

ABSTRACT
The role of NF-κB in influenza A virus (IAV) infection does not reveal a coherent picture, as pro- and also antiviral functions of this transcription factor have been described. To address this issue, we used clustered regularly interspaced short palindromic repeat with Cas9 (CRISPR-Cas9)-mediated genome engineering to generate murine MLE-15 cells lacking two essential components of the NF-κB pathway. Cells devoid of either the central NF-κB essential modulator (NEMO) scaffold protein and thus defective in IκB kinase (IKK) activation or cells not expressing the NF-κB DNA-binding and transactivation subunit p65 were tested for propagation of the SC35 virus, which has an avian host range, and its mouse-adapted variant, SC35M. While NF-κB was not relevant for replication of SC35M, the absence of NF-κB activity increased replication of the nonadapted SC35 virus. This antiviral effect of NF-κB was most prominent upon infection of cells with low virus titers as they usually occur during the initiation phase of IAV infection. The defect in NF-κB signaling resulted in diminished IAV-triggered phosphorylation of interferon regulatory factor 3 (IRF3) and expression of the antiviral beta interferon (IFN-β) gene. To identify the viral proteins responsible for NF-κB dependency, reassortant viruses were generated by reverse genetics. SC35 viruses containing the SC35M segment encoding neuraminidase (NA) were completely inert to the inhibitory effect of NF-κB, emphasizing the importance of the viral genotype for susceptibility to the antiviral functions of NF-κB.
IMPORTANCE This study addresses two different issues. First, we investigated the role of the host cell transcription factor NF-κB in IAV replication by genetic manipulation of IAVs by reverse genetics combined with targeted genome engineering of host cells using CRISPR-Cas9. The analysis of these two highly defined genetic systems indicated that the IAV genotype can influence whether NF-κB displays an antiviral function and thus might in part explain incoherent results from the literature. Second, we found that perturbation of NF-κB function greatly improved the growth of a nonadapted IAV, suggesting that NF-κB may contribute to the maintenance of the host species barrier.
INTRODUCTION
Wild aquatic birds comprise the natural reservoir for influenza A viruses (IAV), which occur in different strains defined by 16 hemagglutinin (HA) and 9 neuraminidase (NA) subtypes (1). The high degree of genomic plasticity of IAV results in constant change of the antigenic HA and NA (antigenic drift), leading to annual epidemics. Furthermore, the reassortment of the segmented viral genome (antigenic shift) enables them to adapt to new species, such as terrestrial birds and mammals, and to give rise to new virus lineages. This crossing of host species barriers can cause occasional pandemics, which are a major threat for human health (2, 3). All pandemics were caused by animal viruses or reassortant viruses carrying animal virus genes, emphasizing the need to study the mechanisms responsible for species barrier perforation (4). Early IAV infection is characterized by the induction of innate immune responses, which leads to the induction of cytokines, such as the antiviral type I interferons (IFNs), which can clear the infection (5, 6).
IAVs have a segmented genome consisting of eight negative-strand RNAs encoding at least 10 viral proteins (7). The viral RNA (vRNA) together with the three subunits of the heterotrimeric RNA-dependent RNA polymerase (RdRp) complex (PB1, PB2, and PA) and the nucleocapsid protein (NP) form the viral replication and transcriptionally active ribonucleoprotein complexes (vRNPs). After infection, the vRNPs enter the nucleus, where viral genome replication and transcription take place. After completion of viral replication, newly synthesized vRNPs are exported from the nucleus to the cytoplasm, where they reach the viral assembly sites at the plasma membrane. From here, assembled virus particles bud into the extracellular lumen (8).
A well-studied model system to investigate IAV adaptation to new host species is provided by the two H7N7-type IAV strains: SC35 and its mouse-adapted descendant, SC35M. SC35 was derived from the original virus isolate A/Seal/Massachusetts/1/80 (H7N7) by serial passages in chicken embryo cells, thereby acquiring a multibasic cleavage site in its HA and becoming 100% lethal for chickens. The mouse-adapted variant SC35M was yielded by repeated serial passages in mouse lungs. Thereby SC35M became highly pathogenic for mice, and infection leads to severe hemorrhagic pneumonia and death of the animals (9). The segmented genomes of SC35 and SC35M differ at nine positions. These are predominantly located in all three subunits (PB2, PB1, and PA) of the RdRp (10).
IAV infection triggers various host cell responses, which either facilitate or antagonize virus propagation. The antiviral response is exemplified by the IFN system, which is often counteracted by different viral mechanisms (9, 11). Expression of beta interferon (IFN-β) requires the induction of signaling cascades ultimately leading to the activation and cooperative assembly of the transcription factors interferon regulatory factor 3 (IRF3), IRF7, and NF-κB at the IFN-β enhancer (9, 12). Each of these transcription factors is important to achieve full expression of the IFN-β gene.
IAV infection causes the activation of the inducible transcription factor NF-κB by a complex signaling cascade. Binding of viral RNA to RIG-I (retinoic acid-inducible gene I) leads to a conformational change of this sensor protein, which is required for downstream signal activation (13, 14). Activated RIG-I then activates the adaptor protein MAVS (mitochondrial antiviral-signaling protein), which in turn leads to NF-κB activation via the IκB kinase (IKK) complex. This complex consists of the kinases IKKα and IKKβ and the scaffold protein NEMO (15). The activated IKK complex phosphorylates the inhibitory IκB protein, thus leading to its subsequent ubiquitination and degradation. The DNA-binding subunits (typically composed as a dimer of p50 and the transactivating subunit p65) then migrate to the nucleus, where they contact their genomic binding sites and activate gene expression (16, 17).
The function of NF-κB for IAV propagation does not reveal a coherent picture. A number of studies showed IAV supporting functions of NF-κB. Various IKK inhibitors such as BAY11-7085, BAY11-7082, or SC75741 severely impaired IAV infection of human lung carcinoma cell lines (18). Also the inhibition of NF-κB by expression of a nondegradable IκBα mutant or a dominant-negative IKKβ mutant resulted in reduced IAV replication in lung A549 cells, further indicating that NF-κB activity promotes efficient IAV production (19). On the other hand, several reports noted an antiviral function of NF-κB in vivo. Mice lacking the NF-κB inhibitory A20 protein show as expected exaggerated NF-κB activation after IAV infection but are protected against lethal IAV infection (20). This antiviral function of NF-κB most probably relies on its ability to induce the expression of inflammatory and antiviral mediators. In support of this notion, pretreatment of mice with 5′ triphosphate RNA (5′ppp RNA) to trigger the RIG-I-mediated induction of inflammatory and IFN-stimulated genes protects the animals from a subsequent infection with IAVs (21).
In order to clarify the role of NF-κB in IAV propagation and adaptation to new species, we engineered MLE-15 mouse lung epithelial cells using clustered regularly interspaced short palindromic repeats with Cas9 (CRISPR-Cas9) to delete two central components of the NF-κB system. Cells had deletion of either the scaffold protein NEMO to block the activity of the entire IKK complex or the strongly transactivating p65 DNA-binding subunit. While propagation of the mouse-adapted SC35M virus was not affected by NF-κB, deletion of either NEMO or p65 significantly increased the growth of the nonadapted SC35 virus, highlighting the role of the IAV genotype in the antiviral function of NF-κB.
MATERIALS AND METHODS
Antibodies, plasmids, and reagents.
The antibodies, plasmids, and reagents used in this study are listed in Table 1. The oligonucleotides and their sequences are listed in Table 2.
TABLE 1.
