

Abstract
Multiple sclerosis (MS) frequently starts near the lateral ventricles, which are lined by subventricular zone (SVZ) progenitor cells that can migrate to lesions and contribute to repair. Since MS-induced inflammation may decrease SVZ proliferation and thus limit repair, we studied the role of galectin-3 (Gal-3), a pro-inflammatory protein. Gal-3 expression was increased in periventricular regions of human MS in post-mortem brain samples and was also upregulated in periventricular regions in a murine MS model, Theiler’s murine encephalomyelitis virus (TMEV) infection. Whereas TMEV increased SVZ chemokine (CCL2, CCL5, CCL8 and CXCL10) expression in wild type (WT) mice, this was inhibited in Gal-3−/− mice. Though numerous CD45+ immune cells entered the SVZ of WT mice after TMEV infection, their numbers were significantly diminished in Gal-3−/− mice. TMEV also reduced neuroblast and proliferative SVZ cell numbers in WT mice but this was restored in Gal-3−/− mice and was correlated with increased numbers of doublecortin+ neuroblasts in the corpus callosum. In summary, our data showed that loss of Gal-3 blocked chemokine expression, reduced immune cell migration into the SVZ, reestablished SVZ proliferation and increased the number of progenitors in the corpus callosum. These results suggest Gal-3 plays a central role in modulating the SVZ neurogenic niche’s response to this model of MS.
Graphical Abstract
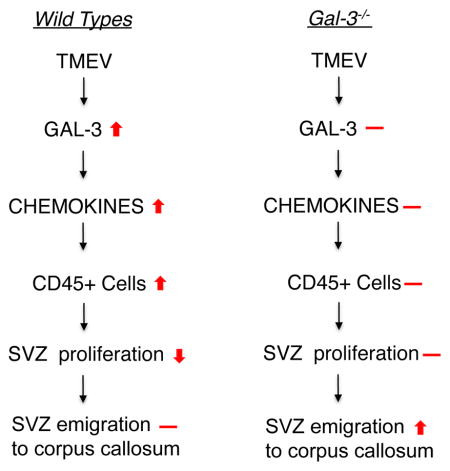
Introduction
Multiple sclerosis (MS) is an inflammatory demyelinating disease of the central nervous system with poor prognosis. A few therapeutically beneficial drugs decrease inflammation associated with relapsing–remitting MS, but there are no effective treatments for primary or secondary progressive MS. Autologous stem cells may provide neuroprotection and offer a novel yet incompletely validated therapeutic strategy (Connick et al. 2012; Xu et al. 2011; Yamout et al. 2010). Importantly, exogenous human and rodent subventricular zone (SVZ) progenitor cells decrease inflammation and provide neuroprotection in models of MS and stroke (Jin et al. 2010; Pluchino et al. 2009; Pluchino et al. 2003; Pluchino et al. 2005). The periventricular white matter surrounding the lateral ventricles and the SVZ stem cell niche is an area of intense inflammation where lesions have lower levels of remyelination than in deep white matter (Dawson 1916; Patrikios et al. 2006). SVZ progenitors actively migrate to demyelinated regions, differentiate into oligodendrocytes and participate in myelin repair (Jablonska et al. 2010; Menn et al. 2006; Nait-Oumesmar et al. 1999; Nait-Oumesmar et al. 2007; Picard-Riera et al. 2002), yet the effects of inflammation on this process is poorly understood.
