

Abstract
Nonalcoholic fatty liver disease (NAFLD) is a major cause of chronic liver disease and it encompasses a spectrum from simple steatosis to steatohepatitis, fibrosis, or cirrhosis. The mechanisms involved in the occurrence of NAFLD and its progression are probably due to a metabolic profile expressed within the context of a genetic predisposition and is associated with a higher energy intake. The metabolic syndrome (MS) is a cluster of metabolic alterations associated with an increased risk for the development of cardiovascular diseases and diabetes. NAFLD patients have more than one feature of the MS, and now they are considered the hepatic components of the MS. Several scientific advances in understanding the association between NAFLD and MS have identified insulin resistance (IR) as the key aspect in the pathophysiology of both diseases. In the multi parallel hits theory of NAFLD pathogenesis, IR was described to be central in the predisposition of hepatocytes to be susceptible to other multiple pathogenetic factors. The recent knowledge gained from these advances can be applied clinically in the prevention and management of NAFLD and its associated metabolic changes. The present review analyses the current literature and highlights the new evidence on the metabolic aspects in the adult patients with NAFLD.
Keywords: Nonalcoholic fatty liver disease, Nonalcoholic steatohepatitis, Insulin resistance, Obesity, Metabolic syndrome
Core tip: In the recent years it has been reported that nonalcoholic fatty liver disease (NAFLD) pathogenesis is related with a multi hits theory where insulin resistance has a central role and renders hepatocytes susceptible to other pathogenetic factors including oxidative stress, dysregulation of cytokines production; increased intestinal permeability, intestinal dysbiosis, inflammation and fibrosis. In this context, numerous metabolic alterations are associated with NAFLD in the adult patients.
INTRODUCTION
The term non-alcoholic fatty liver disease (NAFLD) was first introduced in 1986 by Schaffner and Thaler to define the spectrum of histological and clinical manifestations, ranging from simple steatosis, to nonalcoholic steatohepatitis (NASH), fibrosis and cirrhosis[1]. NAFLD distribution is global and affects both developed and developing world populations. The prevalence of NAFLD in Western world, is reported between 20%-40% in the general adult population, and between 10%-20% in the Asian area, indeed it is increasing with the time[2,3]. However, the real prevalence is unknown and difficult to define, considering that NAFLD is often undiagnosed and that most patients have normal blood examinations, and therefore the clinicians do not suspect the potential presence of NAFLD.
The mechanisms involved in the occurrence of NAFLD and its progression are due to a genetic predisposition expressed in metabolic derangement context, and associated with an higher energy intake[4,5].
NAFLD has frequently been associated with many metabolic disorders and, in particular with insulin resistance (IR), type 2 diabetes mellitus (T2DM), hyperlipidemia and obesity which are the main features of the metabolic syndrome (MS)[6,7]. In particular obesity, if associated with central fat mass, is characterized by a subclinical systemic inflammation. This low-grade inflammatory status, influences the metabolism of lipids and induces adipokine changes production by adipose tissue, that interfere with normal insulin function and thereby induce IR. Not all the obese people develop NAFLD. It has been reported that about 30% of the obese male and 40% of the obese female have NAFLD. Nevertheless, if we consider only the obese patients with IR, and/or T2DM, the reported prevalence is increased and ranges from 30% to 100%[8,9].
Considering the epidemic border of NAFLD and the increased associated healthcare costs, better understanding of the metabolic aspects related to NAFLD is of great interest not only for the researchers and physicians, but also to improve the public health policy[10]. In this article, we have reviewed the current literature data, to highlight the newest evidences on metabolic profile of adult patients with NAFLD (Figure 1).
Figure 1.
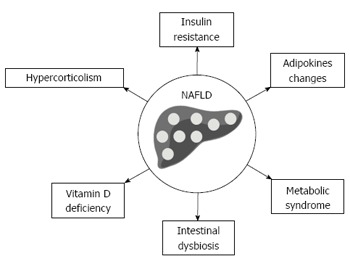
Metabolic changes associated with nonalcoholic fatty liver disease in adult patients. NAFLD: Nonalcoholic fatty liver disease.
INSULIN RESISTANCE AND HEPATIC FAT ACCUMULATION
Hepatic fat accumulation results from an imbalance between triglycerides (TG) accumulation and its removal, and represents the safest way to store the free fatty acids (FFA) in the hepatocytes[11]. The excess of TG in the liver, derives from several sources including dietary content of FFA, peripheral lipolysis secondary to IR in the adipose tissue, and increased hepatic de novo lipogenesis subsequent to hyperinsulinemia[12]. The major pathogenetic determinant of the NAFLD occurrence, is systemic IR that is independent of weight, percentage of body fat and visceral fat mass[13]. The reduction of lipid production through very low-density lipoprotein (VLDL) cholesterol, and the reduced oxidation of FFA, are also involved in the hepatocellular accumulation of the fat[14]. The NASH development within the context of simple hepatic steatosis, has been explained by the occurrence of multi-step pathogenetic mechanism. Day and James firstly proposed a direct pathogenetic link between IR and NAFLD, characterized by a “two hits” hypothesis[15]. The “first hit” is characterized by hepatic TG accumulation resulting in steatosis which increases susceptibility to the “second hit” represented by oxidative stress that causes the inflammation and necrosis. In the last years, this hypothesis has been revised and developed in a “multi parallel hits” theory, where IR the “central hit” in the development of NAFLD, renders susceptible the hepatocytes to other pathogenetic factors, including: free radicals produced by the oxidation of FFA; dysregulation of cytokines production; inflammation triggered by endotoxins due to the increased intestinal permeability of small intestine, and the changes in the gut microbiota composition; mitochondrial dysfunction, and hepatic stellate cells (HSC) activation[16]. Approximately 30% of simple fatty liver progresses to NASH, and about 20% of these develops cirrhosis.
IR is an usual condition in the patients with the MS[17]. Indeed, fasting and post-prandial hyperglycemia, are directly related to the liver fat accumulation. Otherwise, NAFLD worsens the state of IR that may lead on to T2DM in the predisposed subjects[18]. IR is clinically defined a condition in which the body becomes less sensitive to insulin and subsequently higher levels of this essential hormone, are requested to play the same metabolic functions. If additional insulin levels are insufficient, starts a dysregulation of the metabolism of lipids[19,20].
Various studies have shown that cell-derived extracellular vesicles are the cell-to-cell messengers, able to transfer bioactive molecules into the target cells, modulating the occurrence and evolution of NAFLD[21]. In particular, literature have shown that these mediators are involved in some of the key steps described in the pathogenetic mechanism of NAFLD, and in particular in lipotoxicity-associated inflammation and fibrosis[21].
The genetic background provides the basis where environmental factors express their potential pathological effects[22]. Evidence indicates that hepatic steatosis and fibrosis are both of heritable traits[23]. In this way, epidemiological studies have described an increasing gradient of steatosis clearly linked with ethnicity and, in particular: the African Americans < Caucasians < Hispanics < Asian-Indians[24].
