

ABSTRACT
Interferon regulatory factor 3 (IRF3) is a transcription factor involved in the activation of type I alpha/beta interferon (IFN-α/β) in response to viral infection. Upon viral infection, the IRF3 monomer is activated into a phosphorylated dimer, which induces the transcription of interferon genes in the nucleus. Viruses have evolved several ways to target IRF3 in order to subvert the innate immune response. Pestiviruses, such as classical swine fever virus (CSFV), target IRF3 for ubiquitination and subsequent proteasomal degradation. This is mediated by the viral protein Npro that interacts with IRF3, but the molecular details for this interaction are largely unknown. We used recombinant Npro and IRF3 proteins and show that Npro interacts with IRF3 directly without additional proteins and forms a soluble 1:1 complex. The full-length IRF3 but not merely either of the individual domains is required for this interaction. The interaction between Npro and IRF3 is not dependent on the activation state of IRF3, since Npro binds to a constitutively active form of IRF3 in the presence of its transcriptional coactivator, CREB-binding protein (CBP). The results indicate that the Npro-binding site on IRF3 encompasses a region that is unperturbed by the phosphorylation and subsequent activation of IRF3 and thus excludes the dimer interface and CBP-binding site.
IMPORTANCE The pestivirus N-terminal protease, Npro, is essential for evading the host's immune system by facilitating the degradation of interferon regulatory factor 3 (IRF3). However, the nature of the Npro interaction with IRF3, including the IRF3 species (inactive monomer versus activated dimer) that Npro targets for degradation, is largely unknown. We show that classical swine fever virus Npro and porcine IRF3 directly interact in solution and that full-length IRF3 is required for interaction with Npro. Additionally, Npro interacts with a constitutively active form of IRF3 bound to its transcriptional cofactor, the CREB-binding protein. This is the first study to demonstrate that Npro is able to bind both inactive IRF3 monomer and activated IRF3 dimer and thus likely targets both IRF3 species for ubiquitination and proteasomal degradation.
INTRODUCTION
The hallmark of the innate immune response against viruses is the activation of type I alpha/beta interferon (IFN-α/β) signaling in immune cells (1, 2). IFN-α/β synthesis is triggered by several of the interferon regulatory factors (IRFs) (3–5). IRF3 is expressed constitutively in various cell types. In uninfected cells, IRF3 exists as an inactive monomer in a latent state in the cytoplasm. Upon viral infection, IRF3 is activated by phosphorylation by cellular kinases, such as TBK-1/IκB kinase ε (IKKε), through engagement of pattern recognition receptors (PRRs) in the immune cells (6). PRRs recognize anomalous non-self motifs called pathogen-associated molecular patterns (PAMPs), such as the double-stranded (dsRNA) intermediates and 5′-triphosphorylated RNA formed during viral RNA replication. Two groups of PRRs activate type I IFN signaling following RNA virus infection. The Toll-like receptors recognize dsRNA in endosomal compartments, and helicases like RIG-1/MDA5 recognize cytosolic dsRNA intermediates (7, 8). Both pathways result in the phosphorylation and activation of IRF3.
IRF3 is an ∼50-kDa protein with two functional domains, the N-terminal DNA-binding domain (DBD) and the C-terminal IRF association domain (IAD), also called the regulatory domain (9). The IAD carries the dimerization interface, the autoinhibitory region, and the serine-rich region (10) (Fig. 1A). Phosphorylation of IRF3 occurs at two serine clusters in the serine-rich region. In the proposed model of IRF3 activation, phosphorylation induces a conformational change in the autoinhibitory domain, releasing a buried hydrophobic surface to enable dimerization of IRF3 (11–13). The activated IRF3 dimer then translocates from the cytoplasm to the nucleus, where the IRF3 dimer binds to the transcriptional coactivator CREB-binding protein (CBP) and subsequently to the DNA promoter to activate transcription of beta interferon genes (14). IRF3 activation is essential for the host to mount innate and adaptive antiviral responses (15).
FIG 1.

Npro forms a complex with the full-length IRF3 monomer. (A) Schematic of IRF3 domains. IRF3 consists of the DNA-binding domain (DBD; green), a linker (gray), and the IRF association domain (IAD; yellow). The IAD contains the autoinhibitory region along with the C-terminal phosphorylation sites, sites I and II (blue). Residues in the phosphorylation (P) sites that were mutated to glutamic acid to generate the phosphomimetic mutant IRF3-5E/E are indicated. (B) Gel shift assay of IRF3 in the presence of full-length and Δ17N Npro. (Left) IRF3 alone and IRF3 mixed with either full-length Npro (lane WT [wild type]) or the N-terminal deletion mutant (lane Δ17N Npro) were loaded onto native polyacrylamide gels. The gels were spliced, indicated by a space between lanes, to facilitate viewing. Npro-IRF3 complexes (indicated by † and ‡) were eluted for SDS-PAGE analysis. (Right) SDS-PAGE of the Δ17N Npro-IRF3 complex (lane 1) and full-length Npro-IRF3 complex (lane 4) eluted from native polyacrylamide gels, along with that of purified IRF3 (lanes 2 and 5), Δ17N Npro (lane 3), and full-length Npro (lane 6). (C) (Left) Elution profile from size exclusion chromatography of IRF3 and Δ17N Npro protein solution. The absorbance at 280 nm was monitored. mAU, milli-absorbance units. (Right) SDS-PAGE of the fractions from the two peaks. Peak 1 contains both IRF3 and Δ17N Npro coeluting from the column. Excess unbound Npro in the protein mix eluted at a higher volume in peak 2. (D) Sedimentation (sed) velocity profiles of IRF3 alone and the Δ17N Npro-IRF3 complex. IRF3 is a monomer with a molecular mass of 42 kDa. The Npro and IRF3 mixture shows peaks corresponding to free Npro (∼19 kDa) and the bound 1:1 complex of IRF3 and Npro (∼57 kDa). The numbers to the left and right of the SDS-PAGE gels are molecular masses (in kilodaltons).