SVZ progenitors have been shown to migrate into the demyelinated corpus callosum during Theiler’s murine encephalomyelitis virus induced demyelinating disease (TMEV-IDD) (Goings et al. 2008; Mecha et al. 2013b). The TMEV model provides insights into the mechanisms of progressive MS and allows comparison of susceptible to resistant mouse strains (McCarthy et al. 2012; Mecha et al. 2013a). In TMEV-IDD, neuronal loss precedes demyelination and expands over time (Tsunoda et al. 2003). MS is associated with substantial neuronal/axonal loss and this is equally important to oligodendrocyte loss and demyelination (Bo et al. 2006; Bruck 2005; DeLuca et al. 2004). We showed that the SVZ was targeted early and was the most reproducibly inflamed of all brain regions after TMEV infection (Goings et al. 2008). Inflammation may dampen endogenous progenitor repair by decreasing proliferation in adult neurogenic niches (Ekdahl et al. 2003; Kokaia et al. 2012; Monje et al. 2003). Therefore understanding and controlling molecular mechanisms leading to SVZ inflammation may be of benefit when considering the use of endogenous progenitors for therapeutic treatments.
The pro-inflammatory protein, Galectin-3 (Gal-3) is expressed by astrocytes in the healthy SVZ where it regulates neuroblast migration (Comte et al. 2011). It is also expressed by activated glia during CNS inflammation (Walther et al. 2000; Young et al. 2014). Gal-3 is able to regulate inflammation, neurogenesis and migration in multiple systems and could modulate responses to MS (Liu and Rabinovich 2005). For example, glial cells exposed to recombinant Gal-3 showed induction of proinflammatory molecules (Jeon et al. 2010). Conversely, loss of Gal-3 decreased both macrophage infiltration and proinflammatory cytokines in experimental autoimmune encephalomyelitis (Jiang et al. 2009). A recent study showed that cuprizone-induced demyelination was not altered in Gal-3−/− mice but that remyelination was significantly slower (Hoyos et al. 2014). Cuprizone causes less inflammation and immune cell infiltration compared to TMEV. Thus, Gal-3’s role in demyelination and remyelination seems to depend on the nature of the demyelination. It may also be the case that the cell source of Gal-3 makes a difference - the predominantly activated cell type in cuprizone demyelination is microglia, whereas in this TMEV study astrocytes are implicated. Gal-3 expression is enhanced in active human MS lesions (Stancic et al. 2011), but whether Gal-3 is increased in human periventricular regions, corpus callosum or the SVZ in MS was unknown
In both human MS and the murine TMEV MS model the adult SVZ shows marked inflammation, which is characterized by florid CD45+ immune cell infiltration (Goings et al. 2008). We previously showed that TMEV-IDD induced SVZ inflammation occurs early, by 2–3 weeks post infection and before the onset of motor symptoms (Goings et al. 2008). In this study we have show that Gal-3 acts to enhance SVZ inflammation during the onset of MS like disease in adult mice, via up-regulation of the potent macrophage chemoattractant molecules CCL2, CCL5, CCL8 and CXCL10. We also show that blocking this response in Gal-3 knockout mice is associated with a restoration of SVZ proliferation and numbers of Dcx+ neuroblasts in the SVZ and corpus callosum. Our work identifies Gal-3 as a rational therapeutic target in neuroinflammation through its ability to upregulate macrophage chemoattractant molecule expression in the SVZ.
Materials and Methods
Human Sections
Human post-mortem material from controls (n=8, 3s females/5 males) and from MS cases (n=7, 5 females/2 males) was obtained from the Oxford Willis Brain Bank (Oxford Brain Bank or The Thomas Willis Oxford Brain Collection) and collected with informed consent per HTA guidelines (Table 1). The average age and post-mortem interval (PMI) of the control and MS cases did not differ significantly (average age: controls=31.0±7.6 years, MS=43.4±5.4 years; PMI: controls=57.0±9.0 hrs, MS=50.4±10.8 hrs). These differences were not significantly different; the p-values comparing the ages and PMI between the groups are 0.207753 and 0.648451, respectively, by t-test. The controls had no discernable pathology as rated by qualified neuropathologists. The MS cases were selected on the basis of having active inflammatory demyelination (Diaz-Sanchez et al. 2006) in periventricular regions. Controls died due to respiratory arrest, acute cardiac failure, lung carcinoma, septicaemia and unknown causes. Causes of death for MS cases were pneumonia, hypostatic pneumonia, bronchopneumonia, pulmonary embolism (n=2), respiratory arrest and cardiac failure.
