

ABSTRACT
This study was conducted to investigate whether functions encoded in the metagenome could improve our ability to understand the link between microbial community structures and functions in activated sludge. By analyzing data sets from six industrial and six municipal wastewater treatment plants (WWTPs), covering different configurations, operational conditions, and geographic regions, we found that wastewater influent composition was an overriding factor shaping the metagenomic composition of the activated sludge samples. Community GC content profiles were conserved within treatment plants on a time scale of years and between treatment plants with similar influent wastewater types. Interestingly, GC contents of the represented phyla covaried with the average GC contents of the corresponding WWTP metagenome. This suggests that the factors influencing nucleotide composition act similarly across taxa and thus the variation in nucleotide contents is driven by environmental differences between WWTPs. While taxonomic richness and functional richness were correlated, shotgun metagenomics complemented taxon-based analyses in the task of classifying microbial communities involved in wastewater treatment systems. The observed taxonomic dissimilarity between full-scale WWTPs receiving influent types with varied compositions, as well as the inferred taxonomic and functional assignment of recovered genomes from each metagenome, were consistent with underlying differences in the abundance of distinctive sets of functional categories. These conclusions were robust with respect to plant configuration, operational and environmental conditions, and even differences in laboratory protocols.
IMPORTANCE This work contributes to the elucidation of drivers of microbial community assembly in wastewater treatment systems. Our results are significant because they provide clear evidence that bacterial communities in WWTPs assemble mainly according to influent wastewater characteristics. Differences in bacterial community structures between WWTPs were consistent with differences in the abundance of distinctive sets of functional categories, which were related to the metabolic potential that would be expected according to the source of the wastewater.
INTRODUCTION
Revealing the mechanisms that drive microbial community assembly in activated sludge is critical for improving the reliability of wastewater treatment management and helping to develop a conceptual framework that could be applied to microbial communities in other environmental biotechnology processes. Current knowledge of microbial community assembly in biological wastewater treatment systems has greatly expanded in the past few years, because of the extended use of massive sequencing technologies and advances in microbial ecological theory (1). Because of the recognized influence of biodiversity on ecosystem performance and stability, particular attention has been given to elucidating the biogeographic patterns and environmental factors that shape the structure of bacterial communities in activated sludge (2, 3).
Ample experimental evidence has provided support for both stochastic and niche-based species-sorting processes in the assembly of bacterial communities in wastewater treatment facilities (4–9). An early quantitative survey of bacteria in industrial activated sludge from our laboratory showed that the distribution of the most abundant bacteria fitted a geometric distribution, suggesting that resource competition was a primary factor determining the assembly characteristics of those populations (10). Several other 16S rRNA-based surveys of community composition indicated that, apart from being dominated by high-abundance taxa, microbial communities in environmental biotechnology processes were characterized by the presence of large numbers of rare taxa, which appeared to contribute considerably to the overall function, providing the high level of genetic diversity needed to maintain system performance (11, 12). The use of high-density microarrays targeting universal 16S rRNA genes (13) and several amplicon sequencing studies (2, 14–16) revealed that municipal activated sludge contained a bacterial core that was largely dominated by the phylum Proteobacteria, followed by members of Firmicutes, Bacteroidetes, and Actinobacteria. Interestingly, this core appeared to be distributed with similar proportions across municipal wastewater treatment plants (WWTPs) regardless of reactor size, configuration, and operation mode and was consistent across continents (13). In contrast, industrial activated sludge exhibited more varied patterns, quite different from those found in municipal activated sludge, suggesting that influent wastewater characteristics have a strong influence on bacterial community composition (17, 18). According to a meta-analysis of microbial communities of 78 anaerobic digester samples, differences in substrate type also have prevailing effects on the phylogenetic structure, which largely exceed the effects caused by variations in any other operating conditions (19).
Because studies based on the 16S rRNA gene do not necessarily reflect the metabolic capabilities of populations, we reasoned that the molecular functions encoded in the metagenome could improve our ability to understand how taxonomic profiles are shaped by environmental variables and operational parameters in activated sludge. Although challenging because of the large volume of data and the reliance on incomplete databases available for annotation, shotgun metagenomics offers the possibility of revealing unknown genetic content of complex microbial communities in an unprecedented way. From one of the first reports that characterized the genetic diversity of a wastewater treatment plant using 454 sequencing technology (20) to the more recent studies that employed ultradeep Illumina sequencing (21, 22), the shotgun metagenomics approach has allowed cataloging of many functional capabilities of activated sludge, including components of relevant catabolic pathways that are key in the wastewater treatment process (23, 24). However, none of the previous studies was designed specifically to discriminate the different samples according to the observed molecular functional traits.
The ability to relate variations in gene category abundance to differences in environmental conditions was previously demonstrated in metagenomic comparisons of widely different biomes (25, 26). It is unclear, however, whether shotgun sequencing data sets could be valuable for classifying communities from ecosystems that are functionally similar, such as activated sludge ecosystems. This is a major question, given the recent suggestion that functional classification does not provide more discriminatory power than that obtained from taxonomic profiles derived via amplicon sequencing analysis of human microbiome data (27).
Our previous investigation showed that industrial activated sludge samples exhibited unique bacterial community compositions at high taxonomic ranks (17). In the present study, massive shotgun sequencing was performed with activated sludge samples from four full-scale industrial wastewater treatment plants (WWTPs) and two municipal WWTPs, to determine the functional potential encoded in sludge metagenomes. To strengthen our analysis, we included six metagenomic data sets from activated sludge samples obtained by other laboratories. The aims of this work were (i) to test whether shotgun metagenomics can complement taxon-based analyses in the task of discriminating microbial communities involved in wastewater treatment systems and (ii) to determine which functional traits could be used to elucidate drivers of microbial community assembly in activated sludge.
MATERIALS AND METHODS
Sample description.
Samples were taken from the end of the aeration basins of six well-performing full-scale wastewater treatment plants located in Argentina, including four industrial WWTPs (textile dyeing, petroleum refining, whey processing, and polymer synthesis) and two sewage treatment plants. Data on plant configuration, population equivalent, and time of sampling are given in Table S1 in the supplemental material. The six activated sludge plants included in this study were previously analyzed in our amplicon sequencing studies (17, 28) and were chosen on the basis of the different characteristics of the influent receiving streams. Although qualitative in nature, information regarding the raw materials used in the manufacturing processes of the specific industries describes crucial characteristics of the type of wastewater being treated by each WWTP. Petroleum refinery wastewater (labelled P1) contained aliphatic and aromatic hydrocarbons, phenolic compounds, and high ammonia concentrations (on the order of 100 mg/liter). The polymer wastewater treatment plant (labelled P2) received wastewater containing monomers (alkanes, alkenes, and other aliphatic hydrocarbons) from the manufacturing of basic plastics (polyethylene) and performance plastics (polyurethane). Wastewater from textile dyeing (labelled T) was characterized by the presence of unreacted dyestuffs and auxiliary chemicals such as organic acids, fixing reagents, antifoaming agents, and redox reagents, as well as high salinity due to neutralization of the high levels of sodium hydroxide. The milk whey processing plant (labelled W) discharged wastewater containing mainly lactose and milk serum proteins from the automated cleaning-in-place (CIP) systems used for cleaning of equipment and pipelines for the filtering process. Features of the WWTPs, including the plant capacity, chemical oxygen demand (COD) and biochemical oxygen demand (BOD) loading rates, sludge age, mixed liquor concentration, and type of process, are presented in Table S2 in the supplemental material. The concentration of dissolved oxygen (DO) was obtained in each WWTP from the online monitoring and recording equipment. Operational parameters are given as ranges or averages for the month prior to sampling, because it is expected that bacterial communities are shaped over a long time. For the particular case of dissolved oxygen, we preferred the use of an average value over the range between minimum and maximum values, because the latter form of computing would have been considerably biased by nonrepresentative short-term fluctuations.
