

Abstract
Colorectal cancer (CRC) is a type of cancer with high morbidity and mortality rates worldwide and has become a global health problem. The conventional radiotherapy and chemotherapy regimen for CRC not only has a low cure rate but also causes side effects. Many studies have shown that adequate intake of fruits and vegetables in the diet may have a protective effect on CRC occurrence, possibly due to the special biological protective effect of the phytochemicals in these foods. Numerous in vitro and in vivo studies have demonstrated that phytochemicals play strong antioxidant, anti-inflammatory and anti-cancer roles by regulating specific signaling pathways and molecular markers to inhibit the occurrence and development of CRC. This review summarizes the progress on CRC prevention using the phytochemicals sulforaphane, curcumin and resveratrol, and elaborates on the specific underlying mechanisms. Thus, we believe that phytochemicals might provide a novel therapeutic approach for CRC prevention, but future clinical studies are needed to confirm the specific preventive effect of phytochemicals on cancer.
Keywords: Phytochemicals, Sulforaphane, Curcumin, Resveratrol, Colorectal cancer, Chemopreventive effects, Molecular mechanism
Core tip: This review summarizes the in vitro and in vivo studies on the phytochemicals sulforaphane, curcumin and resveratrol, and elaborates on their chemopreventive effects on colorectal cancer as well as their underlying molecular mechanisms. We hypothesize that phytochemicals might provide a new choice for colorectal cancer prevention and have great development potential.
INTRODUCTION
Nowadays, colorectal cancer (CRC) is one of the most common types of cancer worldwide, accounting for 10.0% of the total cancer cases in men and 9.2% of the total in women. There is no effective early detection method for CRC, which is already at an advanced stage when detected, making the treatment often passive. Therefore, CRC prevention prior to its onset has an important public health implication. Many studies have shown that a variety of fruits and vegetables are rich in phytochemicals, which may play strong chemopreventive roles in the prevention of CRC[1-4]. Thus, phytochemicals are expected to become the new choice of CRC prevention and treatment. A large number of studies have indicated that sulforaphane (SFN), curcumin, resveratrol and other phytochemicals can act on specific targeting molecules through regulating different signal transduction pathways in order to play chemopreventive roles in CRC, as detailed below.
SULFORAPHANE
SFN (4-methylsulfinylbutyl isothiocyanate) is abundant in cruciferous vegetables and is an important isothiocyanate derived from the hydrolysis of its precursor glucoraphanin[5]. SFN may act on multiple targets to interfere with early cancer development and progression[6,7]. Currently, a few mechanisms of the preventive role of SFN have been proposed:
Inhibiting phase I metabolic enzymes to reduce their roles in carcinogen activation
A series of metabolic reactions occur after carcinogens enter into the body, among which is phase I metabolism which includes oxidation, reduction and hydrolysis that increases the solubility and oxidation activity of carcinogens. Metabolites of the carcinogens then bind to key intracellular macromolecules (DNA, RNA and proteins) and become more damaging. Cytochrome P450 (CYP) is a major component of phase I metabolic enzymes and can convert xenobiotics into more electrophilic, reactive, mutagenic or even carcinogenic bioactive compounds. CYP3A4 is mainly expressed in the liver and intestine and can mediate the metabolism of carcinogens, making them more carcinogenic.
Previous studies have shown that SFN competitively inhibited or covalently modified some CYP isoforms and reduced their expression, thereby inhibiting DNA-adduct and chemical carcinogenesis[8,9]. Mahéo et al have found that SFN reduced the activity of CYP1A1, 2B1/2 and 3A4 in rat liver cells[10,11], possibly by inhibiting human pregnane X receptor (hPXR)-mediated CYP3A4 expression. In another study, SFN was found to significantly reduce the expression of CYP3A4 mRNA in Caco-2 cells, and CYP3A4 mRNA expression level was not increased in response to the hPXR-inducer rapamycin[5]. These studies suggest that the combined use of SFN and hPXR antagonists might have a synergistic effect, thereby reducing drug dosage and decreasing adverse drug reactions.
Inducing phase II metabolic enzymes to enhance their roles in carcinogen suppression
Phase II metabolism is composed mainly of binding reactions, wherein carcinogens and their metabolite reactive oxygen species (ROS) bind to endogenous ligands such as glutathione and glucuronic acid and form macromolecules to be excreted, thus avoiding further damage to DNA molecules. Currently, many studies have confirmed that SFN induces the expression of a number of phase II metabolic enzymes in CRC cells, including glutathione reductase (GR), glutathione S-transferase (GST), aldehyde reductase (AR), NAD[P]H:quinone oxidoreductase (NQO) and uridine 5’-diphospho (UDP)-glucuronosyltransferase (UGT)[12-21]. GR converts the active and oxidized glutathione to stable and reduced glutathione (GSH). GST catalyzes the binding of electrophilic substrate with GSH to induce its excretion. AR and NQO catalyze the conversion of metabolically active aldehydes and quinones to relatively stable alcohols and hydroquinones respectively, reducing their activities and damaging effects. UGT catalyzes the transferring of β-glucuronic acid from UDP-glucuronic acid to active substrates, thereby increasing their solubility and facilitating their excretion.
A recent research focus is the induction effect of SFN on UGT. Human colon has complex UGT1A loci expression patterns, in which UGT1A8 and 1A10 are specifically expressed in human gut and play an important role in the metabolism of carcinogens[22]. Our previous studies have shown that decreased activity in UGT1A and its isoforms 1A8 and 1A10 was one pathogenic mechanism of colon cancer[23,24]. When Caco-2 cells were treated with 10-30 μmol/L of SFN, UGT1A expression was induced in a dose-dependent manner. Treatment of Caco-2 cells with 25 μmol/L SFN for 24 h significantly improved mRNA and protein expression levels of UGT1A1, 1A8 and 1A10 as well as of their enzymatic activity[18]. It has also been shown that SFN and SFN-GSH significantly improved mRNA and protein expression levels of UGT1A1, mRNA expression level of GSTA1 and activity of bilirubin in HepG2 and HT-29 cells[25].
The underlying mechanism by which SFN regulates the activity of phase II metabolic enzymes is thought to be the Keap1-Nrf2-ARE signaling pathway[6,26]. There is a cis-regulatory structure (5’-A/GTGANNNGCA/G-3’) in the promoter region of phase II metabolic enzymes, known as antioxidant response element (ARE). Among many transcription factors that bind to ARE, Nrf2 was proposed to play a key role and to be induced by external factors to activate the transcription of antioxidant enzymes and detoxification enzymes[24,27]. Nrf2 normally binds to Keap1 and localizes in the cytoplasm, but is activated and translocated into the nucleus to activate the expression of phase II metabolic enzymes following SFN treatment or ROS attack. The Keap1-Nrf2-ARE signaling pathway is regulated by upstream phosphatidylinositol 3-kinase (PI3K), protein kinase C (PKC) and mitogen-activated protein kinases (MAPKs) which consists of extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK) and p38[6,15,28] (Figure 1). Our previous studies have shown that the expression of UGT1A in CRC tissue was down-regulated compared to that in normal tissue and colorectal adenoma tissue, while the expression of Nrf2 in CRC tissue was up-regulated compared to that in normal adenoma tissue, and Nrf2 was only expressed in the cytoplasm of cells in adenomas and CRC tissues[23]. SFN increased Nrf2 mRNA expression in Caco-2 cells and induced the nuclear translocation of Nrf2 in response to SFN treatment, as confirmed by confocal microscopy[18]. These results suggest that SFN might have a chemoprotective effect on CRC through this signaling pathway. In addition, SFN was found to increase the activity of NQO and GST in intestinal cells from wild-type Nrf2+/+ mice[29] and to induce the expression of Nrf2-dependent GST, NQO1 and carboxyl esterase. SFN also significantly improved mRNA expression of GST, NQO1, UGT1A6 and GR, and enhanced the activity of phase II enzymes including GST, NQO1, UDP-glucose dehydrogenase[30]. These effects of SFN were significantly decreased in mutant Nrf2-/- mice, suggesting that the Nrf2 pathway is one of the key mechanisms by which SFN exerts a chemoprevention effect.
Figure 1.
