy was investigated using an M94 mutant virus. In this study, a reverse strategy was used that incorporated the Tet-On transactivator into the recombinant virus, which also carried an M94 knockout. The complementing cell line encoded the M94 gene, which was induced by transactivator expression during MCMV infection. A similar strategy could have been employed for the GPCMV GP85 mutant, especially since a GP85 deletion mutant was also generated as part of the initial study. A potential downside of this approach would have been viral expression of the transactivator in the host animal. However, the M94 mutant vaccine strategy was safe even in innate immune response knockout mice and highly protective against challenge with a pathogenic strain of MCMV because of an induced T cell response (CD4+ and CD8+) as well as effective neutralizing antibodies (85).
In the GPCMV study, a newly developed gamma interferon ELISPOT assay demonstrated that the GP85 DISC vaccine evoked a T cell response to the homolog pp65 protein (GP83). Specific regions within the GP83 protein were identified that produced a heightened response. Additional studies are required to better define if the GP83 response is mainly CD4+ or CD8+ based. A previous GPCMV study demonstrated that the GP83 antigen was partially protective against congenital CMV infection. This previous study employed a defective alphavirus system that encoded GP83, and both CD4+ and CD8+ responses were induced, as shown by flow cytometry (61). Mouse monoclonal antibodies to guinea pig CD3, CD4, and CD8 markers are available (86), and so further characterization of the T cell response to GP83 is possible. However, an evaluation of the T cell response to other homologous T cell target antigens (e.g., IE1 and IE2) is also a high priority for future studies with GPCMV. Currently, splenocytes are necessary to perform this new guinea pig ELISPOT assay. Therefore, it was not possible to correlate the level of T cell response to the pp65 antigen and protection against congenital infection, since animals were required to carry their pregnancy to term, at which point the challenge virus would have complicated the results for any ELISPOT assay. Consequently, this aspect could not be evaluated for the congenital vaccine study but remains an important aspect that in this study is only partially defined.
Both mouse and rhesus macaque CMV animal model studies have demonstrated the importance of a T cell response to the virus to prevent virus dissemination, and vaccine studies suggest that other target antigens in addition to pp65 are important targets (87, 88). However, in the context of congenital CMV infection, the lack of an MCMV congenital infection model prevents any evaluation. Technical issues and additional expense have limited the number of RhCMV studies, but a recent congenital RhCMV study suggested that a CD4 response does have a controlling role in protection against congenital CMV infection but that the effect impacts both CD8+ cells and the antibody response (89).
It is likely that an important component of protection against congenital CMV infection is neutralizing antibodies directed to critical viral complexes necessary for cell entry. Importantly, antibodies can cross the placenta, and therefore anti-CMV antibodies can function in both the maternal and fetal compartments (90). In an earlier study, we characterized the GPCMV gH/gL/gO and gM/gN homologous glycoprotein complexes and demonstrated their essential nature and their importance as immune targets via newly developed ELISAs (36). Overall, the DISC vaccine strategy generated a high anti-GPCMV antibody titer, and the vaccine regimen with 2 booster shots resulted in an antibody response similar to that of natural immunity in hyperimmunized animals. The DISC vaccine induced antibody responses to gB and gH/gL, with similar titers as those found in animals convalescent for natural infection, whereas the immune response to the gM/gN complex was weaker against natural infection. The neutralizing antibody titer was on average lower than that seen in convalescent animals. Unfortunately, other current GPCMV vaccine studies have not included advanced glycoprotein-specific ELISAs, and therefore it is difficult to evaluate previous reports alongside this current research. Recent studies based on live attenuated GPCMV vaccines have demonstrated a robust anti-GPCMV antibody titer and a specific immune response to gB based on Western blot analysis. In our present study, two vaccine regimens were explored for the DISC virus in guinea pigs, and it appeared that a two-booster strategy dramatically increased the immune response to the DISC virus compared to a single-booster strategy. A two-booster DISC vaccine strategy induced an antibody response comparable to that seen for live attenuated GPCMV (anti-GPCMV and gB titers), and a single-booster DISC strategy resulted in lower titers. In contrast, GPCMV gB-specific vaccine strategies produced antibody titers higher than seen for natural convalescent immunity (51, 77, 79, 91), but these gB vaccine strategies were less successful in protecting against congenital infection than was the DISC vaccine. The most effective gB-based vaccine strategies reduced transmission rates from 82 to 45% (gB recombinant baculovirus/GSK adjuvant), 77 to 41% (gB DNA vaccine), and 79 to 59% (MVA gB) (76, 77, 79). Potentially, a failing of these gB vaccine strategies was that the gB lacked a C-terminal domain and therefore there was an inability of the recombinant gB to multimerize, as seen in natural infection. Consequently, a large amount of the gB immune response was potentially directed to the linear, homologous AD-1 domain (92) rather than other gB antigen targets. However, a more recent study by Cardin et al. (82), which used a recombinant lymphocytic choriomeningitis virus vector that encoded gB with a C-terminal domain but lacked a transmembrane domain, did not increase protection against transmission (reduced from 83% to 60%) despite generation of high titers of antibody to gB.
