

ABSTRACT
The K1 gene product of Kaposi's sarcoma-associated herpesvirus (KSHV) is encoded by the first open reading frame (ORF) of the viral genome. To investigate the role of the K1 gene during the KSHV life cycle, we constructed a set of recombinant viruses that contained either wild-type (WT) K1, a deleted K1 ORF (KSHVΔK1), stop codons within the K1 ORF (KSHV-K15×STOP), or a revertant K1 virus (KSHV-K1REV). We report that the recombinant viruses KSHVΔK1 and KSHV-K15×STOP displayed significantly reduced lytic replication compared to WT KSHV and KSHV-K1REV upon reactivation from latency. Additionally, cells infected with the recombinant viruses KSHVΔK1 and KSHV-K15×STOP also yielded smaller amounts of infectious progeny upon reactivation than did WT KSHV- and KSHV-K1REV-infected cells. Upon reactivation from latency, WT KSHV- and KSHV-K1REV-infected cells displayed activated Akt kinase, as evidenced by its phosphorylation, while cells infected with viruses deleted for K1 showed reduced phosphorylation and activation of Akt kinase. Overall, our results suggest that K1 plays an important role during the KSHV life cycle.
IMPORTANCE Kaposi's sarcoma-associated herpesvirus (KSHV) is the etiological agent of three human malignancies, and KSHV K1 is a signaling protein that has been shown to be involved in cellular transformation and to activate the phosphatidylinositol 3-kinase (PI3K)/Akt/mTOR pathway. In order to investigate the role of the K1 protein in the life cycle of KSHV, we constructed recombinant viruses that were deficient for K1. We found that K1 deletion viruses displayed reduced lytic replication compared to the WT virus and also yielded smaller numbers of infectious progeny. We report that K1 plays an important role in the life cycle of KSHV.
INTRODUCTION
Kaposi's sarcoma (KS)-associated herpesvirus (KSHV), also known as human herpesvirus 8, is the causative agent of KS, a vascular neoplasm of endothelial cell origin (1). KSHV infection is linked to two B cell lymphoproliferative disorders: primary effusion lymphoma (PEL) and the plasmablastic variant of multicentric Castleman's disease (MCD) (2–4). KSHV predominantly displays a latent state in infected cells and in KSHV-associated tumors, but a small percentage of KSHV-infected cells undergo reactivation, which is thought to be important for KS tumorigenesis (5). KSHV reactivation can occur through multiple events (6, 7), and the KSHV replication and transcription activator (RTA) protein is the only viral protein that is both necessary and sufficient to reactivate KSHV. The expression of RTA leads to the activation of downstream lytic genes and ultimately the production of progeny virions (8–12).
K1 is a transmembrane glycoprotein encoded by the first open reading frame (ORF) in the KSHV genome (13). Although K1 is highly upregulated during the lytic cycle, it has also been shown to be expressed at lower levels during latency (14). K1 is expressed in KS lesions and primary effusion lymphoma cell lines (13–17). The K1 protein contains an immunoreceptor tyrosine-based activation motif (ITAM) in its cytoplasmic tail, which is involved in activating signal transduction pathways (4, 17–20). K1 transforms mouse fibroblasts (21) and immortalizes primary human endothelial cells (22). The transforming activity of K1 is thought to be conferred through phosphatidylinositol 3-kinase (PI3K)/Akt/mTOR signaling. The PI3K/Akt/mTOR pathway is activated by K1 in B and endothelial cells (18, 22), and this provides a survival advantage to K1-expressing cells (4, 18, 22, 23). K1-expressing cells also secrete increased amounts of vascular endothelial growth factor (VEGF), which is an angiogenic factor that promotes vascularization (24). The K1 ITAM is required for signal transduction in B and endothelial cells (18, 21, 22, 25, 26).
The role of K1-mediated activation of signaling pathways during KSHV reactivation from latency has been studied in the context of exogenous expression of K1 (26, 27). However, thus far, a K1 mutant virus has not been made and tested for its ability to affect KSHV lytic replication in the context of the whole virus.
In this study, we employed bacterial artificial chromosome (BAC) technology to construct a set of recombinant KSHVs: wild-type (WT) K1 (WT KSHV K1), wild-type FLAG-tagged K1 (KSHV-K1FLAG), a K1 deletion virus removing the entire K1 ORF (KSHVΔK1), and a K1 mutant virus (KSHV-K15×STOP) in which stop codons were inserted into the K1 ORF to prevent the expression of the protein. A K1 revertant virus (KSHV-K1REV), in which a FLAG-tagged K1 coding sequence was restored, was also constructed. These different viruses were used to assess the effect of K1 on the KSHV life cycle. We found that deletion of the K1 gene or prevention of K1 expression in KSHVΔK1 or KSHV-K15×STOP, respectively, resulted in decreased production of infectious virus following reactivation of the virus from latency. Moreover, these mutant viruses exhibited less phosphorylation of Akt in infected cells following reactivation. Akt phosphorylation and virus production were restored in K1 revertant virus-infected cells compared to K1 mutant virus-infected cells. Our results suggest that K1 plays an important role in lytic reactivation and replication as well as the activation of the Akt pathway in infected cells.
MATERIALS AND METHODS
Cell culture and recombinant virus construction.
BAC16 and iSLK-RTA cells were kindly provided by Jae U. Jung (28). Vero and HEK293T cells harboring WT KSHV, KSHV-K1FLAG, KSHV-K15×STOP, KSHV-K1REV, and KSHVΔK1 as well as iSLK-RTA and iSLK cells harboring these viruses were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. iSLK-RTA cells were cultured in the presence of 1 μg/ml puromycin and 250 μg/ml G418. HEK293T cells harboring WT KSHV, KSHV-K1FLAG, KSHV-K15×STOP, KSHV-K1REV, and KSHVΔK1 were constructed and maintained in the presence of 100 μg/ml hygromycin. iSLK cells harboring WT KSHV, and the recombinant viruses KSHV-K1FLAG, KSHV-K15×STOP, KSHV-K1REV, and KSHVΔK1 were established and maintained in the presence of 1 μg/ml puromycin, 250 μg/ml G418, and 1,200 μg/ml hygromycin B.
Construction of KSHV-K1FLAG, KSHV-K15×STOP, KSHV-K1REV, and KSHVΔK1.