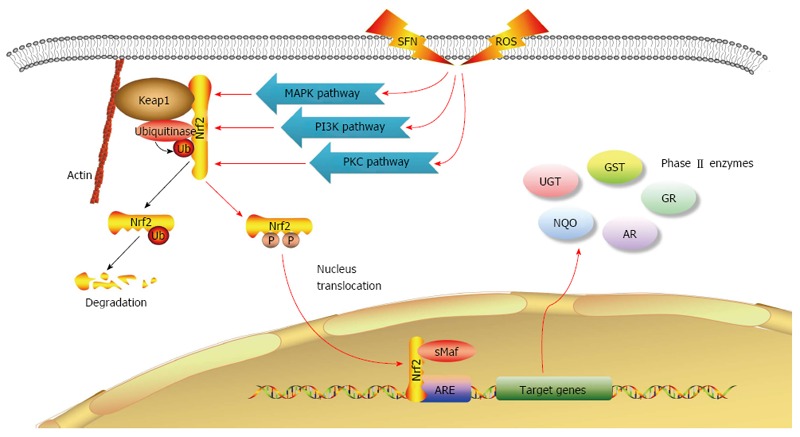
Sulforaphane regulates Keap1-Nrf2-ARE signaling pathway and induces phase II metabolic enzyme expression. Blue arrows indicate that under normal circumstances, Nrf2 binds to Keap1 in the cytoplasm and undergoes ubiquitin-mediated degradation. Red arrows indicate that under SFN treatment or ROS attack, Nrf2 is activated though the MAPK, PIK3 and PKC signaling pathways, and translocated to the nucleus, where it binds to promoter region ARE of the target genes and activates the expression of phase II metabolic enzymes. ARE: Antioxidant response element; MAPKs: Mitogen-activated protein kinases; PI3K: Phosphatidylinositol 3-kinase; PKC: Protein kinase C; ROS: Reactive oxygen species; SFN: Sulforaphane.
Inducing apoptosis
Many studies have shown that SFN treatment inhibited the viability of HT-29, Caco-2, SW620, KM12, SNU-1040, DLD-1 and other CRC cells[18,31-33], induced typical apoptotic morphology[5,32,34], increased caspase-3 activity[32] and mediated the cleavage of poly (ADP-ribose) polymerase (PARP), which is vital in DNA mismatch repair[33,34]. Meanwhile, SFN showed little effect on normal cells[33,35].
Previous studies on the mechanisms underlying SFN-induced apoptosis have shown that large-dose SFN induced apoptosis in CRC cells though reducing the expression of anti-apoptotic Bcl-2 and inducing the expression of pro-apoptotic Bax[18,34,36,37]. Gamet-Payrastre et al[34] have found that after the treatment of SFN, there was no detected expression of Bcl-2 and Bax gradually increasing with the occurrence of apoptosis in HT-29 cells. When Bax is partially translocated into mitochondria, it induces cytochrome C, releasing into the cytoplasm and triggering the caspase cascade, and PARP is then hydrolyzed, indicating that SFN can induce Bax-dependent apoptosis. In addition, Rudolf et al[37] have found that in p53 knock-out HCT116 cells, SFN inhibited the expression of Bcl-2, up-regulated the expression of Bax and promoted the release of cytochrome C. Their studies have also shown that SFN mediated DNA damage, activated the JNK pathway and changed the permeability of early lysosomal membranes to induce cathepsin B and D release, thus exerting a dose-dependent effect on apoptosis.
SFN can also induce apoptosis through the MAPK pathway. Many studies have shown that SFN induced apoptosis, arrested cell cycle or induced phase II metabolic enzyme expression via activation of JNK and p38 signaling pathways[33,37,38]. Hu et al[39] have found that phenethyl isothiocyanate, which has a structure similar to that of SFN, induced the phosphorylation and activation of three MAPKs in HT-29 cells and that inhibitors of JNK significantly inhibited the SFN-mediated apoptosis. Therefore, activation of JNK signaling pathway may play an important role in phenethyl isothiocyanate-mediated apoptosis.
Cell cycle arrest
Previous studies have shown that SFN arrested cell cycle in many types of CRC cells. For example, SFN arrested HT-29 cells in the G0/G1[31,40] and G2/M phases[34,41], arrested Caco-2 cells in the G1/G2[18] and G2/M phases[15], and arrested HCT116 cells in the G2/M phase[42]. SFN also augmented the expression of p21, which exerted inhibitory activity on cyclin-dependent kinase (CDK), and regulated the levels of many cell cycle proteins. While arresting HT-29 cells in the G0/G1 phase, SFN significantly induced the expression of p21 and reduced the expression level of some regulatory proteins, which is vital for G1 phase, such as cyclin A, cyclin D1 and c-myc[40]. While arresting HT-29 cells in the G2/M phase, SFN also up-regulated the expression of p21[41] and induced the expression of cyclin A and B1 that promote cell arrest at the G2 phase[34,41].
Recent studies have shown that activation of MAPK cascade might play a significant role in SFN-induced cycle arrest in CRC cells[33,40]. SFN can activate JNK and p38 pathways, arrest cell cycle in the G2/M phase and significantly inhibit the viability of many types of CRC cells. It is possible that p38 pathway activates MAP kinase-activated protein kinase 2 (MAPKAPK2) to phosphorylate the cell division cycle protein 2 (CDC2), thereby inhibiting its ability to dephosphorylate CDK1, and activates the CDK1-cyclin B complex to arrest cell cycle in the G2/M phase[33]. Addition of specific CDC2 kinase inhibitors retarded the effect of SFN on arresting cells in the G2/M phase and destroyed its apoptosis induction effect[41]. These results suggest that SFN may induce cell cycle arrest and apoptosis through JNK orp38-MK2-CDC2-CDK1 pathways.
SFN can also induce cell cycle arrest by interfering with microtubule formation. Byun et al[33] have recently reported that SFN interfered with microtubule polymerization in CRC cells and reduced microtubule levels in HCT116 and HT-29 cells, thereby arresting cells in the G2/M phase.
Interestingly, SFN can increase the level of ROS in cells while arresting cell cycle progression, thus participating in the induction of apoptosis. Through activation of JNK and p38 pathways, SFN can arrest cell cycle in the G2/M phase, and augment intracellular ROS level while decreasing GSH level. Excess ROS causes apoptosis or death. Addition of antioxidant N-acetylcysteine and catalase in CRC cells reversed the effect of SFN on cell viability and cell cycle arrest but no significant change in normal colorectal cells[33]. A similar study also confirmed that GSH blocked SFN-induced activation of MAPK pathway and inhibited the expression of p21 and cyclin D1[40]. Therefore, SFN may induce ROS synthesis to selectively kill tumor cells, one possible mechanism by which SFN plays a chemopreventive role in CRC.
In addition, SFN can also affect the expression of genes that regulate cell cycle and apoptosis through epigenetic mechanisms. SFN reduced the activity of histone deacetylase (HDAC) in HCT116 cells[42-45], and raised the levels of acetylated histone H3, H4, p21 and tubulin[43,45,46]. Long-term treatment with SFN also inhibited tumor development while increasing the acetylation level of histone on the promoter regions of the genes that specifically bind to p21 and Bax in colon polyps[46]. Nrf2 pathway is likely to play a role in the inhibition of HDAC by SFN. Rajendran et al[47] have found that carcinogen 1,2-dimethylhydrazine more likely induced CRC in wild-type Nrf2+/+ mice than in mutant Nrf2-/+ mice and that HDAC level in wild-type mice was significantly increased as compared to that in Nrf2-/+ mice. SFN treatment had a more profound effect on improving tumors in wild-type mice than in Nrf2-/+ mice. Other studies have shown that SFN inhibited the activity of HDAC by promoting its degradation in the cytoplasm. When HCT116 cells were continuously treated with SFN, HDAC mainly degraded after entering into the cytoplasm and binding to Pin1, and arrested cell cycle at the G2/M phase. However, at 24 h after the removal of SFN, HDAC bound to 14-3-3 and was translocated into the nucleus, allowing arrested cells to enter normal cell cycle[42].
Inducing autophagy
Many studies have found that SFN induced autophagy in human prostate cancer[48], colon cancer[36] and breast cancer[49]. We have found that SFN induced Caco-2 cell autophagy in a dose- and time-dependent manner. At 5 μmol/L to 25 μmol/L, cell autophagy augmented with the increase in SFN concentration. At 25 μmol/L, cell autophagy in Caco-2 cells became enhanced with the SFN treatment time increasing from 6-36 h[5]. Nishikawa et al[36] have shown that SFN inhibited the proliferation of WiDr cells, with high-dose (80 μmol/L) inducing apoptosis and medium- to low-dose (20 μmol/L to 40 μmol/L) inducing autophagy. Autophagy inhibitors suppressed autophagy and enhanced SFN-mediated apoptosis. Therefore, SFN in combination with autophagy inhibitors might restrain cell autophagy and promote apoptosis, thus having a better role in cancer prevention.