Previous congenital vaccine studies based on live attenuated GPCMV included an RNA-dependent protein kinase (PKR) mutant virus (78), a major histocompatibility complex class I-downregulated mutant (80), and a GP83 knockout strain (81). The vaccine strategies were able to reduce transmission by various levels: from 65% to 33% (78), 83% to 57% (80), and 70% to 17% (81), respectively. Surprisingly, the pp65 homolog (GP83) knockout mutant appears to be the most effective live vaccine strategy despite a previous vaccine strategy that used a defective alphavirus encoding GP83 and demonstrated the importance of the T cell response to GP83 in protecting against congenital infection (transmission reduced from 85% to 47%) (61). Currently, only the GP83 knockout mutant vaccine strategy is more effective than the DISC vaccine in reducing the congenital transmission rate. However, the GP83 mutant virus has near-normal growth kinetics in tissue culture, as does an HCMV pp65 mutant (30, 93). Consequently, a live HCMV vaccine based on a pp65 knockout might present an unacceptable risk compared to a DISC vaccine strategy, which would have a greater safety factor due to the DISC virus's inability to replicate outside the supporting cell line.
Unfortunately, the more-effective DISC vaccine regimen was evaluated after the congenital vaccine protection study had been initiated with a single vaccine booster approach. Nonetheless, the DISC vaccine was highly effective in protecting against congenital CMV infection, and importantly, high anti-GPCMV titers were not necessarily an indicator of effective protection against congenital infection. As noted above, it is likely that the T cell response to various target antigens was also important in the prevention of congenital infection in the setting of the current vaccine, and the immune response to the GP83 was an indicator of a positive T cell response to the DISC vaccine. However, a key component missing from the current DISC vaccine was the homologous pentameric complex. Clinical strains of HCMV, unlike lab-adapted strains, encode a pentameric glycoprotein complex (gH/gL/UL128-130-131) for virus entry into epithelial and endothelial cells via an alternative endocytic pathway of cell entry rather than a gB- plus gH/gL-mediated cell membrane fusion approach (94–99). The pentameric complex is considered an important neutralizing target for HCMV on epithelial/endothelial cells and presumably for congenital infection, given the epithelial/endothelial structure of the placenta. The importance of the endocytic pathway in virus infection of cells is underscored by the fact that a gB subunit HCMV vaccine, despite generating a high-titer neutralizing immune response on fibroblasts, lacks an effective ability to neutralize infection of endothelial and epithelial cells compared to convalescent-phase sera (41, 52–55). With RhCMV, the pentameric complex has been demonstrated to be an important pathogenicity factor as well as a neutralizing target antigen (100, 101). In GPCMV, a homologous UL128-131 locus has been identified, and the ability to form a pentameric complex has been investigated (28, 58). We recently demonstrated that this complex is important for virus tropism to epithelial cells, pathogenicity, and congenital infection (103). Importantly, the homolog pentameric complex is highly immunogenic (102). Virus serially passaged as salivary gland stock in animals is stably maintained, but virus on fibroblast cells rapidly undergoes mutations or deletions in this region. Our current DISC vaccine strategy is based on a first-generation GPCMV BAC which carries a deletion in the homologous UL128-131 locus (15, 26). Consequently, the DISC vaccine is incapable of generating an immune response to the pentameric complex, which is potentially an important component of the immune response to GPCMV in convalescent animals. Importantly, we recently identified the pentameric complex as an important target for neutralization of virus infection of epithelial cells (102). Therefore, introduction of the pentameric complex in a second-generation DISC vaccine is likely to have a significant impact on the efficacy of a future DISC vaccine for congenital CMV protection. It is important to note that none of the live GPCMV attenuated vaccine strategies evaluated to date have included a viable pentameric complex; modification of these strains could potentially enhance the efficacy of these vaccine strategies. Indeed, a new GP83 knockout mutant virus that also encodes a pentameric complex was highly effective in inducing a robust immune response and reducing congenital transmission (102).
In conclusion, the first-generation DISC vaccine against congenital GPCMV is a safer strategy than the use of a live attenuated virus, as the new vaccine lacks the ability to produce progeny virus and disseminate in the host. Importantly, the virus evokes an immune response similar to natural infection, which includes an antibody response to viral glycoprotein complexes necessary for cell entry and a T cell response to the pp65 homolog. The vaccine strategy was successful in fully preventing pup deaths and greatly reduced the level and incidence of pups with congenital CMV infection compared to a nonvaccinated control group. This promising vaccine strategy presents potential opportunities for improvement via the inclusion of the homolog pentameric complex, which is considered an important complex for virus infection of epithelial cells (103) and a variety of other cell types. The potential addition of neutralizing antibodies to this complex will greatly reduce congenital transmission.
ACKNOWLEDGMENTS
We thank Jim Choi and Darijana Horvat for their excellent technical assistance. We are grateful to Hubert Schaefer (Robert Koch Institute, Germany) for providing the guinea pig interferon-gamma monoclonal antibodies.