Table 1.
Human cases examined
 Case |
Age | Gender | PMI (hrs) | |
|---|---|---|---|---|
| MS Cases | Diagnosis (cause of death) | |||
| B6653 | 32 | M | 24 | not certain if PPMS or SPMS based on information in post-mortem report (hypostatic pneumonia) |
| B6187 | 67 | F | 48 | SPMS (inactive) (bronchopneumonia) |
| B561 | 51 | F | 35 | RRMS (active) (pulmonary embolism) |
| B133 | 39 | M | 70 | SPMS (active) (pneumonia) |
| B5781 | 37 | F | 48 | RRMS (active) (pulmonary embolism) |
| B1413 | 25 | F | 104 | RRMS (active) (respiratory arrest) |
| B9860 | 53 | F | 24 | RRMS (active) (cardiac failure) |
| Controls | ||||
| 137/05 | 16 | M | 72 | Normal brain (unknown cause of death but some evidence of oedema) |
| 1149/00 | 69 | F | 96 | Normal brain (lung carcinoma) |
| 1144/94 | 7 | M | 72 | Normal brain (unknown cause of death but clinical evidence of epilepsy) |
| 1134/91 | 21 | F | 24 | Normal brain (acute cardiac failure) |
| 1105/98 | 13 | M | 72 | Normal brain (unknown cause of death) |
| C1754 | 45 | M | 48 | No specific pathology. (unknown cause of death) |
| C3279 | 50 | M | 48 | None of the features of Alzheimer’s disease or any other specific pathology. (septicaemia) |
| RI1125/99 | 27 | F | 24 | Normal brain. (unknown cause of death) |
Animals and TMEV injection
Gal-3−/− mice on a C57BL/6 background were purchased from Jackson Laboratories (Bar Harbor, ME). Gal-3−/− mice on a 129sv background had been previously generated by targeted mutation and homologous recombination (Colnot, 1998). To generate Gal-3−/− mice on a SJL/J background, female 129sv Gal-3+/− mice were backcrossed with male SJL/J WT’s for seven generations. All mice were housed in IVC units at the University of Oxford animal housing facility and were provided with unlimited access to standard laboratory chow and water. 6–8 week old female mice were used for experiments. For in vivo experiments, both TMEV and sham mice were anaesthetised with 4% isoflurane. 3x106 PFU BeAn 8386 virus, suspended in sterile 30 μL Hanks buffered saline solution (HBSS), was injected into the right posterior cortex through a 27 gauge needle. This was fitted with a needle guard to prevent penetration beyond 3.5 mm ventral to the skull. Sham mice received intracerebral injections of 30 μL HBSS. For proliferation experiments, animals received a single intraperitoneal injection of BrdU (100 mg/kg body weight) 2 hrs before sacrifice.
Virus production
The BeAn strain of TMEV virus was propagated and titered in BHK cells grown in Dulbecco’s modified Eagle medium supplemented with 10% foetal bovine serum. Viral titres and multiplicity of infection (MOI) were determined by plaque assay.