Inhibiting tumor angiogenesis
Kim et al[50] have found that SFN significantly reduced the expression level of vascular endothelial growth factor (VEGF) and hypoxia inducible factor-1α (HIF-1α), both of which are associated with angiogenesis and tumor metastasis, and inhibited tumor metastasis in a dose-dependent manner in hypoxia-treated HCT116 cells. Therefore, SFN might inhibit the expression of VEGF and HIF-1α to prevent CRC cancer progression and tumor angiogenesis. In addition, previous studies have shown that SFN decreased the expression level of VEGF, HIF-1α and c-myc in hypoxia-treated vascular endothelial cells in a time- and dose-dependent manner, inhibited VEGF receptor expression and affected endothelial cell basal membrane integrity[51]. SFN also arrested vascular endothelial cells at the G2/M phase, interfered with normal polymerization of mitotic microtubules[52] and induced autophagy. SFN in combination with autophagy inhibitors enhanced its pro-apoptotic effect[53]. Therefore, SFN might play a significant role in cancer prevention by inhibiting tumor angiogenesis and remodeling.
Enhancing the efficacy of chemotherapy drugs
Studies have shown that both SFN and antineoplastic agent oxaliplatin could inhibit the proliferation of Caco-2 cells in a dose-dependent manner. Combined use of SFN and oxaliplatin synergistically activated endogenous and exogenous apoptotic signaling pathways in Caco-2 cells, which exhibited typical apoptotic morphology and became necrotic with increased duration and concentration[54]. Pretreatment of HT-29 cells with 2.5 μmol/L SFN activated the cancer drug PR-104A and reduced its EC50 by 3.6-fold. Similar results were observed with SW620 cells. However, SFN had little effect on normal colon cells[55]. Therefore, SFN combined with traditional chemotherapy drugs might reduce drug resistance and enhance drug efficacy. Reducing drug dose while ensuring drug efficacy might reduce the incidence of dose-related adverse reactions, which is one of the future development directions of SFN clinical applications.
The potential chemopreventive effect of SFN on CRC has also been confirmed by in vivo studies. Animal experiments have shown that 2-6 h after oral gavage of 5-20 μmol/L SFN, the highest level of SFN was detected in small intestine, prostate, kidney and lung (in a descending order)[56]. Therefore, SFN might exert a chemopreventive effect by being a target for prevention of CRC. SFN also significantly reduced the size of xenograft tumor in HCT116 cell-transplanted nude mice in a dose-dependent manner[33] and decreased the number of aberrant crypt foci (especially the multi-crypt foci) in azoxymethane-treated rats[57].
Future studies will determine whether SFN in food might play a chemopreventive role through the above-discussed molecular mechanisms, as well as determine its effective concentration, bioavailability and interactions with other dietary components. Understanding the distribution, metabolism and excretion of SFN in vivo could become a significant avenue of research. Large-scale clinical cohort studies are critical and required to confirm the chemopreventive effects of SFN on CRC.
CURCUMIN
Curcumin (diferuloylmethane) is the primary component of turmeric, which is a common spice plant belonging to the ginger family[58,59]. Curcumin can regulate multiple signaling pathways and mediate the activities of target molecules in CRC cells, thereby inhibiting cancer initiation, promotion and progression.
Inhibiting cell proliferation and inducing apoptosis
β-catenin is a key factor in the regulation of cell proliferation in CRC. It stimulated the expression of cell proliferation-related target genes, such as c-myc and cyclin D1, and was regulated by the Wnt signaling pathways[60]. Curcumin and its derivatives suppressed β-catenin response transcription without altering the intracellular level of β-catenin, reduced the expression of Wnt/β-catenin pathway transcriptional coactivator p300 and inhibited β-catenin-related transcription, thus playing a role in inhibiting CRC cell growth and proliferation[61]. Recent studies have found that curcumin blocked Wnt/β-catenin pathway-mediated c-myc expression to disrupt cell-cell adhesion in HCT116 cells, arrested cell cycle at the G2/M phase and induced apoptosis[62]. Thus, the Wnt/β-catenin pathway might represent a key mechanism by which curcumin suppresses cell growth and induces apoptosis.
The adenosine monophosphate (AMP)-activated protein kinase (AMPK)-cyclooxygenase-2 (COX-2) cascade might also be an important mechanism by which curcumin restrains CRC cells proliferation. It has been reported that curcumin arrested HT-29 cells in the G1 phase and induced apoptosis, while reducing the expression of COX-2 and apoptosis-related kinase pAkt and up-regulating the expression level of p-AMPK. Treatment of HT-29 cells with AMPK inhibitor compound C following curcumin inhibited AMPK expression, increased COX-2 and pAkt expression and promoted cell proliferation, but did not induce apoptosis[63]. As such, AMPK might be the key molecule for curcumin-induced apoptosis in CRC. The pAkt-AMPK-COX-2 or AMPK-pAkt-COX-2 pathway might be a vital signaling transduction pathway that regulates CRC cell proliferation or apoptosis.
Cell cycle arrest
Curcumin arrested cell cycle in CRC cells possibly by up-regulating the expression level of p53 and p21[64]. When there was DNA damage in the cells, curcumin increased the expression of p53 to further induce p21 expression and cell apoptosis[65,66]. However, some studies have also shown that curcumin arrested cells in the G2/M phase and induced apoptosis in the absence of p53 and p21 expression[62]. Curcumin also regulated the effect of cyclin and CDK. Irving et al[64] have shown that curcumin arrested cells in the G1/S phase, down-regulated cyclin D and blocked CDK (in particular, CDK4 and CDK6), which may be due to the decreased activity of telomerase and growth factor receptors mediated by curcumin.
Affecting epigenetics
Aberrant DNA methylation has been identified as one of the mechanisms of CRC development[67-72], including silent transcription caused by local hypermethylation of promoter CpG islands, chromosomal instability caused by DNA global hypomethylation and formation of aneuploidy[73-76]. Some researchers have pointed out that different from the DNA methyltransferase inhibitor 5-aza-29-deoxycytidine-induced DNA global hypermethylation in CRC cells, curcumin might induce DNA local hypermethylation in a time-dependent manner, and then induce or reduce the expression of a number of related genes[77]. Thus, curcumin might have a chemopreventive effect on CRC by selectively inducing hypermethylation in some genes; however, the specific mechanism needs further exploration.
Enhancing the efficacy of chemotherapy drugs
Recent studies have shown that curcumin combined with traditional chemotherapy drugs can delay or reduce drug resistance. In vitro experiments have indicated that curcumin down-regulated the expression of insulin-like growth factor-1 receptor, AKT, COX-2 and cyclin D1 to significantly reduce the resistance to 5-fluorouracil (5-FU) plus oxaliplatin in CRC cells[78]. In vivo and in vitro studies have also demonstrated that curcumin enhanced the efficacy of oxaliplatin in drug-resistant CRC cells[79]. Curcumin enhanced the sensitivity of 5-FU-resistant CRC cells and suppressed cancer stem cells[80]. These results suggest that curcumin in combination with conventional chemotherapy drugs can reduce viability of drug-resistant CRC cells and thus plays a role in enhancing the effectiveness of chemotherapy drugs.
A recent clinical trial with 126 CRC patients has shown that oral uptake of 1.08 g curcumin per day (n = 63) decreased the serum TNF-α level, but up-regulated the level of p53 and apoptotic cancer cells in tumor tissue, and increased the weight of the patients at last. Therefore, curcumin might up-regulate the expression level of p53 in tumor cells to accelerate tumor cell apoptosis and enhance the health of CRC patients[81]. Carroll et al[82] selected 41 patients with eight or more aberrant crypt foci to take 2.0 g (n = 22) or 4.0 g (n = 19) curcumin orally per day and found no significant change in patients with daily oral administration of 2.0 g curcumin, but a 40% reduction of the total number of colonic crypt foci was observed in those who received 4.0 g curcumin daily. There are limited clinical trials on curcumin and large-scale studies are needed in the future to explore the specific effective dose and mechanisms by which curcumin prevents CRC.