Because replication of full-scale WWTPs is rarely feasible and the goal was to obtain a descriptive metagenomic data set for each WWTP, rather than to interpret detailed individual snapshots, each WWTP was sampled at two different times over a range of 1 to 4 years. This alternative approach to biological replicates allowed us to assess the variability of bacterial communities within each treatment system. The two samplings at each WWTP were performed in different seasons (see Table S1 in the supplemental material). Mixed liquor temperatures in the four industrial WWTPs were largely unaffected by the time of sampling.
In addition, two technical replicates were analyzed for each time point, to evaluate the variability in sampling and sequencing, yielding a total of 24 metagenomic samples (i.e., six WWTPs, two time points, and two technical replicates). Six additional data sets taken from the literature were included in our downstream analysis, making, in conjunction with our data, a total of 32 metagenomic samples. These data sets corresponded to all metagenomes from industrial and municipal full-scale WWTPs that had been sequenced with the Illumina HiSeq platform and were publicly available at the time of the analysis. Two WWTP samples corresponded to a petrochemical complex in western India that had been sampled at two different times (29), and four samples were from Chinese municipal facilities. One of the latter was published previously (30), whereas the other three are available in the NCBI SRA database (see Table S1 in the supplemental material).
Composite samples from local WWTPs were subjected to determination of total proteins with the Bradford assay, total carbohydrates with the anthrone reagent (31), and total volatile fatty acids with the titration method described by Ripley et al. (32). These measurements can be considered representative of the typical influent composition received by each WWTP, although it must be noted that they were not performed at the time of the sampling but at a later time.
DNA extraction and sequencing.
Sludge samples were transported to the laboratory in plastic flasks with a large air chamber and were stored at −20°C until further processing. One milliliter of sludge was centrifuged, and the pellet was washed in 1 ml of TE buffer (Tris 10 mM, EDTA 1 mM, pH 8.0). DNA extraction was performed with the FastDNA Spin kit for soil (MP Biomedicals, Inc.), as described by the manufacturer.
Each DNA sample was split into two technical replicates and sent to the Instituto de Agrobiotecnología Rosario (Rosario, Argentina) for Nextera DNA library preparation (fragment lengths, 300 to 1,000 bp), cluster generation, and sequencing. A rapid run was carried out in two lanes of the Illumina HiSeq 1500 system, yielding 319 million reads (150 bp long; 47.9 Gbp), with 8.7 million to 16.3 million reads per sample (1.30 to 2.44 Gbp) (see Table S3 in the supplemental material).
Data analysis.
Fastq format files were uploaded to the Metagenome Rapid Annotation using Subsystem Technology (MG-RAST) server (see the identification numbers in Table S3 in the supplemental material) and analyzed with its standard quality filtering and annotation pipeline, using default parameters (33). Results for separate paired-end reads exhibited minimal differences. Therefore, the results for forward (R1) unassembled reads are shown. Briefly, artificial replicates and low-quality sequences were removed according to a minimum Phred score of 15 and a maximum number of 5 bases with the minimum Phred score. Preliminary organism abundance profiles were inferred from all metagenomic shotgun reads using the best hit classification alignment procedure against the M5 nonredundant protein database (M5nr) (34). A minimal alignment of 15 nucleotides, with an E value of <1 × 10−5 and at least 60% identity, was set as threshold.
The GC content per sequence was measured on the whole set of quality-filtered reads with the infoseq function (http://emboss.open-bio.org), and results were binned in the R environment (version 3.0.2) with the hist function, in the interval range of 2%. To test the hypothesis that nucleotide compositions were affected by environmental differences (35), GC contents were calculated for each individual phylum represented in all samples, and Spearman's test was used for rank correlation of the binned average GC content of each phylum with the average GC content of the metagenomic data set (excluding the reads binning to the corresponding phylum) for each WWTP sludge sample.
Functional diversity was quantified utilizing the hierarchical structure of the SEED (a comparative genomics environment with curated subsystems) (36), particularly level 1 (general metabolic functions) and level 3 (specific metabolic pathways and cellular functions). Functional richness was computed on rarefied samples as the number of level 3 subsystems represented (1,127 categories across the 32 samples, rarefied at 2,018,487 counts).
Reads corresponding to rRNA genes were filtered (minimum length, 100 bp), and 16S rRNA reads were classified with mothur v1.33.3 (37), against the RDP database, training set 9 (38), with a 50% cutoff value. The MG-RAST pipeline identified 1.1 × 105 to 6.9 × 105 reads as probable fragments of the 16S rRNA gene (70% identity), of which only 2.3 × 103 to 21.5 × 103 remained after length filtering. Taxonomic richness was estimated on the basis of the abundance of bacterial genera rarefied at 776 counts (810 genera across the 32 samples).
Differences in taxonomic and functional diversity between municipal and industrial activated sludge communities were verified using unpaired t tests. Principal-component analysis (PCA), principal-coordinate analysis (PCoA), and correspondence analysis (CA) were performed in the R environment with the vegan package (Department of Statistics, Iowa State University, Ames, IA, USA). For Table S5 in the supplemental material, PCA and CA were based on SEED subsystems at level 3. Their corresponding ordination loadings were sorted by PCA1 and CA1 (main contributing axes), and the top 10 and bottom 10 functions were listed in order to retrieve the 20 most relevant drivers of both ordinations. Relative loading values are reported, together with the proportion of variance explained by each main axis.
Comparison of functional traits between metagenomes of samples from municipal and petrochemical WWTPs was performed with STAMP v2.0.9 software (39), using a two-sided t test. Technical replicates and samples belonging to different sampling dates were averaged in this analysis. To correct for potential false-positive results, the Benjamini-Hochberg false discovery rate was used for multiple test correction.
To statistically evaluate the association of operational parameters with communities' functional structures, permutation tests based on 1,000 iterations were performed using the vegan envfit function for level 1 subsystem PCA, using WWTPs for the strata option, which allows the inclusion of replicates. Samples that were not sequenced in this study were not included, because of the incompleteness of the available metadata. In order to avoid overfitting, only variables that had been previously evaluated with respect to their roles in shaping the structure of these activated sludge communities (17) were included in the fitting analysis.
Assembly of metagenomic reads.
Raw read files of technical replicates were merged, and low-quality reads were filtered out with Trimmomatic v0.35 (40), using the recommended parameters for paired-end reads to remove low-quality or undetermined bases (http://www.usadellab.org/cms/index.php?page=trimmomatic). For assembly of paired-end reads, Velvet v1.2.10 (41) was utilized to build de Brujin graphs, which were subsequently decomposed with MetaVelvet v1.2.01 (42), setting the k-mer coverage peaks manually. The average insert length was set at 350 bp, and contigs shorter than 1 kb were excluded. A range of k-mer lengths (51 to 101 bp) was tested for each sample, and the optimal k-mer length was chosen after evaluation of four basic parameters of the final assembly, namely, size (base pairs), number of scaffolds, maximal length, and N50.