REFERENCES
- 1.McGregor A, Choi KY. 2011. Cytomegalovirus antivirals and development of improved animal models. Expert Opin Drug Metab Toxicol 7:1245–1265. doi: 10.1517/17425255.2011.613824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pass RF. 1996. Immunization strategy for prevention of congenital cytomegalovirus infection. Infect Agents Dis 5:240–244. [PubMed] [Google Scholar]
- 3.Ross SA, Boppana SB. 2005. Congenital cytomegalovirus infection: outcome and diagnosis. Semin Pediatr Infect Dis 16:44–49. doi: 10.1053/j.spid.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 4.Griffiths PD, Walter S. 2005. Cytomegalovirus. Curr Opin Infect Dis 18:241–245. doi: 10.1097/01.qco.0000168385.39390.1b. [DOI] [PubMed] [Google Scholar]
- 5.Dollard SC, Grosse SD, Ross DS. 2007. New estimates of the prevalence of neurological and sensory sequelae and mortality associated with congenital cytomegalovirus infection. Rev Med Virol 17:355–363. doi: 10.1002/rmv.544. [DOI] [PubMed] [Google Scholar]
- 6.Manicklal S, Emery VC, Lazzarotto T, Boppana SB, Gupta RK. 2013. The “silent” global burden of congenital cytomegalovirus. Clin Microbiol Rev 26:86–102. doi: 10.1128/CMR.00062-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kimberlin DW, Jester PM, Sanchez PJ, Ahmed A, Arav-Boger R, Michaels MG, Ashouri N, Englund JA, Estrada B, Jacobs RF, Romero JR, Sood SK, Whitworth MS, Abzug MJ, Caserta MT, Fowler S, Lujan-Zilbermann J, Storch GA, DeBiasi RL, Han JY, Palmer A, Weiner LB, Bocchini JA, Dennehy PH, Finn A, Griffiths PD, Luck S, Gutierrez K, Halasa N, Homans J, Shane AL, Sharland M, Simonsen K, Vanchiere JA, Woods CR, Sabo DL, Aban I, Kuo H, James SH, Prichard MN, Griffin J, Giles D, Acosta EP, Whitley RJ, National Institute of Allergy and Infectious Diseases Collaborative Antiviral Study Group. 2015. Valganciclovir for symptomatic congenital cytomegalovirus disease. N Engl J Med 372:933–943. doi: 10.1056/NEJMoa1404599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fowler KB, Stagno S, Pass RF. 2003. Maternal immunity and prevention of congenital cytomegalovirus infection. JAMA 289:1008–1011. doi: 10.1001/jama.289.8.1008. [DOI] [PubMed] [Google Scholar]
- 9.Colugnati FA, Staras SA, Dollard SC, Cannon MJ. 2007. Incidence of cytomegalovirus infection among the general population and pregnant women in the United States. BMC Infect Dis 7:71. doi: 10.1186/1471-2334-7-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cannon MJ. 2009. Congenital cytomegalovirus (CMV) epidemiology and awareness. J Clin Virol 46(Suppl 4):S6–S10. doi: 10.1016/j.jcv.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 11.Nishimura N, Kimura H, Yabuta Y, Tanaka N, Ito Y, Ishikawa K, Suzuki C, Morishima T. 1999. Prevalence of maternal cytomegalovirus (CMV) antibody and detection of CMV DNA in amniotic fluid. Microbiol Immunol 43:781–784. doi: 10.1111/j.1348-0421.1999.tb02470.x. [DOI] [PubMed] [Google Scholar]
- 12.Hansen SG, Strelow LI, Franchi DC, Anders DG, Wong SW. 2003. Complete sequence and genomic analysis of rhesus cytomegalovirus. J Virol 77:6620–6636. doi: 10.1128/JVI.77.12.6620-6636.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rawlinson WD, Farrell HE, Barrell BG. 1996. Analysis of the complete DNA sequence of murine cytomegalovirus. J Virol 70:8833–8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vink C, Beuken E, Bruggeman CA. 2000. Complete DNA sequence of the rat cytomegalovirus genome. J Virol 74:7656–7665. doi: 10.1128/JVI.74.16.7656-7665.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schleiss MR, McGregor A, Choi KY, Date SV, Cui X, McVoy MA. 2008. Analysis of the nucleotide sequence of the guinea pig cytomegalovirus (GPCMV) genome. Virol J 5:139. doi: 10.1186/1743-422X-5-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yue Y, Barry PA. 2008. Rhesus cytomegalovirus a nonhuman primate model for the study of human cytomegalovirus. Adv Virus Res 72:207–226. doi: 10.1016/S0065-3527(08)00405-3. [DOI] [PubMed] [Google Scholar]
- 17.Johnson KP. 1969. Mouse cytomegalovirus: placental infection. J Infect Dis 120:445–450. doi: 10.1093/infdis/120.4.445. [DOI] [PubMed] [Google Scholar]
- 18.Kaufmann P. 2004. Guinea pig (Cavia procellus). In Benirschke K. (ed), Comparative placentation. http://placentation.ucsd.edu/guinea/htm.