The glucokinase regular protein receptor variants, neurocan, protein phosphatase 1 regulatory subunit 3B and lysophospholipase-like 1, have all been related with hepatic fat deposition. However, only patatin-like phospholipase domain-containing 3 (PNPLA3) and transmembrane 6 superfamily member 2 (TM6SF2), have been reported as the risk factors for the NASH progression and severity[25,26]. The PNPLA3 gene encodes a transmembrane polypeptide chain, with a TG hydrolase activity primarily expressed at level of adipocytes and hepatocytes. The variant I148M of PNPLA3, reduces the phospholipase activity leading to defective lipid catabolism, increases synthesis of phosphatidic acid and the leak of retinyl-palmitate lipase activity in the stellate cells[26,27]. All these mechanisms can explain the pathogenetic role played to PNPLA3 polymorphisms and NAFLD. The TM6SF2 activity is required for hepatic VLDL secretion, and the E167K amino-acidic substitution generates a functional impairment that promotes a fatty accumulation in the hepatocytes, as well as NAFLD progression.
The role of the genetic variants of PNPLA3 and TM6SF2 in NAFLD, has extensively been validated for population, however the data are a today insufficient to stratify accurately the risk for single subject. In the recent study by Lin et al[28], it has been shown that the combination of the PNPLA3 rs738409 variant and the GCKR rs780094 genotypes confer NAFLD susceptibility. In particular, the subjects with the GCKR rs780094 TT genotype would have 1997 times higher odds to have NAFLD. In addition, this study has shown a significant association of the GCKR rs780094 genotypes with the higher transaminase levels, TG and cholesterol serum concentration. Further research are required to clarify the full role of the genetic variants, their interaction with the environmental factors, the metabolic implications and the prognostic values in the NAFLD picture.
Adipokines changes
Mammals are two types of adipose tissue: the white adipose tissue (WAT), considered as an endocrine organ secreting different hormones and protein called adipokines, and the brown adipose tissue that transfers the energy from food into heat[29]. In the last two decades, the discovery of adipokines and the definition of their activities and involvement in various pathophysiological functions have challenged the researchers[30-32]. The WAT produces more than 50 cytokines and other molecules with endocrine, paracrine and autocrine actions involved in various physiological processes such as inflammation, immunity, vascular sclerotic processes and insulin sensitivity. Different studies have reported that adipokines are involved in the development of simple liver steatosis and to the progression of NAFLD until cirrhosis and its complications (Figure 2).
Figure 2.
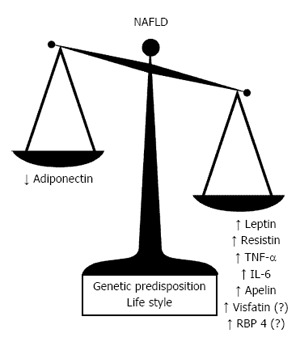
Balance of apipokine changes involved in the pathogenesis and progression of nonalcoholic fatty liver disease. NAFLD: Nonalcoholic fatty liver disease; IL-6: Interleukin 6; TNF-α: Tumor necrosis factor-α; RBP4: Retinol-binding protein 4.
Adiponectin and leptin
Adiponectin is insulin-sensitizing and an anti-inflammatory hormone produced by adipocytes, and its circulating levels are inversely related to visceral adiposity. Its expression is regulated by peroxisome proliferator-activated receptor-χ, a transcription factor also expressed in the adipocytes[33]. Adiponectin is present in the blood circulation, as three oligomeric complexes: trimer, hexamer and high molecular weight (HMW) multimers. In particular, HMW multimers have more biological activity than other two forms. Adiponectin has anti-inflammatory properties, and is able to significantly impair the production of the proinflammatory cytokines as a tumor necrosis factor (TNF)-α and interleukin (IL)-6[34]. Adiponectin has anti-steatotic effects because it increases FFA oxidation and decreases gluconeogenesis, FFA influx and de novo lipogenesis in the liver. Importantly, adiponectin protects hepatocytes from apoptosis, a hallmark in NAFLD[35]. Furthermore, adiponectin exerts an antifibrotic action, achieved via reducing the HSCs activation and proliferation inducing their apoptosis[36,37].
In a recent meta-analysis of 27 studies, which included 2243 individuals (1545 NAFLD patients and 698 controls), Polyzos et al[38] described that lower circulating adiponectin levels were observed in the simple steatosis patients than in the controls, in the NASH patients than in the controls and in NASH than in the simple steatosis patients. When NASH progresses to severe fibrosis and to cirrhosis, circulating adiponectin levels increase. There are two possible mechanisms proposed for this mechanism: (1) the hepatic decrease of adiponectin clearance and/or a compensatory increase toward the uncontrolled secretion of proinflammatory cytokines; (2) adiponectin mediates a reduction to the point of complete hepatic fat loss, in the advanced stages of fibrosis and cirrhosis, independent of metabolic or liver dysfunction (the paradox of burnt-out NASH)[39]. Notable, that serum adiponectin has been included in some noninvasive algorithms for diagnosis and prognosis of NAFLD[40].
Leptin is a peptide hormone mainly expressed in adipose tissues, although its expression has been highlighted in other tissues including skeletal muscle, stomach, placenta and ovaries. It is involved in the regulation of energy homeostasis and in metabolic, reproductive and neuroendocrine functions[41]. Leptin secretion is directly proportional to fat mass and provides anti-obesity signals by the activation of hypothalamic cell populations. Leptin is able to decrease food intake and to increase energy consumption, then to regulate sympathetic tone and energy expenditure in the conditions of energy excess inducing anorexigenic factors and its levels are negatively correlated with glucocorticoids and positively with insulin[42]. Leptin synthesis reflects the body energy stores and acute changes in caloric intake. A systematic review and meta-analysis, which included 2612 individuals (775 controls and 1837 NAFLD patients), reported that circulating leptin levels were observed more in steatosis than in the controls, more in NASH than in the controls and in NASH than in simple steatosis[43]. Leptin serum levels are also high in obese patients with NASH. This finding suggests that these patients are resistant to the action of this adipokine. It is hypothesized that leptin participates in pathogenetic hits of the NASH development, in fact, leptin contributes to the development of IR by the dephosphorylation of the insulin-receptor-substrate (IRS)-1 and the down-regulation of gluconeogenesis inducing liver steatosis. Moreover, leptin has a proinflammatory action; in fact, it is able to enhance the secretion of TNF-α, IL-6, and IL-12 with the consequent amplification of inflammation and the development of liver fibrosis[31,43].
Finally, the leptin/adiponectin ratio has been proposed as a potential surrogate biomarker, more accurate than leptin or adiponectin alone for the diagnosis of cardiometabolic diseases or occurrence of MS[44].
Resistin
Resistin is a dimeric protein secreted by adipose tissue and macrophages[45]. Resistin circulates in two distinct forms, the more predominant high-molecular-mass hexamers and the more bioactive low-molecular-mass complex. Resistin is involved in glucose and lipid metabolism, and it appears to have a central role in the IR development[46]. Moreover, the preclinical studies have shown that resistin has a proinflammatory action, in fact, it stimulates TNF-α and IL-12 in macrophages and regulates the secretion of IL-6 and IL-1β. In human studies, it has been reported that the serum resistin levels are higher in the patients with NAFLD as compared with the controls[47]. It has also been found in the literature that the NAFLD patients have increased circulating resistin and that it is correlated with IR, obesity and histological severity of the disease[48].