Assembled metagenomic data sets were processed through the MG-RAST annotation pipeline in its mode for assembled contigs. For the recovery of population genomes from metagenomic data, binning of contigs was performed on the basis of differential coverage, using samples from two different time points, combined with taxonomic affiliation, followed by tetranucleotide frequency PCA assisted by GC content visualization (43). Estimation of the relative abundance of recovered genomes was assessed by mapping all reads with Bowtie2 (44) (see Table S7 in the supplemental material). Taxonomic classification using bidirectional best hits and functional annotation of each recovered genome were performed on the RAST server (45).
Accession number(s).
Raw sequence files were deposited in the NCBI SRA under accession number SRP060024 (see Table S3 in the supplemental material).
RESULTS
Wastewater treatment plant performance.
At the time of sampling, all WWTPs showed good stable performance, with BOD removal efficiencies in the range of 85 to 95%. None of the WWTPs was designed for nutrient removal. Phosphorus deficiency in the petrochemical WWTPs and nitrogen deficiency in the textile dyeing wastewater were corrected by the addition of phosphoric acid and urea, respectively. The available metadata, in terms of the capacity of all WWTPs, chemical oxygen demand, biochemical oxygen demand, BOD loading rates, sludge age, mixed liquor concentrations, and system configurations, are shown in Tables S1 and S2 in the supplemental material. Data varied substantially among different WWTPs treating similar types of wastewater, such as municipal or petrochemical WWTPs.
Shotgun sequencing overview.
Over 85 to 93% of the reads obtained from the two municipal and four industrial wastewater treatment plants sampled by our laboratory passed the quality control tests (see Table S3 in the supplemental material). The filtered data set contained 34 to 60% predicted proteins with known functions and 27 to 53% reads that were annotated as unknown proteins. Taxonomic classification of unassembled metagenomic reads showed dominance of the domain Bacteria (96.6 to 98.9%), followed by small proportions of reads from Archaea (0.4 to 1.1%), Eukarya (0.6 to 2.3%), and viruses (0.02 to 0.07%). Interestingly, the analysis of the sequence composition of the individual reads in each data set according to GC content indicated that samples from each WWTP, taken at different time points, were distinguished by a characteristic profile, suggesting phylogenetic relatedness of microbial communities over time (Fig. 1). In addition, GC content patterns of samples from WWTPs treating different types of wastewater were very different. Samples from municipal activated sludge displayed two main peaks, with maxima at 35% and 70% GC, whereas samples from activated sludge treating petrochemical wastewater exhibited mostly a dominant peak with a high GC content (Fig. 1). It is clear from Fig. S1 in the supplemental material, representing the empirical cumulative distribution function of GC percentages in metagenomic reads, that municipal and petrochemical WWTPs have very different GC content patterns, with medians of 51 to 61% and 63 to 65%, respectively.
FIG 1.

Histograms showing GC contents per sequence across municipal and industrial activated sludge samples. P1 to P4, petrochemical; T, textile dyeing; W, whey filtering; S1 and S2, municipal. Insets, empirical cumulative frequency distribution (CFD) values. Only WWTPs that were sampled twice are shown. Solid and dotted lines, data from different sampling times (see Table S1 in the supplemental material).
Because the mean GC contents differ among phyla (see Fig. S2 in the supplemental material), the observed differences in nucleotide composition could be explained, in principle, by the observed taxonomic differences between microbial communities from the different WWTPs (17). The GC contents of individual phyla were distributed over various ranges (see Fig. S2), however, and thus the environment could also contribute to the variation in GC contents by selecting bacteria with certain nucleotide compositions within each phylum. To test the latter possibility, the association between the average GC content of each phylum and the average GC content of the entire metagenomic data set, excluding the phylum being evaluated, was computed using Spearman's rank correlation. Table 1 shows that the GC contents of 7 of the 11 most represented phyla followed significantly similar rank orders, compared with the average GC contents of the remaining phyla in each WWTP data set (see Table S4 in the supplemental material). This was also true when the highly dominating Proteobacteria phylum was removed from the metagenomic data set.
TABLE 1.
Spearman rank correlations of the GC contents of each phylum across all samples (n = 12) and the average GC contents of all reads, excluding the reads assigned to the corresponding phylum
| Phylum | ρ | P |
|---|---|---|
| Acidobacteria | 0.47 | 0.13 |
| Actinobacteria | 0.48 | 0.12 |
| Bacteroidetes | 0.48 | 0.12 |
| Chlorobi | 0.55 | 0.06 |
| Chloroflexi | 0.75 | 0.01 |
| Deinococcus-Thermus | 0.87 | <0.01 |
| Euryarchaeota | 0.81 | <0.01 |
| Nitrospirae | 0.63 | 0.03 |
| Planctomycetes | 0.90 | <0.01 |
| Proteobacteria | 0.69 | 0.01 |
| Verrucomicrobia | 0.64 | 0.02 |
Taxonomic and functional richness of activated sludge samples.
The distribution of bacterial taxa determined from shotgun reads corresponding to the bacterial 16S rRNA gene was similar to the distribution previously detected using amplicon sequencing (17). Actinobacteria and candidate phylum TM7 were found at higher percentages, i.e., 31.1% and 7.4%, respectively, in activated sludge samples from the whey processing industry, while Planctomycetes and Verrucomicrobia exhibited relatively high abundance levels, i.e., 8.5% and 8.0%, respectively, in wastewater sludge from the textile dyeing plant. Chlorobi was associated with plants treating petrochemical wastewater, although with low abundance (see Fig. S3A in the supplemental material).
The taxonomic richness of municipal activated sludge was significantly greater than that of industrial activated sludge (t test, P = 0.003) (Fig. 2). Similarly, activated sludge treating municipal sewage exhibited greater functional richness than did sludge treating industrial wastewater (P = 0.001) (Fig. 2). Importantly, the functional richness was significantly correlated with the measured taxonomic richness (Pearson's r = 0.87, P < 0.001) (see Fig. S4 in the supplemental material).
FIG 2.

Bacterial richness of industrial and municipal activated sludge samples. (A) Number of represented SEED subsystems (level 3) per sample (t test, P = 0.001). (B) Number of genera detected per sample, based on reads annotated as 16S rRNA (t test, P = 0.003). Both estimators were rarefied; technical replicates and data from the two different sampling times were averaged for each WWTP. Horizontal bars represent median values. Asterisks indicate significant differences (P < 0.01).
Profiling of activated sludge samples according to functional categories.