BAC16 was kindly provided by Jae U. Jung (28). pcDNA3-WT K1FLAG and pcDNA3-K15×STOP were used as the templates for WT K1 and mutant K1 genes for the construction of recombinant viruses. pcDNA3 WT K1FLAG, which has a flag epitope tag at the N terminus of WT K1 from the BCBL1 genome and was constructed as previously described (24). pcDNA3-K15×STOP was constructed by the insertion of the K15×STOP fragment into the BamHI and EcoRI sites of pcDNA3 WT K1FLAG. The pcDNA3-K15×STOP construct contains 3 stop codons following the start codon of WT K1FLAG and also has 2 TGA stop codons instead of ATG codons at positions 481 and 763 to prevent the expression of WT K1FLAG. The following primers were used: forward 1 (5′-GCAGGATCCATGTAAATAGGTGAGCCCTGCCCGTGACCGCCCTG), reverse 1 (5′-CGATGGAATTCTCAGTACCAATCCACTGGTTGC), forward 2 (5′-GCAGGATCCATGTAAATAGGTGAGCCCTGCCCGTGACCGCCCTG), reverse 2 (5′-GTATAACACCCCTCAGTTTGGTTTCACAGCGTAAAATTATAGTATTTAG), forward 3 (5′-CACTCGTAGCTCTGATAGGAACCTGATGTGGTATCTTAGGAACTATTATC), and reverse 3 (5′-GATAATAGTTCCTAAGATACCACATCAGGTTCCTATCAGAGCTACGAGTG).
DH10B harboring BAC16 was a gift from Jae U. Jung (28). KSHV-K1FLAG, KSHV-K15×STOP, KSHV-K1REV, and KSHVΔK1 recombinant viruses were made by using BAC16. The original K1 gene is located at positions 105 to 959 in the KSHV BAC16 genome (28) (GenBank accession number GQ994935). The recombinant viruses KSHV-K1FLAG, KSHV-K15×STOP, KSHV-K1REV, and KSHVΔK1 were constructed with the Red/ET recombination system, purchased from Gene Bridges Inc., according to the manufacturer's instruction. This system utilizes a dual-antibiotic cassette for the positive selection of kanamycin resistance and negative selection of streptomycin sensitivity rendered by the bacterial ribosomal protein S12 (rpsL+) gene. In brief, a Red/ET expression plasmid was transformed into DH10B cells harboring BAC16 with electroporation. Next, an RpsL-neomycin cassette flanked by homologous nucleotides upstream of the original K1 gene and other homologous nucleotides downstream of K1 was generated by PCR with forward primer 5′-GTGTAAACCTGTCTTTCAGACCTTGTTGGACATCCCGTACAATCAAGGGCCTGGTGATGATGGCGGGATCG and reverse primer 5′-CACATTACAATTATGTTACAGAGAATATTTAGATTATCTTACCTGAATGTCAGAAGAACTCGTCAAGAAGGCG, and the cassette was then electroporated into DH10B-Red/ET cells harboring BAC16. To replace the K1 gene with the linear RpsL-Neo cassette flanked by homologous arms, we used l-arabinose in the growth medium to induce the expression of the Red/ET plasmid and allow for recombination. The transformed bacteria were plated onto LB agar plates and selected with chloramphenicol and kanamycin. Positive colonies growing under chloramphenicol and kanamycin selection were isolated and analyzed by PCR and pulsed-field gel electrophoresis (PFGE). Next, colonies harboring BAC16 containing the RpsL-Neo cassette in place of the K1 gene were subjected to a second recombination step. DNA fragments with homologous arms, including K1FLAG or K15×STOP, were amplified by PCR with forward primer 5′-GTGTAAACCTGTCTTTCAGACCTTGTTGGACATCCCGTACAATCAAGATGGCCCTGCCCGTGACCGCCCTGC and reverse primer CACATTACAATTATGTTACAGAGAATATTTAGATTATCTTACCTGAATGTCAGAAGAACTCGTCAAGAAGGCG for K1FLAG or forward primer GTGTAAACCTGTCTTTCAGACCTTGTTGGACATCCCGTACAATCAAGATGTAAATAGGTGAGCCCTGC and reverse primer ACACATTACAATTATGTTACAGAGAATATTTAGATTATCTTACCTGAATGTCAGTACCAATCCACTGGTTGC for K15×STOP and were transformed into DH10B cells harboring BAC16 with the RpsL-Neo cassette in the K1 location. The bacteria were plated onto plates containing streptomycin and chloramphenicol, and colonies were analyzed by using PCR and PFGE to check for correct clones containing full-length K1FLAG or K15×STOP, etc., in the original K1 location and in place of the RpsL-Neo cassette. To make KSHV-K1REV, we repeated the above-described procedure to replace the K1 mutant gene first with the RpsL-Neo cassette and then replaced the RpsL-Neo cassette with WT K1FLAG. All BAC plasmid DNAs were isolated by the alkaline lysis method for analysis of restriction digestion or with the NucleoBond BAC 100 kit (Clontech) for sequencing.
Establishment of stably infected HEK293T cells and their derivatives.
WT or recombinant virus BAC16 DNA was transfected into HEK293T cells with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Briefly, 5 μg of DNA was transfected into HEK293T cells in one well of a 6-well plate. A total of 50 μg/ml of hygromycin B was added to the medium for selection at 48 h posttransfection. The concentration of hygromycin B was increased up to 100 μg/ml at 96 h posttransfection. Stable infected HEK293T cells were maintained with 100 μg/ml hygromycin B.
Establishment of stably infected iSLK cells and their derivatives.
iSLK cells were cocultured with recombinant virus-infected HEK293T cells. Briefly, a 2:1 ratio of stable recombinant virus-infected HEK293T cells was plated with iSLK-RTA cells so that the total number of cells was 2.5 × 105 cells per well of a 6-well plate. Three milliliters of 2% FBS–DMEM with final concentrations of 25 ng/ml of tetradecanoyl phorbol acetate (TPA) and 0.5 mM sodium butyrate was added to the cells at 24 h postseeding to induce reactivation. Four days later, cells were selected with 250 μg/ml G418, 1 μg/ml puromycin, and 1.2 mg/ml hygromycin B. Next, recombinant virus-infected stable iSLK cells, which were 100% green fluorescent protein (GFP) positive, were reactivated with 3 μg/ml doxycycline and 1 mM sodium butyrate for 3 days. The supernatant was collected, cleared by centrifugation at 950 × g for 10 min, and filtered through a 0.45-μm filter. A new batch of uninfected iSLK-RTA cells was infected with this filtered viral supernatant in the presence of 8 μg/ml of Polybrene and centrifuged for 2 h at 2,500 rpm at 30°C. The cells were then placed into an incubator with 5% CO2 at 37°C. At 48 h postinfection, 1 μg/ml puromycin, 250 μg/ml G418, and 1.2 mg/ml hygromycin were added to the medium to select for stable iSLK-WT KSHV, iSLK-KSHV-K1FLAG, iSLK-KSHV-K15×STOP, KSHV-K1REV, and KSHVΔK1 cell lines. These cell lines were maintained in the presence of 1 μg/ml puromycin, 250 μg/ml G418, and 1.2 mg/ml hygromycin.
Recombinant KSHV DNA purification and analysis.
Viral DNA was isolated from DH10B-BAC cells with the alkaline lysis procedure and from iSLK cells with the DNeasy kit (Qiagen). Purified BAC DNA from alkaline lysis was digested with KpnI for analysis by PFGE. Briefly, 3 ml of BAC16-containing chloramphenicol-resistant bacteria was subjected to alkaline lysis. Purified BAC16 DNA was digested with KpnI and separated on 1% pulsed-field-certified agarose by using PFGE (Chef-Mapper; Bio-Rad) under the following conditions: 6 V/cm for 15 h at 14°C and initial and final switch times of 0.1 and 11.15 s, respectively. Recombinant KSHV DNA purified with the NucleoBond BAC 100 kit was used for sequencing with the 454 GS Junior sequencer. Finished genomic data were submitted to GenBank.