- 19.Carter AM. 2007. Animal models of human placentation: a review. Placenta 28(Suppl A):S41–S47. doi: 10.1016/j.placenta.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 20.Mess A. 2007. The guinea pig placenta: model of placental growth dynamics. Placenta 28:812–815. doi: 10.1016/j.placenta.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 21.Griffith BP, McCormick SR, Fong CK, Lavallee JT, Lucia HL, Goff E. 1985. The placenta as a site of cytomegalovirus infection in guinea pigs. J Virol 55:402–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar ML, Nankervis GA. 1978. Experimental congenital infection with cytomegalovirus: a guinea pig model. J Infect Dis 138:650–654. doi: 10.1093/infdis/138.5.650. [DOI] [PubMed] [Google Scholar]
- 23.Woolf NK. 1991. Guinea pig model of congenital CMV-induced hearing loss: a review. Transplant Proc 23:32–34. [PubMed] [Google Scholar]
- 24.Kern ER. 2006. Pivotal role of animal models in the development of new therapies for cytomegalovirus infections. Antiviral Res 71:164–171. doi: 10.1016/j.antiviral.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 25.McGregor A, McVoy MA, Schleiss MR. 2013. The guinea pig model of congenital cytomegalovirus infection, p 88–118. In Reddehase MJ. (ed), Cytomegaloviruses: from molecular pathogenesis to intervention, vol II Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 26.McGregor A, Schleiss MR. 2001. Molecular cloning of the guinea pig cytomegalovirus (GPCMV) genome as an infectious bacterial artificial chromosome (BAC) in Escherichia coli. Mol Genet Metab 72:15–26. doi: 10.1006/mgme.2000.3102. [DOI] [PubMed] [Google Scholar]
- 27.Cui X, McGregor A, Schleiss MR, McVoy MA. 2008. Cloning the complete guinea pig cytomegalovirus genome as an infectious bacterial artificial chromosome with excisable origin of replication. J Virol Methods 149:231–239. doi: 10.1016/j.jviromet.2008.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamada S, Nozawa N, Katano H, Fukui Y, Tsuda M, Tsutsui Y, Kurane I, Inoue N. 2009. Characterization of the guinea pig cytomegalovirus genome locus that encodes homologs of human cytomegalovirus major immediate-early genes, UL128, and UL130. Virology 391:99–106. doi: 10.1016/j.virol.2009.05.034. [DOI] [PubMed] [Google Scholar]
- 29.Kanai K, Yamada S, Yamamoto Y, Fukui Y, Kurane I, Inoue N. 2011. Re-evaluation of the genome sequence of guinea pig cytomegalovirus. J Gen Virol 92:1005–1020. doi: 10.1099/vir.0.027789-0. [DOI] [PubMed] [Google Scholar]
- 30.McGregor A, Liu F, Schleiss MR. 2004. Molecular, biological, and in vivo characterization of the guinea pig cytomegalovirus (CMV) homologs of the human CMV matrix proteins pp71 (UL82) and pp65 (UL83). J Virol 78:9872–9889. doi: 10.1128/JVI.78.18.9872-9889.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McGregor A, Liu F, Schleiss MR. 2004. Identification of essential and non-essential genes of the guinea pig cytomegalovirus (GPCMV) genome via transposome mutagenesis of an infectious BAC clone. Virus Res 101:101–108. doi: 10.1016/j.virusres.2003.12.030. [DOI] [PubMed] [Google Scholar]
- 32.McGregor A, Choi KY, Cui X, McVoy MA, Schleiss MR. 2008. Expression of the human cytomegalovirus UL97 gene in a chimeric guinea pig cytomegalovirus (GPCMV) results in viable virus with increased susceptibility to ganciclovir and maribavir. Antiviral Res 78:250–259. doi: 10.1016/j.antiviral.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McGregor A, Choi KY, Schleiss MR. 2011. Guinea pig cytomegalovirus GP84 is a functional homolog of the human cytomegalovirus (HCMV) UL84 gene that can complement for the loss of UL84 in a chimeric HCMV. Virology 410:76–87. doi: 10.1016/j.virol.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schleiss MR, Stroup G, Pogorzelski K, McGregor A. 2006. Protection against congenital cytomegalovirus (CMV) disease, conferred by a replication-disabled, bacterial artificial chromosome (BAC)-based DNA vaccine. Vaccine 24:6175–6186. doi: 10.1016/j.vaccine.2006.06.077. [DOI] [PubMed] [Google Scholar]
- 35.McGregor A, Choi KY, Schachtele SJ, Lokensgard JR. 2013. Human herpesviruses and animal models, p 905–925. In Conn PM. (ed), Animal models for the study of human diseases. Elsevier Inc., San Diego, CA. [Google Scholar]
- 36.Coleman S, Hornig J, Maddux S, Choi KY, McGregor A. 2015. Viral glycoprotein complex formation, essential function and immunogenicity in the guinea pig model for cytomegalovirus. PLoS One 10:e0135567. doi: 10.1371/journal.pone.0135567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Britt WJ, Mach M. 1996. Human cytomegalovirus glycoproteins. Intervirology 39:401–412. [DOI] [PubMed] [Google Scholar]