TNF-α and IL-6
TNF-α is an important cytokine characterized by metabolic, inflammatory, and proliferative effects, but also necrotic with enhanced expression in the adipose tissue and liver, thus making it an optimal causative agent for NAFLD[49]. TNF-α is produced by macrophages infiltrated in adipose tissue, hepatocytes, and Kupffer cells, as a response to a chronic inflammatory activity. Is well reported that TNF-α can mediate IR by affecting insulin receptor substrate-1 and insulin receptor kinase in the insulin signal transduction pathways, and can promote decomposition of adipocyte and FFA release. The polymorphisms of TNF-α have been shown by a meta-analysis, to be associated with the high risk of developing NAFLD, especially in Chinese population. TNF-α polymorphisms and in particular the 238 gene polymorphism, have been found to be related with the presence of NAFLD, and the 1031C and 863A polymorphisms are more frequent in the NASH patients[50,51]. Extensive literature reports that TNF-α production is related with presence of IR, enhanced peripheral lipolysis, hepatic fat accumulation, inflammation, necrosis, apoptosis and fibrosis.
IL-6 is an important proinflammatory cytokine secreted by adipocytes, immune system and endothelial cells[52]. The WAT contributes about 30% to the circulating IL-6, particularly the visceral WAT produces higher levels of IL-6 in respect to the subcutaneous WAT. Circulating levels of IL-6 are increased in presence of obesity and IR, and is a predictor factor of T2DM development[53]. IL-6 has a hepatoprotective role, in fact, it is able to suppress the oxidative stress reaction and to prevent mitochondrial dysfunction. However, a long exposure to IL-6 may sensitize the liver to injury and contribute to inflammation[54]. IL-6 circulating levels are increased in patients with NAFLD compared to the control groups, and the higher levels of IL-6 are associated with the advanced histopathology findings. In this way, IL-6 can be useful as a single noninvasive biomarker for distinguishing NASH, from simple steatosis[55]. Literature also reports the data on noninvasive panels of serological biomarkers that include the IL-6 values[56].
Other adipokines
Visfatin is an adipokine with insulin-mimicking effects, able to activate the insulin receptor. It is expressed in skeletal muscles, bone marrow, liver and visceral adipose tissue[32]. The visfatin mRNA levels increase during the adipocyte differentiation and the circulating levels are correlated with the WAT accumulation, and its synthesis is regulated by several factors such as growth hormone, TNF-α and IL-6[57]. The visfatin serum levels increase in association with obesity and T2DM. The human data on visfatin in NAFLD are contradictory today, and most authors have reported similar circulating levels in simple steatosis, NASH and the controls[32,57].
Apelin, a bioactive peptide, correlates positively with the oxidative stress, leptin levels, inflammation status and angiogenesis[58]. Moreover, the hepatic expression of apelin and its receptor increase during the chronic toxic damage. The apelin serum levels were significantly higher and positively correlated with weight and IR in the NAFLD patients[58].
Obestatin is a pleiotropic adipokine which may have a role in the regulation of the β-cell survival and insulin secretion, involved in the glucose and lipid metabolism[59]. It also exhibits regulatory effect on the energy metabolism and the gastrointestinal system. The obestatin levels seem to be decreased in human obesity and negatively associated with IR. However, similar obestatin levels have been observed in the NAFLD patients and the controls, such as in the simple steatosis and NASH patients.
Retinol-binding protein (RBP)-4 is an adipokine predominantly expressed in visceral rather than in the subcutaneous adipose tissue and the liver. Its blood levels are high in the IR states, including obesity and T2DM[60]. The limited data have reported that RBP-4 seems to be positively correlated with the liver fat deposition and inflammation.
Chemerin is an adipokine secreted by the WAT that contributes to the inflammation status which is associated with the TNF-α production and is involved in the enrolment of hepatic Kupffer cells[61]. However, the studies on the role of chemerin in the NAFLD pathogenesis are controversial and it seems that circulating chemerin levels do not reflect the hepatic condition.
Metabolic syndrome
NAFLD is regarded as a hepatic component of the MS[62]. On the basis of the National Cholesterol Education Program Adult Treatment Panel III, MS can be diagnosed in the presence of three out of five criteria which include a high waist circumference, high TG or reduced high-density lipoprotein (HDL)-cholesterol levels, elevated blood pressure, and high fasting-glucose levels or a diagnosis of T2DM[63]. Approximately 90% of the NAFLD patients present more than one component of the MS, and about 33% of the patients meet the criteria of the MS[2].
The prevalence of NAFLD ranges from 57% in the overweight patients attending the outpatient clinics, to 98% in the nondiabetic obese subjects[8]. In the obese people, the median prevalence of NASH is 33%, and ranging from 10% to 56%. The waist circumference, a marker of visceral adipose tissue, is an independent risk factor linked to the MS and NAFLD[64]. However, fat distribution is also the main factor determining metabolic alterations with the NAFLD individuals. The ectopic fat accumulation is defined by the presence of lipid droplets, within the non-adipose tissues that are not rich in lipids[64,65]. The visceral adipose tissue produces FFA and the adipokines involves in the NAFLD pathogenesis. Via adipokines secretion, hypertrophied adipocytes promote the accumulation of macrophages in the visceral fat[65]. These macrophages produce proinflammatory cytokines with subsequent chronic low-grade inflammation, that further exacerbates IR. These data reveal the central role played by obesity in the development and progression of both IR and NAFLD[65,66].
The ectopic accumulation of TG results from the failure of the adipose tissue to expand and store the excess of calories. Once the capacity of the adipocytes to store TG is exceeded, the fat overflows to other tissues, mainly to the muscles and liver[67]. The intracellular TG deposition interferes with insulin signaling and therefore contributes to IR. The liver is the key visceral organ affected by the ectopic fat accumulation[4]. The accumulation of the hepatic fat is due to the increased delivery of the FFA to the liver, the increased VLDL synthesis, decreased TG export through VLDL and reduced FFA β-oxidation. An increase in the liver fat is associated with IR and this is the pathophysiological feature of NAFLD[68].
The hepatic fat accumulation is a factor that induces hypertriglyceridemia associated with visceral obesity. The overproduction of VLDL particles rich in TG is the main mechanism for the increase in the TG concentration associated with the MS[69]. The increased synthesis of the large VLDL particles leads to the decrease of the HDL concentration and to the production of small, dense low-density lipoprotein (LDL). The LDL particles are more atherogenic because they bind less efficiently to the LDL receptors, thereby increasing their residence time and a number in the circulation. In addition, IR worsens the LDL clearance by reducing the insulin ability to stimulate the expression of the LDL receptor, which favors the deposition of LDL particles to the arterial wall[29,69]. Conformational changes in apolipoprotein B on the surface of LDL particles, make them more likely to interact with the surface of endothelial cells lining the arteries which facilitates their entry into the vascular intima. Once inside the arterial wall, the small LDL, particles are sensitive to the chemical modification such as oxidation[70]. The macrophage receptors recognize and take up the oxidized LDL which turns these macrophages into the foam cells, an early step in the development of the atherosclerotic plaque. HDL is more sensitive to the degradation and increased clearance from the blood, and low HDL-cholesterol levels were found in the subjects with the MS[71]. Experimental and clinical data have shown that the increased flux of the FFA from the visceral fat mass, leads to hepatic steatosis and IR[68-70]. Furthermore, the liver fat accumulation is strongly associated with the impaired lipid metabolism, independent of whole body adiposity. Between 57%-68% of the NAFLD patients have disturbed lipid profiles[62,72]. In addition to their central role in TG synthesis and storage, the hepatocytes contain lipid droplets in the lumen of the endoplasmic reticulum, where are assembled the VLDL particles[73]. Subsequently, the liver secretes into the blood the apolipoprotein B-containing VLDL particles. Moreover, NAFLD induces the intrahepatic VLDL production by changes in the rate of the apolipoprotein B synthesis and degradation, or by de novo lipogenesis. Many studies have shown the critical role of visceral obesity as a source of the FFA, that feeds into the portal blood circulation and induces NAFLD[74]. The theory of portal-FFA flux denotes that the visceral fat mass has a high lipolytic activity, and that the FFA derived from it increases the intrahepatocytes fat content[11]. The FFA also appears to induce the c-Jun N-terminal protein kinase activation leading to IR by IRS-1 and IRS-2 serine phosphorylation[75]. Thus, atherogenic dyslipidemia implicated in NAFLD pathogenesis, has a pivotal role also in the development and progression of cardiometabolic disorders through various pathways.