Pestiviruses, such as bovine viral diarrhea virus (BVDV) and classical swine fever virus (CSFV), have two mechanisms to counter the innate immune responses of dendritic cells (DCs) and macrophages (16). One mechanism involves the glycoprotein Erns. The secreted form of Erns is released into the extracellular space, where a role of Erns for degradation of extracellular viral dsRNA and single-stranded RNA molecules has been postulated (17–19). Interestingly, Erns can enter cells by clathrin-dependent endocytosis and digest viral RNA in endolysosomal compartments via its RNase activity before they can trigger type I IFN induction (20). Importantly, Erns is very efficient at preventing activation of plasmacytoid dendritic cells (pDCs) in contact with CSFV-infected cells (21). A second layer of protection against the antiviral response is provided by the N-terminal protease (Npro). The Npro protein is a leader cysteine autoprotease that cleaves itself from the nascent polyprotein during translation of the viral mRNA (22, 23). The released Npro suppresses the transcriptional activation of the IFN-α/β genes by interacting with IRF3 and inducing its ubiquitination and proteasome-dependent degradation (24–27). Npro also interferes with the activity of IRF7 in pDCs, although this interference is probably of minor importance (28). In pigs, Npro contributes to pathogenicity by interfering with the activation of IFN responses at local replication sites (29). This is, however, not essential for the virus to establish infection, although a CSFV isolate with a complete deletion of the Npro gene was shown to be attenuated in pigs (30, 31). The protease activity of Npro is not involved in the degradation of IRF3, since mutation of the catalytic Cys69 had no effect on anti-IFN function (30). An intact zinc-binding site in Npro (C112-C134-D136-C138) is essential for targeting IRF3 for proteasomal degradation, since mutant CSFV or BVDV strains containing individual mutations in the Zn-binding site were unable to inhibit the interferon response in the host cell (30, 32, 33). Interactions between Npro and IRF3/IRF7 were demonstrated using coprecipitation and mammalian two-hybrid assays (25, 26, 28, 32). In particular, mammalian two-hybrid assays in HEK293T cells identified that most of the IRF3 protein, including both the DNA-binding and IRF association domains, is required for Npro-IRF3 and Npro-IRF7 interactions (28).
Recent crystal structures of pestivirus Npro show that Npro has a unique clam shell-like fold consisting of two domains, a cysteine protease domain and a zinc-binding domain (34, 35). The structure establishes the mechanism of autocatalysis and subsequent autoinhibition of Npro but provided little clue as to how Npro binds and mediates IRF3 degradation. Currently, it is not known whether Npro binds IRF3 directly without any other protein and whether the monomeric or dimeric form of IRF3 or both forms interact with Npro. In order to explore this, we studied the interactions between purified recombinant CSFV Npro and porcine IRF3 proteins in vitro. Npro interacts with IRF3 directly without additional proteins and forms a stable 1:1 complex. In addition, Npro recognizes both the inactive IRF3 monomer and the phosphomimetic IRF3 dimer for binding, thus likely targeting all forms of IRF3 species for proteasomal degradation.
MATERIALS AND METHODS
Construction of porcine IRF3 and CBP48 proteins.
The gene coding for porcine IRF3 (GenBank accession number AB116563) was synthesized and cloned into a pJ414 vector that carries the T7 polymerase promoter and ampicillin resistance (pJ414-IRF3; DNA2.0, Menlo Park, CA). All proteins were expressed with an N-terminal 6×His tag. The nucleotide sequence of the gene was optimized for expression in Escherichia coli. The individual domains of IRF3 were designed on the basis of the sequence alignment between the human and porcine IRF3 proteins, which share a sequence identity of 78%. The DNA-binding domain of porcine IRF3 (IRF3-DBD) spans amino acids 1 to 114. Site-directed mutagenesis with the primer pair 5′-GCGTCGGTGACTTCTAACCTGAGCCAGACACC-3′ (forward) and 5′-GGTGTCTGGCTCAGGTTAGAAGTCACCGACGC-3′ (reverse) was used to insert a stop codon in the wild-type IRF3 gene to generate the IRF3-DBD expression plasmid. The C-terminal domain of IRF3 (IRF3-IAD) containing the autoinhibitory region and the serine-rich region spans amino acids 171 to 419. IRF3-IAD was subcloned from full-length porcine IRF3 using the primer pair 5′-GCACATATGTCCCCGAGCGTGGACGCACCGGC-3′ (forward) and 5′-GCACTCGAGTCATTAGAAGTCCATATCTTCCACCAGGTCGCGC-3′ (reverse). The phosphomimetic glutamic acid mutant (IRF3-5E/E) was generated using sequential site-directed mutagenesis on the wild-type pJ414-IRF3 plasmid. Site I amino acid S384 and site II amino acids S394, S396, S400, T402, and S403 were all mutated to glutamic acid. A complete list of the primers used for the site-directed mutagenesis reactions of IRF3-5E/E is given in Table 1. DNA encoding the 46-amino-acid domain of CBP (residues 2065 to 2111), previously identified to be the interferon response binding domain, was synthesized and cloned into pJ411 (DNA2.0) carrying the T7 polymerase promoter and a kanamycin resistance cassette. The final construct has additional two residues (Met and Ser) at the N terminus and is thus named CBP48. The C-terminal deletion mutant of IRF3, IRF3-ΔC, which lacks the terminal autoinhibitory region (amino acids 392 to 419), was generated from the full-length protein using site-directed mutagenesis to insert a stop codon after Leu391 with the primer pair 5′-GAATACCGTGGATCTGCACTAATCAGCAATAGCCATCCTC-3′ (forward) and 5′-GAGGATGGCTATTGCTGATTAGTGCAGATCCACGGTATTC-3′ (reverse). All plasmid constructs were verified by DNA sequencing at the University of Texas Medical Branch's molecular genomics core.
TABLE 1.
Primers used for sequential side-directed mutagenesis to generate the phosphomimetic mutant IRF3-5E/Ea
| Protein | Primer | Sequence |
|---|---|---|
| IRF3-1E | S394E (forward) | CCGTGGATCTGCACATCGAGAATAGCCATCCTCTGAGC |
| S394E (reverse) | GCTCAGAGGATGGCTATTCTCGATGTGCAGATCCACGG | |
| IRF3-2E | S396E (forward) | GGATCTGCACATCGAGAATGAACATCCTCTGAGCCTGACG |
| S396E (reverse) | CGTCAGGCTCAGAGGATGTTCATTCTCGATGTGCAGATCC | |
| IRF3-3E | S400E (forward) | CGAGAATGAACATCCTCTGGAACTGACGAGCGACCAGTAC |
| S400E (reverse) | GTACTGGTCGCTCGTCAGTTCCAGAGGATGTTCATTCTCG | |
| IRF3-5E | T402E/S403E (forward) | GAATGAACATCCTCTGGAACTGGAAGAAGACCAGTACAAGGCGTGTCTGC |
| T402E/S403E (reverse) | GCAGACACGCCTTGTACTGGTCTTCTTCCAGTTCCAGAGGATGTTCATTC | |
| IRF3-5E/E | S384E (forward) | CACGTGACGGTGGTGCAAGCGAATTGGAGAATACCGTGGATCTG |
| S384E (reverse) | CAGATCCACGGTATTCTCCAATTCGCTTGCACCACCGTCACGTG |
Full-length IRF3 was used as the template for the first reaction to mutate Ser394 to Glu (IRF3-1E). Subsequent Glu substitutions were made using the product from the previous mutagenesis as the template.