- 38.Gretch DR, Kari B, Gehrz RC, Stinski MF. 1988. A multigene family encodes the human cytomegalovirus glycoprotein complex gcII (gp47-52 complex). J Virol 62:1956–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huber MT, Compton T. 1998. The human cytomegalovirus UL74 gene encodes the third component of the glycoprotein H-glycoprotein L-containing envelope complex. J Virol 72:8191–8197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gretch DR, Kari B, Rasmussen L, Gehrz RC, Stinski MF. 1988. Identification and characterization of three distinct families of glycoprotein complexes in the envelopes of human cytomegalovirus. J Virol 62:875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pass RF, Zhang C, Evans A, Simpson T, Andrews W, Huang ML, Corey L, Hill J, Davis E, Flanigan C, Cloud G. 2009. Vaccine prevention of maternal cytomegalovirus infection. N Engl J Med 360:1191–1199. doi: 10.1056/NEJMoa0804749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boppana SB, Polis MA, Kramer AA, Britt WJ, Koenig S. 1995. Virus-specific antibody responses to human cytomegalovirus (HCMV) in human immunodeficiency virus type 1-infected persons with HCMV retinitis. J Infect Dis 171:182–185. doi: 10.1093/infdis/171.1.182. [DOI] [PubMed] [Google Scholar]
- 43.Shimamura M, Mach M, Britt WJ. 2006. Human cytomegalovirus infection elicits a glycoprotein M (gM)/gN-specific virus-neutralizing antibody response. J Virol 80:4591–4600. doi: 10.1128/JVI.80.9.4591-4600.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shen S, Wang S, Britt WJ, Lu S. 2007. DNA vaccines expressing glycoprotein complex II antigens gM and gN elicited neutralizing antibodies against multiple human cytomegalovirus (HCMV) isolates. Vaccine 25:3319–3327. doi: 10.1016/j.vaccine.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 45.Revello MG, Gerna G. 2010. Human cytomegalovirus tropism for endothelial/epithelial cells: scientific background and clinical implications. Rev Med Virol 20:136–155. doi: 10.1002/rmv.645. [DOI] [PubMed] [Google Scholar]
- 46.Yamada S, Fukuchi S, Hashimoto K, Fukui Y, Tsuda M, Kataoka M, Katano H, Inoue N. 2014. Guinea pig cytomegalovirus GP129/131/133, homologues of human cytomegalovirus UL128/130/131A, are necessary for infection of monocytes and macrophages. J Gen Virol 95:1376–1382. doi: 10.1099/vir.0.064527-0. [DOI] [PubMed] [Google Scholar]
- 47.Britt WJ, Vugler L, Butfiloski EJ, Stephens EB. 1990. Cell surface expression of human cytomegalovirus (HCMV) gp55-116 (gB): use of HCMV-recombinant vaccinia virus-infected cells in analysis of the human neutralizing antibody response. J Virol 64:1079–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marshall GS, Rabalais GP, Stout GG, Waldeyer SL. 1992. Antibodies to recombinant-derived glycoprotein B after natural human cytomegalovirus infection correlate with neutralizing activity. J Infect Dis 165:381–384. doi: 10.1093/infdis/165.2.381. [DOI] [PubMed] [Google Scholar]
- 49.Chatterjee A, Harrison CJ, Britt WJ, Bewtra C. 2001. Modification of maternal and congenital cytomegalovirus infection by anti-glycoprotein B antibody transfer in guinea pigs. J Infect Dis 183:1547–1553. doi: 10.1086/320714. [DOI] [PubMed] [Google Scholar]
- 50.Schleiss MR. 1994. Cloning and characterization of the guinea pig cytomegalovirus glycoprotein B gene. Virology 202:173–185. doi: 10.1006/viro.1994.1333. [DOI] [PubMed] [Google Scholar]
- 51.Schleiss MR, Bourne N, Stroup G, Bravo FJ, Jensen NJ, Bernstein DI. 2004. Protection against congenital cytomegalovirus infection and disease in guinea pigs, conferred by a purified recombinant glycoprotein B vaccine. J Infect Dis 189:1374–1381. doi: 10.1086/382751. [DOI] [PubMed] [Google Scholar]
- 52.Pass RF, Duliege AM, Boppana S, Sekulovich R, Percell S, Britt W, Burke RL. 1999. A subunit cytomegalovirus vaccine based on recombinant envelope glycoprotein B and a new adjuvant. J Infect Dis 180:970–975. doi: 10.1086/315022. [DOI] [PubMed] [Google Scholar]
- 53.Cui X, Meza BP, Adler SP, McVoy MA. 2008. Cytomegalovirus vaccines fail to induce epithelial entry neutralizing antibodies comparable to natural infection. Vaccine 26:5760–5766. doi: 10.1016/j.vaccine.2008.07.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gerna G, Lilleri D, Rognoni V, Agozzino M, Meloni F, Oggionni T, Pellegrini C, Arbustini E, D'Armini AM. 2009. Preemptive therapy for systemic and pulmonary human cytomegalovirus infection in lung transplant recipients. Am J Transplant 9:1142–1150. doi: 10.1111/j.1600-6143.2009.02616.x. [DOI] [PubMed] [Google Scholar]