Antibodies, plasmids, and reagents used in this study
| Antibody, plasmid, or reagent | Antibody type and speciesa | Source or reference |
|---|---|---|
| Primary antibodies (clone) | ||
| Anti-Flag (M2) | Mouse MAb | Sigma |
| Anti-p65 (C-20) | Rabbit pAb | Santa Cruz |
| Anti-NEMO (FL419) | Rabbit pAb | Santa Cruz |
| Anti-phospho-IκBα (5A5) | Mouse pAb | Cell Signaling |
| Anti-phospho-ERK1/2 (SC7976) | Rabbit pAb | Santa Cruz |
| Anti-phospho-JNK (T183/Y185) | Rabbit pAb | Cell Signaling |
| Anti-NS1 | Mouse MAb | Gift from S. Ludwig, Münster, Germany |
| Anti-IAV NP (immunofluorescence, PA5-32242) | Rabbit pAb | Thermo Scientific |
| Anti-NP | Mouse MAb | S. Ludwig |
| Anti-tubulin (Tub2.1) | Mouse MAb | Sigma |
| Anti-IRF3 (FL-425) | Rabbit pAb | Santa Cruz |
| Anti-phospho-IRF3 (4D4G) | Rabbit MAb | Cell Signaling |
| Plasmids | ||
| px459 | F. Zhang lab (61) | |
| px459-mNEMO | This study | |
| px459-mp65 | This study | |
| pHW2000 plasmids encoding SC35 and SC35 M segments | 10 | |
| Reagents | ||
| Roti-Fect | Carl Roth GmbH | |
| Puromycin | Invitrogen | |
| TNF | Peprotech | |
| PHA-408 | Axon Medchem | |
| Hoechst 33342 | Invitrogen | |
| Mouse IFN-β | PBL Assay Science | |
| BSA | PAA Laboratories | |
| Avicel | FMC BioPolymer | |
| PFA | Carl Roth GmbH | |
| Triton X-100 | Carl Roth GmbH |
MAb, monoclonal antibody; pAb, polyclonal antibody.
TABLE 2.
Oligonucleotide primer sequences
| Primer namea | Sequence (5′ to 3′) |
|---|---|
| mNEMO-CRISPR-f | CACCGAGACCCTCCAGCGCTGCC |
| mNEMO-CRISPR-r | AAACGGCAGCGCTGGAGGGTCTC |
| mp65-CRISPR-f | CACCGCGATTCCGCTATAAATGCG |
| mp65-CRISPR-r | AAACCGCATTTATAGCGGAATCGC |
| mIL6-f | TGGATGCTACCAAACTGGAT |
| mIL6-r | GGACTCTGGTTTGTCTTTC |
| mIFN-beta-f | ATGAACGCTACACACTGCATC |
| mIFN-beta-r | CCATCCTTTTGCCAGTTCCTC |
| mTBP-f | GGGGAGCTGTGATGTGAAGT |
| mTBP-r | CCAGGAAATAATTCTGGCTCAT |
f, forward; r, reverse.
Cell culture and transfections.
Murine MLE-15 lung epithelial cells, 293T cells, and MDCK-II cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal calf serum (FCS) and 1% (wt/vol) penicillin-streptomycin at 37°C and 5% CO2. Dishes were seeded with cells, and the cells were transfected using the transfection reagent Roti-Fect (Carl Roth GmbH). After several pipettings up and down, complex formation occurred in serum- and antibiotic-free DMEM during 20 min at room temperature. After the transfection mixture was added to antibiotic-free DMEM containing FCS, the cells were incubated for 4 h before the medium was changed.
Viruses.
Influenza A virus SC35 was propagated in embryonated chicken eggs, and SC35M was propagated in MDCK-II cells. Titers of the virus stocks were determined as described below. The chicken-adapted virus SC35 was originally derived from the seal isolate A/Seal/Massachusetts/1/80 (H7N7) by serial passages in chicken embryo cells (22), and SC35M was obtained from SC35 by sequential passages in mouse lung (10).
Generation of recombinant SC35 and SC35M.
The pHW2000 plasmids containing the cloned segments of SC35 and SC35M were described before (10). Recombinant viruses were generated upon transfection of the respective 8 plasmids into a coculture of 293T and MDCK-II cells (3:1 ratio) using Lipofectamine 2000 (Invitrogen) as previously described (10). The cells were then incubated for 12 h at 37°C in the presence of 5% CO2. At the next day, the transfection media were replaced with 2 ml of Opti-MEM (containing penicillin-streptomycin and 0.2% bovine serum albumin [BSA]), and the cell cultures were incubated for 48 h. Then cell culture supernatant was harvested, and cell debris was removed by centrifugation. A 500-μl aliquot of each supernatant was used to inoculate MDCK-II cells, which were then incubated for 72 h. The viruses rescued from these cells were stored at −80°C until further use.
IAV titration.
MDCK-II cells grown in 96-well plates were infected with 20-μl virus samples serially diluted in PBS/BA (phosphate-buffered saline containing 0.2% BSA, 1 mM MgCl2, 0.9 mM CaCl2, and penicillin-streptomycin) from 10−1 to 10−7 for 1 h at room temperature. The inoculum was aspirated off, 150 μl Avicel medium (MEM supplemented with 1% penicillin-streptomycin, 0.3% BSA, 0.3% NaHCO3, 1.5% MC, 0.01% DEAE dextran, 1.25% Avicel) was added, and cells were further incubated at 37°C in 5% CO2 for 24 h. To detect foci of infected cells resulting from an infectious particle, cells were fixed and permeabilized with 150 μl fixing solution (4% paraformaldehyde [PFA], 1% Triton X-100 (Carl Roth GmbH, Germany) in PBS++ (PBS containing 1 mM MgCl2, 0.9 mM CaCl2) and incubated at 4°C for 1 h. The solution was then discarded, and cells were washed three times with PBS++ containing 0.05% Tween 20. Next, the cells were incubated with 50 μl/well primary antibody recognizing IAV NP diluted 1:100 in PBS++ containing 3% BSA for 1 h at room temperature. Then cells were washed three times with PBS++ containing 0.05% Tween 20 and incubated with 50 μl/well horseradish peroxidase-coupled secondary antibody diluted 1:1,000 in PBS++ containing 3% BSA for 1 h at room temperature. Cells were then washed three times with PBS++ containing 0.05% Tween 20 and incubated with 40 μl/well 3-amino-9-ethylcarbazole (AEC) staining solution for 40 min at 37°C in the dark. After staining, the substrate was removed, and cells were washed twice with H2O to remove salts. To detect and quantify foci, the 96-well plates were scanned at a resolution of 1,200 dpi using the Epson Perfection V500Photo scanner and analyzed using the Photoshop software package (Adobe). Results represent the averages from three independent experiments.
Cell lysis and Western blotting.
Cells were washed once with 1× PBS, harvested by scraping, and collected by centrifugation. The pellet was resuspended in NP-40 buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM phenylmethylsulfonyl fluoride, 10 mM NaF, 0.5 mM sodium orthovanadate, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1% NP-40, 10% glycerol) and incubated on ice for 20 min. The lysate was cleared by centrifugation for 10 min at 16,000 × g. The supernatant was then mixed with 5× SDS sample buffer and boiled at 95°C for 5 min, and proteins were separated via SDS-PAGE, followed by semidry blotting to a polyvinylidene difluoride membrane (Millipore) as previously described (23).
Generation and characterization of CRISPR knockout cells.
Oligonucleotides targeting the first exon of the NEMO or p65 gene were cloned into pX459 (Addgene). These plasmids (or the empty vector pX459 as a control) were transfected into MLE-15 cells, and cells were selected with puromycin (1 μg/ml) for 3 days, followed by growth of the surviving cells to colonies. Individual cell clones were picked and further expanded. Expression of p65, NEMO, and Cas9 was tested by Western blotting. Cas9-mediated mutations were characterized by isolation of genomic DNA using the NucleoSpin tissue kit according to the protocol of the manufacturer (Macherey-Nagel). Fifty nanograms of genomic DNA was used to amplify the genomic region encompassing the expected mutation using Phusion high-fidelity DNA polymerase and specific primers as specified in Table 2. The PCR product was excised from an agarose gel, and one of the PCR primers was directly used for sequencing of the PCR product.
Real-time qPCR.