Immunohistochemistry
Animals were perfused with 4% paraformaldehyde and postfixed for 24 hrs. Tissue was cryoprotected in 30% sucrose for 72 hrs before sectioning into 30 micron sections and storage at −20°C. Immunohistochemistry was carried out as in (Goings et al. 2008; Goings et al. 2006). Sections were washed in 0.1 M phosphate buffered saline (PBS) 3 x 10 min and then blocked with 50 mM glycine in PBS to reduce autofluorescence of paraformaldehyde-fixed tissue. For BrdU immunohistochemistry, sections were incubated 20 min in DNAse buffer, followed by 30 min incubation in DNAse (2 mg/mL, 569 Kunitz Units/mg, Sigma-Aldrich) at 36.5°C before blocking. Sections were washed in PBS a further 3 x, before blocking for 1 hr in PBS containing 0.1% Triton X-100 and 10% Donkey Serum, DS (Sigma), (PBS+). Double and triple immunohistochemistry were carried out using primary antibodies produced in different host species in which sections were co-incubated overnight at 4°C. After overnight incubation, sections were washed in PBS 3 x 10 min before incubation with species appropriate secondary antibodies for 1 hr at RT. The secondary antibodies were conjugated to Alexa-488, -568 or -647 (Invitrogen Paisley, Renfrewshire, UK; 1:500), Cy3 or Cy5 (JacksonImmunoResearch, West Grove, PA, USA; 1:500) fluorophores. After washing in PBS 3 x 10 mins, sections were incubated for 10 min in 4′,6-diamidino-2-phenylindole (DAPI, 10 μg/mL, Sigma-Aldrich) to counterstain cell nuclei and rinsed 3 x 10 min in 0.1 M phosphate buffer.
Primary antibodies used in mouse studies: BrdU (1:400, sheep, Abcam Ab1893), Caspase-3 (1:500, rabbit, Cell Signaling 9661), CD3 (1:200, rat, Santa Cruz sc-18843), CD45 (1:400, rat, Millipore 05-1416), doublecortin (1:200, goat, Santa Cruz sc-8066), Iba1 (1:200, goat, Abcam), Galectin-3 (1:200, rabbit, Santa Cruz, sc-20157), GFAP (1:500, rabbit, Sigma Aldrich G9269), GFAP (1:1000, mouse, Millipore 557355), NeuN (1:400, mouse, Millipore MAB377), PECAM (1:200, rat, BD 557355), Phi3 (1:400, rabbit, Millipore 06-570).
For human immunohistochemistry, 10 micron paraffin embedded sections were stained according to the avidin-biotinylated peroxidase (ABC) method. Sections were rehydrated and placed in water bath (80°C for 10 min) for antigen retrieval. Endogenous peroxidase was inactivated by 0.3% H2O2 in PBS (10 min RT) followed by an extensive rinse in PBS. Sections were pretreated with normal donkey serum diluted to 10% in PBS for 90 min to block non-specific binding of antibodies. This and following steps were followed by PBS rinses (5 min, RT). Antibodies used in human study: galectin-3 (1:400, rabbit, Santa Cruz sc-20157) or (1:200, rat, Santa Cruz sc-23938), GFAP (1:500, mouse, Millipore GA5,MAB360), CD68 (1:50, mouse, DAKO PGM1), PLP (1:1000, rabbit AbD Serotec (MCA8399). Primary antibodies were diluted in PBS containing 0.1% Triton X-100. Biotinylated anti-rabbit, anti-rat or anti-mouse antibodies (Vector Laboratories, CA, 1:1000, 1:400, 1:400, respectively) were applied for 3 hr at RT, followed by ABC complex (Vector ABC Elite Kit, Vector Laboratories, 30 min, RT) exposure. To visualize the immunohistochemical labeling, DAB Substrate Kit (SK-4100, Vector Laboratories) was used. The peroxidase-reaction was visually monitored and stopped by replacing the DAB solution with PBS. The sections were counterstained by haematoxylin, dehydrated and mounted onto glasses coverslipped by DePeX.
Astrocyte cultures
Astrocyte cultures were produced from the SVZ or cerebral cortex isolated by microdissection from 6 day old SJL/J pups. Tissue was digested in 0.25% trypsin before being plated on poly-d-lysine and laminin coated plates and grown in high glucose DMEM with 10% FBS. Both cultures were grown in the same manner according to previously published protocols (McCarthy and de Vellis 1980). After 21 days in culture, microglia and OPCs were removed by shaking for 24 hrs at 300 rpm. Astrocytes at passage 2 were plated in triplicate (50,000 cells in 6 cm dishes) and left to adhere for 24 hrs. Cultures were treated with 0.5 mL of either plain media (controls) or TMEV at MOI of 1 or 10 for 1 hr at RT before being brought up to normal volume with complete media and returned to 37°C incubator for a further 24 hrs. Cells were counted using trypan blue, which also verified that TMEV did not cause cell death.