Expression and purification of Npro and IRF3 proteins.
Recombinant IRF3 proteins were expressed in E. coli BL21(DE3) cells (Stratagene) grown in Terrific broth supplemented with 50 μg/ml of ampicillin. Cells were grown at 37°C to an optical density at 600 nm of 0.8, and protein expression was induced by the addition of 0.5 mM IPTG (isopropyl-β-d-thiogalactopyranoside), with growth being continued overnight at 18°C. The cell pellet from a 2-liter culture was resuspended in 50 ml of lysis buffer (50 mM sodium phosphate, pH 8.0, 300 mM NaCl) and lysed by sonication. Protein in the soluble fraction of the lysate was loaded onto Talon (Clontech) metal-affinity chromatography resin preequilibrated in lysis buffer. Bound IRF3 was eluted using a gradient of 5 to 150 mM imidazole in wash buffer (50 mM sodium phosphate, pH 7.1, 300 mM NaCl). Fractions containing IRF3 were pooled and concentrated to a final volume of 1 ml using an Amicon Ultra centrifugal filter device (Millipore). The sample was then loaded onto a Superdex 200 size exclusion column (GE Healthcare) that had been preequilibrated with buffer A (20 mM Tris-HCl, pH 7.5, 200 mM NaCl, 5 mM β-mercaptoethanol) in order to separate IRF3 from degradation products. Following size exclusion chromatography (SEC), the protein was further purified using anion-exchange chromatography. Pooled protein from size exclusion chromatography was exchanged into buffer B (20 mM Tris-HCl, pH 7.5, 50 mM NaCl) and loaded onto MonoQ anion-exchange column (GE Healthcare) equilibrated in buffer B. A salt gradient from 50 mM to 400 mM NaCl was used to elute pure IRF3 from the column. IRF3-DBD and IRF-IAD were similarly purified but without the anion-exchange chromatography step.
To express IRF3-5E/E and CBP48 together, the IRF3-5E/E- and CBP48-expressing plasmids were cotransformed into E. coli BL21(DE3) cells, and the transformants were selected for dual antibiotic resistance to ampicillin and kanamycin. The two proteins were then coexpressed under conditions similar to those used for wild-type IRF3 expression. IRF3-5E/E and CBP48 were then coextracted from the cell lysate using metal-affinity chromatography on Talon resin. IRF3-5E/E and CBP48 formed a tight complex that was further purified from the degradation products and low-molecular-mass contaminants using size exclusion chromatography in buffer A. The IRF3-5E/E–CBP48 complex was buffer exchanged into 20 mM Tris, pH 7.5, and 100 mM NaCl and subjected to anion-exchange chromatography on the MonoQ column. A gradient between 100 mM and 1 M NaCl was used to elute the pure complex from the column. Similarly, the IRF3-ΔC–CBP48 protein complex was purified using Talon metal affinity chromatography followed by size exclusion chromatography on Superdex 200. Full-length CSFV Npro and the N-terminal deletion mutant of CSFV Npro that lacked the first 17 amino acids (the Δ17N mutant) were expressed and purified as previously described (32, 34). SDS-polyacrylamide gel electrophoresis (PAGE) analysis showed that the purified proteins were greater than 95% pure.
Gel shift assay.
Binding of Npro to full-length IRF3 and individual IRF3 domains (DBD and IAD) was tested by native PAGE using a GE PhastGel mini-electrophoresis system (GE Healthcare Life Sciences). Native PAGE was run using precast gradient gels, either 4 to 15% or 10 to 15%, following the manufacturer's recommendations. PhastGel native buffer strips were used to maintain the buffer composition during electrophoresis at 0.88 M l-alanine and 0.25 M Tris, pH 8.8. Prior to electrophoresis all protein samples were prepared in 20 mM Tris buffer at pH 7.5 and 200 mM NaCl. IRF3 constructs were either incubated with Npro or diluted to the same volume with buffer before loading on the gel. NativeMark, a colored native gel protein marker from Invitrogen, was used as a reference for the electrophoresis run. To extract proteins, native PAGE analysis was repeated using Mini-Protean TGX precast gels from Bio-Rad. Following electrophoresis, the gels were stained using colloidal Coomassie G-250 in water, and the individual protein bands were excised. Proteins were eluted into 20 μl of 20 mM Tris buffer, pH 7.5, containing 200 mM NaCl and loaded onto SDS-containing polyacrylamide gels for identification.
Size exclusion chromatography of Npro-IRF3 complexes.
Purified Npro and IRF3 proteins (full-length IRF3, the IRF3-5E/E–CBP48 complex, the IRF3-ΔC–CBP48 complex, and IRF3-DBD) were mixed and loaded onto a Superdex 200 size exclusion chromatography column (GE Healthcare Life Sciences) preequilibrated in buffer A. IRF3-IAD was incubated with Npro and loaded onto a Superdex 75 size exclusion chromatography column. A light precipitate was visible in the IRF3-IAD and Npro mixture and removed by centrifugation prior to application of the mixture to the column. Protein peaks on the chromatogram were analyzed by SDS-PAGE to verify coelution and, by extension, complex formation in the protein mixtures.
Analytical ultracentrifugation of the Npro-IRF3 complex.
IRF3 and the mixture of Npro and IRF3 were analyzed by sedimentation velocity using a Beckman Coulter XL-A analytical ultracentrifuge. Following buffer exchange against buffer A, the protein solution was diluted with the buffer to yield an absorbance at 280 nm of 0.3. A 400-μl aliquot was loaded into the sample compartment of a double-sector cell, assembled with 1.2-cm charcoal-Epon centerpiece and quartz windows. Dialysis buffer was placed in the reference compartment. The protein samples were sedimented at 45,000 rpm at 20°C. A total of 400 scans were collected. The resulting data set was analyzed with SEDFIT software, version 9.4, using the c(s) continuous size distribution model, allowing the frictional ratio to float (36).
RESULTS
CSFV Npro binds to the IRF3 monomer directly and forms a 1:1 complex in solution.