It is also interesting to note that the overweight/obese NAFLD patients have a reduced basal whole-body and hepatic fat oxidation, inversely related to severity of the hepatic disease, independently of the visceral fat mass and body mass index (BMI)[76]. This observation suggests that the reduced fat oxidation may contribute to the liver steatosis development, and may induce IR the cornerstone in the pathogenesis of NAFLD[76].
INTESTINAL DYSBIOSIS
Gastrointestinal diseases, such as inflammatory bowel disease, celiac disease and gut impairment, like short bowel syndrome and bypass surgery for obesity, have been associated with the NAFLD development[77]. The gut harbours microbiota as an integrated ecosystem of at least 1013-1014 microbial cells characterized by four main phyla of bacteria: Firmicutes (65%), Bacteroidetes (16%), Proteobacteria (9%) and Actinobacteria (5%), and they present more than 100 times the number of the overall genes of human genomes[77,78]. An innovative hypothesis recently proposed involves the gut-liver axis as the central pathogenetic component, that influences the differences in the body weight, insulin sensitivity, and other metabolic risk factors. In particular, recent data in the field suggest that, the increased consumption of obesogenic foods especially if enriched with fructose and fat, may unbalance the gut microbiota and alter the intestinal barrier function with the predisposition to a metabolic endotoxemia and a subclinical inflammatory status[9]. The gut microbiota is involved in the development and progression of NAFLD via several mechanisms: (1) increasing the intestinal permeability that promotes endotoxemia[77]; (2) enhancing the energy harvest with weight gain[79]; (3) causing choline deficiency with lower secretion of VLDL[80]; (4) producing endogenous ethanol; and (5) decreasing the secondary bile acid production and increasing the primary bile acid concentration that promote dietary lipid emulsification, digestion and absorption[77,80].
The derangement of the microbiota, in particular a small intestinal bacterial overgrowth, occurs in patients with chronic liver disease, and in particular in the NASH patients with a prevalence of 50%[77]. The microbiota of obese people presents a different microbial diversity if compared to the lean controls. Indeed these subjects have less Bacteroides and more Firmicutes, with a higher intestinal Firmicutes/Bacteroidetes ratio[81]. The obesogenic diet, changes the proportion of Gram-negative to Gram-positive bacteria, and results in the increased synthesis of lipopolysaccharide, with a higher liver exposure to endotoxins via the portal vein with the consequent hepatic inflammation[76]. Within this metabolic context, the obese patients have an increased ability to extract energy from food. The bacterial enzymes extract the calories from otherwise indigestible dietary polysaccharides. The enteric bacteria suppress the synthesis and secretion of a small intestinal fasting-induced adipocyte factor, and resulting in the activation of lipoprotein lipase and increased of TG accumulation in the hepatocytes[82]. The existing literature data have reported that the NAFLD patients compared with the healthy controls have increased Lactobacillus and a decreased family of Ruminococcaceae (both phylum: Firmicutes). On the association with Lactobacillus, it is interesting to note that many species from this genus are used to produce probiotic formulations[76,77]. In particular, Lactobacillus is a lactic acid bacterium that can enhance the intestinal barrier function, modulate immune response with subsequent inhibition of pathogens, all skills that protect to the pathogenesis of NAFLD[83]. However, Lactobacillus may be associated with the synthesis of volatile organic compounds, such as acetate and ethanol, involved in the pathogenesis of obesity and liver steatosis. The genus of Lactobacillus comprises over 180 species, while some can make lactic acid from the fermentation of sugars, others can also produce ethanol. On the other hand, the decrease of the Ruminococcaceae family induces a decrease in the production of a short chain fatty acid, as a butyrate, that has been associated with NASH. Some species of Ruminococcaceae are pro-inflammatory and can produce ethanol, two mechanisms involved in the progression of NAFLD[84]. The NAFLD patients have also shown an increased percentage of bacteria from the Escherichia genera (phylum: Proteobacteria), and the Streptococcus genera (phylum: Firmicutes), both known to be involved in the persistent inflammation of the gut mucosa.
VITAMIN D DEFICIENCY
Vitamin D is a fat-soluble vitamin which exists in several forms, but two forms, ergocalciferol and cholecalciferol, are most expressed[85]. The different vitamins D mediate their signals via vitamin D receptor (VDR), which is expressed in the hepatocytes. It is known that this binding between vitamins D and their receptor leads to the regulation of over 200 genes involved in the lipid and glucose metabolism[85,86]. Several studies have shown that vitamin D deficiencies, and in particular the low levels of cholecalciferol, are strongly associated with MS and may increase the risk of cardiometabolic outcomes such as T2DM, hypertension and cardiovascular diseases, as well as with an increased mortality risk[87]. A recent systematic review has found that the cholecalciferol levels higher than 25 ng/mL, are associated with 43% lower risk of T2DM, if compared to the levels less than 14 ng/mL, and that the vitamin D treatment can improve IR in the patients with baseline IR[88]. A meta-analysis has shown that the supplementation of vitamin D, improves slightly IR if compared to placebo. Another meta-analysis has reported that the NAFLD patients are 26% more likely to be deficient of vitamin D, compared to controls[89]. In particular, it has been showed that the concentrations of cholecalciferol is lower in the NAFLD subjects compared to the lean controls, and that these levels predict the histological severity of the disease[89].
The experimental evidence has established different mechanisms by which vitamin D may influence NAFLD development. First, it has been proposed that the liver VDR expression is inversely related with the histological spectrum of NAFLD on histopathology, independently from other metabolic features such as BMI[90,91]. Vitamin D presents antiproliferative and antifibrotic properties and has an important role in the regulation of extracellular matrix in the liver by VDR signaling that can suppress the fibrotic gene expression, as the TNF-α and transforming growth factor β, and it can inhibit the HSC proliferation[92]. Second, the role of vitamin D in the adipokines modulation is an active research topic. In this way, a direct association between cholecalciferol and adiponectin serum levels, independently of the BMI, has been reported[93].
HYPERCORTICOLISM
Hypercortisolism shares metabolic features, and in particular liver steatosis and IR. It Is known that cortisol, impairs insulin sensitivity by interfering with the receptor pathway of insulin, and by stimulating lipolysis and proteolysis, with subsequent increasing of amino acid and FFA release[94]. The prevalence of NAFLD in patients with Cushing syndrome, is estimated around 20%. The hypothesis is that the enzyme 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1), expressed in adipose tissue and liver, increases intracellular glucocorticoid levels, by converting inert cortisone to active cortisol, which is able to promote metabolic alterations[95]. In fact, in obese people, the increased regeneration of cortisone to cortisol, is present mediated by the activity of 11β-HSD1. However, the studies on the activity of 11β-HSD1 in obese subjects are contradictory. In the pathogenesis of NAFLD in course of Cushing syndrome, another possible factor involved, is the decreased clearance of cortisol via the A-ring reductases (5α- and 5β-reductase). The role of the A-ring reductase has recently been explored. In the early stages of NAFLD, the 5α-reductase activity is increased to protect the liver from cortisol exposure, with increasing cortisol clearance[94,95]. A the same time, 11β-HSD1 activity is decreased, and the decreasing cortisol production produces the activation of hypothalamic-pituitary-adrenal axis, with increase in urinary free cortisol concentrations. To the contrary in patients with NASH, there is an increase in the expression of 11β-HSD1, with elevation of intrahepatic glucocorticoid levels[95]. The over expression of receptor α- and the reduced activity of 5α-reductase, produces also lipid accumulation in the hepatocytes.