We investigated whether functional traits detected among the shotgun sequencing reads could provide useful discriminatory power to differentiate activated sludge samples. To this end, relative abundances of SEED subsystems at level 1 were first subjected to principal-component analysis (Fig. 3). Each set of technical replicates clustered closely together, indicating that sequencing was reproducible and that the distributions of gene fragments were not distorted by random sampling from metagenomic data sets with low coverage. The first principal component, which accounted for 55% of the total variance, separated samples (including data sets generated in laboratories in India and China) according to wastewater type. A t test of the first principal component scores showed that there were significant differences between samples from municipal and petrochemical WWTPs (P = 0.003). Sludge samples from textile dyeing and whey processing wastewater treatment plants were located on the opposite side of the biplot. The two most influential subsystems in the ordination were carbohydrates and metabolism of aromatic compounds, which were associated mainly with the activated sludge of whey processing and petrochemical plants, in accordance with the typical chemical contents of the respective influent wastewater (see Table S2 in the supplemental material). A third important category was protein metabolism, which was associated with the whey processing plant in the first principal component and with the sewage facilities in the second principal component (Fig. 3). This result was consistent with the high protein contents in such wastewater (see Table S2).
FIG 3.
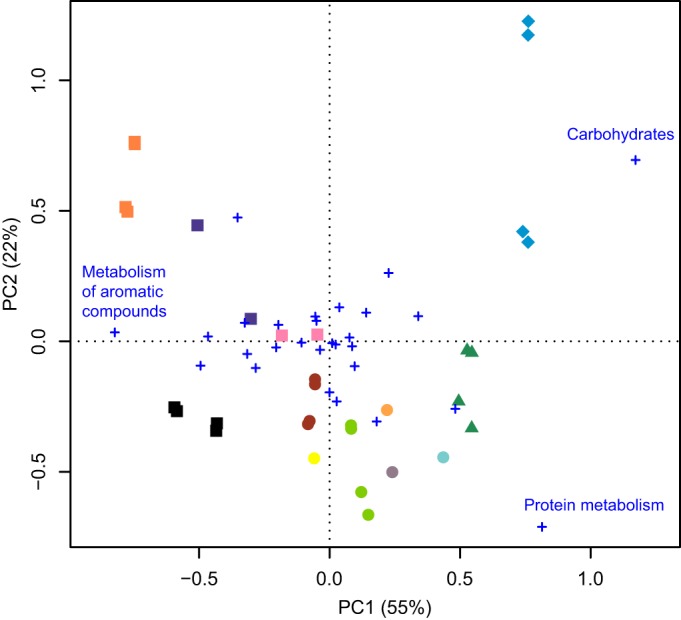
Principal-component analysis of the abundance of SEED subsystems at level 1. Activated sludge samples are labeled according to the WWTP (different colors) and the type of wastewater treated (circles, municipal; squares, petrochemical; triangles, textile dyeing; diamonds, whey processing). Functional categories (crosses) were labeled only in cases in which PC1 was at least |0.5|. Technical replicates and data from the two different sampling times were included separately.
In order to investigate which functional traits were responsible for the separation along the first axis, we searched among the highest loadings of both the principal-component analysis and the correspondence analysis, based on level 3 SEED subsystems. We found that sugar utilization genes, such as those for lactose and galactose uptake and utilization, were more represented at the whey processing treatment facility (PCA1 of >0 and CA1 of >0), whereas acetophenone carboxylase 1 and n-phenylalkanoic acid degradation, both part of the metabolism of aromatic compounds, had greater prevalence at wastewater treatment plants from petrochemical facilities (PCA1 of <0 and CA1 of <0) (see Table S5 in the supplemental material).
Five operational variables (temperature, pH, mixed liquor suspended solids [MLSS], solids retention time [SRT], and dissolved oxygen [DO]), which were selected on the basis of the important roles that these factors play in shaping the structures of activated sludge microbial communities, were tested for their associations with the functional profiles of activated sludge. A procedure involving permutations to fit the environmental variables to the subsystem PCA at level 1 showed a significant P value only for dissolved oxygen (see Table S6 in the supplemental material).
Taxa exhibited more variations than did functions across all WWTPs (see Fig. S3 in the supplemental material). Although the magnitude of the differences between WWTPs was lower at the functional level than at the taxonomic level, functional categories were able to achieve clear discrimination between activated sludge communities according to influent wastewater type. Subsystem-based PCoA clustered samples in a manner similar to that of 16S rRNA-based ordination (Mantel test, R = 0.76, P < 0.0001), except that the analysis based on functional genes was better able to separate municipal plants from petrochemical WWTPs and explained a greater relative variance in the two most relevant axes (see Fig. S5 in the supplemental material).
We focused particularly on samples from municipal and petrochemical wastewater treatment plants, because the numbers of plants receiving each type of wastewater in our data set (six plants and four plants, respectively) allowed us to make statistical comparisons of functional profiles between the two groups of WWTPs. Functional annotation (level 1 of SEED subsystems) showed that, among all categories, seven were found to be significantly different. Most of the annotations displaying significantly greater proportions in samples from municipal WWTPs were related to nucleotide and protein metabolism, whereas samples from petrochemical WWTPs were relatively enriched in genes associated with membrane transport and cofactor synthesis (Fig. 4). At level 3, of 21 categories exhibiting significant differences, 14 were characteristic for petrochemical activated sludge, including sulfur and benzoate metabolism-related genes; the other 7 were diverse traits related to general cellular functions that were represented more in municipal activated sludge (see Fig. S6 in the supplemental material).
FIG 4.

Comparison of functional traits (level 1 SEED subsystems; cutoff value, 0.5% relative abundance) between metagenomes of petrochemical (n = 4) (blue) and municipal (n = 6) (red) activated sludge samples. Technical replicates and data from the two different sampling times were averaged for each WWTP. Pr., prosthetic.
The quantitative analysis derived from unassembled reads may be affected by the low reliability of annotation afforded by such short reads. In order to validate the accuracy of our approach, we compared the relative abundance of functional categories across assembled metagenomes, normalized using Z-scores (Fig. 5). Clear differences were observed for key functional categories related to wastewater composition. The genes involved in the metabolism of aromatic compounds, fatty acids, lipids, and isoprenoids, sulfur metabolism, and the stress response were enriched in activated sludge communities from the local petrochemical WWTPs. In contrast, pathways related to the metabolism of carbohydrates were augmented in the WWTPs from the whey processing plant and, less prominently, the textile dyeing industry. These results are in accordance with the higher concentrations of hydrocarbons and carbohydrates, respectively, in influent wastewater (see Table S2 in the supplemental material).
FIG 5.

Comparison of subsystems (level 1) among assembled metagenomic reads. Relative abundance values of functional categories at the two sampling points were transformed to Z-scores and averaged. W, whey filtering; T, textile dyeing; S1 and S2, municipal; P1 and P2, petrochemical; Pr., prosthetic. Error bars, standard deviations.
In silico function-based analysis of recovered genomes.
Additional insight confirming the previous observations was obtained from the recovered genome sequences extracted from metagenomic data. The assembly statistics for the extracted genomes are presented in Table S7 in the supplemental material. A detailed analysis of genomes recovered in all data sets is beyond the scope of this work; we highlight the most relevant issues here.