RT-PCR arrays
SVZ tissue was microdissected from 1 mm thick coronal sections of brains from C57BL/6 and SJL/J WT mice with a control group and TMEV 7 days post infection (dpi) group (n=6/group). Total RNA of virus-infected brains or sham infected was extracted from the SVZ using the RNAeasy minikit (Qiagen, Carlsbad, CA) and Qiashredder column according to manufacturer’s instructions. 0.7μg RNA was reverse transcribed for each sample using RT2 First Strand Kit and applied to Inflammation Response and Autoimmunity RT-PCR array plates (PAM-077A, SABiosciences). Each sample was run in triplicate on three separate plates. Plates were processed in an Applied Biosystems 7500 Real-Time PCR system, using the software’s automated baseline and threshold cycle detection. Relative fold changes were calculated using SABiosciences web-based PCR array data analysis tool using the ΔΔCT method. All genes were compared to 5 housekeeping genes: β-actin, β-2 microglobulin, GAPDH, Glucuronidase-β and HSP-90.
ELISAs
For analysis of tissue protein levels, TMEV infected or sham mice were perfused with ice cold PBS at 7 dpi before microdissection of the SVZ and cortex. SVZ cells were lysed on ice with lysis buffer (25 mM Tris-HCL pH 7.4, 150 mM NaCL, 1 mM EDTA, 1% NP-40 and 5% glycerol, 1 mM PMSF) for 15 min. Protein levels were measured using a standard Bradford assay protocol (Bio-Rad, Hemel Hempstead). For all protein experiments, 0.1 mg of tissue was used per sample and run in triplicate. For each experiment, tissue from 6 animals per group was pooled and replicated in three independent experiments. For analysis of supernatants, 100 μl of appropriate fluid dilutions were used (galectin-3 1:100). Cytokines were assayed from samples using respective ELISA kit according to the manufacturers instructions (R&D DuoSets).
Primary Mouse Macrophage Isolation
Male C57BL/6 mice were injected intraperitoneally with 1 ml of 4%w/v Biogel P100 polyacrylamide beads (Bio-Rad, Hemel Hempstead) in PBS. Four days later mice were sacrificed and the peritoneum lavaged with 10 ml ice cold PBS containing 2 mM EDTA. The peritoneal exudates of 2 or 3 mice were pooled, centrifuged at 250G for 5 min, resuspended in 10 ml chemotaxis buffer (RPMI, 0.2% BSA, 25 mM Hepes pH 7.4) and plated in bacterial petri dishes (Greiner, Stonehouse) overnight in 5% CO2 at 37°C. The following day plates were washed with PBS (10 ml) and enriched macrophages were lifted with PBS containing 10 mM EDTA. Cells were harvested by centrifugation 250G for 5 min, resuspended in chemotaxis buffer and analysed by flow cytometry using F4:80 antibody (Adb Serotech) and Ly6B (7/4 BD Pharmingen).
xCELLigence real-time chemotaxis assay
Mouse macrophage chemotaxis was measured using CIM-16 plates in an ACEA xCELLigence RTCA-DP instrument as described (Iqbal et al. 2013). Briefly, proteins (CCL2 10 nM, CCL5 10 nM, CCL8 10 nM, CXCL10 nM and human Gal-3 300 nM, (Peprotech, London) were loaded into the lower chamber before the upper chamber was attached. A gold electrode embedded membrane with 8 micron diameter pores was equilibrated with chemotaxis buffer for 15 min prior to addition of macrophages (200,000 cells/well). Migration was assessed every 5 sec over four hrs. Data were normalized to wells that received chemotaxis buffer alone (vehicle) and analysis of slope and max-min was performed using RCTA Software Version1.2.1, and is directly related to how many macrophages migrated onto and adhered to the gold plated filter. Slope of the real time chemotaxis readout was used as a measure of initial rate of macrophage chemotaxis and was calculated between 15 to 60 min, as described previously (Iqbal et al. 2013). Cell Index is an arbitrary value generated by the ACEA xCELLigence machine software based on the measured cell electrode impedance using a calculation described in (Sun et al. 2012).