CONCLUSION
In the last years, epidemiology supports the idea that NAFLD is the most common cause of a chronic liver disease worldwide, and the progression from simple steatosis to steatohepatitis and cirrhosis presents an emerging and urgent problem for the global public health[96]. It is important to highlight that one of the major hallmarks of NAFLD is the association with various metabolic features of the MS that is the keystone physiopathological mechanism of the IR. In this way, the growing body of evidence suggests that NAFLD should be considered the hepatic border of the MS[6]. Recently several scientific advances in understanding the association between NAFLD and metabolic changes have been observed. We believe that the knowledge gained owing to this progress can be applied in the clinical practice to improve the prevention of the condition, to help the management and its metabolic sequelae and to discover the new potential therapies for NAFLD.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Italy
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B, B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Conflict-of-interest statement: The authors confirm that this article content has no conflict of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: May 11, 2016
First decision: May 30, 2016
Article in press: June 28, 2016
P- Reviewer: Lee HC, Tarantino G S- Editor: Gong ZM L- Editor: A E- Editor: Wang CH
References
- 1.Schaffner F, Thaler H. Nonalcoholic fatty liver disease. Prog Liver Dis. 1986;8:283–298. [PubMed] [Google Scholar]
- 2.Masarone M, Federico A, Abenavoli L, Loguercio C, Persico M. Non alcoholic fatty liver: epidemiology and natural history. Rev Recent Clin Trials. 2014;9:126–133. doi: 10.2174/1574887109666141216111143. [DOI] [PubMed] [Google Scholar]
- 3.Sayiner M, Koenig A, Henry L, Younossi ZM. Epidemiology of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis in the United States and the Rest of the World. Clin Liver Dis. 2016;20:205–214. doi: 10.1016/j.cld.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 4.Petta S, Muratore C, Craxì A. Non-alcoholic fatty liver disease pathogenesis: the present and the future. Dig Liver Dis. 2009;41:615–625. doi: 10.1016/j.dld.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 5.Nascimbeni F, Pais R, Bellentani S, Day CP, Ratziu V, Loria P, Lonardo A. From NAFLD in clinical practice to answers from guidelines. J Hepatol. 2013;59:859–871. doi: 10.1016/j.jhep.2013.05.044. [DOI] [PubMed] [Google Scholar]
- 6.Tarantino G, Finelli C. What about non-alcoholic fatty liver disease as a new criterion to define metabolic syndrome? World J Gastroenterol. 2013;19:3375–3384. doi: 10.3748/wjg.v19.i22.3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.European Association for the Study of the Liver (EASL) Electronic address: easloffice@easloffice.eu; European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J Hepatol. 2016;64:1388–1402. doi: 10.1016/j.jhep.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 8.Almeda-Valdes P, Aguilar-Olivos N, Uribe M, Méndez-Sánchez N. Common features of the metabolic syndrome and nonalcoholic fatty liver disease. Rev Recent Clin Trials. 2014;9:148–158. doi: 10.2174/1574887109666141216103908. [DOI] [PubMed] [Google Scholar]
- 9.Machado MV, Cortez-Pinto H. Diet, Microbiota, Obesity, and NAFLD: A Dangerous Quartet. Int J Mol Sci. 2016;17:481. doi: 10.3390/ijms17040481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Younossi ZM, Henry L. Economic and Quality-of-Life Implications of Non-Alcoholic Fatty Liver Disease. Pharmacoeconomics. 2015;33:1245–1253. doi: 10.1007/s40273-015-0316-5. [DOI] [PubMed] [Google Scholar]
- 11.Gaggini M, Morelli M, Buzzigoli E, DeFronzo RA, Bugianesi E, Gastaldelli A. Non-alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance, dyslipidemia, atherosclerosis and coronary heart disease. Nutrients. 2013;5:1544–1560. doi: 10.3390/nu5051544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Byrne CD, Targher G. NAFLD: a multisystem disease. J Hepatol. 2015;62:S47–S64. doi: 10.1016/j.jhep.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 13.Berk PD, Verna EC. Nonalcoholic Fatty Liver Disease: Lipids and Insulin Resistance. Clin Liver Dis. 2016;20:245–262. doi: 10.1016/j.cld.2015.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gambino R, Bugianesi E, Rosso C, Mezzabotta L, Pinach S, Alemanno N, Saba F, Cassader M. Different Serum Free Fatty Acid Profiles in NAFLD Subjects and Healthy Controls after Oral Fat Load. Int J Mol Sci. 2016;17:479. doi: 10.3390/ijms17040479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 16.Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52:1836–1846. doi: 10.1002/hep.24001. [DOI] [PubMed] [Google Scholar]
- 17.Abenavoli L, Milic N, Peta V, Alfieri F, De Lorenzo A, Bellentani S. Alimentary regimen in non-alcoholic fatty liver disease: Mediterranean diet. World J Gastroenterol. 2014;20:16831–16840. doi: 10.3748/wjg.v20.i45.16831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walle P, Takkunen M, Männistö V, Vaittinen M, Lankinen M, Kärjä V, Käkelä P, Ågren J, Tiainen M, Schwab U, et al. Fatty acid metabolism is altered in non-alcoholic steatohepatitis independent of obesity. Metabolism. 2016;65:655–666. doi: 10.1016/j.metabol.2016.01.011. [DOI] [PubMed] [Google Scholar]
- 19.Barbarroja N, Lopez-Pedrera C, Garrido-Sanchez L, Mayas MD, Oliva-Olivera W, Bernal-Lopez MR, El Bekay R, Tinahones FJ. Progression from high insulin resistance to type 2 diabetes does not entail additional visceral adipose tissue inflammation. PLoS One. 2012;7:e48155. doi: 10.1371/journal.pone.0048155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abenavoli L, Bellentani S. Milk thistle to treat non-alcoholic fatty liver disease: dream or reality? Expert Rev Gastroenterol Hepatol. 2013;7:677–679. doi: 10.1586/17474124.2013.842893. [DOI] [PubMed] [Google Scholar]