Immunoprecipitation, coimmunofluorescence, and mammalian two-hybrid assays have all indicated that wild-type pestivirus Npro interacts with IRF3 in vivo (26, 30). This interaction is responsible for the subsequent ubiquitination and degradation of IRF3 by the proteasome. IRF3 undergoes multiple conformational changes during activation, but it is not known to which species of IRF3 Npro binds, inducing the proteasomal degradation of IRF3. In the mammalian two-hybrid assay used to show the interaction between Npro and IRF3, the IRF3 protein pulled down with Npro is likely to be a monomer, since IRF3 is expressed from a plasmid and is not activated. These assays, however, do not show whether Npro and IRF3 interact directly without additional proteins. We thus used recombinant Npro and IRF3 proteins to test if they interact directly in vitro using a native gel shift assay and size exclusion chromatography (SEC). To this end, we expressed recombinant CSFV Npro and porcine IRF3 in bacteria. Full-length Npro and the N-terminal deletion mutant lacking the first 17 amino acids (the Δ17N mutant) were used to test their interaction with IRF3. The Δ17N construct, whose high-resolution crystal structure was determined previously (34), was chosen because it is less prone to degradation than the full-length protein. Full-length and Δ17N Npro have been shown to be functionally identical with respect to their protease and interferon antagonistic activities (20, 30, 34). Native gel shift assays showed that in the presence of either full-length or Δ17N Npro, the IRF3 protein band shifted to a higher molecular mass (Fig. 1B, left). Npro is a highly basic protein and hence migrated in the opposite direction, toward the cathode (see Fig. 4A, left). In order to verify that the higher-molecular-mass product seen in the gel shift assay corresponds to the Npro-IRF3 protein complex, the higher-molecular-mass bands were excised and proteins were eluted from the gel. The eluted proteins were then analyzed by denaturing SDS-PAGE. SDS-PAGE showed the presence of both IRF3 and the respective Npro proteins (Fig. 1B, right). The result indicates that Npro and IRF3 interact directly and the first 17 amino acids of Npro are not required for this interaction. Because of the superior stability of Δ17N Npro, further studies on the interaction of Npro with IRF3 were carried out using this shorter construct.
FIG 4.
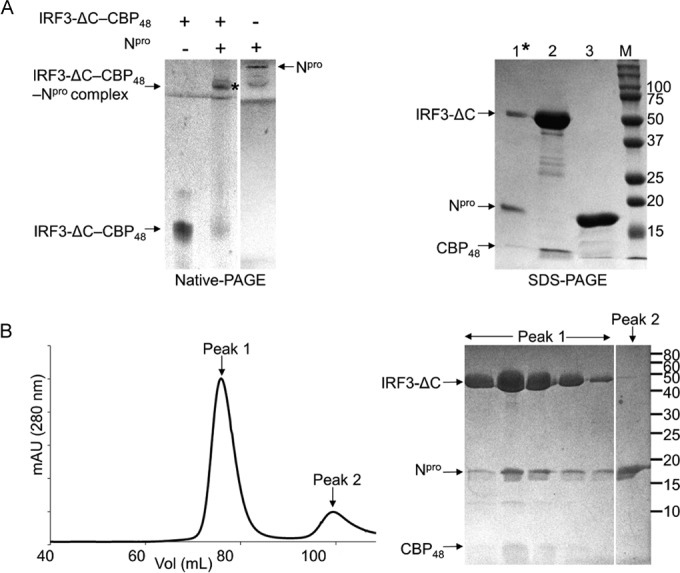
Npro interacts with the IRF3-ΔC–CBP48 complex. (A) (Left) Gel shift assay of the IRF3-ΔC–CBP48 complex in the presence of Npro. IRF3-ΔC–CBP48, Npro, and the mixture of IRF3-ΔC–CBP48 and Npro were loaded onto a native polyacrylamide gel. The immobile band indicated by an asterisk was eluted for SDS-PAGE analysis (right). Note that Npro migrates in the opposite direction from the loading position, toward the cathode. (Right) SDS-PAGE of the Npro and IRF3-ΔC–CBP48 complex (lane 1) along with IRF3-ΔC–CBP48 and Npro (lanes 2 and 3, respectively) is shown. (B) (Left) Elution profile from SEC of IRF3-ΔC–CBP48 and Npro on a Superdex 200 column. (Right) SDS-PAGE shows that Npro coelutes with the IRF3-ΔC–CBP48 complex in peak 1. The numbers to the right of the gels are molecular masses (in kilodaltons).
We further analyzed the formation of the Npro and IRF3 complex using SEC. The individual Npro and IRF3 proteins eluted as monomers, as determined by SEC. When the mixture of Npro and IRF3 at a 2:1 molar ratio was loaded onto the SEC column, two major peaks were observed (Fig. 1C, left). Peak 1 contained both Npro (17 kDa) and IRF3 (49 kDa) proteins, as identified by SDS-PAGE (Fig. 1C, right). Thus, Npro and IRF3 interact directly and form a stable complex in solution, consistent with the native PAGE result. Excess free Npro was seen in the later lower-molecular-mass peak (peak 2).
The stoichiometry of Npro and IRF3 in the Npro-IRF3 complex was next determined using analytical ultracentrifugation (Fig. 1D). IRF3 alone or as a mixture with Npro was subjected to sedimentation velocity experiments. Consistent with the SEC result, IRF3 alone was a monomer in its native form with a sedimentation coefficient of 3.1S, corresponding to a molecular mass of 42 kDa. The calculated molecular mass of IRF3 is 49 kDa. The Npro and IRF3 mixture (1.5:1 ratio) shows two species at 1.7S and 3.8S, which correspond to the predicted molecular masses of 19 and 57 kDa, respectively (Fig. 1D). The 19-kDa peak would correspond to the Npro monomer, and the 57-kDa peak would correspond to the Npro-IRF3 complex; the calculated molecular masses of Npro and the Npro-IRF3 complex are 17 and 66 kDa, respectively. Thus, Npro and IRF3 form a 1:1 complex in solution.
Full-length IRF3 is required for stable interaction with Npro.
IRF3 consists of two domains, the N-terminal DNA-binding domain (DBD) and the C-terminal IRF association domain (IAD) (Fig. 1A). To localize the region of IRF3 required for interaction with Npro, we designed constructs expressing the two IRF3 domains, IRF3-DBD (residues 1 to 114) and IRF3-IAD (residues 171 to 419), separately. Binding between Npro and each IRF3 domain was tested using a gel shift assay and SEC. IRF3-DBD runs as a mixture of species seen as a smear on a native gel (Fig. 2A). Addition of Npro to IRF3-DBD did not result in a shift of IRF3-DBD, indicating that the two proteins did not interact in vitro (Fig. 2A). SEC was also used to determine the interaction between Npro and IRF3-DBD. Because of the similarity in the sizes of Npro (17 kDa) and IRF3-DBD (15 kDa), the elution profile of Npro and the IRF3-DBD protein mixture showed one broad peak (Fig. 2B). However, SDS-PAGE showed two different concentration profiles corresponding to the two proteins in the peak, suggesting that the proteins did not interact with each other (Fig. 2B). Coelution resulting from stable complex formation would have resulted in a concomitant increase or decrease in their concentrations during elution. Thus, the results of both the native gel shift assay and SEC indicate that Npro does not interact with IRF3-DBD.