Preparation of viruses.
A total of 5 × 105 iSLK-WT and recombinant virus-infected cells were plated into one well of a 6-well plate overnight, after which cells were reactivated with 3 μg/ml doxycycline in DMEM with 2% FBS for 72 h. The supernatant was collected and centrifuged at 950 × g for 10 min. The supernatant was used for infection studies. Genomic DNA of recombinant KSHVs was isolated from the supernatant or infected cells with the DNeasy blood and tissue kit (Qiagen) for the detection of KSHV genomic copies by quantitative PCR (qPCR).
Infectivity assay.
A total of 1.9 × 104 Vero cells or primary human umbilical vein endothelial cells (HUVECs) per well in a 96-well plate were seeded in triplicate for each sample. The next day, 100 μl of fresh supernatant containing WT KSHV or recombinant viruses was added to naive Vero cells with 8 μg/ml Polybrene. The plate was centrifuged at 2,500 rpm at 30°C for 2 h for Vero cells and for 1 h for primary HUVECs. Twenty-four hours later, the supernatant was removed and washed with DMEM for Vero cells or EBM-2 (endothelial cell basal medium 2) for primary HUVECs, and 100 μl fresh medium was added. GFP-positive cells were counted at 72 h postinfection. For flow cytometry analysis, a similar protocol was performed. A total of 1 × 105 Vero cells were plated into each well of a 24-well plate, and 500 μl of fresh supernatant containing WT KSHV or recombinant viruses was added to naive Vero cells. Cells were collected following trypsinization at 72 h postinfection, washed with Dulbecco's phosphate-buffered saline (DPBS) containing 5% bovine serum albumin (BSA), and analyzed by flow cytometry.
Genomic DNA for qPCR was isolated with the DNeasy blood and tissue kit (Qiagen) 72 h after infection of Vero cells with WT or recombinant virus.
Analysis of Akt activation.
Cells harboring WT KSHV, KSHV-K1FLAG, KSHV-K15×STOP, KSHV-K1REV, and KSHVΔK1 were serum starved for 48 h with DMEM. The cells were then treated with 1.5 μg/ml doxycycline for 24, 48, and 72 h. Cells were washed with ice-cold phosphate-buffered saline (PBS) containing 1 mmol/liter Na3VO4 and a protease inhibitor cocktail (Roche) and then lysed in Triton–NP-40 lysis buffer. Western blot analyses were performed with the indicated antibodies, which were purchased from Cell Signaling Technology.
Real-time PCR for viral load determination.
For real-time PCR for viral load determinations, cells were treated with 1 μg/ml doxycycline for 72 h to reactivate the virus. The supernatant was harvested and divided into two aliquots. For DNase-resistant KSHV genome samples, one aliquot of the supernatant was treated with 1 U of DNase at 37°C for 10 min. Ten micrograms of salmon sperm DNA was added to 200 μl of the clarified supernatant prior to purification of viral genomic DNA with the DNeasy kit (Qiagen) according to the manufacturer's protocol. Cell pellets were also harvested, and DNA was purified. SYBR green real-time PCR was performed in a 384-well format by using an ABI Prism 7900 sequence detection system (Applied Biosystems Inc., Foster City, CA). To build a standard curve for the cycle threshold (CT) versus the genome copy number, plasmid pcDNA3-ORF57 was serially diluted to known concentrations in the range of 4 × 102 to 4 × 107 plasmid molecules/μl. Each PCR mixture contained 4 μl of viral DNA isolated from the supernatant or 1 μl of viral DNA isolated from the cell pellet, 7.5 μl of SYBR green 2× PCR mix (Applied Biosystems), and 0.05 μl each 100 μM ORF57 primer, and the final volume of 15 μl was standardized by the addition of DNase- and RNase-free water (Sigma). Primers for amplification of the ORF57 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) genes were ORF57 forward primer 5′-CAGGATGACGACGTCAGAC, ORF57 reverse primer 5′-GAGCGGTGATATCCCTGTCC-3′, GAPDH forward primer 5′-GAAGGTGAAGGTCGGAGT, and GAPDH reverse primer 5′-GAAGATGGTGATGGGATTTC.
Western blot analysis.
A total of 5 × 105 cells per well were seeded into a 6-well plate. The next day, cells were treated with 1.5 μg/ml doxycycline for 24, 48, and 72 h. The cells were scraped and lysed, and the lysate was mixed with 2× Laemmli buffer. Forty micrograms of each protein sample was subjected to SDS-PAGE and transferred onto a nitrocellulose membrane. Western blot analyses were performed with rabbit viral interleukin-6 (vIL-6) polyclonal antibody (1:1,000 dilution; ABI), rabbit anti-pAkt-S473 antibody (1:1,000; Cell Signaling), horseradish peroxidase-conjugated FLAG M2 monoclonal antibody (1:500 dilution; Sigma), mouse anti-K8α monoclonal antibody (1:1,000; Santa Cruz Biotechnology), mouse anti-KSHV ORF45 monoclonal antibody (1:1,000; Thermo Scientific), and antitubulin horseradish peroxidase (HRP)-conjugated polyclonal antibody.
Accession numbers.
The sequence data for the recombinant viruses have been submitted to GenBank under accession numbers KX189626, KX189627, KX189628, and KX189629.
RESULTS
Construction of KSHV-K1FLAG, KSHV-K15×STOP, KSHV-K1REV, and KSHVΔK1 BAC clones derived from WT KSHV BAC (BAC16).
BAC16 is a KSHV BAC clone derived from the KSHV JSC1 strain as previously described (28). BAC16 has green fluorescent protein under the control of the EF-1α promoter to indicate KSHV infection. To assess the role of K1 in the KSHV genome, we either deleted the K1 ORF by replacing it with an RpsL-Neo cassette or replaced the WT K1 ORF with a FLAG-tagged K1 gene or a K1 gene with five stop codons. Using BAC mutagenesis, we made KSHVΔK1, KSHV-K1FLAG, KSHV-K15×STOP, and a revertant virus, KSHV-K1REV. KSHV-K15×STOP has a mutant K15×STOP gene that contains 5 stop codons, 3 of which are inserted following the first start codon of K1 and the other 2 of which replace two downstream ATG codons at positions 481 and 763 in the K1 gene. Hence, KSHV-K15×STOP is not able to express any full-length K1 protein or any one of the possible shorter ORFs within K1. A schematic diagram of WT K1 and the modified or mutant K1 genes in the recombinant viruses is shown in Fig. 1A. The recombinant virus KSHV-K1REV has a WT K1FLAG ORF replacing the mutant K1 ORF.
FIG 1.