- 55.Fouts AE, Chan P, Stephan JP, Vandlen R, Feierbach B. 2012. Antibodies against the gH/gL/UL128/UL130/UL131 complex comprise the majority of the anti-cytomegalovirus (anti-CMV) neutralizing antibody response in CMV hyperimmune globulin. J Virol 86:7444–7447. doi: 10.1128/JVI.00467-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Harrison CJ, Britt WJ, Chapman NM, Mullican J, Tracy S. 1995. Reduced congenital cytomegalovirus (CMV) infection after maternal immunization with a guinea pig CMV glycoprotein before gestational primary CMV infection in the guinea pig model. J Infect Dis 172:1212–1220. doi: 10.1093/infdis/172.5.1212. [DOI] [PubMed] [Google Scholar]
- 57.Schleiss MR. 2002. Animal models of congenital cytomegalovirus infection: an overview of progress in the characterization of guinea pig cytomegalovirus (GPCMV). J Clin Virol 25:37–49. doi: 10.1016/S1386-6532(02)00100-2. [DOI] [PubMed] [Google Scholar]
- 58.Auerbach M, Yan D, Fouts A, Xu M, Estevez A, Austin CD, Bazan F, Feierbach B. 2013. Characterization of the guinea pig CMV gH/gL/GP129/GP131/GP133 complex in infection and spread. Virology 441:75–84. doi: 10.1016/j.virol.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 59.Harari A, Zimmerli SC, Pantaleo G. 2004. Cytomegalovirus (CMV)-specific cellular immune responses. Hum Immunol 65:500–506. doi: 10.1016/j.humimm.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 60.Plotkin S. 2015. The history of vaccination against cytomegalovirus. Med Microbiol Immunol 204:247–254. doi: 10.1007/s00430-015-0388-z. [DOI] [PubMed] [Google Scholar]
- 61.Schleiss M, Lacayo J, Belkaid Y, McGregor A, Stroup G, Rayner J, Alterson K, Chulay J, Smith J. 2007. Preconceptual administration of an alphavirus replicon UL83 (pp65 homolog) vaccine induces humoral and cellular immunity and improves pregnancy outcome in the guinea pig model of congenital cytomegalovirus infection. J Infect Dis 195:789–798. doi: 10.1086/511982. [DOI] [PubMed] [Google Scholar]
- 62.Wills MR, Mason GM, Sissons JGP. 2013. Adaptive cellular immunity to human cytomegalovirus, p 142–172. In Reddehase MJ. (ed), Cytomegaloviruses: from molecular pathogenesis to intervention, vol II Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 63.Gibson W, Bogner E. 2013. Morphogenesis of the cytomegalovirus virion and subviral particles, p 230–246. In Reddehase MJ. (ed), Cytomegaloviruses: from molecular pathogenesis to intervention, vol I Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 64.Urlinger S, Baron U, Thellmann M, Hasan MT, Bujard H, Hillen W. 2000. Exploring the sequence space for tetracycline-dependent transcriptional activators: novel mutations yield expanded range and sensitivity. Proc Natl Acad Sci U S A 97:7963–7968. doi: 10.1073/pnas.130192197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 66.Schafer H, Klippert K, Meuer P, Borsdorf B, Kiderlen AF, Burger R. 2007. Biologic activity of guinea pig IFN-gamma in vitro. J Interferon Cytokine Res 27:305–315. doi: 10.1089/jir.2006.0108. [DOI] [PubMed] [Google Scholar]
- 67.Jeevan A, McFarland CT, Yoshimura T, Skwor T, Cho H, Lasco T, McMurray DN. 2006. Production and characterization of guinea pig recombinant gamma interferon and its effect on macrophage activation. Infect Immun 74:213–224. doi: 10.1128/IAI.74.1.213-224.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cho H, McMurray D. 2007. Recombinant guinea pig TNF-alpha enhances antigen-specific type 1 T lymphocyte activation in guinea pig splenocytes. Tuberculosis 87:87–93. doi: 10.1016/j.tube.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 69.Anthony DD, Lehmann PV. 2003. T-cell epitope mapping using the ELISPOT approach. Methods 29:260–269. doi: 10.1016/S1046-2023(02)00348-1. [DOI] [PubMed] [Google Scholar]
- 70.Hoffmeister B, Kiecker F, Tesfa L, Volk HD, Picker LJ, Kern F. 2003. Mapping T cell epitopes by flow cytometry. Methods 29:270–281. doi: 10.1016/S1046-2023(02)00349-3. [DOI] [PubMed] [Google Scholar]
- 71.Farrell HE, McLean CS, Harley C, Efstathiou S, Inglis S, Minson AC. 1994. Vaccine potential of a herpes simplex virus type 1 mutant with an essential glycoprotein deleted. J Virol 68:927–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Brown JC, Newcomb WW. 2011. Herpesvirus capsid assembly: insights from structural analysis. Curr Opin Virol 1:142–149. doi: 10.1016/j.coviro.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fong CK, Bia F, Hsiung GD, Madore P, Chang PW. 1979. Ultrastructural development of guinea pig cytomegalovirus in cultured guinea pig embryo cells. J Gen Virol 42:127–140. doi: 10.1099/0022-1317-42-1-127. [DOI] [PubMed] [Google Scholar]