FIG 2.

Npro does not interact with individual domains of IRF3. (A) Gel shift assays of the DNA-binding domain of IRF3 (IRF3-DBD) in the presence of Npro. Left lane, IRF3-DBD alone; right lane, IRF3-DBD mixed with Npro. Note the lack of a shift in the position of IRF3-DBD in the presence of Npro. (B) (Left) Elution profile from SEC of an IRF3-DBD and Npro solution on a Superdex 200 column. (Right) SDS-PAGE of the fractions from the elution peak on the chromatogram. Bands corresponding to IRF3-DBD and Npro are indicated. (C) Gel shift assays of the IRF association domain (IRF3-IAD) in the presence of Npro. Left lane, IRF3-IAD; right lane, IRF3-IAD mixed with Npro. (D) (Left) Elution profile from SEC of an IRF3-IAD and Npro solution on a Superdex 75 column. (Right) SDS-PAGE of the fractions from the two elution peaks. Positions of IRF3-IAD and Npro in the gel are indicated. The numbers to the right of the gels are molecular masses (in kilodaltons).
IRF3-IAD (25 kDa) mediates the homo- and heterodimeric interactions of IRF3 and binding to its transcriptional cofactors (11, 12) (Fig. 1A). On the native gel, IRF3-IAD showed a single band. Addition of Npro to IRF3-IAD (at a 1:1 or 2:1 ratio) did not induce any shift in the IRF3-IAD protein band (Fig. 2C), suggesting that Npro did not form a complex with IRF3-IAD. When SEC was used to determine the interaction, however, a small amount of Npro coeluted with IRF3-IAD (Fig. 2D, right). SDS-PAGE analysis of the higher-molecular-mass peak (peak 1) showed that IRF3-IAD was present at a much higher concentration than Npro in the coeluted fractions and the majority of Npro eluted in a separate lower-molecular-mass peak (peak 2). Although the SEC result suggests a weak interaction between Npro and IRF3-IAD, when the results were taken together with the results of the gel shift assay, we concluded that IRF3-IAD is not sufficient to form a stable complex with Npro.
Npro binds to the phosphomimetic IRF3-5E/E monomer and dimer in the presence of CBP.
Phosphorylation of IRF3 in the cytoplasm leads to its activation and dimerization, following which it translocates into the nucleus. In the nucleus, the activated dimer of IRF3 associates with the CREB-binding protein (CBP), among other cofactors, to form the transcription complex that regulates the IFN-α/β gene promoters (4, 37). We thus tested whether Npro binds the phosphorylated IRF3 dimer using a phosphomimetic mutant, IRF3-5E/E. Phosphorylation of IRF3 occurs at two highly conserved Ser/Thr clusters, viz., site I and site II, in the serine-rich region of IRF3 (Fig. 1A). Site I is constituted by S383 and S384, and site II is formed by S394, S396, S400, T402, and S403 (37–40). It was shown that the IRF3 phosphomimetic mutant, in which all five Ser/Thr residues at site II were replaced with Asp or Glu residues (IRF3-5D and IRF3-5E, respectively), forms a stable IRF3 dimer that constitutively binds DNA (10, 41, 42). In the case of IRF3-5D expressed in insect cells, an additional phosphorylation in site I was observed, suggesting that the phosphorylation at site I may also be important for IRF3 activation (41). We thus generated IRF3-5E/E, in which all five Ser/Thr residues in site II were mutated to Glu (S394E, S396E, S400E, T402E, and S403E) with an additional S384E substitution at site I. Because S385 in human IRF3 was reported to have an autoinhibitory function rather than an activating function (38), the corresponding S383 in porcine IRF3 was not changed to Glu. IRF3-5E/E was found to be both a monomer and a dimer in solution, but the construct was unstable and degraded rather rapidly. To stabilize the IRF3 dimer, we coexpressed a CBP peptide along with IRF3-5E/E. Although CBP is a large ∼250-kDa protein, its interaction with IRF3 is mediated by the small interferon response binding domain (IBiD; 46 residues) (43). The binding of IBiD to the phosphomimetic mutants of IRF3 has been shown to induce the dimerization of IRF3 in solution (38). Thus, a 48-amino-acid domain of CBP (referred to here as CBP48) was used for coexpression. CBP binds to both the monomeric and dimeric 5E/E forms of IRF3. Accordingly, IRF3-5E/E–CBP48 complexes eluted as two peaks in SEC. The peak with the higher molecular mass is likely the heterotetramer of the IRF3-5E/E dimer and two CBP48 proteins, and the peak with the lower molecular mass is the heterodimer of the IRF3-5E/E monomer and CBP48 (Fig. 3).
FIG 3.

Npro interacts with the IRF3-5E/E–CBP48 complex. (A) Gel shift assay of IRF3-5E/E–CBP48 in the presence of Npro. Left lane, IRF3-5E/E–CBP48 complex; right lane, IRF3-5E/E–CBP48 complex mixed with Npro. Phosphomimetic mutant IRF3-5E/E and CBP48 form a heterotetramer (IRF3-5E/E dimer and two CBP48 proteins) and a heterodimer (IRF3-5E/E monomer and one CBP48 protein) (left lane), and Npro interacts with both IRF3-5E/E–CBP48 forms (right lane). Arrows, positions of monomeric and dimeric IRF3-5E/E bound to CBP48 and their Npro complexes. (B) (Left) Elution profile from SEC of IRF3-5E/E–CBP48 and Npro solution on a Superdex 200 column. (Right) SDS-PAGE of the peak fractions shows that both the CBP48-bound monomer and the CBP48-bound dimer of IRF3-5E/E coelute with Npro. Excess unbound Npro eluted in the third peak. The numbers to the right of the gel are molecular masses (in kilodaltons).
With IRF3-5E/E–CBP48 we tested whether the phosphomimetic IRF3 mutant interacts with Npro using a gel shift assay. In the presence of Npro, both the activated monomer and dimer species of IRF3 were shifted toward a higher molecular mass in the native gel (Fig. 3A). This clearly indicates that Npro binds the constitutively active form of IRF3 even in the presence of its cofactor, CBP. Next, IRF3-5E/E–CBP48 and Npro were mixed and separated by SEC. The SEC profile showed three peaks (Fig. 3B, left). Even though distinct peaks were observed for the IRF3-5E/E–CBP48 heterodimer and heterotetramer, SEC could not resolve the free IRF3-5E/E–CBP48 and the complexed IRF3-5E/E–CBP48–Npro species. SDS-PAGE analysis identified that the first two peaks (peaks 1 and 2) contained Npro, IRF3-5E/E, and CBP48, confirming that Npro interacts with both the IRF3-5E/E monomer and the IRF3-5E/E dimer in the presence of CBP. Thus, Npro and CBP do not appear to compete for IRF3 binding, indicating that the Npro-binding site on IRF3 and the CBP-binding site on IRF3-IAD are two distinct sites.