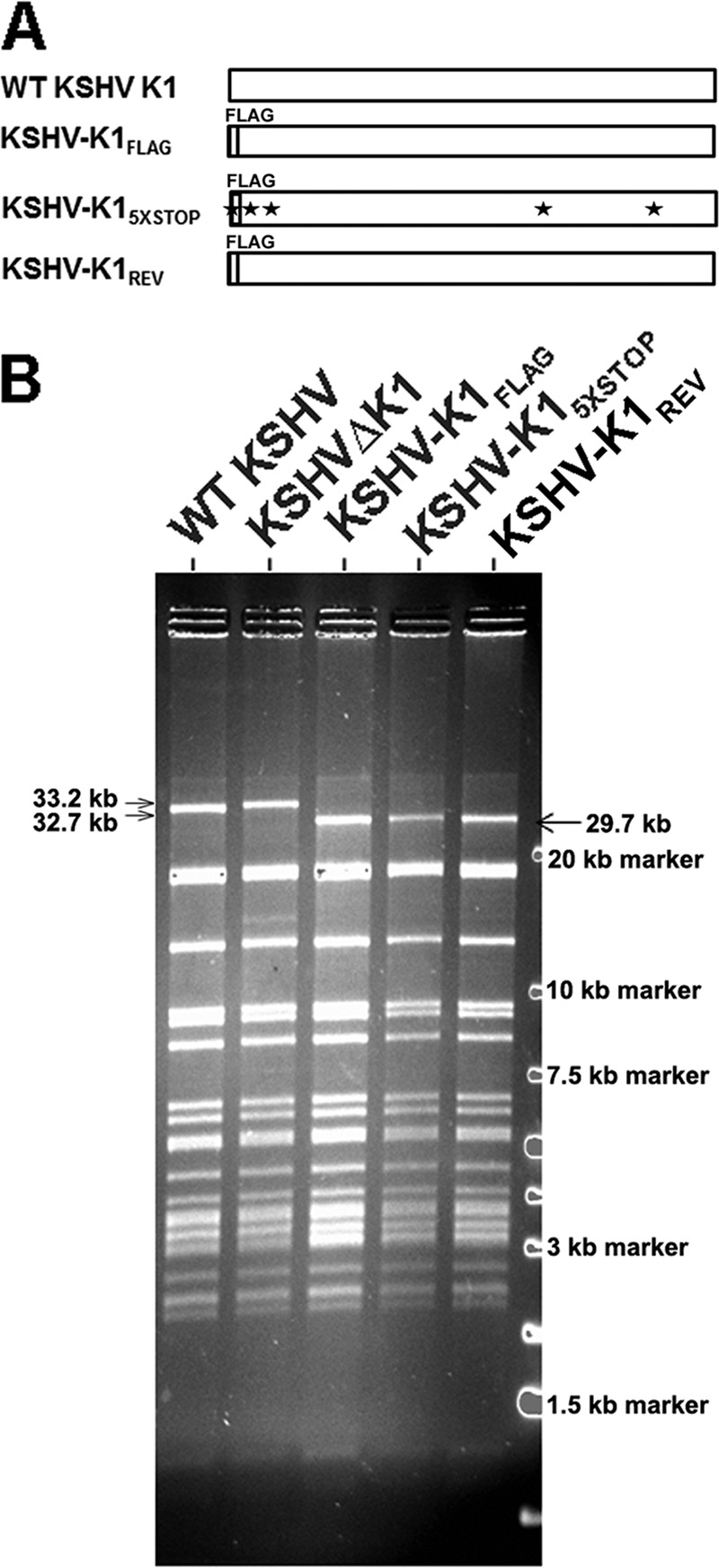
Construction of wild-type or mutant K1 recombinant viruses. (A) Schematic diagram of WT K1 and mutant K1 genes. The gray bar represents a FLAG epitope tag, and each star represents a stop codon. (B) PFGE analysis of KpnI-digested DNA of BAC16 and its derivatives. DNAs of WT KSHV (BAC16) and its derivatives KSHV-K1FLAG, KSHV-K15×STOP, KSHV-K1REV, and KSHVΔK1 were digested with KpnI and analyzed by PFGE. The 29.7-kb fragment is the result of replacing the original WT K1 gene with FLAG-tagged K1 and the presence of an additional KpnI site in the FLAG-tagged K1 gene compared to the WT K1 gene. The 33.2-kb fragment in the KSHVΔK1 virus is due to the replacement of the K1 ORF with the Kan-Neo (RpsL-Neo) cassette.
Analysis of the genomic integrity of candidate clones.
Ten to twenty candidates for each clone were picked for PCR and PFGE analysis. After WT KSHV K1FLAG, mutant KSHV K15×STOP, or the RpsL-Neo cassette was identified to be present in the candidate clone genomes by PCR (data not shown), the PCR products of K1, K1FLAG, K15×STOP, and the RpsL-Neo cassette were amplified from WT KSHV, KSHV-K1FLAG, KSHV-K15×STOP, and KSHVΔK1 candidate clones, respectively. These products were sequenced to confirm their sequences and matched to the previously reported sequences (GenBank accession numbers GQ994935 and U86667). The integrity, sequence, and position of these genes were confirmed by next-generation whole-genome sequencing. All viruses (WT KSHV, KSHV-K1FLAG, KSHV-K15×STOP, KSHV-K1REV, and KSHVΔK1) matched sequences in the database.
For PFGE analysis, digestion of BAC16 and each candidate virus with KpnI revealed various sizes of KSHV DNA fragments, which matched the predicted digestion pattern and fragment sizes based on the previously reported BAC16 sequence (Fig. 1B). A high-molecular-weight fragment of 32.7 kb appears in WT KSHV, and a 33.2-kb fragment appears in the KSHVΔK1 sample containing the RpsL-Neo cassette. In the KSHV-K1FLAG, KSHV-K1REV, and KSHV-K15×STOP constructs, a 29.7-kb fragment appears because a new KpnI site at position 196 of KSHV-K1FLAG, KSHV-K1REV, and KSHV-K15×STOP BAC16 was created due to the fact that WT K1FLAG or K15×STOP was substituted in place of the original K1 gene of BAC16. The original K1 gene and RpsL-Neo cassette have no KpnI site. PFGE analysis demonstrated the integrity of the candidate viral genomes.
K1 is required for Akt phosphorylation.
We next investigated the biology of the K1 mutant viruses in the doxycycline-inducible KSHV-infected iSLK line, which was described by Myoung and Ganem (29). Previously, we reported that the PI3K/Akt/mTOR pathway is activated in cells expressing K1 (18, 22). To examine this pathway in cells infected with the recombinant viruses described above, we investigated phosphorylation of Akt by Western blotting. We found that Akt was phosphorylated at serine 473 (S473) at 72 h postreactivation (with 1.5 μg/ml of doxycycline) in cells latently infected with WT KSHV and KSHV-K1FLAG but not in cells infected with the recombinant viruses KSHVΔK1 and KSHV-K15×STOP (Fig. 2A). In KSHV-K1REV-infected cells, we found that the level of phosphorylated Akt at S473 was restored after reactivation from latency (Fig. 2A). As a control for efficient reactivation, we also performed Western blotting for KSHV vIL-6 and found that cells infected with WT KSHV and KSHV-K1FLAG expressed much higher levels of vIL-6 than did cells infected with the recombinant viruses KSHVΔK1 and KSHV-K15×STOP (Fig. 2A).