The RNeasy minikit (Qiagen) was used to extract total RNA from cells according to the instructions of the manufacturer. Oligo(dT)20 primers and the Superscript II first-strand synthesis system (Invitrogen) were used to synthesize cDNA. Real-time quantitative PCR (qPCR) was performed using Absolute SYBR green ROX mix (Thermo Scientific) with specific primers (Table 1). Gene expression was determined using an Applied Biosystems 7300 real-time PCR system, all experiments were performed in triplicate, and quantification was done using the comparative threshold cycle (ΔΔCT) method. For quantification, data were normalized to the TBP housekeeping gene, and the resulting ΔCT values were compared to that of a sample that was chosen as a calibrator. The relative expression level was then calculated according to the following formula: R = 2−ΔΔCT.
Indirect immunofluorescence.
Cells were grown in 12-well plates on coverslips and infected with SC35 or SC35M. Cells were washed twice with 1× PBS and fixed for 10 min with 4% PFA at room temperature. The fixing solution was aspirated off, and cells were washed with 1× PBS and permeabilized with 0.5% Triton X-100 for 7 min. The cells were then washed with PBS and blocked for 60 min with 1× PBS containing 10% (vol/vol) BSA. After incubation with the primary antibody diluted in 1× PBS containing 1% (vol/vol) BSA overnight at 4°C, the cells were washed three times for 5 min with 1× PBS and incubated with the Cy3-coupled secondary antibody diluted in PBS containing 1% (vol/vol) BSA for 2 h in the dark. The incubation was followed by three washing steps for 5 min with 1× PBS. Nuclear DNA was stained by incubating the cells with Hoechst 33324 for 7 min. Cells were again washed three times for 5 min and then mounted on microscope slides with IS mounting medium (Dianova) and sealed with Roti-Seal (Carl Roth GmbH). The stained proteins were analyzed using a confocal laser scanning microscope (Leica TCS-SP5). Only intact interphase cells were analyzed.
RESULTS
Generation of NF-κB-defective MLE-15 cells.
Murine MLE-15 lung cells (representing the distal bronchiolar and alveolar epithelium) are easy to grow, represent a widely used model system in the study of IAV infection (24, 25), and are also suitable for genomic engineering. In order to reveal the role of NF-κB for IAV propagation and surmounting of species barriers, we eliminated two critical components of the canonical NF-κB activation pathway using CRISPR-Cas9. On the one hand, we targeted the NEMO protein, an essential component of the IKK complex, which is absolutely required for the canonical NF-κB activation pathway (26). As IKKs also display NF-κB-independent functions upon phosphorylation of different additional cytoplasmic and nuclear substrate proteins (27), we also targeted the critical DNA-binding subunit p65. Cell clones were analyzed for expression of NF-κB p65 and NEMO by Western blotting (Fig. 1A and B). Cell clones neither expressing p65 or NEMO nor showing any Cas9 expression were then further characterized by DNA sequencing. The NEMO mutation inserted a frameshift after amino acid 53, thus ensuring that all functionally relevant domains are not expressed (28; data not shown). The p65 mutation destroyed the start codon, leading to the complete absence of the p65 protein (data not shown). It was then important to ensure that IAV-induced NF-κB activation is defective after deletion of p65 or NEMO. Control MLE-15 cells or NEMO-deficient (NEMO−) or p65-deficient (p65−) cells were infected with SC35 or SC35M, followed by analysis of expression of the IL-6 gene, which is a prototypical NF-κB-dependent gene that contains functional p65 binding sites in its promoter (29, 30). The analysis of relative mRNA levels by qPCR showed that deficiency in NEMO or p65 was sufficient to prevent inducible IL-6 expression (Fig. 1C).
FIG 1.

Generation and characterization of NF-κB-defective MLE-15 cells. (A) Cells were transfected with the vector px459 or px459-mp65. Transfected cells were selected by puromycin treatment, and surviving clones were grown to colonies. A fraction of the cells was lysed, and equal amounts of protein were tested by immunoblotting for expression levels of p65, Cas9, and tubulin with specific antibodies. (B) The experiment was done as in panel A, with the difference that cells were transfected with px459-mNEMO and extracts from cell clones were tested for the expression of NEMO. (C) The indicated cells were infected with SC35 or SC35M (MOI of 1), and IL-6 gene expression was quantified by qPCR 24 h p.i. Error bars display standard errors of the means (SEM) derived from two independent experiments performed in triplicate. Student's t test was used for statistical analysis. **, P < 0.01. All other differences had P values of <0.001. ns, not significant.
NF-κB inactivation facilitates propagation of nonadapted SC35 viruses.
MLE-15 control cells and their NF-κB-defective derivatives were infected with SC35M or SC35 viruses. The adapted SC35M viruses grew to high titers irrespective of the functionality of the NF-κB system. In contrast, the poor propagation of SC35 viruses was strongly augmented upon deletion of either NEMO or p65 (Fig. 2A). To investigate whether the contribution of NF-κB to SC35 propagation is influenced by the virus load, control or CRISPR knockout cells were infected at different multiplicities of infection (MOI), and virus titers were determined 24 h postinfection (p.i.). The antiviral effects of NF-κB were most prominent at a low MOI of 0.001, but this effect was significantly reduced at higher virus concentrations, which resulted only in minor differences lacking biological significance (Fig. 2B). This result suggests that the antiviral effect of NF-κB against the nonadapted SC35 leading to reduced viral titers can be partially counteracted by increasing viral replication through higher infection rates.
FIG 2.
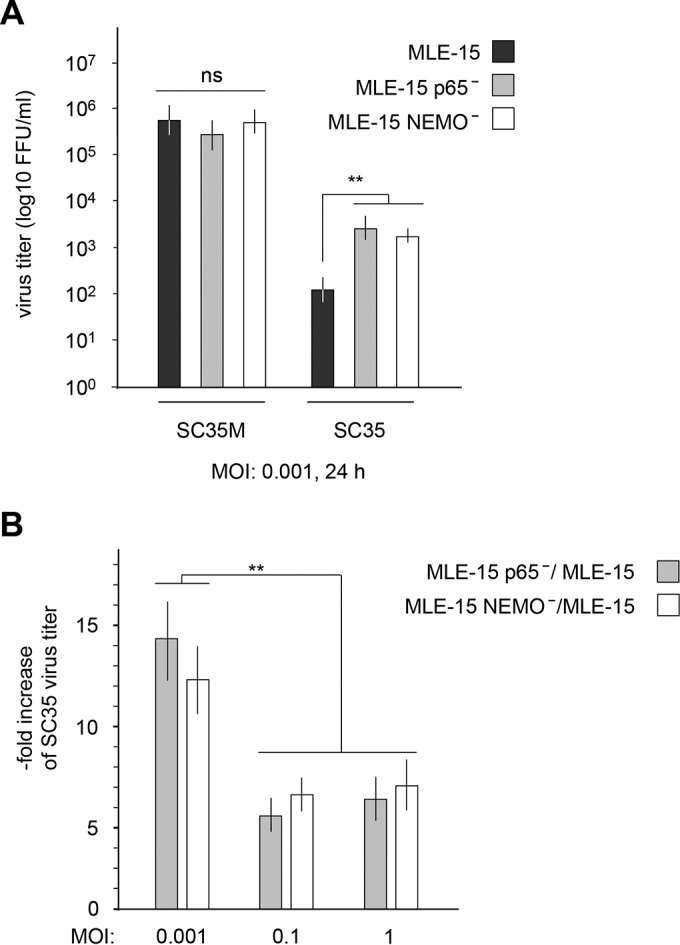
Inhibition of NF-κB allows increased production of SC35. (A) The indicated MLE-15 cells were infected with SC35 or SC35M (MOI of 0.001). The plaque titers were determined 24 h p.i. on MDCK-II cells. The virus titer (focus-forming units [FFU] per milliliter) is indicated; error bars show SEM obtained from three independent experiments performed in triplicate. (B) MLE-15 cells and their NEMO- and p65-deficient derivatives were infected with different titers of SC35. The plaque titers were determined 24 h p.i. on MDCK-II cells, and the ratio between virus numbers in knockout cells and those in control cells is displayed on the y axis. Error bars show SEMs obtained from five independent experiments performed in triplicate. **, P < 0.01. ns, not significant.