Several partial genomes recovered from petrochemical facilities belonged to bacteria specialized in either the metabolism of aromatic compounds or sulfur metabolism. Among the former, both petrochemical WWTPs contained bacteria classified in the family Comamonadaceae (such as Alicycliphilus denitrificans), which are capable of degrading alicyclic or aromatic compounds. In accordance, a gene encoding phenol monooxygenase (46) was annotated in the A. denitrificans genome. Gene sequences coding for functions clustered in the subsystem involving fatty acids, lipids, and isoprenoids were also overrepresented in the assembled genome of A. denitrificans. Partial genomes recovered from the petroleum refinery also included Mycobacterium vanbaalenii and Aromatoleum aromaticum. For the former, the presence of two genes annotated in a dioxygenase pathway is in agreement with the reported ability of members of this species to degrade polycyclic aromatic hydrocarbons (47). A. aromaticum is an aromatic compound-degrading bacterium belonging to the Azoarcus-Thauera cluster within the Betaproteobacteria. Its recovered partial genome contained the last enzyme of the pathway for the conversion of ethylbenzene to benzoyl-CoA (48). Another genome recovered from the petroleum refinery metagenome matched the autotrophic sulfur-oxidizing Proteobacteria species Thiobacillus denitrificans, a species known to perform sulfur compound oxidation (49).
Most of the recovered genomes (16/20 genomes) from the WWTP in the whey processing industry had greater than average percentages in the category related to the metabolism of carbohydrates (SEED subsystem level 1). This is consistent with the high levels of carbohydrates in the influent wastewater (see Table S2 in the supplemental material). Bidirectional best-hit classification revealed that five of the extracted genomes belonged to the genus Propionibacterium. Finally, a number of recovered genomes in the Planctomycetes-Verrucomicrobia-Chlamydiae superphylum distinguished the WWTP treating textile dyeing wastewater (see Table S7 in the supplemental material). Pedosphaera parvula Ellin514 (50) shares the trait of members of Verrucomicrobia of being specialized in the utilization of sugars. Interestingly, the genome of Delftia acidovorans, a bacterium that can degrade linear alkylbenzenesulfonate surfactants (51), was also recovered from the metagenome of the textile dyeing WWTP.
DISCUSSION
Wastewater type is a major determinant of the unique composition of bacterial communities of activated sludge. In this work, we showed that shotgun metagenomics complemented taxon-based analyses in the task of discriminating microbial communities involved in wastewater treatment systems. Importantly, differences in bacterial community structure between WWTPs were consistent with underlying differences in the abundance of distinctive sets of functional categories.
The issue of representativeness was an essential criterion of our experimental design. Sampling each WWTP at two different times, separated by 1 to 4 years, allowed us to assess the variability of bacterial communities within each treatment system, providing a feasible alternative to the use of actual biological replicates. Additionally, the use of technical replicates confirmed that shotgun sequencing was highly reproducible, with no bias resulting from differences in fragment distributions related to random sampling of data sets with low coverage.
The taxonomic and functional richness of municipal activated sludge was significantly greater than that of industrial activated sludge, an observation that is in agreement with our previous conclusions drawn from amplicon sequencing data (17). Based on a metagenomic analysis of 16S rRNA amplicon and mRNA shotgun reads from 10 full-scale wastewater treatment plants in Switzerland, Johnson et al. (22) reported a positive association between the number of bacterial taxa and the number of functional categories and argued that the result was consistent with the ecological theory prediction that richer communities encompass a broader range of functions (52). It is unlikely that the greater diversity observed in sludge from sewage treatment facilities could result from the presence of nongrowing species introduced into the system through immigration from sewage, compared to industrial wastewater, because this fraction makes only a small contribution to bacterial richness (53). Rather, we speculate that municipal sewage contains a much wider range of chemical constituents, in comparison to industrial wastewater; therefore, microbial richness is strongly influenced by the diversity of carbon substrates (54). However, some caution should be exercised in deriving conclusions on richness from the annotation of metagenomic sequence data alone, owing to the fact that activated sludge community compositions vary among WWTPs and the potentially uneven representation of taxa in databases used for massive annotation could lead to biased richness estimation.
Community GC contents exhibited characteristic profiles, which were conserved within treatment plants on a time scale of years and between treatment plants sharing similar influent wastewater. In contrast, GC contents of activated sludge from WWTPs receiving different wastewater types were clearly distinct. It has been suggested that the base composition of bacterial genomes may be subject to selection at many sites (55), and the nucleotide composition of complex microbial communities appears to be largely influenced by both phylogeny and environment (56). As previously observed from shotgun sequencing metagenomic data sets taken from many different types of environments (35), the GC contents of each phylum were distributed over different ranges across all WWTP data sets. Therefore, we followed the hypothesis that, if environmental forces influence the nucleotide composition, then each phylum would be ranked according to GC contents in a correlated manner across all sludge communities (35). The GC contents of 7 of the 11 most represented phyla were correlated significantly with the average GC contents of the remaining phyla in each WWTP data set, indicating that the factors affecting nucleotide composition acted similarly across taxa; therefore, the variation in nucleotide contents is driven by environmental differences between WWTPs (35). Since the GC contents appeared to be related to wastewater type, with petrochemical WWTPs showing the highest values (see Fig. S7 in the supplemental material), we propose that the characteristics of influent wastewater likely represent one of the main environmental drivers of the variations in nucleotide composition. The presence of harsh environmental conditions found in specific industrial wastewater (10) could potentially be added to the list of environmental factors, such as UV exposure, high temperatures, and aerobiosis, which were assumed to drive changes in the base composition of microbial populations (57). However, mechanisms explaining how or why these environments exert pressure that would favor changes in GC contents are still lacking.
Taxa exhibited more variation than did functions across WWTPs. Similar behavior has already been revealed by comparative metagenomics for several body habitats from different human individuals (58). This is consistent with the existence of a large core of genes that are essential for cellular and community functions (i.e., encoding proteins involved in metabolic pathways). Although the magnitude of the differences between WWTPs was smaller at the functional level than at the taxonomic level, functional categories were able to achieve clear discrimination between activated sludge communities according to influent wastewater type.
All samples, including data sets from municipal and petrochemical sludge samples generated in laboratories from India and China (29, 30), clustered according to wastewater type. The capacity of SEED subsystem-based annotations to detect differential traits in related but distinct microbial ecosystems reported in this work for activated sludge samples has been recognized in previous studies. Lamendella and coworkers identified unique functional elements that distinguished the gut of pigs from the gut of other animals, such as cows, chickens, and fish (59). Differences were also reported within separate environments of the human body (58), where low-abundance pathways were consistently represented only at particular body sites. In permafrost soil, a metagenomic analysis allowed the detection of rapid shifts in functional genes in response to thaw (60). This work demonstrates that shotgun sequencing can also be used to classify functionally similar ecosystems.
Process and environmental variables can be also important factors in shaping the bacterial community structure. In particular, the influence of oxygen concentrations on the process performance and community structure of biological treatment systems has been addressed previously (17, 61, 62). Chapman et al. hypothesized that deeper oxygen penetration inside the floc at high dissolved oxygen concentrations (∼6 mg/liter) could sustain greater numbers of viable microorganisms within the system (63). In this work, we found that dissolved oxygen concentrations were significantly associated with functional profiles of activated sludge but could not be used to group samples into meaningful bins.