To conclude, the protect role of curcumin as a phytochemical agent against CRC is now well supported, but a larger sample size is needed in the future research to investigate its bioavailability in the body, plasma concentration and clinical efficacy.
RESVERATROL
Resveratrol (trans-3,4’,5-trihydroxystilbene) belongs to the stilbene family of phenolic compounds and is found in nuts, pines and berries, and especially in the skin of red grapes[83]. Resveratrol can inhibit lipid peroxidation thrombocyte aggregation, thus playing anti-oxidation, anti-inflammatory and vasodilation roles[84,85]. Numerous studies have confirmed that resveratrol might also exert a chemopreventive effect on CRC, and the underlying molecular biological mechanisms are detailed below.
Inhibiting cell proliferation and inducing apoptosis
Resveratrol can regulate cell energy metabolism to play an interesting role in suppressing cancer cell growth and inducing apoptosis. Studies have shown that resveratrol inhibited the pentose phosphate pathway to further inhibit focal adhesion complex proteins, such as talin and focal adhesion kinase, then suppressed cell growth and induced apoptosis in CRC cells[86]. Calorie restriction by resveratrol has been recently proposed[87]. Fouad et al[88] have confirmed that resveratrol significantly reduced the levels of glycolytic enzymes in Caco-2 cells, thus enhancing the activity of citrate synthase and reducing the consumption of glucose. More importantly, resveratrol decreased the expression of leptin, c-myc and VEGF, and activated apoptotic markers caspases 3 and 8 by increasing the ratio of Bax/Bcl-2. These results indicate that resveratrol might induce apoptosis and inhibit angiogenesis via the caloric restriction pathway.
Cell cycle arrest
Recent studies have shown that resveratrol arrested cells in the G1/S phase and induced apoptosis via cyclin-CDK pathway-dependent caspase in HCT116 and Caco-2 cells[89]. Another study has also shown that resveratrol inhibited the growth of Caco-2 cells and suppressed the expression level of cyclin D1 and CDK-4. Low-dose resveratrol arrested cells in the S/G2 phase, while high-dose resveratrol induced cell apoptosis[90]. Similarly, it has been found that resveratrol inhibited the survival of Caco-2 cells, arrested cell cycle in the S/G2 phase and reduced the activity of ornithine decarboxylase, which is essential for polyamine synthesis, without inducing apoptosis[91]. These results are similar with others; for instance, Wolter et al[92] have proposed that resveratrol may reduce polyamine biosynthesis by interfering with the activity of ornithine decarboxylase, thereby inhibiting CRC cell proliferation.
Anti-inflammatory effect
With greater anatomic extent and severity of colitis, the risk of CRC occurrence and progression increase[93,94]. A number of inflammation-related genes were up-regulated in intestinal inflammatory mucosa and tumors[95]. Many inflammatory mediators, such as pro-inflammatory cytokines, prostaglandins and nitric oxide (NO), are associated with the occurrence of cancer and play a role in the progression of cancer. Recent studies have confirmed that resveratrol interfered with NF-κB-dependent signaling mechanisms, reduced mRNA and protein expression of inducible NO synthase and inhibited NO production. Resveratrol also reduced the expression of Toll-like receptor-4 and reduced lipopolysaccharide-mediated inflammatory response in Caco-2 and SW480 cells[96]. Similarly, in the dextran sulfate sodium colitis mouse model, resveratrol significantly increased the anti-inflammatory score, reduced the proportion of neutrophils in some lymphoid tissue and down-regulated the levels of inducible NO synthase, COX-2 and TNF-α[97]. These studies confirm the anti-inflammatory effect of resveratrol and suggest that resveratrol might play a role in inflammation-related tumor resistance.
Improving the effectiveness of chemotherapy drugs
Some researchers have explored the effect of resveratrol on decreasing CRC drug resistance and enhancing chemotherapy efficacy. Resveratrol has been reported to induce apoptosis in chemotherapy drug etoposide-resistant HT-29 cells via the AMPK pathway. It also increased the level of phosphorylated acetyl-CoA carboxylase and phosphorylated and activated downstream AMPK, thereby inhibiting cell growth, up-regulating ROS level and inducing apoptosis. AMPK inhibitor compound C decreased the cytotoxicity induced by the combined use of resveratrol and etoposide[98]. In addition, resveratrol in combination with chemotherapeutic drug 1,3-Bis (2-chloroethyl)-1-nitrosourea induced the apoptosis of 5-FU-resistant HCT116 cells[99], and resveratrol sulfate mixture enhanced effectiveness of the anticancer drug oxaliplatin[100]. However, it has also been reported that resveratrol in combination of oxaliplatin did not have an antichemosensitizing effect[101]. Thus, large-scale studies are required to confirm the efficacy of resveratrol in improving CRC cell sensitivity to chemotherapy drugs.
Metabolic kinetic studies have indicated a high clearance rate in circulation and low bioavailability of resveratrol[87,102,103], thus its use for CRC prevention has been highly controversial. But, some studies have suggested that the possible reason for low level of resveratrol in the circulatory system is because resveratrol can quickly induce the phase II metabolic enzymes UGT and sulfotransferase to produce resveratrol glucuronides and resveratrol sulfates[104-107]. A recent clinical trial has shown that the level of three resveratrol sulfate conjugates was high in 8 patients with oral administration of 0.5 g or 1.0 g resveratrol daily prior to surgery and that the proliferation of tumor cells was significantly suppressed in these patients, suggesting that daily oral uptake of 0.5 g or 1.0 g resveratrol might play a key role in cancer prevention[108]. Other researchers have used various resveratrol precursors, sulfates and other derivatives to treat CRC cells and found that they could be used alone or synergistically to suppress cancer cell proliferation, stimulate apoptosis, arrest cell cycle, induce cell differentiation and enhance effectiveness of anticancer drugs[100,109,110]. These studies have suggested that despite low resveratrol bioavailability in the body, resveratrol might have an anticancer effect through its relevant metabolites; large-scale clinical trials are required to confirm this hypothesis.
Resveratrol might play a chemoprotective effect on CRC via its metabolites and derivatives. Further clinical studies are necessary to explore the effective concentration and specific mechanism of resveratrol.