NF-κB deficiency increases the expression levels of SC35 NS1 and leads to an earlier nuclear export of SC35 NP.
It was then interesting to investigate whether the increased replication of SC35 viruses in NF-κB-deficient cells is also reflected at the level of virus-encoded NS1 protein. Infection of cells with SC35M allowed NS1 detection 6 h p.i. in cells with and without functional NF-κB (Fig. 3A). Infection of control wild-type cells with SC35 resulted in poor NS1 expression after 6 h p.i., while NEMO− and p65− cells displayed earlier and stronger expression of this viral protein (Fig. 3A). To investigate the impact of NF-κB signaling on IAV-encoded proteins by a complementary experimental approach, we investigated the intracellular localization of the NP protein by indirect immunofluorescence. The intracellular localization of NP is a convenient marker for the stage of IAV infection. In the early stage (0 to 4 h p.i.) of the viral replication cycle, the NP accumulates in the nucleus to support viral genome replication as part of the RNP complex. At later stages (4 to 8 h p.i.), the RNPs (containing the NP) are exported into the cytoplasm to be transported to the cell membrane, where they are packaged into progeny virions budding from the cell surface (31). In order to investigate whether NF-κB activity can influence the intracellular localization of NP, control cells as well as NEMO− and p65− cells were infected with SC35 for 6 h. Indirect immunofluorescence showed that some NP is still found in the nucleus of most SC35-infected control MLE-15 cells, while in cells lacking NEMO or p65, the NP protein predominantly localized in the cytosol (Fig. 3B). Also the amount of immunoreactive NP protein was increased in NEMO- or p65-deficient cells, supporting the notion that infection is already more advanced in NF-κB-deficient cells. No changes in NP localization or abundance were seen at later stages of infection or in SC35M-infected cells (data not shown). Collectively, these data suggest that the effect of NF-κB on IAV propagation is already seen at the earlier stages of virus replication prior to their release from the host cell.
FIG 3.

NF-κB deficiency affects expression and localization of IAV-encoded proteins. (A) The indicated cells were infected with SC35 or SC35M (MOI of 3) for the indicated periods. Cells were harvested, and cell extracts were analyzed by immunoblotting for the occurrence of the viral NS1 protein and the loading control tubulin. (B) MLE-15, MLE-15 NEMO−, and MLE-15 p65− cells were infected with SC35 (MOI of 1). Cells were fixed 6 h p.i., and the amount and localization of NP were determined by indirect immunofluorescence. The left part shows pictures from cells representing typical localization of NP. Nuclear DNA was stained by Hoechst. The scale bar is 10 μm. The right part shows a quantitative analysis of NP localization in 100 cells.
IKKβ inhibition results in increased SC35 replication in MLE-15 cells.
While these data obtained with genetically altered cells show an antiviral function of NF-κB, previous reports using small molecule IKK inhibitors mainly suggested a proviral function of this transcription factor (32–34). It was thus interesting to investigate whether the antiviral function of NF-κB is also seen by an independent experimental approach using an IKK inhibitor with less off-target effects and a higher specificity, such as the second-generation selective IKKβ inhibitor PHA-408 (35). To determine the optimal concentration of PHA-408 required for efficient blockage of NF-κB activation in MLE-15 cells, cells were pretreated with increasing concentrations of this IKK inhibitor and subsequently stimulated with the NF-κB-activating cytokine tumor necrosis factor (TNF). A concentration of 3 μM almost completely inhibited inducible phosphorylation of IκBα (Fig. 4A) and IL-6 expression (Fig. 4B). Inhibition of IKK activity by PHA-408 increased production of SC35, while it only slightly interfered with the propagation of SC35M (Fig. 4C). We also used this inhibitor to test its effects on IAV replication in human A549 lung cells. Pretreatment of A549 cells with PHA-408 resulted in slightly augmented replication of SC35 and SC35M, which were statistically and biologically not significant (Fig. 5). Of note, the genetic knockout or inhibitor-mediated blocking of target proteins will result in different outcomes, due to differences in the kinetics and strength of inhibition. In addition, most commonly used kinase inhibitors affect more than one target (36), thus raising the necessity to reveal the importance of NF-κB in further species via CRISPR-Cas9 perturbations rather than by small molecule inhibitors.
FIG 4.
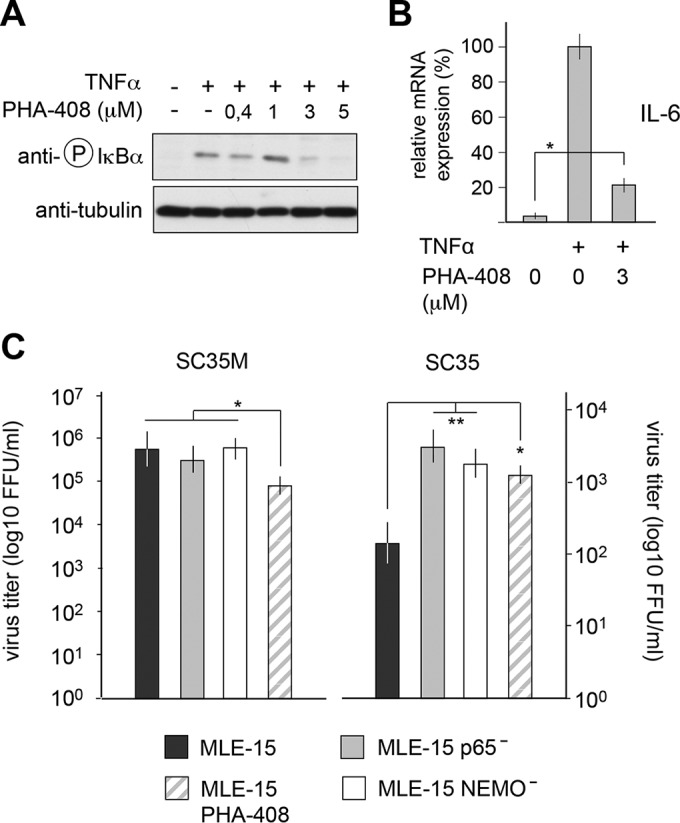
Effect of PHA-408 on replication of SC35 and SC35M viruses. (A) MLE-15 cells were preincubated for 1 h with the indicated concentrations of PHA-408 or vehicle. Cells were then stimulated for 20 min with TNF-α and analyzed by immunoblotting for IκBα phosphorylation. (B) MLE-15 cells were preincubated for 1 h with PHA-408 and stimulated for 6 h with TNF. The expression of IL-6 mRNA was quantified by qPCR. Error bars show SEM obtained from two independent experiments performed in triplicate. *, P < 0.05. All other differences had P values of <0.01. (C) MLE-15 cells were treated with 3 μM PHA-408 or vehicle. These cells and MLE-15 NEMO− and MLE-15 p65− cells were infected with SC35 and SC35M (MOI of 0.001). Virus titers were determined 24 h p.i. Error bars show SEM from two independent experiments performed in triplicate. The P values are indicated by asterisks: *, P < 0.05; **, P < 0.01.
FIG 5.
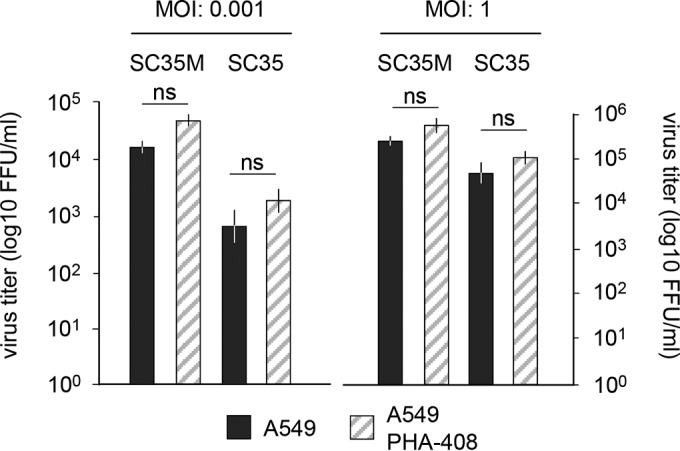
Effect of PHA-408 on A549 cells. A549 cells were treated with 3 mM PHA-408 or vehicle. These cells were infected with SC35 and SC35M (MOI of 0.001 or 1). Virus titers were determined after 24 h. Error bars show SEM from three independent experiments performed in triplicate. ns, not significant.