By performing a statistical comparison between municipal and petrochemical sludge samples, we observed that the former were enriched in genes related to nucleotide and protein metabolism, which may indicate the characteristic need for bacteria in municipal WWTPs to continuously process a larger variety of biological products. In contrast, the enrichment of genes associated with membrane transport in samples from petrochemical WWTPs may reflect the fact that the passage of hydrophobic substrates across the cell membrane is a first step in the biodegradation of hydrocarbons (64). We initially used unassembled reads to extract ecological information from the data sets. It might be argued that the confidence in the annotation accuracy is greatly diminished when the annotation is based on such short sequences, so the data would be valid only at a very general level. To address this concern, confirmation of the observed differences between wastewater treatment plants receiving influent of different compositions was obtained by a qualitative comparison of assembled metagenomic sequences generated from local WWTPs (Fig. 5). Subsystems related to the metabolism of aromatic compounds, fatty acids, lipids, and isoprenoids, sulfur metabolism, and the stress response were enriched in communities from petrochemical wastewaters, whereas communities from the whey processing plant invested heavily in the metabolism of carbohydrates.
Results obtained from the comparison of unassembled reads (Fig. 4) and from the comparison of assembled contigs (Fig. 5) were in reasonable agreement. The comparison of assembled reads indicated that communities from local municipal WWTPs were enriched in genes related to nucleotide metabolism and protein metabolism. In contrast, petrochemical WWTP communities contained a greater abundance of genes related to cofactors, vitamins, prosthetic groups, and pigments and a lesser representation of genes related to protein metabolism. The few conflicting results could be attributed to the fact that many reads either were not included in contigs or were included in contigs shorter than 1 kb. In addition, frequency information is partially missed when multiple reads are assembled into a contig (33). A third source of discrepancy could be the fact that unassembled reads were compared using all municipal and petrochemical samples, whereas assembled contigs were generated only from metagenomic DNA extracted from local WWTPs. Thus, Fig. 5 showed that only one of the two local petrochemical WWTPs displayed increased abundance of the membrane transport category. Analogously, assembled reads matching the categories of the stress response and sulfur metabolism showed clear differences between local petrochemical and municipal plants, although the differences were not significant when the complete unassembled data set was used for comparison between groups. Still, the metagenomes of petrochemical activated sludge samples did show a significantly larger proportion of genes matching sulfur oxidation (level 3 of the SEED hierarchy), compared with samples from municipal plants (see Fig. S6 in the supplemental material).
The inferred taxonomic and functional assignment of population genomes recovered from each metagenome, using differential coverage as the primary binning method (43), could also be related to the metabolic potential that would be expected according to the source of the wastewater. Thus, petrochemical WWTPs contained a large number of recovered genomes with hydrocarbon and sulfur metabolism potential, whereas metagenomes from the whey processing WWTP were dominated by lactose-using Propionibacterium genomes, and the textile dyeing WWTP was characterized by genomes belonging to the Planctomycetes-Verrucomicrobia-Chlamydiae superphylum. The recovered scaffolds included relatively abundant microbial populations, corresponding to 0.5% to 15.3% of relative metagenome abundance (see Table S7 in the supplemental material). A sequencing coverage higher than the one used in this study would be required to assess the role of less abundant members of the community.
We concluded that wastewater influent is an overriding factor shaping the metagenomic composition of activated sludge. Although this study included a limited number of WWTPs, these findings are particularly exciting, given that the studied wastewater treatment plants have very different configurations and some of the data were obtained in different laboratories and sequenced under similar but not identical conditions. Our work highlights the sensitivity of comparative shotgun metagenomics to achieve considerable levels of discrimination of complex microbial consortia having related functions in environmental biotechnology processes. We expect that the availability of more WWTP metagenomes, together with richer and more comprehensive databases and the continuing improvement in annotation methods, will allow a more complete interpretation of metagenomic data, which in turn will lead to a more thorough understanding of microbial assembly in biotechnological processes.
Supplementary Material
ACKNOWLEDGMENTS
We thank WWTP staff members who kindly provided us with samples and the information in Tables S1 and S2 in the supplemental material. We are also grateful to the research groups that made their metagenomic data publicly available.
F.M.I. is a fellow and E.L.M.F. and L.E. are researchers from the Consejo Nacional de Investigaciones Científicas y Técnicas of Argentina.
We have no conflicts of interest to declare.
Funding Statement
Funding was provided to Leonardo Erijman by MINCyT | Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT) under grant number PICT 2012 1877 and by Universidad de Buenos Aires (UBA) (UBACyT 2012-2015). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00916-16.
REFERENCES
- 1.Zhou J, He Z, Yang Y, Deng Y, Tringe SG, Alvarez-Cohen L. 2015. High-throughput metagenomic technologies for complex microbial community analysis: open and closed formats. mBio 6:e02288-14. doi: 10.1128/mBio.02288-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang T, Shao MF, Ye L. 2012. 454 pyrosequencing reveals bacterial diversity of activated sludge from 14 sewage treatment plants. ISME J 6:1137–1147. doi: 10.1038/ismej.2011.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao D, Huang R, Zeng J, Yu Z, Liu P, Cheng S, Wu QL. 2014. Pyrosequencing analysis of bacterial community and assembly in activated sludge samples from different geographic regions in China. Appl Microbiol Biotechnol 98:9119–9128. doi: 10.1007/s00253-014-5920-3. [DOI] [PubMed] [Google Scholar]
- 4.Ayarza JM, Guerrero LD, Erijman L. 2010. Nonrandom assembly of bacterial populations in activated sludge flocs. Microb Ecol 59:436–444. doi: 10.1007/s00248-009-9581-1. [DOI] [PubMed] [Google Scholar]
- 5.Ayarza JM, Erijman L. 2011. Balance of neutral and deterministic components in the dynamics of activated sludge floc assembly. Microb Ecol 61:486–495. doi: 10.1007/s00248-010-9762-y. [DOI] [PubMed] [Google Scholar]
- 6.Lee SH, Kang HJ, Park HD. 2015. Influence of influent wastewater communities on temporal variation of activated sludge communities. Water Res 73:132–144. doi: 10.1016/j.watres.2015.01.014. [DOI] [PubMed] [Google Scholar]