CONCLUSION
This review summarizes the chemopreventive effect of phytochemicals sulforaphane, curcumin and resveratrol on CRC and the underlying molecular mechanisms. Phytochemicals are safe and their raw materials are readily available, which contributes to making them promising new therapeutic options for CRC. Early marketing of phytochemicals can provide supplemental diet to better prevent or treat high-risk groups and cancer patients, reduce CRC incidence and mortality, and benefit mankind.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: China
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): 0
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Conflict-of-interest statement: We declare that all the authors have no financial and personal relationships with other people or organization that can inappropriately influence our work. Additionally, there is no conflict of interests of any product, service or company to declare.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: March 28, 2016
First decision: May 12, 2016
Article in press: June 15, 2016
P- Reviewer: Cerwenka HR S- Editor: Ma YJ L- Editor: Filipodia E- Editor: Ma S
References
- 1.Jaganathan SK, Vellayappan MV, Narasimhan G, Supriyanto E, Octorina Dewi DE, Narayanan AL, Balaji A, Subramanian AP, Yusof M. Chemopreventive effect of apple and berry fruits against colon cancer. World J Gastroenterol. 2014;20:17029–17036. doi: 10.3748/wjg.v20.i45.17029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Macdonald RS, Wagner K. Influence of dietary phytochemicals and microbiota on colon cancer risk. J Agric Food Chem. 2012;60:6728–6735. doi: 10.1021/jf204230r. [DOI] [PubMed] [Google Scholar]
- 3.Madka V, Rao CV. Anti-inflammatory phytochemicals for chemoprevention of colon cancer. Curr Cancer Drug Targets. 2013;13:542–557. doi: 10.2174/15680096113139990036. [DOI] [PubMed] [Google Scholar]
- 4.Westergaard D, Li J, Jensen K, Kouskoumvekaki I, Panagiotou G. Exploring mechanisms of diet-colon cancer associations through candidate molecular interaction networks. BMC Genomics. 2014;15:380. doi: 10.1186/1471-2164-15-380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang M, Zhu JY, Chen S, Qing Y, Wu D, Lin YM, Luo JZ, Han W, Li YQ. Effects of co-treatment with sulforaphane and autophagy modulators on uridine 5’-diphospho-glucuronosyltransferase 1A isoforms and cytochrome P450 3A4 expression in Caco-2 human colon cancer cells. Oncol Lett. 2014;8:2407–2416. doi: 10.3892/ol.2014.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Juge N, Mithen RF, Traka M. Molecular basis for chemoprevention by sulforaphane: a comprehensive review. Cell Mol Life Sci. 2007;64:1105–1127. doi: 10.1007/s00018-007-6484-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheung KL, Kong AN. Molecular targets of dietary phenethyl isothiocyanate and sulforaphane for cancer chemoprevention. AAPS J. 2010;12:87–97. doi: 10.1208/s12248-009-9162-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang CS, Smith TJ, Hong JY. Cytochrome P-450 enzymes as targets for chemoprevention against chemical carcinogenesis and toxicity: opportunities and limitations. Cancer Res. 1994;54:1982s–1986s. [PubMed] [Google Scholar]
- 9.Zhang Y, Talalay P. Anticarcinogenic activities of organic isothiocyanates: chemistry and mechanisms. Cancer Res. 1994;54:1976s–1981s. [PubMed] [Google Scholar]
- 10.Zhou C, Poulton EJ, Grün F, Bammler TK, Blumberg B, Thummel KE, Eaton DL. The dietary isothiocyanate sulforaphane is an antagonist of the human steroid and xenobiotic nuclear receptor. Mol Pharmacol. 2007;71:220–229. doi: 10.1124/mol.106.029264. [DOI] [PubMed] [Google Scholar]
- 11.Mahéo K, Morel F, Langouët S, Kramer H, Le Ferrec E, Ketterer B, Guillouzo A. Inhibition of cytochromes P-450 and induction of glutathione S-transferases by sulforaphane in primary human and rat hepatocytes. Cancer Res. 1997;57:3649–3652. [PubMed] [Google Scholar]
- 12.Talalay P. Chemoprotection against cancer by induction of phase 2 enzymes. Biofactors. 2000;12:5–11. doi: 10.1002/biof.5520120102. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y, Talalay P, Cho CG, Posner GH. A major inducer of anticarcinogenic protective enzymes from broccoli: isolation and elucidation of structure. Proc Natl Acad Sci USA. 1992;89:2399–2403. doi: 10.1073/pnas.89.6.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prochaska HJ, Santamaria AB, Talalay P. Rapid detection of inducers of enzymes that protect against carcinogens. Proc Natl Acad Sci USA. 1992;89:2394–2398. doi: 10.1073/pnas.89.6.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jakubíková J, Sedlák J, Mithen R, Bao Y. Role of PI3K/Akt and MEK/ERK signaling pathways in sulforaphane- and erucin-induced phase II enzymes and MRP2 transcription, G2/M arrest and cell death in Caco-2 cells. Biochem Pharmacol. 2005;69:1543–1552. doi: 10.1016/j.bcp.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 16.Yanaka A, Zhang S, Tauchi M, Suzuki H, Shibahara T, Matsui H, Nakahara A, Tanaka N, Yamamoto M. Role of the nrf-2 gene in protection and repair of gastric mucosa against oxidative stress. Inflammopharmacology. 2005;13:83–90. doi: 10.1163/156856005774423863. [DOI] [PubMed] [Google Scholar]
- 17.Banning A, Deubel S, Kluth D, Zhou Z, Brigelius-Flohé R. The GI-GPx gene is a target for Nrf2. Mol Cell Biol. 2005;25:4914–4923. doi: 10.1128/MCB.25.12.4914-4923.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang M, Chen S, Wang S, Sun D, Chen J, Li Y, Han W, Yang X, Gao HQ. Effects of phytochemicals sulforaphane on uridine diphosphate-glucuronosyltransferase expression as well as cell-cycle arrest and apoptosis in human colon cancer Caco-2 cells. Chin J Physiol. 2012;55:134–144. doi: 10.4077/CJP.2012.BAA085. [DOI] [PubMed] [Google Scholar]
- 19.Krehl S, Loewinger M, Florian S, Kipp AP, Banning A, Wessjohann LA, Brauer MN, Iori R, Esworthy RS, Chu FF, et al. Glutathione peroxidase-2 and selenium decreased inflammation and tumors in a mouse model of inflammation-associated carcinogenesis whereas sulforaphane effects differed with selenium supply. Carcinogenesis. 2012;33:620–628. doi: 10.1093/carcin/bgr288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Veeranki OL, Bhattacharya A, Marshall JR, Zhang Y. Organ-specific exposure and response to sulforaphane, a key chemopreventive ingredient in broccoli: implications for cancer prevention. Br J Nutr. 2013;109:25–32. doi: 10.1017/S0007114512000657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang ZQ, Chen C, Yang B, Hebbar V, Kong AN. Differential responses from seven mammalian cell lines to the treatments of detoxifying enzyme inducers. Life Sci. 2003;72:2243–2253. doi: 10.1016/s0024-3205(03)00101-2. [DOI] [PubMed] [Google Scholar]
- 22.Wang M, Sun DF, Wang S, Qing Y, Chen S, Wu D, Lin YM, Luo JZ, Li YQ. Polymorphic expression of UDP-glucuronosyltransferase UGTlA gene in human colorectal cancer. PLoS One. 2013;8:e57045. doi: 10.1371/journal.pone.0057045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang M, Qi YY, Chen S, Sun DF, Wang S, Chen J, Li YQ, Han W, Yang XY. Expression of UDP-glucuronosyltransferase 1A, nuclear factor erythroid-E2-related factor 2 and Kelch-like ECH-associated protein 1 in colonic mucosa, adenoma and adenocarcinoma tissue. Oncol Lett. 2012;4:925–930. doi: 10.3892/ol.2012.850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang M, Qi YY, Chen S, Liu L, Chen J, Li YQ. [The UGT1A, Nrf2 and Keap1 protein expression and significance in colon tumor] Zhonghua Nei Ke Zazhi. 2012;51:531–535. [PubMed] [Google Scholar]