- 21.Povero D, Feldstein AE. Novel Molecular Mechanisms in the Development of Non-Alcoholic Steatohepatitis. Diabetes Metab J. 2016;40:1–11. doi: 10.4093/dmj.2016.40.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Macaluso FS, Maida M, Petta S. Genetic background in nonalcoholic fatty liver disease: A comprehensive review. World J Gastroenterol. 2015;21:11088–11111. doi: 10.3748/wjg.v21.i39.11088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dongiovanni P, Romeo S, Valenti L. Genetic Factors in the Pathogenesis of Nonalcoholic Fatty Liver and Steatohepatitis. Biomed Res Int. 2015;2015:460190. doi: 10.1155/2015/460190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kalia HS, Gaglio PJ. The Prevalence and Pathobiology of Nonalcoholic Fatty Liver Disease in Patients of Different Races or Ethnicities. Clin Liver Dis. 2016;20:215–224. doi: 10.1016/j.cld.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 25.Xia MF, Ling Y, Bian H, Lin HD, Yan HM, Chang XX, Li XM, Ma H, Wang D, Zhang LS, et al. I148M variant of PNPLA3 increases the susceptibility to non-alcoholic fatty liver disease caused by obesity and metabolic disorders. Aliment Pharmacol Ther. 2016;43:631–642. doi: 10.1111/apt.13521. [DOI] [PubMed] [Google Scholar]
- 26.Chen LZ, Hua-Xiang Xia H, Xin YN, Lin ZH, Xuan SY. TM6SF2 E167K Variant, a Novel Genetic Susceptibility Variant, Contributing to Nonalcoholic Fatty Liver Disease. J Clin Transl Hepatol. 2015;3:265–270. doi: 10.14218/JCTH.2015.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan HL, Mohamed R, Mohamed Z, Zain SM. Phosphatidylethanolamine N-methyltransferase gene rs7946 polymorphism plays a role in risk of nonalcoholic fatty liver disease: evidence from meta-analysis. Pharmacogenet Genomics. 2016;26:88–95. doi: 10.1097/FPC.0000000000000193. [DOI] [PubMed] [Google Scholar]
- 28.Lin YC, Chang PF, Chang MH, Ni YH. Genetic variants in GCKR and PNPLA3 confer susceptibility to nonalcoholic fatty liver disease in obese individuals. Am J Clin Nutr. 2014;99:869–874. doi: 10.3945/ajcn.113.079749. [DOI] [PubMed] [Google Scholar]
- 29.Tchernof A, Després JP. Pathophysiology of human visceral obesity: an update. Physiol Rev. 2013;93:359–404. doi: 10.1152/physrev.00033.2011. [DOI] [PubMed] [Google Scholar]
- 30.Stojsavljević S, Gomerčić Palčić M, Virović Jukić L, Smirčić Duvnjak L, Duvnjak M. Adipokines and proinflammatory cytokines, the key mediators in the pathogenesis of nonalcoholic fatty liver disease. World J Gastroenterol. 2014;20:18070–18091. doi: 10.3748/wjg.v20.i48.18070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bekaert M, Verhelst X, Geerts A, Lapauw B, Calders P. Association of recently described adipokines with liver histology in biopsy-proven non-alcoholic fatty liver disease: a systematic review. Obes Rev. 2016;17:68–80. doi: 10.1111/obr.12333. [DOI] [PubMed] [Google Scholar]
- 32.Abenavoli L, Peta V. Role of adipokines and cytokines in non-alcoholic fatty liver disease. Rev Recent Clin Trials. 2014;9:134–140. doi: 10.2174/1574887109666141216102458. [DOI] [PubMed] [Google Scholar]
- 33.Polyzos SA, Kountouras J, Mantzoros CS. Adipokines in nonalcoholic fatty liver disease. Metabolism. 2016;65:1062–1079. doi: 10.1016/j.metabol.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 34.Kaser S, Moschen A, Cayon A, Kaser A, Crespo J, Pons-Romero F, Ebenbichler CF, Patsch JR, Tilg H. Adiponectin and its receptors in non-alcoholic steatohepatitis. Gut. 2005;54:117–121. doi: 10.1136/gut.2003.037010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heiker JT, Kosel D, Beck-Sickinger AG. Molecular mechanisms of signal transduction via adiponectin and adiponectin receptors. Biol Chem. 2010;391:1005–1018. doi: 10.1515/BC.2010.104. [DOI] [PubMed] [Google Scholar]
- 36.Medina-Urrutia A, Posadas-Romero C, Posadas-Sánchez R, Jorge-Galarza E, Villarreal-Molina T, González-Salazar Mdel C, Cardoso-Saldaña G, Vargas-Alarcón G, Torres-Tamayo M, Juárez-Rojas JG. Role of adiponectin and free fatty acids on the association between abdominal visceral fat and insulin resistance. Cardiovasc Diabetol. 2015;14:20. doi: 10.1186/s12933-015-0184-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park PH, Sanz-Garcia C, Nagy LE. Adiponectin as an anti-fibrotic and anti-inflammatory adipokine in the liver. Curr Pathobiol Rep. 2015;3:243–252. doi: 10.1007/s40139-015-0094-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Polyzos SA, Toulis KA, Goulis DG, Zavos C, Kountouras J. Serum total adiponectin in nonalcoholic fatty liver disease: a systematic review and meta-analysis. Metabolism. 2011;60:313–326. doi: 10.1016/j.metabol.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 39.van der Poorten D, Samer CF, Ramezani-Moghadam M, Coulter S, Kacevska M, Schrijnders D, Wu LE, McLeod D, Bugianesi E, Komuta M, et al. Hepatic fat loss in advanced nonalcoholic steatohepatitis: are alterations in serum adiponectin the cause? Hepatology. 2013;57:2180–2188. doi: 10.1002/hep.26072. [DOI] [PubMed] [Google Scholar]
- 40.Younossi ZM, Jarrar M, Nugent C, Randhawa M, Afendy M, Stepanova M, Rafiq N, Goodman Z, Chandhoke V, Baranova A. A novel diagnostic biomarker panel for obesity-related nonalcoholic steatohepatitis (NASH) Obes Surg. 2008;18:1430–1437. doi: 10.1007/s11695-008-9506-y. [DOI] [PubMed] [Google Scholar]
- 41.Coppari R, Bjørbæk C. Leptin revisited: its mechanism of action and potential for treating diabetes. Nat Rev Drug Discov. 2012;11:692–708. doi: 10.1038/nrd3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meek TH, Morton GJ. The role of leptin in diabetes: metabolic effects. Diabetologia. 2016;59:928–932. doi: 10.1007/s00125-016-3898-3. [DOI] [PubMed] [Google Scholar]
- 43.Polyzos SA, Aronis KN, Kountouras J, Raptis DD, Vasiloglou MF, Mantzoros CS. Circulating leptin in non-alcoholic fatty liver disease: a systematic review and meta-analysis. Diabetologia. 2016;59:30–43. doi: 10.1007/s00125-015-3769-3. [DOI] [PubMed] [Google Scholar]
- 44.López-Jaramillo P, Gómez-Arbeláez D, López-López J, López-López C, Martínez-Ortega J, Gómez-Rodríguez A, Triana-Cubillos S. The role of leptin/adiponectin ratio in metabolic syndrome and diabetes. Horm Mol Biol Clin Investig. 2014;18:37–45. doi: 10.1515/hmbci-2013-0053. [DOI] [PubMed] [Google Scholar]
- 45.Bokarewa M, Nagaev I, Dahlberg L, Smith U, Tarkowski A. Resistin, an adipokine with potent proinflammatory properties. J Immunol. 2005;174:5789–5795. doi: 10.4049/jimmunol.174.9.5789. [DOI] [PubMed] [Google Scholar]
- 46.Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, Patel HR, Ahima RS, Lazar MA. The hormone resistin links obesity to diabetes. Nature. 2001;409:307–312. doi: 10.1038/35053000. [DOI] [PubMed] [Google Scholar]