The C-terminal autoinhibitory region of IRF3 is not required for interaction with Npro.
Since Npro binds to the IRF3 monomer and phosphomimetic IRF3-5E/E monomer and dimer, Npro likely recognizes the structural features that are shared in both the IRF3 monomer and dimer. Phosphorylation of IRF3 results in the release of the C-terminal autoinhibitory region, exposing the dimerization interface and the cofactor (here, CBP) binding region on IRF3. Since phosphorylation, dimerization, and subsequent CBP binding could involve major rearrangements in the autoinhibitory region at the C terminus (9, 13), this region is not likely responsible for the physical interaction of IRF3 with Npro. We thus tested whether the C-terminal autoinhibitory region of IRF3 is required for the interaction of IRF3 with Npro. To this end we constructed a C-terminal deletion mutant, IRF3-ΔC, that lacks 27 amino acids at the C terminus, including the second phosphorylation site, site II (residues 392 to 419) (Fig. 1A). Because the C-terminal deletion in IRF3-ΔC would expose the CBP-binding site, we coexpressed IRF3-ΔC with CBP48. IRF3-ΔC indeed formed a complex with CBP48 that could be purified using SEC. In the native gel shift assay, the mixture of the purified IRF3-ΔC–CBP48 complex and Npro was predominantly localized in the well and remained immobile (Fig. 4A, left). In order to determine the composition of the immobile product in the native gel, proteins were eluted from the band and loaded onto SDS-polyacrylamide gels. The product contained IRF3-ΔC, CBP48, and Npro proteins (Fig. 4A, right). Thus, Npro binds to the IRF3-ΔC–CBP48 complex in solution. The IRF3-ΔC–CBP48 complex was also mixed with Npro and loaded onto an SEC column. All three proteins, Npro, IRF3-ΔC, and CBP48, coeluted as a single peak, as shown by the chromatogram and SDS-PAGE analysis (Fig. 4B). This indicates that the C-terminal 27 residues that contain phosphorylation site II or the CBP-binding site on IRF3 are not involved in Npro binding. Thus, Npro recognizes the structural features of IRF3 that do not undergo a conformational change during IRF3 activation and dimerization, in line with our results showing that Npro is able to recognize both the IRF3 monomer and the IRF3 dimer.
DISCUSSION
We have shown that Npro forms a stable soluble complex with wild-type full-length IRF3 in vitro. This is the first report that conclusively demonstrates a direct one-to-one interaction between the two proteins in solution. We further show that Npro cannot form a complex with the individual domains of IRF3, the N-terminal DBD or the C-terminal IAD; thus, the Npro-IRF3 interaction requires both IRF3 domains. Previously, a mammalian two-hybrid assay was used to show that Npro's interaction with IRF3 requires both the DBD and the IAD of IRF3 (28). Either the DBD or the IAD alone had only 5 to 10% of the Npro-binding ability compared to that of full-length IRF3. The largest IRF3 construct tested, consisting of residues 1 to 272 containing the DBD and partial IAD of IRF3, had ∼20% of the Npro-binding ability of full-length IRF3 (28). The findings of the present in vitro interaction studies using the recombinant proteins are consistent with those of the mammalian two-hybrid assay. Additionally, we have shown that Npro interacts with IRF3-ΔC, which lacks the C-terminal 27 amino acids, and thus, the C-terminal phosphorylation site in IRF3 is not required for Npro binding. Furthermore, the binding interaction between Npro and IRF3 is independent of IRF3's activation state, and Npro is able to form a complex with the inactive IRF3 monomer as well as the constitutively active monomer and dimer (IRF3-5E/E). Thus, Npro likely induces the degradation of the inactive IRF3 monomer and the activated phosphorylated IRF3 dimer in the cytoplasm. Finally, the presence of CBP, a cofactor of IRF3, does not interfere with its interaction with Npro in vitro. These data support the findings of previous in vivo studies that Npro does not disrupt activation of IRF3 or its translocation into the nucleus (26).
The Npro-binding site on IRF3 is currently unknown. Since Npro does not recognize the individual IRF3 domains, it is likely that the Npro-binding site on IRF3 is composed of the two IRF3 domains, including the ∼60-amino-acid linker region in between (Fig. 1A). However, the full-length IRF3 structure is unavailable, and it is therefore not known how the DBD and the IAD are arranged in the IRF3 monomer and dimer (11, 44, 45). The linker region is shown to be partially helical in the full-length IRF3 protein, while it is not structured when either of the domains is absent (46). Thus, the intact linker region may also be involved in Npro binding. Npro interacts with the IRF3 monomer and phosphomimetic dimer, suggesting that Npro recognizes a structural feature of IRF3 that is preserved during IRF3 activation from a monomer to a dimer. Since the C-terminal phosphorylation sites on IRF3 undergo significant structural changes and expose the CBP-binding site during IRF3 activation, Npro is unlikely to recognize the phosphorylation sites or the CBP-binding site for interaction. Indeed, Npro interacts with IRF3-ΔC in complex with CBP, indicating that the C-terminal phosphorylation sites or the CBP-binding site of IRF3 is not involved in Npro interaction.
CSFV Npro interacts with the phosphomimetic IRF3 dimer and CBP-bound form of IRF3, the predominant forms in the nucleus. Since Npro's function of degrading IRF3 has been shown only in the cytoplasm, Npro is likely to induce the degradation of the IRF3 dimer by the proteasome when the IRF3 dimer is formed or exported from the nucleus. It is currently not clear whether Npro has an additional role in the nucleus. Npro is shown to diffuse into the nucleus (47, 48), wherein it would interact with the IRF3 dimer or the CBP-bound form of the IRF3 dimer. In the nucleus, the IRF3 dimer binds to the transcriptional coactivator CBP and subsequently to the promoter region of beta interferon genes to activate transcription of interferon genes. Interaction of Npro with IRF3 in the nucleus could thus interfere with its role as a transcription factor, facilitating viral replication further. BVDV Npro has been shown to inhibit IRF3 from binding to DNA even when the proteasomal degradation of IRF3 is blocked (27). Whether CSFV Npro also inhibits the transcriptional activity of IRF3 in the nucleus is not known and requires further investigation.