NF-κB-dependent IRF phosphorylation and IFN-β expression contribute to its antiviral function.
The antiviral function of NF-κB might rely on its ability to control intracellular events, such as the differential regulation of caspase 3 activation or vRNA synthesis (37, 38). Alternatively, NF-κB may contribute to the synthesis of virus-regulating cytokines such as IFN-β. To test the impact of NF-κB on IFN-β gene expression, control cells and NF-κB-defective cells were infected with SC35 or SC35M, and mRNA levels were determined by qPCR. The SC35 infection led to a stronger IFN-β expression compared to SC35M infection (Fig. 6A), which is consistent with the previous notion of a blunted cytokine response in SC35M cells (9). IFN-β expression in SC35M cells was only partially impaired in the absence of NEMO or p65, suggesting the existence of compensatory mechanisms. In contrast, SC35-induced IFN-β expression was fully dependent on functional NF-κB signaling. As NF-κB and IRF pathways show mutual cross-regulation (39, 40), it was interesting to investigate whether the defect of NF-κB function might affect phosphorylation and thus activation of IRF3. MLE-15 control cells and their NEMO- or p65-deficient derivatives were infected with SC35 viruses, and IRF3 phosphorylation was monitored at various time points p.i. with phospho-specific antibodies. These experiments revealed that IRF3 phosphorylation is partially diminished in NEMO− cells and strongly impaired in the absence of p65 (Fig. 6B). The derogated IRF3 phosphorylation is consistent with reduced IFN-β gene expression in the NF-κB-defective cells and might be explained by lack of NF-κB-derived signals required for full activation of the IRF3 kinases IKKε and TBK1. To test whether increased virus titers from infected NF-κB-defective cells can be attributed to reduced expression of IFN-β, we preincubated control or NEMO− and p65− cells with recombinant IFN and measured the impact on virus replication. Preincubation with IFN-β only allowed us to measure SC35 virus replication in control cells at MOI of >0.001 (data not shown) and diminished the difference in virus replication between NF-κB-defective cells and the MLE-15 controls in a dose-dependent manner (Fig. 6C). These data indicate that a substantial part of the antiviral effect of NF-κB is due to its contribution to IFN expression.
FIG 6.

The antiviral effect of NF-κB partially depends on its ability to trigger IFN-β expression. (A) The indicated MLE-15 cell lines were infected with SC35 or SC35M (MOI of 0.001) for 8 h. Cells were harvested, and expression of the IFN-β gene was quantified by qPCR. Error bars show SEM from three independent experiments performed in triplicate. (B) The indicated cells were infected with SC35 or SC35M (MOI of 3) for the indicated periods. Cells were harvested, and cell extracts were analyzed by immunoblotting for the occurrence and phosphorylation of IRF3 with specific antibodies. Error bars show SEM (n = 3). (C) Control cells or p65− and NEMO− cells were preincubated for 14 h with recombinant IFN-β at the indicated concentrations. Subsequently cells were infected with SC35 (MOI of 0.05), and virus titers were determined after 24 h p.i. The ratio between virus numbers in knockout cells and those in control cells is displayed on the y axis. Error bars show SEM (n = 3). The P values are indicated by asterisks: *, P < 0.05; **, P < 0.01. ns, not significant.
The IAV genotype is decisive for the antiviral function of NF-κB.
The SC35 and SC35M viruses differ in nine amino acid exchanges, with three changes occurring in PB2, two changes in PB1, and one change in PA, NP, HA, and neuraminidase (NA), respectively (10). In order to identify the virus-encoded proteins responsible for NF-κB sensitivity, distinct segments of SC35M were used to replace the respective segments of SC35 using reverse genetics (41). Various plasmid combinations were transfected into 293T and MDCK cocultured cells, and the resulting reassortant viruses were used to infect control cells or their NEMO- and p65-deficient derivatives. As expected, a recombinant SC35 virus expressing all six genes from SC35M lost its sensitivity to antiviral NF-κB activity (Fig. 7). An SC35 virus containing the NP segment from SC35M (designated SC35-NPSC35M) was still sensitive to NF-κB inhibition (Fig. 7). Similarly, also the individual exchange of PA, PB1, PB2, and HA did not diminish the sensitivity to NF-κB function. Intriguingly, the expression of NA from SC35M was fully sufficient to render the resulting SC35-NASC35M virus completely inert to any NF-κB effect. An SC35 virus expressing the HA segment from SC35M or the combined exchange of several segments resulted in viruses showing a strongly increased sensitivity to the antiviral activity of NF-κB (Fig. 7). These data are also displayed as the NF-κB-dependent fold increase of viral titers in Fig. 8 and show that various combinations of viral proteins differentially affect the sensitivity to the antiviral function of NF-κB.
FIG 7.
Impact of viral proteins on the antiviral function of NF-κB. Plasmids encoding the indicated SC35M segments were combined with plasmids encoding the other SC35 segments to produce reassortant viruses. These chimeric viruses and the SC35 and SC35M controls were then used to infect control MLE-15 cells, MLE-15 NEMO− cells, or MLE-15 p65− cells (MOI of 0.001), and virus titers were determined 24 h p.i. Mean values from three independent experiments are shown. Error bars indicate SEM. The P values are indicated by asterisks: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
FIG 8.
Impact of viral proteins on NF-κB sensitivity. Plasmids encoding the indicated SC35M segments were combined with plasmids encoding the other SC35 segments to produce reassortant viruses. These chimeric viruses and the SC35 and SC35M controls were then used to infect control MLE-15 cells, MLE-15 NEMO− cells, or MLE-15 p65− cells at an MOI of 0.001, and virus titers were determined after 1 day. The y axis shows the ratio between the indicated NF-κB-deficient cells and MLE-15 controls. Mean values from several independent experiments are shown; error bars indicate SEM. **, P < 0.01. All other differences had P values of >0.05.
DISCUSSION
Here we show that genetic inhibition of NF-κB activation by two independent approaches renders mouse cells more susceptible to IAV infection by a nonadapted SC35 virus. In contrast, the mouse-adapted SC35M virus was not affected by NF-κB status. Recombinant SC35 viruses expressing the NA protein from SC35M were not affected by the antiviral function of NF-κB, but the molecular mechanisms underlying this effect need to be studied in the future. On the other hand, the replication of a reassortant SC35 expressing the HA segment from SC35M was enhanced >10,000-fold upon deletion of the p65 protein, emphasizing that the role of this transcription factor in IAV spread depends on the viral genotype. This may also explain in part the conflicting results assigning either pro- or antiviral functions for NF-κB, as discussed in detail in a recent review (42). It will thus be very relevant in future studies to carefully consider the contribution of virus genetics to the specific effect of the NF-κB system on IAV replication in a given host (cells). This study employed widely used murine MLE-15 lung cells to reveal the antiviral function of NF-κB. It will be important in the future to reveal the impact of NF-κB perturbation systematically in human cells and also in species important for the transmission of IAVs. We also found that the role of NF-κB was most pronounced after infection with low virus titers, as they typically occur during the onset of the disease. We suggest that NF-κB is antiviral during this initial phase because it activates antiviral IFNs and further chemokines serving to attract neutrophils and monocytes (43, 44). This in turn allows uninfected cells to detect IAV infection with higher sensitivity and to produce even larger amounts of antiviral IFNs. We found an important contribution NF-κB p65 to the IAV-triggered phosphorylation of IRF3, thus identifying a novel facet of cross-regulation between NF-κB and IRF pathways (39, 40). The impaired IRF3 phosphorylation might be attributable to defective expression or activation of the IRF kinase IKKε, which is induced in response to inflammatory insults (45). While one part of the antiviral NF-κB response is mediated by IFNs, further antiviral functions of this transcription factor are already seen at the earlier stages of virus replication, as revealed by effects on the synthesis rate of NS1 and the intracellular localization of NP. To counteract these antiviral functions, IAVs employ several mechanisms to dampen NF-κB activity. The NS1 protein antagonizes IAV-induced activation of the canonical NF-κB activation pathway by constitutive association with IKKα and IKKβ and inhibition of their catalytic activities, and it also inhibits the noncanonical NF-κB activation pathway (46, 47). Conceivable, therefore, evolution of IAVs not only will be directed at inhibiting the signaling steps leading to IFN production but will also adapt the virus to avoid excessive NF-κB activation early on. Therefore, not only is SC35M replication unaffected by NF-κB, but the virus may also have evolved to restrict NF-κB activity to reduce expression of proinflammatory cytokines such as IL-6 (Fig. 1C).