- 7.Ju F, Xia Y, Guo F, Wang Z, Zhang T. 2014. Taxonomic relatedness shapes bacterial assembly in activated sludge of globally distributed wastewater treatment plants. Environ Microbiol 16:2421–2432. doi: 10.1111/1462-2920.12355. [DOI] [PubMed] [Google Scholar]
- 8.Ju F, Zhang T. 2015. Bacterial assembly and temporal dynamics in activated sludge of a full-scale municipal wastewater treatment plant. ISME J 9:683–695. doi: 10.1038/ismej.2014.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ofiteru ID, Lunn M, Curtis TP, Wells GF, Criddle CS, Francis CA, Sloan WT. 2010. Combined niche and neutral effects in a microbial wastewater treatment community. Proc Natl Acad Sci U S A 107:15345–15350. doi: 10.1073/pnas.1000604107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Figuerola EL, Erijman L. 2007. Bacterial taxa abundance pattern in an industrial wastewater treatment system determined by the full rRNA cycle approach. Environ Microbiol 9:1780–1789. doi: 10.1111/j.1462-2920.2007.01298.x. [DOI] [PubMed] [Google Scholar]
- 11.Lawson CE, Strachan BJ, Hanson NW, Hahn AS, Hall ER, Rabinowitz B, Mavinic DS, Ramey WD, Hallam SJ. 2015. Rare taxa have potential to make metabolic contributions in enhanced biological phosphorus removal ecosystems. Environ Microbiol 17:4979–4993. doi: 10.1111/1462-2920.12875. [DOI] [PubMed] [Google Scholar]
- 12.Shade A, Jones SE, Caporaso JG, Handelsman J, Knight R, Fierer N, Gilbert JA. 2014. Conditionally rare taxa disproportionately contribute to temporal changes in microbial diversity. mBio 5:e01371-14. doi: 10.1128/mBio.01371-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xia S, Duan L, Song Y, Li J, Piceno YM, Andersen GL, Alvarez-Cohen L, Moreno-Andrade I, Huang CL, Hermanowicz SW. 2010. Bacterial community structure in geographically distributed biological wastewater treatment reactors. Environ Sci Technol 44:7391–7396. doi: 10.1021/es101554m. [DOI] [PubMed] [Google Scholar]
- 14.Wang X, Hu M, Xia Y, Wen X, Ding K. 2012. Pyrosequencing analysis of bacterial diversity in 14 wastewater treatment systems in China. Appl Environ Microbiol 78:7042–7047. doi: 10.1128/AEM.01617-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ranasinghe PD, Satoh H, Oshiki M, Oshima K, Suda W, Hattori M, Mino T. 2012. Revealing microbial community structures in large- and small-scale activated sludge systems by barcoded pyrosequencing of 16S rRNA gene. Water Sci Technol 66:2155–2161. doi: 10.2166/wst.2012.428. [DOI] [PubMed] [Google Scholar]
- 16.Ju F, Zhang T. 2015. 16S rRNA gene high-throughput sequencing data mining of microbial diversity and interactions. Appl Microbiol Biotechnol 99:4119–4129. doi: 10.1007/s00253-015-6536-y. [DOI] [PubMed] [Google Scholar]
- 17.Ibarbalz FM, Figuerola EL, Erijman L. 2013. Industrial activated sludge exhibit unique bacterial community composition at high taxonomic ranks. Water Res 47:3854–3864. doi: 10.1016/j.watres.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 18.van der Gast CJ, Ager D, Lilley AK. 2008. Temporal scaling of bacterial taxa is influenced by both stochastic and deterministic ecological factors. Environ Microbiol 10:1411–1418. doi: 10.1111/j.1462-2920.2007.01550.x. [DOI] [PubMed] [Google Scholar]
- 19.Zhang W, Werner JJ, Agler MT, Angenent LT. 2014. Substrate type drives variation in reactor microbiomes of anaerobic digesters. Bioresour Technol 151:397–401. doi: 10.1016/j.biortech.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 20.Sanapareddy N, Hamp TJ, Gonzalez LC, Hilger HA, Fodor AA, Clinton SM. 2009. Molecular diversity of a North Carolina wastewater treatment plant as revealed by pyrosequencing. Appl Environ Microbiol 75:1688–1696. doi: 10.1128/AEM.01210-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ju F, Guo F, Ye L, Xia Y, Zhang T. 2014. Metagenomic analysis on seasonal microbial variations of activated sludge from a full-scale wastewater treatment plant over 4 years. Environ Microbiol Rep 6:80–89. doi: 10.1111/1758-2229.12110. [DOI] [PubMed] [Google Scholar]
- 22.Johnson DR, Lee TK, Park J, Fenner K, Helbling DE. 2015. The functional and taxonomic richness of wastewater treatment plant microbial communities are associated with each other and with ambient nitrogen and carbon availability. Environ Microbiol 17:4851–4860. doi: 10.1111/1462-2920.12429. [DOI] [PubMed] [Google Scholar]
- 23.Fang H, Cai L, Yu Y, Zhang T. 2013. Metagenomic analysis reveals the prevalence of biodegradation genes for organic pollutants in activated sludge. Bioresour Technol 129:209–218. doi: 10.1016/j.biortech.2012.11.054. [DOI] [PubMed] [Google Scholar]
- 24.More RP, Mitra S, Raju SC, Kapley A, Purohit HJ. 2014. Mining and assessment of catabolic pathways in the metagenome of a common effluent treatment plant to induce the degradative capacity of biomass. Bioresour Technol 153:137–146. doi: 10.1016/j.biortech.2013.11.065. [DOI] [PubMed] [Google Scholar]
- 25.Fierer N, Leff JW, Adams BJ, Nielsen UN, Bates ST, Lauber CL, Owens S, Gilbert JA, Wall DH, Caporaso JG. 2012. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc Natl Acad Sci U S A 109:21390–21395. doi: 10.1073/pnas.1215210110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dinsdale EA, Edwards RA, Hall D, Angly F, Breitbart M, Brulc JM, Furlan M, Desnues C, Haynes M, Li L, McDaniel L, Moran MA, Nelson KE, Nilsson C, Olson R, Paul J, Brito BR, Ruan Y, Swan BK, Stevens R, Valentine DL, Thurber RV, Wegley L, White BA, Rohwer F. 2008. Functional metagenomic profiling of nine biomes. Nature 452:629–632. doi: 10.1038/nature06810. [DOI] [PubMed] [Google Scholar]
- 27.Xu Z, Malmer D, Langille MG, Way SF, Knight R. 2014. Which is more important for classifying microbial communities: who's there or what they can do? ISME J 8:2357–2359. doi: 10.1038/ismej.2014.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ibarbalz FM, Perez MV, Figuerola EL, Erijman L. 2014. The bias associated with amplicon sequencing does not affect the quantitative assessment of bacterial community dynamics. PLoS One 9:e99722. doi: 10.1371/journal.pone.0099722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yadav TC, Pal RR, Shastri S, Jadeja NB, Kapley A. 2015. Comparative metagenomics demonstrating different degradative capacity of activated biomass treating hydrocarbon contaminated wastewater. Bioresour Technol 188:24–32. doi: 10.1016/j.biortech.2015.01.141. [DOI] [PubMed] [Google Scholar]
- 30.Ye L, Zhang T, Wang T, Fang Z. 2012. Microbial structures, functions, and metabolic pathways in wastewater treatment bioreactors revealed using high-throughput sequencing. Environ Sci Technol 46:13244–13252. doi: 10.1021/es303454k. [DOI] [PubMed] [Google Scholar]
- 31.Yemm EW, Willis AJ. 1954. The estimation of carbohydrates in plant extracts by anthrone. Biochem J 57:508–514. doi: 10.1042/bj0570508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ripley L, Boyle W, Converse J. 1986. Improved alkalimetric monitoring for anaerobic digestion of high-strength wastes. J Water Pollut Control Fed 58:406–411. [Google Scholar]