- 25.Basten GP, Bao Y, Williamson G. Sulforaphane and its glutathione conjugate but not sulforaphane nitrile induce UDP-glucuronosyl transferase (UGT1A1) and glutathione transferase (GSTA1) in cultured cells. Carcinogenesis. 2002;23:1399–1404. doi: 10.1093/carcin/23.8.1399. [DOI] [PubMed] [Google Scholar]
- 26.Yang L, Palliyaguru DL, Kensler TW. Frugal chemoprevention: targeting Nrf2 with foods rich in sulforaphane. Semin Oncol. 2016;43:146–153. doi: 10.1053/j.seminoncol.2015.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maruyama A, Nishikawa K, Kawatani Y, Mimura J, Hosoya T, Harada N, Yamamato M, Itoh K. The novel Nrf2-interacting factor KAP1 regulates susceptibility to oxidative stress by promoting the Nrf2-mediated cytoprotective response. Biochem J. 2011;436:387–397. doi: 10.1042/BJ20101748. [DOI] [PubMed] [Google Scholar]
- 28.Svehlíková V, Wang S, Jakubíková J, Williamson G, Mithen R, Bao Y. Interactions between sulforaphane and apigenin in the induction of UGT1A1 and GSTA1 in CaCo-2 cells. Carcinogenesis. 2004;25:1629–1637. doi: 10.1093/carcin/bgh169. [DOI] [PubMed] [Google Scholar]
- 29.McMahon M, Itoh K, Yamamoto M, Chanas SA, Henderson CJ, McLellan LI, Wolf CR, Cavin C, Hayes JD. The Cap’n’Collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res. 2001;61:3299–3307. [PubMed] [Google Scholar]
- 30.Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002;62:5196–5203. [PubMed] [Google Scholar]
- 31.Gamet-Payrastre L, Lumeau S, Gasc N, Cassar G, Rollin P, Tulliez J. Selective cytostatic and cytotoxic effects of glucosinolates hydrolysis products on human colon cancer cells in vitro. Anticancer Drugs. 1998;9:141–148. doi: 10.1097/00001813-199802000-00005. [DOI] [PubMed] [Google Scholar]
- 32.Andelová H, Rudolf E, Cervinka M. In vitro antiproliferative effects of sulforaphane on human colon cancer cell line SW620. Acta Medica (Hradec Kralove) 2007;50:171–176. [PubMed] [Google Scholar]
- 33.Byun S, Shin SH, Park J, Lim S, Lee E, Lee C, Sung D, Farrand L, Lee SR, Kim KH, et al. Sulforaphene suppresses growth of colon cancer-derived tumors via induction of glutathione depletion and microtubule depolymerization. Mol Nutr Food Res. 2016;60:1068–1078. doi: 10.1002/mnfr.201501011. [DOI] [PubMed] [Google Scholar]
- 34.Gamet-Payrastre L, Li P, Lumeau S, Cassar G, Dupont MA, Chevolleau S, Gasc N, Tulliez J, Tercé F. Sulforaphane, a naturally occurring isothiocyanate, induces cell cycle arrest and apoptosis in HT29 human colon cancer cells. Cancer Res. 2000;60:1426–1433. [PubMed] [Google Scholar]
- 35.Zeng H, Trujillo ON, Moyer MP, Botnen JH. Prolonged sulforaphane treatment activates survival signaling in nontumorigenic NCM460 colon cells but apoptotic signaling in tumorigenic HCT116 colon cells. Nutr Cancer. 2011;63:248–255. doi: 10.1080/01635581.2011.523500. [DOI] [PubMed] [Google Scholar]
- 36.Nishikawa T, Tsuno NH, Okaji Y, Shuno Y, Sasaki K, Hongo K, Sunami E, Kitayama J, Takahashi K, Nagawa H. Inhibition of autophagy potentiates sulforaphane-induced apoptosis in human colon cancer cells. Ann Surg Oncol. 2010;17:592–602. doi: 10.1245/s10434-009-0696-x. [DOI] [PubMed] [Google Scholar]
- 37.Rudolf E, Cervinka M. Sulforaphane induces cytotoxicity and lysosome- and mitochondria-dependent cell death in colon cancer cells with deleted p53. Toxicol In Vitro. 2011;25:1302–1309. doi: 10.1016/j.tiv.2011.04.019. [DOI] [PubMed] [Google Scholar]
- 38.Kim BR, Hu R, Keum YS, Hebbar V, Shen G, Nair SS, Kong AN. Effects of glutathione on antioxidant response element-mediated gene expression and apoptosis elicited by sulforaphane. Cancer Res. 2003;63:7520–7525. [PubMed] [Google Scholar]
- 39.Hu R, Kim BR, Chen C, Hebbar V, Kong AN. The roles of JNK and apoptotic signaling pathways in PEITC-mediated responses in human HT-29 colon adenocarcinoma cells. Carcinogenesis. 2003;24:1361–1367. doi: 10.1093/carcin/bgg092. [DOI] [PubMed] [Google Scholar]
- 40.Shen G, Xu C, Chen C, Hebbar V, Kong AN. p53-independent G1 cell cycle arrest of human colon carcinoma cells HT-29 by sulforaphane is associated with induction of p21CIP1 and inhibition of expression of cyclin D1. Cancer Chemother Pharmacol. 2006;57:317–327. doi: 10.1007/s00280-005-0050-3. [DOI] [PubMed] [Google Scholar]
- 41.Parnaud G, Li P, Cassar G, Rouimi P, Tulliez J, Combaret L, Gamet-Payrastre L. Mechanism of sulforaphane-induced cell cycle arrest and apoptosis in human colon cancer cells. Nutr Cancer. 2004;48:198–206. doi: 10.1207/s15327914nc4802_10. [DOI] [PubMed] [Google Scholar]
- 42.Rajendran P, Delage B, Dashwood WM, Yu TW, Wuth B, Williams DE, Ho E, Dashwood RH. Histone deacetylase turnover and recovery in sulforaphane-treated colon cancer cells: competing actions of 14-3-3 and Pin1 in HDAC3/SMRT corepressor complex dissociation/reassembly. Mol Cancer. 2011;10:68. doi: 10.1186/1476-4598-10-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Myzak MC, Karplus PA, Chung FL, Dashwood RH. A novel mechanism of chemoprotection by sulforaphane: inhibition of histone deacetylase. Cancer Res. 2004;64:5767–5774. doi: 10.1158/0008-5472.CAN-04-1326. [DOI] [PubMed] [Google Scholar]
- 44.Rajendran P, Kidane AI, Yu TW, Dashwood WM, Bisson WH, Löhr CV, Ho E, Williams DE, Dashwood RH. HDAC turnover, CtIP acetylation and dysregulated DNA damage signaling in colon cancer cells treated with sulforaphane and related dietary isothiocyanates. Epigenetics. 2013;8:612–623. doi: 10.4161/epi.24710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dickinson SE, Rusche JJ, Bec SL, Horn DJ, Janda J, Rim SH, Smith CL, Bowden GT. The effect of sulforaphane on histone deacetylase activity in keratinocytes: Differences between in vitro and in vivo analyses. Mol Carcinog. 2015;54:1513–1520. doi: 10.1002/mc.22224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Myzak MC, Dashwood WM, Orner GA, Ho E, Dashwood RH. Sulforaphane inhibits histone deacetylase in vivo and suppresses tumorigenesis in Apc-minus mice. FASEB J. 2006;20:506–508. doi: 10.1096/fj.05-4785fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rajendran P, Dashwood WM, Li L, Kang Y, Kim E, Johnson G, Fischer KA, Löhr CV, Williams DE, Ho E, et al. Nrf2 status affects tumor growth, HDAC3 gene promoter associations, and the response to sulforaphane in the colon. Clin Epigenetics. 2015;7:102. doi: 10.1186/s13148-015-0132-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Herman-Antosiewicz A, Johnson DE, Singh SV. Sulforaphane causes autophagy to inhibit release of cytochrome C and apoptosis in human prostate cancer cells. Cancer Res. 2006;66:5828–5835. doi: 10.1158/0008-5472.CAN-06-0139. [DOI] [PubMed] [Google Scholar]
- 49.Kanematsu S, Uehara N, Miki H, Yoshizawa K, Kawanaka A, Yuri T, Tsubura A. Autophagy inhibition enhances sulforaphane-induced apoptosis in human breast cancer cells. Anticancer Res. 2010;30:3381–3390. [PubMed] [Google Scholar]
- 50.Kim DH, Sung B, Kang YJ, Hwang SY, Kim MJ, Yoon JH, Im E, Kim ND. Sulforaphane inhibits hypoxia-induced HIF-1α and VEGF expression and migration of human colon cancer cells. Int J Oncol. 2015;47:2226–2232. doi: 10.3892/ijo.2015.3200. [DOI] [PubMed] [Google Scholar]
- 51.Bertl E, Bartsch H, Gerhäuser C. Inhibition of angiogenesis and endothelial cell functions are novel sulforaphane-mediated mechanisms in chemoprevention. Mol Cancer Ther. 2006;5:575–585. doi: 10.1158/1535-7163.MCT-05-0324. [DOI] [PubMed] [Google Scholar]
- 52.Jackson SJ, Singletary KW, Venema RC. Sulforaphane suppresses angiogenesis and disrupts endothelial mitotic progression and microtubule polymerization. Vascul Pharmacol. 2007;46:77–84. doi: 10.1016/j.vph.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 53.Nishikawa T, Tsuno NH, Okaji Y, Sunami E, Shuno Y, Sasaki K, Hongo K, Kaneko M, Hiyoshi M, Kawai K, et al. The inhibition of autophagy potentiates anti-angiogenic effects of sulforaphane by inducing apoptosis. Angiogenesis. 2010;13:227–238. doi: 10.1007/s10456-010-9180-2. [DOI] [PubMed] [Google Scholar]