At later stages of an established infection, this situation changes as the massively released virus progeny will on balance cause an exaggerated NF-κB activation that further supports IAV replication by ill-defined mechanisms (19, 48). However, this overshooting NF-κB response underlies the excessive proinflammatory response during influenza virus pneumonia (49–51). IAVs cause pneumonia in humans, with progression to lung failure and fatal outcome. Dysregulated release of cytokines leads to autocrine and NF-κB-dependent induction of the proapoptotic factor TNF-related apoptosis-inducing ligand (TRAIL), which in turn triggers cell death of alveolar epithelial cells (52). IAV-infected patients suffering from acute respiratory distress syndrome (ARDS) are characterized by local and systemic increases in cytokines (IL-6, IL-10, IL-15, and TNF) and reactive oxygen intermediates (49). The causative effects of elevated cytokines on lung injury suggest that interference with exaggerated innate immunity responses will be of therapeutic use (53). It is clear that interference with the exaggerated production of inflammatory cytokines will protect from tissue damage and is an important goal to control mortality by IAV infection. On the other hand, it should be considered that early inflammation during the infection phase has antiviral and beneficial effects. Thus, we are in need of drugs that selectively interfere with signaling pathways contributing to exacerbated production of inflammatory mediators causing lung injury, while maintaining the ability of host cells to mount an antiviral response.
The NF-κB pathway will be compromised not only in patients taking anti-inflammatory drugs such as steroids (54) but also in individuals where mutations or epigenetic events affect key components of the core NF-κB module. These germ line or somatic mutations include point mutations and deletions (NEMO, c-REL, NFKB2, IKBA, and CYLD), chromosomal translocations (p65 and BCL-3), and also gene amplifications (e.g., c-REL) (55–58). NF-κB mutations typically result in the early onset of cancer and inflammatory diseases but may also increase the susceptibility to IAV infection. One naturally occurring mutation in the NEMO gene leads to an in-frame splicing event and the exclusive production of a shortened protein. A patient carrying this mutation shows a strongly diminished RNA-induced induction of the IFN-β response and an increased susceptibility to viral infections (59). Another report describes the synonymous mutation in the gene encoding NEMO that enhances alternative splicing and leads to the production of a shorter protein (Δ-NEMO). Cultivated cells from these individuals showed an impaired association of Δ-NEMO with TANK-binding kinase 1 (TBK1) and an incomplete RNA-induced nuclear translocation of NF-κB. Concomitantly cells expressing Δ-NEMO displayed impaired RNA-induced IFN-β production and permitted increased virus propagation (60). These observations are in line with the results reported here using CRISPR-Cas9-mediated gene engineering of NEMO and p65. Systematic application of this approach will now allow determination of the individual contributions of host cell factors from the NF-κB system to IAV infection in unprecedented detail in future studies.
ACKNOWLEDGMENTS
We thank H.-D. Klenk (Marburg) and G. Gabriel (Hamburg) for the SC35/SC35M virus reverse genetics system and Christin Müller for help with confocal microscopy. We are also grateful to S. Ludwig (Münster) for the gift of anti-NP and anti-NS1 antibodies and Feng Zhang (Boston) for the plasmid px459 (Addgene 62988).
Funding Statement
The work of M.L.S. is supported by grants from the Deutsche Krebshilfe (111447) and Deutsche Forschungsgemeinschaft (SCHM 1417/8-3 and SCHM 1417/9-1). The work of M.K. is supported by the Deutsche Forschungsgemeinschaft (Kr1143/5-3 and Kr1143/7-3). The work of M.K. and M.L.S. is further supported by the Excellence Cluster Cardio-Pulmonary System (ECCPS) (SFB/TRR81 and SFB1021). The work of S.P. is supported by DFG-funded grants SFB1021 and SFB/TR84 and by the BMBF-funded DZIF, partner site Giessen, Germany (TTU Emerging Infections). Sharmistha Dam received funding from the Deutsche Forschungsgemeinschaft (DFG) (SFB1021).
REFERENCES
- 1.Olsen B, Munster VJ, Wallensten A, Waldenstrom J, Osterhaus AD, Fouchier RA. 2006. Global patterns of influenza A virus in wild birds. Science 312:384–388. doi: 10.1126/science.1122438. [DOI] [PubMed] [Google Scholar]
- 2.Mänz B, Schwemmle M, Brunotte L. 2013. Adaptation of avian influenza A virus polymerase in mammals to overcome the host species barrier. J Virol 87:7200–7209. doi: 10.1128/JVI.00980-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tscherne DM, Garcia-Sastre A. 2011. Virulence determinants of pandemic influenza viruses. J Clin Invest 121:6–13. doi: 10.1172/JCI44947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ludwig S, Zell R, Schwemmle M, Herold S. 2014. Influenza, a One Health paradigm—novel therapeutic strategies to fight a zoonotic pathogen with pandemic potential. Int J Med Microbiol 304:894–901. doi: 10.1016/j.ijmm.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 5.Garcia-Sastre A, Biron CA. 2006. Type 1 interferons and the virus-host relationship: a lesson in detente. Science 312:879–882. doi: 10.1126/science.1125676. [DOI] [PubMed] [Google Scholar]
- 6.Haller O, Kochs G, Weber F. 2006. The interferon response circuit: induction and suppression by pathogenic viruses. Virology 344:119–130. doi: 10.1016/j.virol.2005.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McGeoch D, Fellner P, Newton C. 1976. Influenza virus genome consists of eight distinct RNA species. Proc Natl Acad Sci U S A 73:3045–3049. doi: 10.1073/pnas.73.9.3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rossman JS, Lamb RA. 2011. Influenza virus assembly and budding. Virology 411:229–236. doi: 10.1016/j.virol.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gabriel G, Klingel K, Planz O, Bier K, Herwig A, Sauter M, Klenk HD. 2009. Spread of infection and lymphocyte depletion in mice depends on polymerase of influenza virus. Am J Pathol 175:1178–1186. doi: 10.2353/ajpath.2009.090339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gabriel G, Dauber B, Wolff T, Planz O, Klenk HD, Stech J. 2005. The viral polymerase mediates adaptation of an avian influenza virus to a mammalian host. Proc Natl Acad Sci U S A 102:18590–18595. doi: 10.1073/pnas.0507415102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stetson DB, Medzhitov R. 2006. Type I interferons in host defense. Immunity 25:373–381. doi: 10.1016/j.immuni.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 12.Panne D, Maniatis T, Harrison SC. 2007. An atomic model of the interferon-beta enhanceosome. Cell 129:1111–1123. doi: 10.1016/j.cell.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peisley A, Wu B, Yao H, Walz T, Hur S. 2013. RIG-I forms signaling-competent filaments in an ATP-dependent, ubiquitin-independent manner. Mol Cell 51:573–583. doi: 10.1016/j.molcel.2013.07.024. [DOI] [PubMed] [Google Scholar]