- 47.Kang YE, Kim JM, Joung KH, Lee JH, You BR, Choi MJ, Ryu MJ, Ko YB, Lee MA, Lee J, et al. The Roles of Adipokines, Proinflammatory Cytokines, and Adipose Tissue Macrophages in Obesity-Associated Insulin Resistance in Modest Obesity and Early Metabolic Dysfunction. PLoS One. 2016;11:e0154003. doi: 10.1371/journal.pone.0154003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shen C, Zhao CY, Wang W, Wang YD, Sun H, Cao W, Yu WY, Zhang L, Ji R, Li M, et al. The relationship between hepatic resistin overexpression and inflammation in patients with nonalcoholic steatohepatitis. BMC Gastroenterol. 2014;14:39. doi: 10.1186/1471-230X-14-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Valenti L, Fracanzani AL, Dongiovanni P, Santorelli G, Branchi A, Taioli E, Fiorelli G, Fargion S. Tumor necrosis factor alpha promoter polymorphisms and insulin resistance in nonalcoholic fatty liver disease. Gastroenterology. 2002;122:274–280. doi: 10.1053/gast.2002.31065. [DOI] [PubMed] [Google Scholar]
- 50.Wang JK, Feng ZW, Li YC, Li QY, Tao XY. Association of tumor necrosis factor-α gene promoter polymorphism at sites -308 and -238 with non-alcoholic fatty liver disease: a meta-analysis. J Gastroenterol Hepatol. 2012;27:670–676. doi: 10.1111/j.1440-1746.2011.06978.x. [DOI] [PubMed] [Google Scholar]
- 51.Cheng Y, An B, Jiang M, Xin Y, Xuan S. Association of Tumor Necrosis Factor-alpha Polymorphisms and Risk of Coronary Artery Disease in Patients With Non-alcoholic Fatty Liver Disease. Hepat Mon. 2015;15:e26818. doi: 10.5812/hepatmon.26818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tilg H. The role of cytokines in non-alcoholic fatty liver disease. Dig Dis. 2010;28:179–185. doi: 10.1159/000282083. [DOI] [PubMed] [Google Scholar]
- 53.Wolf J, Rose-John S, Garbers C. Interleukin-6 and its receptors: a highly regulated and dynamic system. Cytokine. 2014;70:11–20. doi: 10.1016/j.cyto.2014.05.024. [DOI] [PubMed] [Google Scholar]
- 54.Tarantino G, Conca P, Pasanisi F, Ariello M, Mastrolia M, Arena A, Tarantino M, Scopacasa F, Vecchione R. Could inflammatory markers help diagnose nonalcoholic steatohepatitis? Eur J Gastroenterol Hepatol. 2009;21:504–511. doi: 10.1097/MEG.0b013e3283229b40. [DOI] [PubMed] [Google Scholar]
- 55.Grigorescu M, Crisan D, Radu C, Grigorescu MD, Sparchez Z, Serban A. A novel pathophysiological-based panel of biomarkers for the diagnosis of nonalcoholic steatohepatitis. J Physiol Pharmacol. 2012;63:347–353. [PubMed] [Google Scholar]
- 56.Jamali R, Arj A, Razavizade M, Aarabi MH. Prediction of Nonalcoholic Fatty Liver Disease Via a Novel Panel of Serum Adipokines. Medicine (Baltimore) 2016;95:e2630. doi: 10.1097/MD.0000000000002630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Genc H, Dogru T, Kara M, Tapan S, Ercin CN, Acikel C, Karslioglu Y, Bagci S. Association of plasma visfatin with hepatic and systemic inflammation in nonalcoholic fatty liver disease. Ann Hepatol. 2013;12:548–555. [PubMed] [Google Scholar]
- 58.Ercin CN, Dogru T, Tapan S, Kara M, Haymana C, Karadurmus N, Karslioglu Y, Acikel C. Plasma apelin levels in subjects with nonalcoholic fatty liver disease. Metabolism. 2010;59:977–981. doi: 10.1016/j.metabol.2009.10.019. [DOI] [PubMed] [Google Scholar]
- 59.Estep M, Abawi M, Jarrar M, Wang L, Stepanova M, Elariny H, Moazez A, Goodman Z, Chandhoke V, Baranova A, et al. Association of obestatin, ghrelin, and inflammatory cytokines in obese patients with non-alcoholic fatty liver disease. Obes Surg. 2011;21:1750–1757. doi: 10.1007/s11695-011-0475-1. [DOI] [PubMed] [Google Scholar]
- 60.Chang X, Yan H, Bian H, Xia M, Zhang L, Gao J, Gao X. Serum retinol binding protein 4 is associated with visceral fat in human with nonalcoholic fatty liver disease without known diabetes: a cross-sectional study. Lipids Health Dis. 2015;14:28. doi: 10.1186/s12944-015-0033-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hatziagelaki E, Herder C, Tsiavou A, Teichert T, Chounta A, Nowotny P, Pacini G, Dimitriadis G, Roden M. Serum Chemerin Concentrations Associate with Beta-Cell Function, but Not with Insulin Resistance in Individuals with Non-Alcoholic Fatty Liver Disease (NAFLD) PLoS One. 2015;10:e0124935. doi: 10.1371/journal.pone.0124935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fotbolcu H, Zorlu E. Nonalcoholic fatty liver disease as a multi-systemic disease. World J Gastroenterol. 2016;22:4079–4090. doi: 10.3748/wjg.v22.i16.4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation. 2002;106:3143–3421. [PubMed] [Google Scholar]
- 64.Bugianesi E, McCullough AJ, Marchesini G. Insulin resistance: a metabolic pathway to chronic liver disease. Hepatology. 2005;42:987–1000. doi: 10.1002/hep.20920. [DOI] [PubMed] [Google Scholar]
- 65.Motamed N, Sohrabi M, Ajdarkosh H, Hemmasi G, Maadi M, Sayeedian FS, Pirzad R, Abedi K, Aghapour S, Fallahnezhad M, et al. Fatty liver index vs waist circumference for predicting non-alcoholic fatty liver disease. World J Gastroenterol. 2016;22:3023–3030. doi: 10.3748/wjg.v22.i10.3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Abenavoli L, DI Renzo L, Guzzi PH, Pellicano R, Milic N, DE Lorenzo A. Non-alcoholic fatty liver disease severity, central fat mass and adinopectin: a close relationship. Clujul Med. 2015;88:489–493. doi: 10.15386/cjmed-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Walker GE, Marzullo P, Ricotti R, Bona G, Prodam F. The pathophysiology of abdominal adipose tissue depots in health and disease. Horm Mol Biol Clin Investig. 2014;19:57–74. doi: 10.1515/hmbci-2014-0023. [DOI] [PubMed] [Google Scholar]
- 68.Abenavoli L, Luigiano C, Guzzi PH, Milic N, Morace C, Stelitano L, Consolo P, Miraglia S, Fagoonee S, Virgilio C, et al. Serum adipokine levels in overweight patients and their relationship with non-alcoholic fatty liver disease. Panminerva Med. 2014;56:189–193. [PubMed] [Google Scholar]
- 69.Byrne CD, Targher G. Ectopic fat, insulin resistance, and nonalcoholic fatty liver disease: implications for cardiovascular disease. Arterioscler Thromb Vasc Biol. 2014;34:1155–1161. doi: 10.1161/ATVBAHA.114.303034. [DOI] [PubMed] [Google Scholar]
- 70.Smith U. Abdominal obesity: a marker of ectopic fat accumulation. J Clin Invest. 2015;125:1790–1792. doi: 10.1172/JCI81507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Byrne CD. Ectopic fat, insulin resistance and non-alcoholic fatty liver disease. Proc Nutr Soc. 2013;72:412–419. doi: 10.1017/S0029665113001249. [DOI] [PubMed] [Google Scholar]