- 54.Kaminski BM, Weigert A, Brüne B, Schumacher M, Wenzel U, Steinhilber D, Stein J, Ulrich S. Sulforaphane potentiates oxaliplatin-induced cell growth inhibition in colorectal cancer cells via induction of different modes of cell death. Cancer Chemother Pharmacol. 2011;67:1167–1178. doi: 10.1007/s00280-010-1413-y. [DOI] [PubMed] [Google Scholar]
- 55.Erzinger MM, Bovet C, Hecht KM, Senger S, Winiker P, Sobotzki N, Cristea S, Beerenwinkel N, Shay JW, Marra G, et al. Sulforaphane Preconditioning Sensitizes Human Colon Cancer Cells towards the Bioreductive Anticancer Prodrug PR-104A. PLoS One. 2016;11:e0150219. doi: 10.1371/journal.pone.0150219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Clarke JD, Hsu A, Williams DE, Dashwood RH, Stevens JF, Yamamoto M, Ho E. Metabolism and tissue distribution of sulforaphane in Nrf2 knockout and wild-type mice. Pharm Res. 2011;28:3171–3179. doi: 10.1007/s11095-011-0500-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chung FL, Conaway CC, Rao CV, Reddy BS. Chemoprevention of colonic aberrant crypt foci in Fischer rats by sulforaphane and phenethyl isothiocyanate. Carcinogenesis. 2000;21:2287–2291. doi: 10.1093/carcin/21.12.2287. [DOI] [PubMed] [Google Scholar]
- 58.Das RK, Kasoju N, Bora U. Encapsulation of curcumin in alginate-chitosan-pluronic composite nanoparticles for delivery to cancer cells. Nanomedicine. 2010;6:153–160. doi: 10.1016/j.nano.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 59.Goel A, Kunnumakkara AB, Aggarwal BB. Curcumin as “Curecumin”: from kitchen to clinic. Biochem Pharmacol. 2008;75:787–809. doi: 10.1016/j.bcp.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 60.Chung MY, Lim TG, Lee KW. Molecular mechanisms of chemopreventive phytochemicals against gastroenterological cancer development. World J Gastroenterol. 2013;19:984–993. doi: 10.3748/wjg.v19.i7.984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ryu MJ, Cho M, Song JY, Yun YS, Choi IW, Kim DE, Park BS, Oh S. Natural derivatives of curcumin attenuate the Wnt/beta-catenin pathway through down-regulation of the transcriptional coactivator p300. Biochem Biophys Res Commun. 2008;377:1304–1308. doi: 10.1016/j.bbrc.2008.10.171. [DOI] [PubMed] [Google Scholar]
- 62.Jaiswal AS, Marlow BP, Gupta N, Narayan S. Beta-catenin-mediated transactivation and cell-cell adhesion pathways are important in curcumin (diferuylmethane)-induced growth arrest and apoptosis in colon cancer cells. Oncogene. 2002;21:8414–8427. doi: 10.1038/sj.onc.1205947. [DOI] [PubMed] [Google Scholar]
- 63.Kovacic P, Somanathan R. Multifaceted approach to resveratrol bioactivity: Focus on antioxidant action, cell signaling and safety. Oxid Med Cell Longev. 2010;3:86–100. doi: 10.4161/oxim.3.2.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Irving GR, Karmokar A, Berry DP, Brown K, Steward WP. Curcumin: the potential for efficacy in gastrointestinal diseases. Best Pract Res Clin Gastroenterol. 2011;25:519–534. doi: 10.1016/j.bpg.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 65.Basile V, Ferrari E, Lazzari S, Belluti S, Pignedoli F, Imbriano C. Curcumin derivatives: molecular basis of their anti-cancer activity. Biochem Pharmacol. 2009;78:1305–1315. doi: 10.1016/j.bcp.2009.06.105. [DOI] [PubMed] [Google Scholar]
- 66.Howells LM, Mitra A, Manson MM. Comparison of oxaliplatin- and curcumin-mediated antiproliferative effects in colorectal cell lines. Int J Cancer. 2007;121:175–183. doi: 10.1002/ijc.22645. [DOI] [PubMed] [Google Scholar]
- 67.Issa JP, Baylin SB, Belinsky SA. Methylation of the estrogen receptor CpG island in lung tumors is related to the specific type of carcinogen exposure. Cancer Res. 1996;56:3655–3658. [PubMed] [Google Scholar]
- 68.Issa JP. DNA methylation as a therapeutic target in cancer. Clin Cancer Res. 2007;13:1634–1637. doi: 10.1158/1078-0432.CCR-06-2076. [DOI] [PubMed] [Google Scholar]
- 69.Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer. 2003;3:253–266. doi: 10.1038/nrc1045. [DOI] [PubMed] [Google Scholar]
- 70.Link A, Balaguer F, Goel A. Cancer chemoprevention by dietary polyphenols: promising role for epigenetics. Biochem Pharmacol. 2010;80:1771–1792. doi: 10.1016/j.bcp.2010.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nagasaka T, Goel A, Notohara K, Takahata T, Sasamoto H, Uchida T, Nishida N, Tanaka N, Boland CR, Matsubara N. Methylation pattern of the O6-methylguanine-DNA methyltransferase gene in colon during progressive colorectal tumorigenesis. Int J Cancer. 2008;122:2429–2436. doi: 10.1002/ijc.23398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sporn MB, Suh N. Chemoprevention: an essential approach to controlling cancer. Nat Rev Cancer. 2002;2:537–543. doi: 10.1038/nrc844. [DOI] [PubMed] [Google Scholar]
- 73.Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, Leonhardt H, Jaenisch R. Induction of tumors in mice by genomic hypomethylation. Science. 2003;300:489–492. doi: 10.1126/science.1083558. [DOI] [PubMed] [Google Scholar]
- 74.Goel A, Nagasaka T, Arnold CN, Inoue T, Hamilton C, Niedzwiecki D, Compton C, Mayer RJ, Goldberg R, Bertagnolli MM, et al. The CpG island methylator phenotype and chromosomal instability are inversely correlated in sporadic colorectal cancer. Gastroenterology. 2007;132:127–138. doi: 10.1053/j.gastro.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 75.Karpf AR, Matsui S. Genetic disruption of cytosine DNA methyltransferase enzymes induces chromosomal instability in human cancer cells. Cancer Res. 2005;65:8635–8639. doi: 10.1158/0008-5472.CAN-05-1961. [DOI] [PubMed] [Google Scholar]
- 76.Rodriguez J, Frigola J, Vendrell E, Risques RA, Fraga MF, Morales C, Moreno V, Esteller M, Capellà G, Ribas M, et al. Chromosomal instability correlates with genome-wide DNA demethylation in human primary colorectal cancers. Cancer Res. 2006;66:8462–9468. doi: 10.1158/0008-5472.CAN-06-0293. [DOI] [PubMed] [Google Scholar]
- 77.Link A, Balaguer F, Shen Y, Lozano JJ, Leung HC, Boland CR, Goel A. Curcumin modulates DNA methylation in colorectal cancer cells. PLoS One. 2013;8:e57709. doi: 10.1371/journal.pone.0057709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Patel BB, Gupta D, Elliott AA, Sengupta V, Yu Y, Majumdar AP. Curcumin targets FOLFOX-surviving colon cancer cells via inhibition of EGFRs and IGF-1R. Anticancer Res. 2010;30:319–325. [PMC free article] [PubMed] [Google Scholar]
- 79.Howells LM, Sale S, Sriramareddy SN, Irving GR, Jones DJ, Ottley CJ, Pearson DG, Mann CD, Manson MM, Berry DP, et al. Curcumin ameliorates oxaliplatin-induced chemoresistance in HCT116 colorectal cancer cells in vitro and in vivo. Int J Cancer. 2011;129:476–486. doi: 10.1002/ijc.25670. [DOI] [PubMed] [Google Scholar]
- 80.Shakibaei M, Buhrmann C, Kraehe P, Shayan P, Lueders C, Goel A. Curcumin chemosensitizes 5-fluorouracil resistant MMR-deficient human colon cancer cells in high density cultures. PLoS One. 2014;9:e85397. doi: 10.1371/journal.pone.0085397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.He ZY, Shi CB, Wen H, Li FL, Wang BL, Wang J. Upregulation of p53 expression in patients with colorectal cancer by administration of curcumin. Cancer Invest. 2011;29:208–213. doi: 10.3109/07357907.2010.550592. [DOI] [PubMed] [Google Scholar]