- 14.Peisley A, Wu B, Xu H, Chen ZJ, Hur S. 2014. Structural basis for ubiquitin-mediated antiviral signal activation by RIG-I. Nature 509:110–114. doi: 10.1038/nature13140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hinz M, Scheidereit C. 2014. The IkappaB kinase complex in NF-kappaB regulation and beyond. EMBO Rep 15:46–61. doi: 10.1002/embr.201337983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayden MS, Ghosh S. 2012. NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev 26:203–234. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oeckinghaus A, Hayden MS, Ghosh S. 2011. Crosstalk in NF-kappaB signaling pathways. Nat Immunol 12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 18.Nimmerjahn F, Dudziak D, Dirmeier U, Hobom G, Riedel A, Schlee M, Staudt LM, Rosenwald A, Behrends U, Bornkamm GW, Mautner J. 2004. Active NF-kappaB signalling is a prerequisite for influenza virus infection. J Gen Virol 85:2347–2356. doi: 10.1099/vir.0.79958-0. [DOI] [PubMed] [Google Scholar]
- 19.Wurzer WJ, Ehrhardt C, Pleschka S, Berberich-Siebelt F, Wolff T, Walczak H, Planz O, Ludwig S. 2004. NF-kappaB-dependent induction of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and Fas/FasL is crucial for efficient influenza virus propagation. J Biol Chem 279:30931–30937. doi: 10.1074/jbc.M403258200. [DOI] [PubMed] [Google Scholar]
- 20.Maelfait J, Roose K, Bogaert P, Sze M, Saelens X, Pasparakis M, Carpentier I, van Loo G, Beyaert R. 2012. A20 (Tnfaip3) deficiency in myeloid cells protects against influenza A virus infection. PLoS Pathog 8:e1002570. doi: 10.1371/journal.ppat.1002570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goulet ML, Olagnier D, Xu Z, Paz S, Belgnaoui SM, Lafferty EI, Janelle V, Arguello M, Paquet M, Ghneim K, Richards S, Smith A, Wilkinson P, Cameron M, Kalinke U, Qureshi S, Lamarre A, Haddad EK, Sekaly RP, Peri S, Balachandran S, Lin R, Hiscott J. 2013. Systems analysis of a RIG-I agonist inducing broad spectrum inhibition of virus infectivity. PLoS Pathog 9:e1003298. doi: 10.1371/journal.ppat.1003298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li SQ, Orlich M, Rott R. 1990. Generation of seal influenza virus variants pathogenic for chickens, because of hemagglutinin cleavage site changes. J Virol 64:3297–3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moreno R, Sobotzik JM, Schultz C, Schmitz ML. 2010. Specification of the NF-kappaB transcriptional response by p65 phosphorylation and TNF-induced nuclear translocation of IKK epsilon. Nucleic Acids Res 38:6029–6044. doi: 10.1093/nar/gkq439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Le Goffic R, Arshad MI, Rauch M, L‘Helgoualc'h A, Delmas B, Piquet-Pellorce C, Samson M. 2011. Infection with influenza virus induces IL-33 in murine lungs. Am J Respir Cell Mol Biol 45:1125–1132. doi: 10.1165/rcmb.2010-0516OC. [DOI] [PubMed] [Google Scholar]
- 25.Wikenheiser KA, Vorbroker DK, Rice WR, Clark JC, Bachurski CJ, Oie HK, Whitsett JA. 1993. Production of immortalized distal respiratory epithelial cell lines from surfactant protein C/simian virus 40 large tumor antigen transgenic mice. Proc Natl Acad Sci U S A 90:11029–11033. doi: 10.1073/pnas.90.23.11029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamaoka S, Courtois G, Bessia C, Whiteside ST, Weil R, Agou F, Kirk HE, Kay RJ, Israel A. 1998. Complementation cloning of NEMO, a component of the IkappaB kinase complex essential for NF-kappaB activation. Cell 93:1231–1240. doi: 10.1016/S0092-8674(00)81466-X. [DOI] [PubMed] [Google Scholar]
- 27.Chariot A. 2009. The NF-kappaB-independent functions of IKK subunits in immunity and cancer. Trends Cell Biol 19:404–413. doi: 10.1016/j.tcb.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 28.Clark K, Nanda S, Cohen P. 2013. Molecular control of the NEMO family of ubiquitin-binding proteins. Nat Rev Mol Cell Biol 14:673–685. doi: 10.1038/nrm3644. [DOI] [PubMed] [Google Scholar]
- 29.Libermann TA, Baltimore D. 1990. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol Cell Biol 10:2327–2334. doi: 10.1128/MCB.10.5.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Handschick K, Beuerlein K, Jurida L, Bartkuhn M, Muller H, Soelch J, Weber A, Dittrich-Breiholz O, Schneider H, Scharfe M, Jarek M, Stellzig J, Schmitz ML, Kracht M. 2014. Cyclin-dependent kinase 6 is a chromatin-bound cofactor for NF-kappaB-dependent gene expression. Mol Cell 53:193–208. doi: 10.1016/j.molcel.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 31.Whittaker G, Bui M, Helenius A. 1996. Nuclear trafficking of influenza virus ribonucleoproteins in heterokaryons. J Virol 70:2743–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ehrhardt C, Ruckle A, Hrincius ER, Haasbach E, Anhlan D, Ahmann K, Banning C, Reiling SJ, Kuhn J, Strobl S, Vitt D, Leban J, Planz O, Ludwig S. 2013. The NF-kappaB inhibitor SC75741 efficiently blocks influenza virus propagation and confers a high barrier for development of viral resistance. Cell Microbiol 15:1198–1211. doi: 10.1111/cmi.12108. [DOI] [PubMed] [Google Scholar]
- 33.Haasbach E, Reiling SJ, Ehrhardt C, Droebner K, Ruckle A, Hrincius ER, Leban J, Strobl S, Vitt D, Ludwig S, Planz O. 2013. The NF-kappaB inhibitor SC75741 protects mice against highly pathogenic avian influenza A virus. Antiviral Res 99:336–344. doi: 10.1016/j.antiviral.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 34.Mazur I, Wurzer WJ, Ehrhardt C, Pleschka S, Puthavathana P, Silberzahn T, Wolff T, Planz O, Ludwig S. 2007. Acetylsalicylic acid (ASA) blocks influenza virus propagation via its NF-kappaB-inhibiting activity. Cell Microbiol 9:1683–1694. doi: 10.1111/j.1462-5822.2007.00902.x. [DOI] [PubMed] [Google Scholar]
- 35.Mbalaviele G, Sommers CD, Bonar SL, Mathialagan S, Schindler JF, Guzova JA, Shaffer AF, Melton MA, Christine LJ, Tripp CS, Chiang PC, Thompson DC, Hu Y, Kishore N. 2009. A novel, highly selective, tight binding IkappaB kinase-2 (IKK-2) inhibitor: a tool to correlate IKK-2 activity to the fate and functions of the components of the nuclear factor-kappaB pathway in arthritis-relevant cells and animal models. J Pharmacol Exp Ther 329:14–25. doi: 10.1124/jpet.108.143800. [DOI] [PubMed] [Google Scholar]