- 74.Abate DA, Watanabe S, Mocarski ES. 2004. Major human cytomegalovirus structural protein pp65 (ppUL83) prevents interferon response factor 3 activation in the interferon response. J Virol 78:10995–11006. doi: 10.1128/JVI.78.20.10995-11006.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Godard B, Gazagne A, Gey A, Baptiste M, Vingert B, Pegaz-Fiornet B, Strompf L, Fridman WH, Glotz D, Tartour E. 2004. Optimization of an ELISPOT assay to detect cytomegalovirus-specific CD8+ T lymphocytes. Hum Immunol 65:1307–1318. doi: 10.1016/j.humimm.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 76.Schleiss MR, Bourne N, Bernstein DI. 2003. Preconception vaccination with a glycoprotein B (gB) DNA vaccine protects against cytomegalovirus (CMV) transmission in the guinea pig model of congenital CMV infection. J Infect Dis 188:1868–1874. doi: 10.1086/379839. [DOI] [PubMed] [Google Scholar]
- 77.Schleiss M, Choi KY, Anderson J, Mash J, Wettendorff M, Mossman S, Van Damme M. 2014. Glycoprotein B (gB) vaccines adjuvanted with AS01 or AS02 protect female guinea pigs against cytomegalovirus (CMV) viremia and offspring mortality in a CMV-challenge model. Vaccine 32:2756–2762. doi: 10.1016/j.vaccine.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schleiss MR, Bierle CJ, Swanson EC, McVoy MA, Wang JB, Al-Mahdi Z, Geballe AP. 2015. Vaccination with a live attenuated cytomegalovirus devoid of a protein kinase R inhibitory gene results in reduced maternal viremia and improved pregnancy outcome in a guinea pig congenital infection model. J Virol 89:9727–9738. doi: 10.1128/JVI.01419-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Swanson EC, Gillis P, Hernandez-Alvarado N, Fernandez-Alarcon C, Schmit M, Zabeli JC, Wussow F, Diamond DJ, Schleiss MR. 2015. Comparison of monovalent glycoprotein B with bivalent gB/pp65 (GP83) vaccine for congenital cytomegalovirus infection in a guinea pig model: inclusion of GP83 reduces gB antibody response but both vaccine approaches provide equivalent protection against pup mortality. Vaccine 33:4013–4018. doi: 10.1016/j.vaccine.2015.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Crumpler MM, Choi KY, McVoy MA, Schleiss MR. 2009. A live guinea pig cytomegalovirus vaccine deleted of three putative immune evasion genes is highly attenuated but remains immunogenic in a vaccine/challenge model of congenital cytomegalovirus infection. Vaccine 27:4209–4218. doi: 10.1016/j.vaccine.2009.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schleiss MR, Buus R, Choi KY, McGregor A. 2013. An Attenuated CMV vaccine with a deletion in tegument protein GP83 (pp65 homolog) protects against placental infection and improves pregnancy outcome in a guinea pig challenge model. Future Virol 8:1151–1160. doi: 10.2217/fvl.13.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cardin R, Bravo F, Pullum D, Orlinger K, Watson E, Aspoeck A, Fuhrmann G, Guirakhoo F, Monath T, Bernstein D. 2016. Replication-defective lymphocytic choriomeningitis virus vectors expressing guinea pig cytomegalovirus gB and pp65 homologs are protective against congenital guinea pig cytomegalovirus infection. Vaccine 34:1993–1999. doi: 10.1016/j.vaccine.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 83.Bia FJ, Griffith BP, Tarsio M, Hsiung GD. 1980. Vaccination for the prevention of maternal and fetal infection with guinea pig cytomegalovirus. J Infect Dis 142:732–738. doi: 10.1093/infdis/142.5.732. [DOI] [PubMed] [Google Scholar]
- 84.Bowman JJ, Lacayo JC, Burbelo P, Fischer ER, Cohen JI. 2011. Rhesus and human cytomegalovirus glycoprotein L are required for infection and cell-to-cell spread of virus but cannot complement each other. J Virol 85:2089–2099. doi: 10.1128/JVI.01970-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mohr CA, Arapovic J, Muhlbach H, Panzer M, Weyn A, Dolken L, Krmpotic A, Voehringer D, Ruzsics Z, Koszinowski U, Sacher T. 2010. A spread-deficient cytomegalovirus for assessment of first-target cells in vaccination. J Virol 84:7730–7742. doi: 10.1128/JVI.02696-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Padilla-Carlin DJ, McMurray DN, Hickey AJ. 2008. The guinea pig as a model of infectious diseases. Comp Med 58:324–340. [PMC free article] [PubMed] [Google Scholar]