- 82.Carroll RE, Benya RV, Turgeon DK, Vareed S, Neuman M, Rodriguez L, Kakarala M, Carpenter PM, McLaren C, Meyskens FL, et al. Phase IIa clinical trial of curcumin for the prevention of colorectal neoplasia. Cancer Prev Res (Phila) 2011;4:354–364. doi: 10.1158/1940-6207.CAPR-10-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rotelli MT, Bocale D, De Fazio M, Ancona P, Scalera I, Memeo R, Travaglio E, Zbar AP, Altomare DF. IN-VITRO evidence for the protective properties of the main components of the Mediterranean diet against colorectal cancer: A systematic review. Surg Oncol. 2015;24:145–152. doi: 10.1016/j.suronc.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 84.Pace-Asciak CR, Hahn S, Diamandis EP, Soleas G, Goldberg DM. The red wine phenolics trans-resveratrol and quercetin block human platelet aggregation and eicosanoid synthesis: implications for protection against coronary heart disease. Clin Chim Acta. 1995;235:207–219. doi: 10.1016/0009-8981(95)06045-1. [DOI] [PubMed] [Google Scholar]
- 85.Shankar S, Singh G, Srivastava RK. Chemoprevention by resveratrol: molecular mechanisms and therapeutic potential. Front Biosci. 2007;12:4839–4854. doi: 10.2741/2432. [DOI] [PubMed] [Google Scholar]
- 86.Vanamala J, Radhakrishnan S, Reddivari L, Bhat VB, Ptitsyn A. Resveratrol suppresses human colon cancer cell proliferation and induces apoptosis via targeting the pentose phosphate and the talin-FAK signaling pathways-A proteomic approach. Proteome Sci. 2011;9:49. doi: 10.1186/1477-5956-9-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov. 2006;5:493–506. doi: 10.1038/nrd2060. [DOI] [PubMed] [Google Scholar]
- 88.Fouad MA, Agha AM, Merzabani MM, Shouman SA. Resveratrol inhibits proliferation, angiogenesis and induces apoptosis in colon cancer cells: calorie restriction is the force to the cytotoxicity. Hum Exp Toxicol. 2013;32:1067–1080. doi: 10.1177/0960327113475679. [DOI] [PubMed] [Google Scholar]
- 89.Liu B, Zhou Z, Zhou W, Liu J, Zhang Q, Xia J, Liu J, Chen N, Li M, Zhu R. Resveratrol inhibits proliferation in human colorectal carcinoma cells by inducing G1/Sphase cell cycle arrest and apoptosis through caspase/cyclinCDK pathways. Mol Med Rep. 2014;10:1697–1702. doi: 10.3892/mmr.2014.2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wolter F, Akoglu B, Clausnitzer A, Stein J. Downregulation of the cyclin D1/Cdk4 complex occurs during resveratrol-induced cell cycle arrest in colon cancer cell lines. J Nutr. 2001;131:2197–2203. doi: 10.1093/jn/131.8.2197. [DOI] [PubMed] [Google Scholar]
- 91.Schneider Y, Vincent F, Duranton B, Badolo L, Gossé F, Bergmann C, Seiler N, Raul F. Anti-proliferative effect of resveratrol, a natural component of grapes and wine, on human colonic cancer cells. Cancer Lett. 2000;158:85–91. doi: 10.1016/s0304-3835(00)00511-5. [DOI] [PubMed] [Google Scholar]
- 92.Wolter F, Ulrich S, Stein J. Molecular mechanisms of the chemopreventive effects of resveratrol and its analogs in colorectal cancer: key role of polyamines? J Nutr. 2004;134:3219–3222. doi: 10.1093/jn/134.12.3219. [DOI] [PubMed] [Google Scholar]
- 93.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 94.Cunningham D, Atkin W, Lenz HJ, Lynch HT, Minsky B, Nordlinger B, Starling N. Colorectal cancer. Lancet. 2010;375:1030–1047. doi: 10.1016/S0140-6736(10)60353-4. [DOI] [PubMed] [Google Scholar]
- 95.Triantafillidis JK, Nasioulas G, Kosmidis PA. Colorectal cancer and inflammatory bowel disease: epidemiology, risk factors, mechanisms of carcinogenesis and prevention strategies. Anticancer Res. 2009;29:2727–2737. [PubMed] [Google Scholar]
- 96.Panaro MA, Carofiglio V, Acquafredda A, Cavallo P, Cianciulli A. Anti-inflammatory effects of resveratrol occur via inhibition of lipopolysaccharide-induced NF-κB activation in Caco-2 and SW480 human colon cancer cells. Br J Nutr. 2012;108:1623–1632. doi: 10.1017/S0007114511007227. [DOI] [PubMed] [Google Scholar]
- 97.Cui X, Jin Y, Hofseth AB, Pena E, Habiger J, Chumanevich A, Poudyal D, Nagarkatti M, Nagarkatti PS, Singh UP, et al. Resveratrol suppresses colitis and colon cancer associated with colitis. Cancer Prev Res (Phila) 2010;3:549–559. doi: 10.1158/1940-6207.CAPR-09-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hwang JT, Kwak DW, Lin SK, Kim HM, Kim YM, Park OJ. Resveratrol induces apoptosis in chemoresistant cancer cells via modulation of AMPK signaling pathway. Ann N Y Acad Sci. 2007;1095:441–448. doi: 10.1196/annals.1397.047. [DOI] [PubMed] [Google Scholar]
- 99.Das D, Preet R, Mohapatra P, Satapathy SR, Kundu CN. 1,3-Bis(2-chloroethyl)-1-nitrosourea enhances the inhibitory effect of resveratrol on 5-fluorouracil sensitive/resistant colon cancer cells. World J Gastroenterol. 2013;19:7374–7388. doi: 10.3748/wjg.v19.i42.7374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Aires V, Limagne E, Cotte AK, Latruffe N, Ghiringhelli F, Delmas D. Resveratrol metabolites inhibit human metastatic colon cancer cells progression and synergize with chemotherapeutic drugs to induce cell death. Mol Nutr Food Res. 2013;57:1170–1181. doi: 10.1002/mnfr.201200766. [DOI] [PubMed] [Google Scholar]
- 101.Park DG. Antichemosensitizing effect of resveratrol in cotreatment with oxaliplatin in HCT116 colon cancer cell. Ann Surg Treat Res. 2014;86:68–75. doi: 10.4174/astr.2014.86.2.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gescher AJ, Steward WP. Relationship between mechanisms, bioavailibility, and preclinical chemopreventive efficacy of resveratrol: a conundrum. Cancer Epidemiol Biomarkers Prev. 2003;12:953–957. [PubMed] [Google Scholar]
- 103.Delmas D, Lin HY. Role of membrane dynamics processes and exogenous molecules in cellular resveratrol uptake: consequences in bioavailability and activities. Mol Nutr Food Res. 2011;55:1142–1153. doi: 10.1002/mnfr.201100065. [DOI] [PubMed] [Google Scholar]
- 104.Lançon A, Hanet N, Jannin B, Delmas D, Heydel JM, Lizard G, Chagnon MC, Artur Y, Latruffe N. Resveratrol in human hepatoma HepG2 cells: metabolism and inducibility of detoxifying enzymes. Drug Metab Dispos. 2007;35:699–703. doi: 10.1124/dmd.106.013664. [DOI] [PubMed] [Google Scholar]
- 105.Wenzel E, Soldo T, Erbersdobler H, Somoza V. Bioactivity and metabolism of trans-resveratrol orally administered to Wistar rats. Mol Nutr Food Res. 2005;49:482–494. doi: 10.1002/mnfr.200500003. [DOI] [PubMed] [Google Scholar]
- 106.Wenzel E, Somoza V. Metabolism and bioavailability of trans-resveratrol. Mol Nutr Food Res. 2005;49:472–481. doi: 10.1002/mnfr.200500010. [DOI] [PubMed] [Google Scholar]
- 107.Boocock DJ, Faust GE, Patel KR, Schinas AM, Brown VA, Ducharme MP, Booth TD, Crowell JA, Perloff M, Gescher AJ, et al. Phase I dose escalation pharmacokinetic study in healthy volunteers of resveratrol, a potential cancer chemopreventive agent. Cancer Epidemiol Biomarkers Prev. 2007;16:1246–1252. doi: 10.1158/1055-9965.EPI-07-0022. [DOI] [PubMed] [Google Scholar]
- 108.Patel KR, Brown VA, Jones DJ, Britton RG, Hemingway D, Miller AS, West KP, Booth TD, Perloff M, Crowell JA, et al. Clinical pharmacology of resveratrol and its metabolites in colorectal cancer patients. Cancer Res. 2010;70:7392–7399. doi: 10.1158/0008-5472.CAN-10-2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.De Maria S, Scognamiglio I, Lombardi A, Amodio N, Caraglia M, Cartenì M, Ravagnan G, Stiuso P. Polydatin, a natural precursor of resveratrol, induces cell cycle arrest and differentiation of human colorectal Caco-2 cell. J Transl Med. 2013;11:264. doi: 10.1186/1479-5876-11-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang J, Cox DG, Ding W, Huang G, Lin Y, Li C. Three new resveratrol derivatives from the mangrove endophytic fungus Alternaria sp. Mar Drugs. 2014;12:2840–2850. doi: 10.3390/md12052840. [DOI] [PMC free article] [PubMed] [Google Scholar]