- 72.Vollenweider P, von Eckardstein A, Widmann C. HDLs, diabetes, and metabolic syndrome. Handb Exp Pharmacol. 2015;224:405–421. doi: 10.1007/978-3-319-09665-0_12. [DOI] [PubMed] [Google Scholar]
- 73.Ponziani FR, Pecere S, Gasbarrini A, Ojetti V. Physiology and pathophysiology of liver lipid metabolism. Expert Rev Gastroenterol Hepatol. 2015;9:1055–1067. doi: 10.1586/17474124.2015.1056156. [DOI] [PubMed] [Google Scholar]
- 74.Cohen DE, Fisher EA. Lipoprotein metabolism, dyslipidemia, and nonalcoholic fatty liver disease. Semin Liver Dis. 2013;33:380–388. doi: 10.1055/s-0033-1358519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kluwe J, Pradere JP, Gwak GY, Mencin A, De Minicis S, Osterreicher CH, Colmenero J, Bataller R, Schwabe RF. Modulation of hepatic fibrosis by c-Jun-N-terminal kinase inhibition. Gastroenterology. 2010;138:347–359. doi: 10.1053/j.gastro.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Croci I, Byrne NM, Choquette S, Hills AP, Chachay VS, Clouston AD, O’Moore-Sullivan TM, Macdonald GA, Prins JB, Hickman IJ. Whole-body substrate metabolism is associated with disease severity in patients with non-alcoholic fatty liver disease. Gut. 2013;62:1625–1633. doi: 10.1136/gutjnl-2012-302789. [DOI] [PubMed] [Google Scholar]
- 77.Abenavoli L, Scarpellini E, Rouabhia S, Balsano C, Luzza F. Probiotics in non-alcoholic fatty liver disease: which and when. Ann Hepatol. 2013;12:357–363. [PubMed] [Google Scholar]
- 78.Abdou RM, Zhu L, Baker RD, Baker SS. Gut Microbiota of Nonalcoholic Fatty Liver Disease. Dig Dis Sci. 2016;61:1268–1281. doi: 10.1007/s10620-016-4045-1. [DOI] [PubMed] [Google Scholar]
- 79.He X, Ji G, Jia W, Li H. Gut Microbiota and Nonalcoholic Fatty Liver Disease: Insights on Mechanism and Application of Metabolomics. Int J Mol Sci. 2016;17:300. doi: 10.3390/ijms17030300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schnabl B, Brenner DA. Interactions between the intestinal microbiome and liver diseases. Gastroenterology. 2014;146:1513–1524. doi: 10.1053/j.gastro.2014.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Houghton D, Stewart CJ, Day CP, Trenell M. Gut Microbiota and Lifestyle Interventions in NAFLD. Int J Mol Sci. 2016;17:447. doi: 10.3390/ijms17040447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wieland A, Frank DN, Harnke B, Bambha K. Systematic review: microbial dysbiosis and nonalcoholic fatty liver disease. Aliment Pharmacol Ther. 2015;42:1051–1063. doi: 10.1111/apt.13376. [DOI] [PubMed] [Google Scholar]
- 83.Ritze Y, Bárdos G, Claus A, Ehrmann V, Bergheim I, Schwiertz A, Bischoff SC. Lactobacillus rhamnosus GG protects against non-alcoholic fatty liver disease in mice. PLoS One. 2014;9:e80169. doi: 10.1371/journal.pone.0080169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Raman M, Ahmed I, Gillevet PM, Probert CS, Ratcliffe NM, Smith S, Greenwood R, Sikaroodi M, Lam V, Crotty P, et al. Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2013;11:868–875.e1-3. doi: 10.1016/j.cgh.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 85.Chung GE, Kim D, Kwak MS, Yang JI, Yim JY, Lim SH, Itani M. The serum vitamin D level is inversely correlated with nonalcoholic fatty liver disease. Clin Mol Hepatol. 2016;22:146–151. doi: 10.3350/cmh.2016.22.1.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Abenavoli L, Milic N, Luigiano C. Low 25-hydroxyvitamin D3 serum levels and non-alcoholic fatty liver disease. Minerva Med. 2014;105:175. [PubMed] [Google Scholar]
- 87.Eliades M, Spyrou E. Vitamin D: a new player in non-alcoholic fatty liver disease? World J Gastroenterol. 2015;21:1718–1727. doi: 10.3748/wjg.v21.i6.1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.George PS, Pearson ER, Witham MD. Effect of vitamin D supplementation on glycaemic control and insulin resistance: a systematic review and meta-analysis. Diabet Med. 2012;29:e142–e150. doi: 10.1111/j.1464-5491.2012.03672.x. [DOI] [PubMed] [Google Scholar]
- 89.Eliades M, Spyrou E, Agrawal N, Lazo M, Brancati FL, Potter JJ, Koteish AA, Clark JM, Guallar E, Hernaez R. Meta-analysis: vitamin D and non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2013;38:246–254. doi: 10.1111/apt.12377. [DOI] [PubMed] [Google Scholar]
- 90.Barchetta I, Carotti S, Labbadia G, Gentilucci UV, Muda AO, Angelico F, Silecchia G, Leonetti F, Fraioli A, Picardi A, et al. Liver vitamin D receptor, CYP2R1, and CYP27A1 expression: relationship with liver histology and vitamin D3 levels in patients with nonalcoholic steatohepatitis or hepatitis C virus. Hepatology. 2012;56:2180–2187. doi: 10.1002/hep.25930. [DOI] [PubMed] [Google Scholar]
- 91.Bril F, Maximos M, Portillo-Sanchez P, Biernacki D, Lomonaco R, Subbarayan S, Correa M, Lo M, Suman A, Cusi K. Relationship of vitamin D with insulin resistance and disease severity in non-alcoholic steatohepatitis. J Hepatol. 2015;62:405–411. doi: 10.1016/j.jhep.2014.08.040. [DOI] [PubMed] [Google Scholar]
- 92.Abramovitch S, Dahan-Bachar L, Sharvit E, Weisman Y, Ben Tov A, Brazowski E, Reif S. Vitamin D inhibits proliferation and profibrotic marker expression in hepatic stellate cells and decreases thioacetamide-induced liver fibrosis in rats. Gut. 2011;60:1728–1737. doi: 10.1136/gut.2010.234666. [DOI] [PubMed] [Google Scholar]
- 93.Marino L, Jornayvaz FR. Endocrine causes of nonalcoholic fatty liver disease. World J Gastroenterol. 2015;21:11053–11076. doi: 10.3748/wjg.v21.i39.11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Candia R, Riquelme A, Baudrand R, Carvajal CA, Morales M, Solís N, Pizarro M, Escalona A, Carrasco G, Boza C, et al. Overexpression of 11β-hydroxysteroid dehydrogenase type 1 in visceral adipose tissue and portal hypercortisolism in non-alcoholic fatty liver disease. Liver Int. 2012;32:392–399. doi: 10.1111/j.1478-3231.2011.02685.x. [DOI] [PubMed] [Google Scholar]
- 95.Tarantino G, Finelli C. Pathogenesis of hepatic steatosis: the link between hypercortisolism and non-alcoholic fatty liver disease. World J Gastroenterol. 2013;19:6735–6743. doi: 10.3748/wjg.v19.i40.6735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.de Silva HJ, Dassanayake AS. Non-alcoholic fatty liver disease: confronting the global epidemic requires better awareness. J Gastroenterol Hepatol. 2009;24:1705–1707. doi: 10.1111/j.1440-1746.2009.06026.x. [DOI] [PubMed] [Google Scholar]