FIG 2.

K1 is required for Akt phosphorylation and lytic gene expression in reactivated cells. (A and B) A total of 5 × 105 WT KSHV-, KSHV-K1FLAG-, KSHV-K15×STOP-, KSHV-K1REV-, and KSHVΔK1-infected iSLK cells were seeded overnight and then treated with 1.5 μg/ml of doxycycline for 72 h (A) or for 24, 48, and 72 h (B). Cells were harvested, lysed, and subjected to Western blot analysis with an anti-FLAG antibody to detect K1, an anti-pAkt (S473) antibody to detect phosphorylated Akt, an anti-vIL-6 antibody to detect vIL-6, an anti-K8α antibody to detect K8α, and an anti-ORF45 antibody to detect ORF45. (C) A total of 2 × 105 cells were seeded overnight and then treated with 3 μg/ml of doxycycline for 72 h. Cells were fixed with 3.7% formaldehyde and subjected to an immunofluorescence assay with primary anti-ORF59 antibody (1:500; Advanced Biotechnologies) and secondary anti-mouse IgG antibody conjugated to tetramethyl rhodamine isocyanate (1:500). (D) Graph depicting the number of quantitated ORF59-positive cells per field following reactivation.
To elucidate our observations further, we compared Akt phosphorylation and viral lytic gene expression 24, 48, and 72 h after reactivation with 1.5 μg/ml of doxycycline. We found that the level of phosphorylated Akt at S473 increased at several time points following reactivation for WT KSHV-, KSHV-K1FLAG-, and KSHV-K1REV-infected cells but not for KSHVΔK1- and KSHV-K15×STOP-infected cells. Following reactivation, the lytic K8α and ORF45 proteins were detectable in WT KSHV-, KSHV-K1FLAG-, and KSHV-K1REV-infected cells but were barely detectable in KSHVΔK1- and KSHV-K15×STOP-infected cells (Fig. 2B).
To further confirm that the K1 gene augments viral lytic replication, we performed an immunofluorescence assay with an antibody against the viral lytic protein ORF59. The number of red fluorescence-labeled ORF59 cells was distinctly higher for WT KSHV-, KSHV-K1FLAG-, and KSHV-K1REV-infected cells than for KSHVΔK1- and KSHV-K15×STOP-infected cells at 72 h postreactivation (Fig. 2C and D).
To determine whether K1 could be detected at the protein level in uninduced iSLK cells, we performed immunoprecipitation from infected cells using anti-FLAG to pull down K1FLAG. Immunoprecipitates using FLAG antibody from iSLK cells latently infected with KSHV-K1FLAG, KSHV-ΔK1, and KSHV-K1REV were run on SDS-PAGE gels and subjected to Western blot analysis with an anti-FLAG antibody. As shown in Fig. 3, we were able to detect K1 expression in cells following immunoprecipitation. Our results are concordant with those reported by Chandriani and Ganem, who reported that K1 can be detected in latently infected cells (14).
FIG 3.
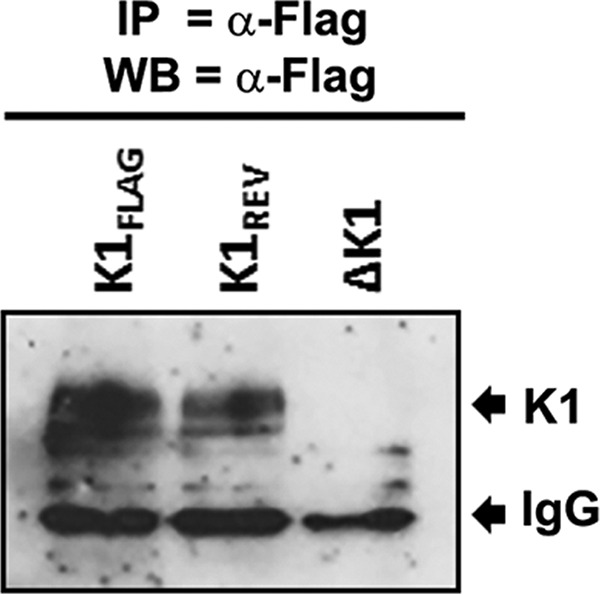
Small amounts of K1 protein are expressed in uninduced KSHV-infected iSLK cells. iSLK cells latently infected with KSHV-K1FLAG, KSHV-K1REV, and KSHVΔK1 were harvested and subjected to immunoprecipitation (IP) with an anti-FLAG antibody to immunoprecipitate FLAG-tagged K1 and then subjected to Western blot (WB) analysis with an anti-FLAG-HRP-conjugated antibody.
K1 deletion from KSHV results in reduced lytic replication of KSHV.
To test the role of K1 during viral replication, we reactivated iSLK cells with doxycycline. We observed greater total GFP fluorescence in cells harboring WT KSHV, KSHV-K1FLAG, and KSHV-K1REV than in KSHVΔK1- and KSHV-K15×STOP-infected iSLK cells following reactivation (Fig. 4A and B). The GFP fluorescence in WT KSHV-, KSHV-K1FLAG-, and KSHV-K1REV-infected cells ranged from 4- to 8-fold higher than that in KSHVΔK1- and KSHV-K15×STOP-infected cells (Fig. 4C).
FIG 4.

KSHV K1 is required for efficient reactivation from latency. (A and B) A total of 5 × 105 stable iSLK cells latently infected with WT KSHV, KSHV-K1FLAG, KSHV-K15×STOP, KSHV-K1REV, and KSHVΔK1 were plated and examined by fluorescence microscopy in the absence of any induction (A) or after being treated with 1 μg/ml of doxycycline in DMEM supplemented with 2% FBS for 72 h (B). Images were taken at a ×4 magnification. hpi, hours postinfection. (C) Graph depicting the number of GFP-positive cells per field that were quantitated following reactivation.
To quantify viral genomes, we extracted DNA from cell-free supernatants and lysates of recombinant virus-infected cells following reactivation. We performed quantitative real-time PCR to determine the CT values and genome copy numbers. In the supernatants, the genome copy numbers of released viruses from induced cells harboring WT KSHV, KSHV-K1FLAG, and KSHV-K1REV were markedly higher than those from cells harboring KSHV-K15×STOP and KSHVΔK1 (Fig. 5A). Following reactivation, WT KSHV, KSHV-K1FLAG, and KSHV-K1REV displayed a 5-fold increase in virion production, as measured by viral genomes, compared to KSHVΔK1 and KSHV-K15×STOP (1 × 105 genome copies versus 2 × 104 genome copies) (Fig. 5A). Similar results were observed for intracellular genome copy numbers in reactivated cell lysates from cells that harbored WT KSHV, KSHV-K1FLAG, and KSHV-K1REV compared to cells harboring KSHV-K15×STOP and KSHVΔK1 (Fig. 5B).