- 87.Holtappels R, Ebert S, Podlech J, Fink A, Bohm V, Lemmermann N, Renzaho A, Thomas D, Reddehase MJ. 2013. Murine model for cytoimmunotherapy of CMV disease after hematopoietic cell transplantation, p 345–381. In Reddehase MJ. (ed), Cytomegaloviruses: from molecular pathogenesis to intervention, vol II Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 88.Fruh K, Malouli D, Oxford KL, Barry PA. 2013. Non-human-primate models of cytomegalovirus infection, prevention, and therapy, p 463–496. In Reddehase MJ. (ed), Cytomegaloviruses: from molecular pathogenesis to intervention, vol II Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 89.Bialas KM, Tanaka T, Tran D, Varner V, Cisneros De La Rosa E, Chiuppesi F, Wussow F, Kattenhorn L, Macri S, Kunz EL, Estroff JA, Kirchherr J, Yue Y, Fan Q, Lauck M, O'Connor DH, Hall AH, Xavier A, Diamond DJ, Barry PA, Kaur A, Permar SR. 2015. Maternal CD4+ T cells protect against severe congenital cytomegalovirus disease in a novel nonhuman primate model of placental cytomegalovirus transmission. Proc Natl Acad Sci U S A 112:13645–13650. doi: 10.1073/pnas.1511526112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pereira L, Tabata T, Petitt M, Fang-Hoover J. 2013. Cytomegalovirus replication in the developing human placenta, p 74–87. In Reddehase MJ. (ed), Cytomegaloviruses: from molecular pathogenesis to intervention, vol II Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 91.Hashimoto K, Yamada S, Katano H, Fukuchi S, Sato Y, Kato M, Yamaguchi T, Moriishi K, Inoue N. 2013. Effects of immunization of pregnant guinea pigs with guinea pig cytomegalovirus glycoprotein B on viral spread in the placenta. Vaccine 31:3199–3205. doi: 10.1016/j.vaccine.2013.04.078. [DOI] [PubMed] [Google Scholar]
- 92.Speckner A, Glykofrydes D, Ohlin M, Mach M. 1999. Antigenic domain 1 of human cytomegalovirus glycoprotein B induces a multitude of different antibodies which, when combined, results in incomplete virus neutralization. J Gen Virol 80(Pt 8):2183–2191. doi: 10.1099/0022-1317-80-8-2183. [DOI] [PubMed] [Google Scholar]
- 93.Schmolke S, Kern HF, Drescher P, Jahn G, Plachter B. 1995. The dominant phosphoprotein pp65 (UL83) of human cytomegalovirus is dispensable for growth in cell culture. J Virol 69:5959–5968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hahn G, Revello MG, Patrone M, Percivalle E, Campanini G, Sarasini A, Wagner M, Gallina A, Milanesi G, Koszinowski U, Baldanti F, Gerna G. 2004. Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J Virol 78:10023–10033. doi: 10.1128/JVI.78.18.10023-10033.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ryckman BJ, Jarvis MA, Drummond DD, Nelson JA, Johnson DC. 2006. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J Virol 80:710–722. doi: 10.1128/JVI.80.2.710-722.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ryckman BJ, Rainish BL, Chase MC, Borton JA, Nelson JA, Jarvis MA, Johnson DC. 2008. Characterization of the human cytomegalovirus gH/gL/UL128-131 complex that mediates entry into epithelial and endothelial cells. J Virol 82:60–70. doi: 10.1128/JVI.01910-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang D, Shenk T. 2005. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc Natl Acad Sci U S A 102:18153–18158. doi: 10.1073/pnas.0509201102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang D, Shenk T. 2005. Human cytomegalovirus UL131 open reading frame is required for epithelial cell tropism. J Virol 79:10330–10338. doi: 10.1128/JVI.79.16.10330-10338.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Patrone M, Secchi M, Fiorina L, Ierardi M, Milanesi G, Gallina A. 2005. Human cytomegalovirus UL130 protein promotes endothelial cell infection through a producer cell modification of the virion. J Virol 79:8361–8373. doi: 10.1128/JVI.79.13.8361-8373.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wussow F, Yue Y, Martinez J, Deere J, Longmate J, Herrmann A, Barry P, Diamond D. 2013. A vaccine based on the rhesus cytomegalovirus UL128 complex induces broadly neutralizing antibodies in rhesus macaques. J Virol 87:1322–1332. doi: 10.1128/JVI.01669-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lilja AE, Shenk T. 2008. Efficient replication of rhesus cytomegalovirus variants in multiple rhesus and human cell types. Proc Natl Acad Sci U S A 105:19950–19955. doi: 10.1073/pnas.0811063106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Choi KY, Horvat D, McGregor A. 2015. Live vaccine strategies demonstrate the potential importance of humoral responses to the pentameric complex in the guinea pig congenital CMV model abstr O22, p 44 5th International Congenital CMV Conference and 15th International CMV/Beta Herpes Virus Workshop, Brisbane, Australia, 20 to 24 April 2015. [Google Scholar]
- 103.Coleman S, Choi KY, Root M, McGregor A. 2016. A homolog pentameric complex dictates viral epithelial tropism, pathogenicity and congenital infection rate in guinea pig cytomegalovirus. PLoS Pathog 12:e1005755 10.1371/journal.ppat.1005755. [DOI] [PMC free article] [PubMed] [Google Scholar]