- 36.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. 2007. The selectivity of protein kinase inhibitors: a further update. Biochem J 408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumar N, Xin ZT, Liang Y, Ly H, Liang Y. 2008. NF-kappaB signaling differentially regulates influenza virus RNA synthesis. J Virol 82:9880–9889. doi: 10.1128/JVI.00909-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wurzer WJ, Planz O, Ehrhardt C, Giner M, Silberzahn T, Pleschka S, Ludwig S. 2003. Caspase 3 activation is essential for efficient influenza virus propagation. EMBO J 22:2717–2728. doi: 10.1093/emboj/cdg279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grumont RJ, Gerondakis S. 2000. Rel induces interferon regulatory factor 4 (IRF-4) expression in lymphocytes: modulation of interferon-regulated gene expression by rel/nuclear factor kappaB. J Exp Med 191:1281–1292. doi: 10.1084/jem.191.8.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iwanaszko M, Kimmel M. 2015. NF-kappaB and IRF pathways: cross-regulation on target genes promoter level. BMC Genomics 16:307. doi: 10.1186/s12864-015-1511-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoffmann E, Neumann G, Kawaoka Y, Hobom G, Webster RG. 2000. A DNA transfection system for generation of influenza A virus from eight plasmids. Proc Natl Acad Sci U S A 97: 6108–6113. doi: 10.1073/pnas.100133697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmitz ML, Kracht M, Saul VV. 2014. The intricate interplay between RNA viruses and NF-kappaB. Biochim Biophys Acta 1843:2754–2764. doi: 10.1016/j.bbamcr.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muramoto Y, Shoemaker JE, Le MQ, Itoh Y, Tamura D, Sakai-Tagawa Y, Imai H, Uraki R, Takano R, Kawakami E, Ito M, Okamoto K, Ishigaki H, Mimuro H, Sasakawa C, Matsuoka Y, Noda T, Fukuyama S, Ogasawara K, Kitano H, Kawaoka Y. 2014. Disease severity is associated with differential gene expression at the early and late phases of infection in nonhuman primates infected with different H5N1 highly pathogenic avian influenza viruses. J Virol 88:8981–8997. doi: 10.1128/JVI.00907-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xiao H, Killip MJ, Staeheli P, Randall RE, Jackson D. 2013. The human interferon-induced MxA protein inhibits early stages of influenza A virus infection by retaining the incoming viral genome in the cytoplasm. J Virol 87:13053–13058. doi: 10.1128/JVI.02220-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bulek K, Liu C, Swaidani S, Wang L, Page RC, Gulen MF, Herjan T, Abbadi A, Qian W, Sun D, Lauer M, Hascall V, Misra S, Chance MR, Aronica M, Hamilton T, Li X. 2011. The inducible kinase IKKi is required for IL-17-dependent signaling associated with neutrophilia and pulmonary inflammation. Nat Immunol 12:844–852. doi: 10.1038/ni.2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gao S, Song L, Li J, Zhang Z, Peng H, Jiang W, Wang Q, Kang T, Chen S, Huang W. 2012. Influenza A virus-encoded NS1 virulence factor protein inhibits innate immune response by targeting IKK. Cell Microbiol 14:1849–1866. doi: 10.1111/cmi.12005. [DOI] [PubMed] [Google Scholar]
- 47.Ayllon J, Garcia-Sastre A. 2015. The NS1 protein: a multitasking virulence factor. Curr Top Microbiol Immunol 386:73–107. doi: 10.1007/82_2014_400. [DOI] [PubMed] [Google Scholar]
- 48.Mühlbauer D, Dzieciolowski J, Hardt M, Hocke A, Schierhorn KL, Mostafa A, Muller C, Wisskirchen C, Herold S, Wolff T, Ziebuhr J, Pleschka S. 2015. Influenza virus-induced caspase-dependent enlargement of nuclear pores promotes nuclear export of viral ribonucleoprotein complexes. J Virol 89:6009–6021. doi: 10.1128/JVI.03531-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bian JR, Nie W, Zang YS, Fang Z, Xiu QY, Xu XX. 2014. Clinical aspects and cytokine response in adults with seasonal influenza infection. Int J Clin Exp Med 7:5593–5602. [PMC free article] [PubMed] [Google Scholar]
- 50.Hagau N, Slavcovici A, Gonganau DN, Oltean S, Dirzu DS, Brezoszki ES, Maxim M, Ciuce C, Mlesnite M, Gavrus RL, Laslo C, Hagau R, Petrescu M, Studnicska DM. 2010. Clinical aspects and cytokine response in severe H1N1 influenza A virus infection. Crit Care 14:R203. doi: 10.1186/cc9324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Julkunen I, Melen K, Nyqvist M, Pirhonen J, Sareneva T, Matikainen S. 2000. Inflammatory responses in influenza A virus infection. Vaccine 19(Suppl 1):S32–S37. doi: 10.1016/S0264-410X(00)00275-9. [DOI] [PubMed] [Google Scholar]
- 52.Högner K, Wolff T, Pleschka S, Plog S, Gruber AD, Kalinke U, Walmrath HD, Bodner J, Gattenlohner S, Lewe-Schlosser P, Matrosovich M, Seeger W, Lohmeyer J, Herold S. 2013. Macrophage-expressed IFN-beta contributes to apoptotic alveolar epithelial cell injury in severe influenza virus pneumonia. PLoS Pathog 9:e1003188. doi: 10.1371/journal.ppat.1003188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ramos I, Fernandez-Sesma A. 2015. Modulating the innate immune response to influenza A virus: potential therapeutic use of anti-inflammatory drugs. Front Immunol 6:361. doi: 10.3389/fimmu.2015.00361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dumont A, Hehner SP, Schmitz ML, Gustafsson JA, Liden J, Okret S, van der Saag PT, Wissink S, van der Burg B, Herrlich P, Haegeman G, De Bosscher K, Fiers W. 1998. Cross-talk between steroids and NF-kappa B: what language? Trends Biochem Sci 23:233–235. doi: 10.1016/S0968-0004(98)01212-2. [DOI] [PubMed] [Google Scholar]
- 55.Courtois G, Gilmore TD. 2006. Mutations in the NF-kappaB signaling pathway: implications for human disease. Oncogene 25:6831–6843. doi: 10.1038/sj.onc.1209939. [DOI] [PubMed] [Google Scholar]
- 56.Parker M, Mohankumar KM, Punchihewa C, Weinlich R, Dalton JD, Li Y, Lee R, Tatevossian RG, Phoenix TN, Thiruvenkatam R, White E, Tang B, Orisme W, Gupta K, Rusch M, Chen X, Li Y, Nagahawhatte P, Hedlund E, Finkelstein D, Wu G, Shurtleff S, Easton J, Boggs K, Yergeau D, Vadodaria B, Mulder HL, Becksfort J, Gupta P, Huether R, Ma J, Song G, Gajjar A, Merchant T, Boop F, Smith AA, Ding L, Lu C, Ochoa K, Zhao D, Fulton RS, Fulton LL, Mardis ER, Wilson RK, Downing JR, Green DR, Zhang J, Ellison DW, Gilbertson RJ. 2014. C11orf95-RELA fusions drive oncogenic NF-kappaB signalling in ependymoma. Nature 506:451–455. doi: 10.1038/nature13109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smahi A, Courtois G, Vabres P, Yamaoka S, Heuertz S, Munnich A, Israel A, Heiss NS, Klauck SM, Kioschis P, Wiemann S, Poustka A, Esposito T, Bardaro T, Gianfrancesco F, Ciccodicola A, D'Urso M, Woffendin H, Jakins T, Donnai D, Stewart H, Kenwrick SJ, Aradhya S, Yamagata T, Levy M, Lewis RA, Nelson DL. 2000. Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. The International Incontinentia Pigmenti (IP) Consortium. Nature 405:466–472. [DOI] [PubMed] [Google Scholar]
- 58.Döffinger R, Smahi A, Bessia C, Geissmann F, Feinberg J, Durandy A, Bodemer C, Kenwrick S, Dupuis-Girod S, Blanche S, Wood P, Rabia SH, Headon DJ, Overbeek PA, Le Deist F, Holland SM, Belani K, Kumararatne DS, Fischer A, Shapiro R, Conley ME, Reimund E, Kalhoff H, Abinun M, Munnich A, Israel A, Courtois G, Casanova JL. 2001. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat Genet 27:277–285. doi: 10.1038/85837. [DOI] [PubMed] [Google Scholar]
- 59.Wessel A, Hsu A, Zilberman-Rudenko J, Goldbach-Mansky R, Siegel RM, Hanson EP. 2013. Inflammatory disease due to dysregulated nuclear factor-kappa B activation and impaired type I interferon response resulting from a de novo human NEMO hypomorphic mutation. Arthritis Rheum 65:S323. [Google Scholar]
- 60.Wessel A, Hsu AZ-R, Goldbach-Mansky R, Siegel RM, Hanson EP. 2013. Inflammatory disease and impaired antiviral immunity due to unbalanced NF-kB and IRF3 activation result from a de novo human NEMO mutation. J Immunol 190(Suppl 1):57.19. [Google Scholar]
- 61.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. 2013. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]