FIG 5.

KSHV K1 is required for efficient viral replication. Stable iSLK cells latently infected with WT KSHV, KSHV-K1FLAG, KSHV-K15×STOP, KSHV-K1REV, and KSHVΔK1 were treated with 1 μg/ml of doxycycline for 72 h. Following reactivation, the supernatants (A and C) and cells (B) were harvested for qPCR to determine viral genome copy numbers. (A) KSHV genome copy numbers in the supernatants of reactivated WT KSHV-, KSHV-K1FLAG-, KSHV-K15×STOP-, KSHV-K1REV-, and KSHVΔK1-infected iSLK cells. (B) Quantification of intracellular viral genome copy numbers present in cells of reactivated WT KSHV-, KSHV-K1FLAG-, KSHV-K15×STOP-, KSHV-K1REV-, and KSHVΔK1-infected iSLK cells. For normalization across samples, intracellular viral genome copy numbers were normalized to the intracellular CT value of GAPDH. (C) KSHV genome copy numbers from DNase-treated and untreated supernatants of reactivated WT KSHV-, KSHV-K1FLAG-, KSHV-K15×STOP-, KSHV-K1REV-, and KSHVΔK1-infected iSLK cells.
In order to measure capsid-protected viral DNA, WT KSHV-, KSHV-K1FLAG-, KSHV-K1REV-, KSHVΔK1-, and KSHV-K15×STOP-infected iSLK cells were reactivated as described above. The supernatants were harvested and subjected to DNase treatment for 1 h, after which viral genomic DNA was harvested and quantitated. Once again, we observed that WT KSHV, KSHV-K1FLAG, and KSHV-K1REV displayed a 5- to 6-fold increase in virion production (as measured by viral genomes) compared to KSHVΔK1 and KSHV-K15×STOP (Fig. 5C).
K1 deletion from KSHV results in decreased production of infectious virions.
To test the infectivity of viruses produced from reactivated cells, cell-free supernatants from reactivated cells were used to infect naive Vero cells in triplicate, as described in Materials and Methods. GFP-positive cells were observed at 72 h postinfection (Fig. 6A). We observed more GFP-positive, i.e., infected, cells from supernatants of WT KSHV-, KSHV-K1FLAG-, and KSHV-K1REV-infected cells than from KSHVΔK1- and KSHV-K15×STOP-infected cells. The number of GFP-positive cells in six random fields for each recombinant virus-infected sample was quantified (Fig. 6B). The numbers of infectious virions from reactivated cells harboring WT KSHV, KSHV-K1FLAG, and KSHV-K1REV were found to be dramatically higher than those from cells harboring KSHV-K15×STOP and KSHVΔK1.
FIG 6.

KSHV K1 is required for efficient infectious virion production. Supernatants from reactivated WT KSHV-, KSHV-K1FLAG-, KSHV-K15×STOP-, KSHV-K1REV-, and KSHVΔK1-infected iSLK cells were used to infect naive Vero cells for 72 h. (A) GFP-positive cells were imaged by fluorescence microscopy at 72 h postinfection (hpi) at a ×20 magnification. (B) Graph depicting numbers of GFP-positive cells per field. (C) Supernatants from reactivated WT KSHV-, KSHV-K1FLAG-, KSHV-K15×STOP-, KSHV-K1REV-, and KSHVΔK1-infected iSLK cells were used to infect naive Vero cells for 72 h, and infected cells were analyzed by flow cytometry for the percentage of GFP-positive cells to quantitate infectivity. (D) Infected Vero cells from panel A were harvested, and the intracellular viral genome copies present in infected cells were quantified by real-time PCR. For normalization across samples, intracellular viral genome copy numbers were normalized to the intracellular CT value of GAPDH. FITC, fluorescein isothiocyanate; SSC-A, side-scattered light area.
To further assess the production of infectious virus and quantify the number of KSHV-infected Vero cells, infected cells were also subjected to flow cytometry analysis. Figure 6C shows that the percentages of GFP-positive Vero cells were 12.5%, 10.8%, 3.0%, 3.2%, and 14.1% for WT KSHV, KSHV-K1FLAG, KSHVΔK1, KSHV-K15×STOP, and KSHV-K1REV, respectively, indicating that, similar to our microscopic quantitation described above, the percentages of KSHV-infected Vero cells were higher with WT KSHV, KSHV-K1FLAG, and KSHV-K1REV viruses than with KSHVΔK1 and KSHV-K15×STOP viruses.
To confirm the optical readout, we harvested infected Vero cells as described in the legend of Fig. 6A, extracted DNA, and performed real-time qPCR for viral genomes. We found that the numbers of viral genomes were higher in reactivated cells harboring WT KSHV, KSHV-K1FLAG, and KSHV-K1REV than in cells harboring KSHV-K15×STOP and KSHVΔK1 viruses (Fig. 6D).
To further assess the infectivity of these viruses on a more relevant cell line, we harvested supernatants from reactivated KSHV-infected iSLK cells and infected primary HUVECs. Similar to the case with Vero cells, the numbers of GFP-positive primary HUVECs were significantly higher following infection with WT KSHV, KSHV-K1FLAG, and KSHV-K1REV than with KSHVΔK1 and KSHV-K15×STOP (Fig. 7A and B).
FIG 7.
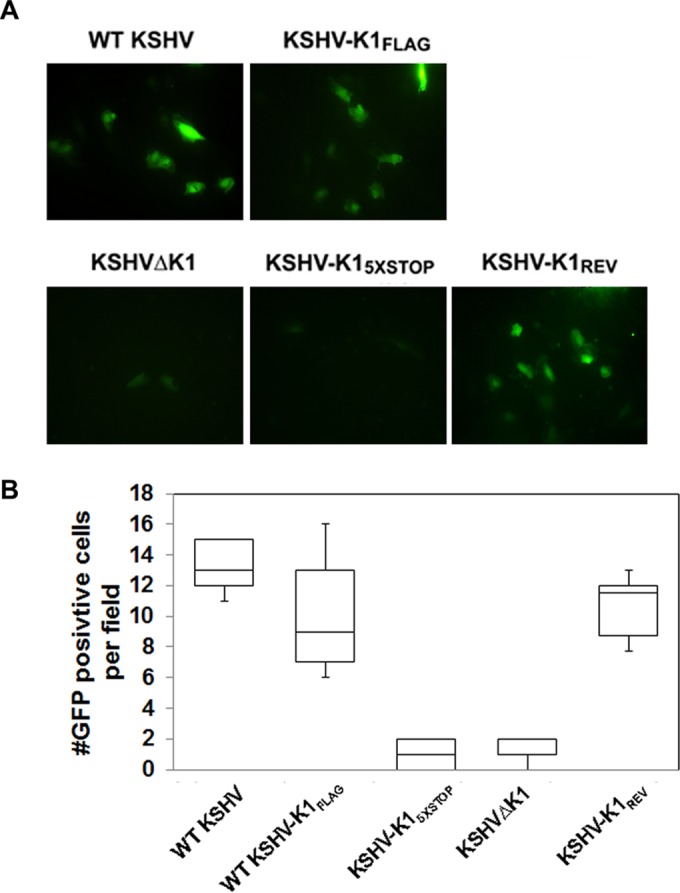
KSHV K1 is required for efficient infectious virion production and infection of primary endothelial cells. Primary HUVECs were infected with supernatants from reactivated WT KSHV-, KSHV-K1FLAG-, KSHV-K15×STOP-, KSHV-K1REV-, and KSHVΔK1-infected iSLK cells for 72 h. (A) Images of GFP-positive infected primary HUVECs were taken by fluorescence microscopy at 72 h postinfection (hpi). (B) The graph depicts the number of GFP-positive infected primary HUVECs per field.