- 33.Meyer F, Paarmann D, D'Souza M, Olson R, Glass EM, Kubal M, Paczian T, Rodriguez A, Stevens R, Wilke A, Wilkening J, Edwards RA. 2008. The metagenomics RAST server: a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9:386. doi: 10.1186/1471-2105-9-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilke A, Harrison T, Wilkening J, Field D, Glass EM, Kyrpides N, Mavrommatis K, Meyer F. 2012. The M5nr: a novel non-redundant database containing protein sequences and annotations from multiple sources and associated tools. BMC Bioinformatics 13:141. doi: 10.1186/1471-2105-13-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reichenberger ER, Rosen G, Hershberg U, Hershberg R. 2015. Prokaryotic nucleotide composition is shaped by both phylogeny and the environment. Genome Biol Evol 7:1380–1389. doi: 10.1093/gbe/evv063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang HY, Cohoon M, de Crecy-Lagard V, Diaz N, Disz T, Edwards R, Fonstein M, Frank ED, Gerdes S, Glass EM, Goesmann A, Hanson A, Iwata-Reuyl D, Jensen R, Jamshidi N, Krause L, Kubal M, Larsen N, Linke B, McHardy AC, Meyer F, Neuweger H, Olsen G, Olson R, Osterman A, Portnoy V, Pusch GD, Rodionov DA, Ruckert C, Steiner J, Stevens R, Thiele I, Vassieva O, Ye Y, Zagnitko O, Vonstein V. 2005. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res 33:5691–5702. doi: 10.1093/nar/gki866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parks DH, Tyson GW, Hugenholtz P, Beiko RG. 2014. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30:3123–3124. doi: 10.1093/bioinformatics/btu494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Namiki T, Hachiya T, Tanaka H, Sakakibara Y. 2012. MetaVelvet: an extension of Velvet assembler to de novo metagenome assembly from short sequence reads. Nucleic Acids Res 40:e155. doi: 10.1093/nar/gks678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Albertsen M, Hugenholtz P, Skarshewski A, Nielsen KL, Tyson GW, Nielsen PH. 2013. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol 31:533–538. doi: 10.1038/nbt.2579. [DOI] [PubMed] [Google Scholar]
- 44.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. 2008. The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oosterkamp MJ, Veuskens T, Saia FT, Weelink SAB, Goodwin LA, Daligault HE, Bruce DC, Detter JC, Tapia R, Han CS, Land ML, Hauser LJ, Langenhoff AAM, Gerritse J, van Berkel WJH, Pieper DH, Junca H, Smidt H, Schraa G, Davids M, Schaap PJ, Plugge CM, Stams AJM. 2013. Genome analysis and physiological comparison of Alicycliphilus denitrificans strains BC and K601T. PLoS One 8:e66971. doi: 10.1371/journal.pone.0066971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim S-J, Kweon O, Jones RC, Edmondson RD, Cerniglia CE. 2008. Genomic analysis of polycyclic aromatic hydrocarbon degradation in Mycobacterium vanbaalenii PYR-1. Biodegradation 19:859–881. doi: 10.1007/s10532-008-9189-z. [DOI] [PubMed] [Google Scholar]
- 48.Rabus R, Kube M, Beck A, Widdel F, Reinhardt R. 2002. Genes involved in the anaerobic degradation of ethylbenzene in a denitrifying bacterium, strain EbN1. Arch Microbiol 178:506–516. doi: 10.1007/s00203-002-0487-2. [DOI] [PubMed] [Google Scholar]
- 49.Beller HR, Chain PS, Letain TE, Chakicherla A, Larimer FW, Richardson PM, Coleman MA, Wood AP, Kelly DP. 2006. The genome sequence of the obligately chemolithoautotrophic, facultatively anaerobic bacterium Thiobacillus denitrificans. J Bacteriol 188:1473–1488. doi: 10.1128/JB.188.4.1473-1488.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kant R, van Passel MW, Sangwan P, Palva A, Lucas S, Copeland A, Lapidus A, del Rio TG, Dalin E, Tice H, Bruce D, Goodwin L, Pitluck S, Chertkov O, Larimer FW, Land ML, Hauser L, Brettin TS, Detter JC, Han S, de Vos WM, Janssen PH, Smidt H. 2011. Genome sequence of “Pedosphaera parvula” Ellin514, an aerobic verrucomicrobial isolate from pasture soil. J Bacteriol 193:2900–2901. doi: 10.1128/JB.00299-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schleheck D, Knepper TP, Fischer K, Cook AM. 2004. Mineralization of individual congeners of linear alkylbenzenesulfonate by defined pairs of heterotrophic bacteria. Appl Environ Microbiol 70:4053–4063. doi: 10.1128/AEM.70.7.4053-4063.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chapin FS, Walker BH, Hobbs RJ, Hooper DU, Lawton JH, Sala OE, Tilman D. 1997. Biotic control over the functioning of ecosystems. Science 277:500–504. doi: 10.1126/science.277.5325.500. [DOI] [Google Scholar]
- 53.Saunders AM, Albertsen M, Vollertsen J, Nielsen PH. 2016. The activated sludge ecosystem contains a core community of abundant organisms. ISME J 10:11–20. doi: 10.1038/ismej.2015.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou J, Xia B, Treves DS, Wu LY, Marsh TL, O'Neill RV, Palumbo AV, Tiedje JM. 2002. Spatial and resource factors influencing high microbial diversity in soil. Appl Environ Microbiol 68:326–334. doi: 10.1128/AEM.68.1.326-334.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hildebrand F, Meyer A, Eyre-Walker A. 2010. Evidence of selection upon genomic GC-content in bacteria. PLoS Genet 6:e1001107. doi: 10.1371/journal.pgen.1001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Foerstner KU, von Mering C, Hooper SD, Bork P. 2005. Environments shape the nucleotide composition of genomes. EMBO Rep 6:1208–1213. doi: 10.1038/sj.embor.7400538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Agashe D, Shankar N. 2014. The evolution of bacterial DNA base composition. J Exp Zool B Mol Dev Evol 322:517–528. doi: 10.1002/jez.b.22565. [DOI] [PubMed] [Google Scholar]
- 58.The Human Microbiome Project Consortium. 2012. Structure, function and diversity of the healthy human microbiome. Nature 486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lamendella R, Domingo JW, Ghosh S, Martinson J, Oerther DB. 2011. Comparative fecal metagenomics unveils unique functional capacity of the swine gut. BMC Microbiol 11:103. doi: 10.1186/1471-2180-11-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mackelprang R, Waldrop MP, DeAngelis KM, David MM, Chavarria KL, Blazewicz SJ, Rubin EM, Jansson JK. 2011. Metagenomic analysis of a permafrost microbial community reveals a rapid response to thaw. Nature 480:368–371. doi: 10.1038/nature10576. [DOI] [PubMed] [Google Scholar]
- 61.Yoon Kim H, Yeon KM, Lee CH, Lee S, Swaminathan T. 2006. Biofilm structure and extracellular polymeric substances in low and high dissolved oxygen membrane bioreactors. Sep Sci Technol 41:1213–1230. doi: 10.1080/01496390600632354. [DOI] [Google Scholar]
- 62.Wells GF, Park H-D, Eggleston B, Francis CA, Criddle CS. 2011. Fine-scale bacterial community dynamics and the taxa-time relationship within a full-scale activated sludge bioreactor. Water Res 45:5476–5488. doi: 10.1016/j.watres.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 63.Chapman T, Matsch L, Zander E. 1976. Effect of high dissolved oxygen concentration in activated sludge systems. J Water Pollut Control Fed 48:2486–2510. [Google Scholar]
- 64.Hearn EM, Patel DR, van den Berg B. 2008. Outer-membrane transport of aromatic hydrocarbons as a first step in biodegradation. Proc Natl Acad Sci U S A 105:8601–8606. doi: 10.1073/pnas.0801264105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.