REFERENCES
- 1.Kawai T, Akira S. 2006. Innate immune recognition of viral infection. Nat Immunol 7:131–137. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- 2.Fensterl V, Sen GC. 2009. Interferons and viral infections. Biofactors 35:14–20. doi: 10.1002/biof.6. [DOI] [PubMed] [Google Scholar]
- 3.Honda K, Taniguchi T. 2006. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol 6:644–658. doi: 10.1038/nri1900. [DOI] [PubMed] [Google Scholar]
- 4.Honda K, Takaoka A, Taniguchi T. 2006. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 25:349–360. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 5.Tamura T, Yanai H, Savitsky D, Taniguchi T. 2008. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol 26:535–584. doi: 10.1146/annurev.immunol.26.021607.090400. [DOI] [PubMed] [Google Scholar]
- 6.Haller O, Kochs G, Weber F. 2006. The interferon response circuit: induction and suppression by pathogenic viruses. Virology 344:119–130. doi: 10.1016/j.virol.2005.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takeuchi O, Akira S. 2009. Innate immunity to virus infection. Immunol Rev 227:75–86. doi: 10.1111/j.1600-065X.2008.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thompson MR, Kaminski JJ, Kurt-Jones EA, Fitzgerald KA. 2011. Pattern recognition receptors and the innate immune response to viral infection. Viruses 3:920–940. doi: 10.3390/v3060920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen W, Royer WE Jr. 2010. Structural insights into interferon regulatory factor activation. Cell Signal 22:883–887. doi: 10.1016/j.cellsig.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin R, Mamane Y, Hiscott J. 1999. Structural and functional analysis of interferon regulatory factor 3: localization of the transactivation and autoinhibitory domains. Mol Cell Biol 19:2465–2474. doi: 10.1128/MCB.19.4.2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qin BY, Liu C, Lam SS, Srinath H, Delston R, Correia JJ, Derynck R, Lin K. 2003. Crystal structure of IRF-3 reveals mechanism of autoinhibition and virus-induced phosphoactivation. Nat Struct Biol 10:913–921. doi: 10.1038/nsb1002. [DOI] [PubMed] [Google Scholar]
- 12.Qin BY, Liu C, Srinath H, Lam SS, Correia JJ, Derynck R, Lin K. 2005. Crystal structure of IRF-3 in complex with CBP. Structure 13:1269–1277. doi: 10.1016/j.str.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 13.Chen W, Lam SS, Srinath H, Jiang Z, Correia JJ, Schiffer CA, Fitzgerald KA, Lin K, Royer WE Jr. 2008. Insights into interferon regulatory factor activation from the crystal structure of dimeric IRF5. Nat Struct Mol Biol 15:1213–1220. doi: 10.1038/nsmb.1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin R, Genin P, Mamane Y, Hiscott J. 2000. Selective DNA binding and association with the CREB binding protein coactivator contribute to differential activation of alpha/beta interferon genes by interferon regulatory factors 3 and 7. Mol Cell Biol 20:6342–6353. doi: 10.1128/MCB.20.17.6342-6353.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iwasaki A, Medzhitov R. 2010. Regulation of adaptive immunity by the innate immune system. Science 327:291–295. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Summerfield A, Ruggli N. 2015. Immune responses against classical swine fever virus: between ignorance and lunacy. Front Vet Sci 2:10. doi: 10.3389/fvets.2015.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Magkouras I, Mätzener P, Rümenapf T, Peterhans E, Schweizer M. 2008. RNase-dependent inhibition of extracellular, but not intracellular, dsRNA-induced interferon synthesis by Erns of pestiviruses. J Gen Virol 89:2501–2506. doi: 10.1099/vir.0.2008/003749-0. [DOI] [PubMed] [Google Scholar]
- 18.Mätzener P, Magkouras I, Rümenapf T, Peterhans E, Schweizer M. 2009. The viral RNase E(rns) prevents IFN type-I triggering by pestiviral single- and double-stranded RNAs. Virus Res 140:15–23. doi: 10.1016/j.virusres.2008.10.015. [DOI] [PubMed] [Google Scholar]
- 19.Iqbal M, Poole E, Goodbourn S, McCauley JW. 2004. Role for bovine viral diarrhea virus Erns glycoprotein in the control of activation of beta interferon by double-stranded RNA. J Virol 78:136–145. doi: 10.1128/JVI.78.1.136-145.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zürcher C, Sauter KS, Mathys V, Wyss F, Schweizer M. 2014. Prolonged activity of the pestiviral RNase Erns as an interferon antagonist after uptake by clathrin-mediated endocytosis. J Virol 88:7235–7243. doi: 10.1128/JVI.00672-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Python S, Gerber M, Suter R, Ruggli N, Summerfield A. 2013. Efficient sensing of infected cells in absence of virus particles by plasmacytoid dendritic cells is blocked by the viral ribonuclease E(rns.). PLoS Pathog 9:e1003412. doi: 10.1371/journal.ppat.1003412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rumenapf T, Stark R, Heimann M, Thiel HJ. 1998. N-terminal protease of pestiviruses: identification of putative catalytic residues by site-directed mutagenesis. J Virol 72:2544–2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gottipati K, Acholi S, Ruggli N, Choi KH. 2014. Autocatalytic activity and substrate specificity of the pestivirus N-terminal protease N(pro). Virology 452–453:303–309. doi: 10.1016/j.virol.2014.01.026. [DOI] [PubMed] [Google Scholar]
- 24.Seago J, Hilton L, Reid E, Doceul V, Jeyatheesan J, Moganeradj K, McCauley J, Charleston B, Goodbourn S. 2007. The Npro product of classical swine fever virus and bovine viral diarrhea virus uses a conserved mechanism to target interferon regulatory factor-3. J Gen Virol 88:3002–3006. doi: 10.1099/vir.0.82934-0. [DOI] [PubMed] [Google Scholar]