DISCUSSION
The KSHV K1 gene has a dramatic impact on cell signaling in single-gene assays and, in the context of herpesvirus saimiri (HVS), can replace the HVS-transforming protein, STP (30). In order to investigate the role of the KSHV K1 ORF in the life cycle of KSHV, we constructed a set of K1 deletion or K1 mutant recombinant viruses, including KSHV-K1FLAG, KSHV-K15×STOP, KSHV-K1REV, and KSHVΔK1. JSC-1-derived recombinant KSHV BAC16 (28) was used as the parental BAC for the construction of K1 deletion or mutant recombinant viruses. The integrity and sequences of all the WT KSHV, KSHV-K1FLAG, KSHV-K15×STOP, KSHV-K1REV, and KSHVΔK1 genomes in this study were identified and verified by multiple independent methods, including whole-genome sequencing. We also epitope tagged the K1 gene with the FLAG epitope in the context of the virus to use it as a tool for investigating K1 function during the KSHV life cycle. Previously, our laboratory and other groups studied K1 signal transduction events (4, 14, 17-20, 22). We reported that K1 activates the PI3K/Akt/mTOR pathway and provides a survival advantage to K1-expressing cells (18, 22). Here, we extended these studies and found that cells that were infected with K1 deletion or mutant viruses (KSHV-K15×STOP and KSHV-ΔK1) displayed lower phospho-Akt levels than cells infected with viruses containing WT K1. We detected low levels of K1 expression in cells infected with KSHV-K1FLAG using coimmunoprecipitation. This finding correlates with data from previous reports suggesting that K1 can be expressed under latency conditions (14, 22). However, we cannot rule out the possibility that the K1 that we are detecting is due to a low level of lytic replication in infected iSLK cells.
All iSLK cells harboring WT KSHV, KSHV-K1FLAG, KSHV-K15×STOP, KSHV-K1REV, or KSHVΔK1 were able to produce infectious virus in response to doxycycline induction; however, viruses lacking the K1 gene, including KSHV-K15×STOP and KSHV-ΔK1, were significantly attenuated for viral reactivation in response to RTA induction. Accumulating evidence suggests that lytic replication may play a prominent role in KS (5, 31–34), and our data suggest that K1 is involved in enhancing lytic reactivation and/or replication. Several groups have investigated the link between K1 signal transduction and KSHV reactivation by the overexpression of K1 or deletion mutants (26, 27). Lagunoff et al. reported that the expression of lytic cycle genes was diminished up to 80% in the presence of a K1 dominant negative mutant, which inhibited wild-type K1 signal transduction in BCBL-1 cells that were induced into reactivation by the ectopic expression of the KSHV ORF50 transactivator (26). Our results demonstrate that deletion of K1 using the deletion virus KSHVΔK1 as well as the recombinant virus KSHV-K15×STOP displayed reduced KSHV reactivation as measured by vIL-6 lytic gene expression, KSHV genome copy numbers, and the production of infectious viruses. In contrast, a revertant virus, where mutant K1 was restored back to WT K1, grew similarly to KSHV-K1FLAG. Collectively, our data indicate that the lack of K1 protein expression leads to decreased production of KSHV infectious virions. However, the exact mechanism by which this occurs is yet to be determined. In conclusion, we have shown that KSHV K1 plays an important role in the KSHV viral life cycle.
ACKNOWLEDGMENTS
We thank Jae Jung for the BAC16 construct and Shogo Misumi for pcDNA3.1D/V5-hGAPDH.
B.D. is a Leukemia and Lymphoma Society Scholar and a Burroughs Wellcome Fund Investigator in Infectious Disease.
Funding Statement
The UNC Vironomics Core is supported by NCI core grant P30 CA01608 to the UNC Lineberger Comprehensive Cancer Center.
REFERENCES
- 1.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 2.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 332:1186–1191. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- 3.Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d'Agay MF, Clauvel JP, Raphael M, Degos L, Sigaux F. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86:1276–1280. [PubMed] [Google Scholar]
- 4.Wen KW, Damania B. 2010. Hsp90 and Hsp40/Erdj3 are required for the expression and anti-apoptotic function of KSHV K1. Oncogene 29:3532–3544. doi: 10.1038/onc.2010.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grundhoff A, Ganem D. 2004. Inefficient establishment of KSHV latency suggests an additional role for continued lytic replication in Kaposi sarcoma pathogenesis. J Clin Invest 113:124–136. doi: 10.1172/JCI200417803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dillon PJ, Gregory SM, Tamburro K, Sanders MK, Johnson GL, Raab-Traub N, Dittmer DP, Damania B. 2013. Tousled-like kinases modulate reactivation of gammaherpesviruses from latency. Cell Host Microbe 13:204–214. doi: 10.1016/j.chom.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gregory SM, West JA, Dillon PJ, Hilscher C, Dittmer DP, Damania B. 2009. Toll-like receptor signaling controls reactivation of KSHV from latency. Proc Natl Acad Sci U S A 106:11725–11730. doi: 10.1073/pnas.0905316106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gradoville L, Gerlach J, Grogan E, Shedd D, Nikiforow S, Metroka C, Miller G. 2000. Kaposi's sarcoma-associated herpesvirus open reading frame 50/Rta protein activates the entire viral lytic cycle in the HH-B2 primary effusion lymphoma cell line. J Virol 74:6207–6212. doi: 10.1128/JVI.74.13.6207-6212.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lukac DM, Kirshner JR, Ganem D. 1999. Transcriptional activation by the product of open reading frame 50 of Kaposi's sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J Virol 73:9348–9361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Renne R, Zhong W, Herndier B, McGrath M, Abbey N, Kedes D, Ganem D. 1996. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat Med 2:342–346. doi: 10.1038/nm0396-342. [DOI] [PubMed] [Google Scholar]