- 25.Chen Z, Rijnbrand R, Jangra RK, Devaraj SG, Qu L, Ma Y, Lemon SM, Li K. 2007. Ubiquitination and proteasomal degradation of interferon regulatory factor-3 induced by Npro from a cytopathic bovine viral diarrhea virus. Virology 366:277–292. doi: 10.1016/j.virol.2007.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bauhofer O, Summerfield A, Sakoda Y, Tratschin JD, Hofmann MA, Ruggli N. 2007. Classical swine fever virus Npro interacts with interferon regulatory factor 3 and induces its proteasomal degradation. J Virol 81:3087–3096. doi: 10.1128/JVI.02032-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hilton L, Moganeradj K, Zhang G, Chen YH, Randall RE, McCauley JW, Goodbourn S. 2006. The NPro product of bovine viral diarrhea virus inhibits DNA binding by interferon regulatory factor 3 and targets it for proteasomal degradation. J Virol 80:11723–11732. doi: 10.1128/JVI.01145-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fiebach AR, Guzylack-Piriou L, Python S, Summerfield A, Ruggli N. 2011. Classical swine fever virus N(pro) limits type I interferon induction in plasmacytoid dendritic cells by interacting with interferon regulatory factor 7. J Virol 85:8002–8011. doi: 10.1128/JVI.00330-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tamura T, Nagashima N, Ruggli N, Summerfield A, Kida H, Sakoda Y. 2014. Npro of classical swine fever virus contributes to pathogenicity in pigs by preventing type I interferon induction at local replication sites. Vet Res 45:47. doi: 10.1186/1297-9716-45-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruggli N, Summerfield A, Fiebach AR, Guzylack-Piriou L, Bauhofer O, Lamm CG, Waltersperger S, Matsuno K, Liu L, Gerber M, Choi KH, Hofmann MA, Sakoda Y, Tratschin JD. 2009. Classical swine fever virus can remain virulent after specific elimination of the interferon regulatory factor 3-degrading function of Npro. J Virol 83:817–829. doi: 10.1128/JVI.01509-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mayer D, Hofmann MA, Tratschin JD. 2004. Attenuation of classical swine fever virus by deletion of the viral N(pro) gene. Vaccine 22:317–328. doi: 10.1016/j.vaccine.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 32.Szymanski MR, Fiebach AR, Tratschin JD, Gut M, Ramanujam VM, Gottipati K, Patel P, Ye M, Ruggli N, Choi KH. 2009. Zinc binding in pestivirus N(pro) is required for interferon regulatory factor 3 interaction and degradation. J Mol Biol 391:438–449. doi: 10.1016/j.jmb.2009.06.040. [DOI] [PubMed] [Google Scholar]
- 33.Kozasa T, Abe Y, Mitsuhashi K, Tamura T, Aoki H, Ishimaru M, Nakamura S, Okamatsu M, Kida H, Sakoda Y. 2015. Analysis of a pair of END+ and END− viruses derived from the same bovine viral diarrhea virus stock reveals the amino acid determinants in Npro responsible for inhibition of type I interferon production. J Vet Med Sci 77:511–518. doi: 10.1292/jvms.14-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gottipati K, Ruggli N, Gerber M, Tratschin JD, Benning M, Bellamy H, Choi KH. 2013. The structure of classical swine fever virus N(pro): a novel cysteine autoprotease and zinc-binding protein involved in subversion of type I interferon induction. PLoS Pathog 9:e1003704. doi: 10.1371/journal.ppat.1003704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zogg T, Sponring M, Schindler S, Koll M, Schneider R, Brandstetter H, Auer B. 2013. Crystal structures of the viral protease Npro imply distinct roles for the catalytic water in catalysis. Structure 21:929–938. doi: 10.1016/j.str.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schuck P. 2000. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys J 78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin R, Heylbroeck C, Pitha PM, Hiscott J. 1998. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol Cell Biol 18:2986–2996. doi: 10.1128/MCB.18.5.2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen W, Srinath H, Lam SS, Schiffer CA, Royer WE Jr, Lin K. 2008. Contribution of Ser386 and Ser396 to activation of interferon regulatory factor 3. J Mol Biol 379:251–260. doi: 10.1016/j.jmb.2008.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mori M, Yoneyama M, Ito T, Takahashi K, Inagaki F, Fujita T. 2004. Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. J Biol Chem 279:9698–9702. doi: 10.1074/jbc.M310616200. [DOI] [PubMed] [Google Scholar]
- 40.Servant MJ, Grandvaux N, ten Oever BR, Duguay D, Lin R, Hiscott J. 2003. Identification of the minimal phosphoacceptor site required for in vivo activation of interferon regulatory factor 3 in response to virus and double-stranded RNA. J Biol Chem 278:9441–9447. doi: 10.1074/jbc.M209851200. [DOI] [PubMed] [Google Scholar]
- 41.Panne D, McWhirter SM, Maniatis T, Harrison SC. 2007. Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. J Biol Chem 282:22816–22822. doi: 10.1074/jbc.M703019200. [DOI] [PubMed] [Google Scholar]
- 42.Dragan AI, Hargreaves VV, Makeyeva EN, Privalov PL. 2007. Mechanisms of activation of interferon regulator factor 3: the role of C-terminal domain phosphorylation in IRF-3 dimerization and DNA binding. Nucleic Acids Res 35:3525–3534. doi: 10.1093/nar/gkm142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin CH, Hare BJ, Wagner G, Harrison SC, Maniatis T, Fraenkel E. 2001. A small domain of CBP/p300 binds diverse proteins: solution structure and functional studies. Mol Cell 8:581–590. doi: 10.1016/S1097-2765(01)00333-1. [DOI] [PubMed] [Google Scholar]
- 44.Takahasi K, Suzuki NN, Horiuchi M, Mori M, Suhara W, Okabe Y, Fukuhara Y, Terasawa H, Akira S, Fujita T, Inagaki F. 2003. X-ray crystal structure of IRF-3 and its functional implications. Nat Struct Biol 10:922–927. doi: 10.1038/nsb1001. [DOI] [PubMed] [Google Scholar]
- 45.Fujii Y, Shimizu T, Kusumoto M, Kyogoku Y, Taniguchi T, Hakoshima T. 1999. Crystal structure of an IRF-DNA complex reveals novel DNA recognition and cooperative binding to a tandem repeat of core sequences. EMBO J 18:5028–5041. doi: 10.1093/emboj/18.18.5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shukla H, Vaitiekunas P, Majumdar AK, Dragan AI, Dimitriadis EK, Kotova S, Crane-Robinson C, Privalov PL. 2012. The linker of the interferon response factor 3 transcription factor is not unfolded. Biochemistry 51:6320–6327. doi: 10.1021/bi300260s. [DOI] [PubMed] [Google Scholar]
- 47.Doceul V, Charleston B, Crooke H, Reid E, Powell PP, Seago J. 2008. The Npro product of classical swine fever virus interacts with IkappaBalpha, the NF-kappaB inhibitor. J Gen Virol 89:1881–1889. doi: 10.1099/vir.0.83643-0. [DOI] [PubMed] [Google Scholar]
- 48.Li Y, Shen L, Li C, Huang J, Zhao B, Sun Y, Li S, Luo Y, Qiu HJ. 2014. Visualization of the Npro protein in living cells using biarsenically labeling tetracysteine-tagged classical swine fever virus. Virus Res 189:67–74. doi: 10.1016/j.virusres.2014.04.018. [DOI] [PubMed] [Google Scholar]