- 11.Cheng F, Weidner-Glunde M, Varjosalo M, Rainio EM, Lehtonen A, Schulz TF, Koskinen PJ, Taipale J, Ojala PM. 2009. KSHV reactivation from latency requires Pim-1 and Pim-3 kinases to inactivate the latency-associated nuclear antigen LANA. PLoS Pathog 5:e1000324. doi: 10.1371/journal.ppat.1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu F, Harada JN, Brown HJ, Deng H, Song MJ, Wu TT, Kato-Stankiewicz J, Nelson CG, Vieira J, Tamanoi F, Chanda SK, Sun R. 2007. Systematic identification of cellular signals reactivating Kaposi sarcoma-associated herpesvirus. PLoS Pathog 3:e44. doi: 10.1371/journal.ppat.0030044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lagunoff M, Ganem D. 1997. The structure and coding organization of the genomic termini of Kaposi's sarcoma-associated herpesvirus. Virology 236:147–154. doi: 10.1006/viro.1997.8713. [DOI] [PubMed] [Google Scholar]
- 14.Chandriani S, Ganem D. 2010. Array-based transcript profiling and limiting-dilution reverse transcription-PCR analysis identify additional latent genes in Kaposi's sarcoma-associated herpesvirus. J Virol 84:5565–5573. doi: 10.1128/JVI.02723-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bowser BS, DeWire SM, Damania B. 2002. Transcriptional regulation of the K1 gene product of Kaposi's sarcoma-associated herpesvirus. J Virol 76:12574–12583. doi: 10.1128/JVI.76.24.12574-12583.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee BS, Connole M, Tang Z, Harris NL, Jung JU. 2003. Structural analysis of the Kaposi's sarcoma-associated herpesvirus K1 protein. J Virol 77:8072–8086. doi: 10.1128/JVI.77.14.8072-8086.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Samaniego F, Pati S, Karp JE, Prakash O, Bose D. 2001. Human herpesvirus 8 K1-associated nuclear factor-kappa B-dependent promoter activity: role in Kaposi's sarcoma inflammation? J Natl Cancer Inst Monogr 2001:15–23. [DOI] [PubMed] [Google Scholar]
- 18.Tomlinson CC, Damania B. 2004. The K1 protein of Kaposi's sarcoma-associated herpesvirus activates the Akt signaling pathway. J Virol 78:1918–1927. doi: 10.1128/JVI.78.4.1918-1927.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lagunoff M, Majeti R, Weiss A, Ganem D. 1999. Deregulated signal transduction by the K1 gene product of Kaposi's sarcoma-associated herpesvirus. Proc Natl Acad Sci U S A 96:5704–5709. doi: 10.1073/pnas.96.10.5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang S, Wang S, Maeng H, Young DP, Prakash O, Fayad LE, Younes A, Samaniego F. 2007. K1 protein of human herpesvirus 8 suppresses lymphoma cell Fas-mediated apoptosis. Blood 109:2174–2182. doi: 10.1182/blood-2006-02-003178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee H, Guo J, Li M, Choi JK, DeMaria M, Rosenzweig M, Jung JU. 1998. Identification of an immunoreceptor tyrosine-based activation motif of K1 transforming protein of Kaposi's sarcoma-associated herpesvirus. Mol Cell Biol 18:5219–5228. doi: 10.1128/MCB.18.9.5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang L, Dittmer DP, Tomlinson CC, Fakhari FD, Damania B. 2006. Immortalization of primary endothelial cells by the K1 protein of Kaposi's sarcoma-associated herpesvirus. Cancer Res 66:3658–3666. doi: 10.1158/0008-5472.CAN-05-3680. [DOI] [PubMed] [Google Scholar]
- 23.Bhatt AP, Damania B. 2012. AKTivation of PI3K/AKT/mTOR signaling pathway by KSHV. Front Immunol 3:401. doi: 10.3389/fimmu.2012.00401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang L, Wakisaka N, Tomlinson CC, DeWire SM, Krall S, Pagano JS, Damania B. 2004. The Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) K1 protein induces expression of angiogenic and invasion factors. Cancer Res 64:2774–2781. doi: 10.1158/0008-5472.CAN-03-3653. [DOI] [PubMed] [Google Scholar]
- 25.Tomlinson CC, Damania B. 2008. Critical role for endocytosis in the regulation of signaling by the Kaposi's sarcoma-associated herpesvirus K1 protein. J Virol 82:6514–6523. doi: 10.1128/JVI.02637-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lagunoff M, Lukac DM, Ganem D. 2001. Immunoreceptor tyrosine-based activation motif-dependent signaling by Kaposi's sarcoma-associated herpesvirus K1 protein: effects on lytic viral replication. J Virol 75:5891–5898. doi: 10.1128/JVI.75.13.5891-5898.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee BS, Paulose-Murphy M, Chung YH, Connlole M, Zeichner S, Jung JU. 2002. Suppression of tetradecanoyl phorbol acetate-induced lytic reactivation of Kaposi's sarcoma-associated herpesvirus by K1 signal transduction. J Virol 76:12185–12199. doi: 10.1128/JVI.76.23.12185-12199.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brulois KF, Chang H, Lee AS, Ensser A, Wong LY, Toth Z, Lee SH, Lee HR, Myoung J, Ganem D, Oh TK, Kim JF, Gao SJ, Jung JU. 2012. Construction and manipulation of a new Kaposi's sarcoma-associated herpesvirus bacterial artificial chromosome clone. J Virol 86:9708–9720. doi: 10.1128/JVI.01019-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Myoung J, Ganem D. 2011. Generation of a doxycycline-inducible KSHV producer cell line of endothelial origin: maintenance of tight latency with efficient reactivation upon induction. J Virol Methods 174:12–21. doi: 10.1016/j.jviromet.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee H, Veazey R, Williams K, Li M, Guo J, Neipel F, Fleckenstein B, Lackner A, Desrosiers RC, Jung JU. 1998. Deregulation of cell growth by the K1 gene of Kaposi's sarcoma-associated herpesvirus. Nat Med 4:435–440. doi: 10.1038/nm0498-435. [DOI] [PubMed] [Google Scholar]
- 31.Kedes DH, Ganem D. 1997. Sensitivity of Kaposi's sarcoma-associated herpesvirus replication to antiviral drugs. Implications for potential therapy. J Clin Invest 99:2082–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martin DF, Kuppermann BD, Wolitz RA, Palestine AG, Li H, Robinson CA. 1999. Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. Roche Ganciclovir Study Group. N Engl J Med 340:1063–1070. [DOI] [PubMed] [Google Scholar]
- 33.Campbell TB, Borok M, Gwanzura L, MaWhinney S, White IE, Ndemera B, Gudza I, Fitzpatrick L, Schooley RT. 2000. Relationship of human herpesvirus 8 peripheral blood virus load and Kaposi's sarcoma clinical stage. AIDS 14:2109–2116. doi: 10.1097/00002030-200009290-00006. [DOI] [PubMed] [Google Scholar]
- 34.Whitby D, Howard MR, Tenant-Flowers M, Brink NS, Copas A, Boshoff C, Hatzioannou T, Suggett FE, Aldam DM, Denton AS, Miller RF, Weller IVD. 1995. Detection of Kaposi sarcoma associated herpesvirus in peripheral blood of HIV-infected individuals and progression to Kaposi's sarcoma. Lancet 346:799–802. doi: 10.1016/S0140-6736(95)91619-9. [DOI] [PubMed] [Google Scholar]