Cell quantification and analysis
Human
We used an Aperio image analysis system to select Gal-3+ cells above predetermined immunohistochemical thresholds in periventricular white matter, MS lesions and lesion cores. We quantified Gal-3 in n=5 controls (1149/00, 1144/94, C1754, C3279, RI1125/99) and n=5 MS cases (B6653, B561, B5781, B1413, B9860).
Mouse
For immunohistochemistry, a set of brain sections from anatomical coordinates Bregma 1.7 to −0.5 mm were examined to include the SVZ, striatum and cortex. Immunohistochemistry was examined and recorded on a Leica DMIRB microscope using Openlab software (Improvision, UK) and on a Zeiss Meta confocal microscope, and analysed in 3-D with Zeiss and Volocity Software (Improvision, UK). Images were composed in Microsoft Powerpoint and Adobe Photoshop. For quantification of CD3+ cells, images of CD3 and DAPI labeled sections were taken at 40x magnification on a Leica DMIRB microscope at 5 viewpoints for SVZ; dorsal-striatal, dorsal-septal, medial-septal, medial-striatal and ventral. For striatum quantification, images were taken at 20x magnification with the SVZ in the left margin of the field of view, and with the SVZ in the right field of view for the septum. Pictures were taken at 20x for the septum as well as the entire length of corpus callosum and azygos pericallosal artery. Images were saved as TIFF files and exported into Volocity software for analysis. The number of CD3+ cells was counted and the corresponding area measured to give a value of number of CD3+ cells/mm2. For SVZ regions, DAPI was used to distinguish the SVZ and only cells within the DAPI dense area were included (ependymal cells included). Doublecortin labeled striatal sections were viewed with a 63x objective lens on a Zeiss confocal microscope. With the SVZ to the left margin of the field of view at 63x, Dcx+ cells in the striatum were counted, with the SVZ in the middle of the field showing the full extent of the CC thickness, Dcx+ cells in the CC were counted as in (Goings et al. 2008). For emigration measurement, cells were only counted if they could be shown to have a DAPI+ nucleus, processes that could not be traced to a cell body were not included. Only cells that had a cell body more than 50 μm from the SVZ were counted as having ‘emigrated’. Images of Dcx+ cells in the dorsolateral SVZ were taken with a 40x objective lens and images counted in Volocity software. For all Dcx quantifications a minimum of 3 sections were counted per mouse including both left and right dlSVZ, N=3–7 for all groups. Images of PHi3 or BrdU and CD45 labeled sections of 4 viewpoints; dorsal- striatal, dorsal-septal, medial-striatal and ventral were taken at 40x on a Zeiss Confocal microscope with a 30 μm stack taken of each channel that allowed co-localisation to be studied in 3-dimensions. Together these regions covered the entirety of the SVZ. The images were saved and exported as Tiff images into Volocity. The number of PHi3+ or BrdU+ cells that were CD45+ or CD45− were counted and averaged for 4 consecutive sections per animal ranging from ~Bregma 0.9 to 0.0. The volumes of the stacks were measured to give values of PHi3+cells/mm3 and PHi3+CD45+ cells/mm3. The number of PHi3+cells/mm3 were then subtracted from PHi3+CD45+ cells/mm3 to give the number of proliferating neural cells. Only cells that had bright, complete labeling in the nuclei were included, cells that showed fragmented staining were excluded. The PHi3/CD45/DAPI stained sections included a cohort of 6 naive mice in order to account for any changes in proliferation induced by the needle injury. All measurements were taken blind to genotype and treatment to avoid quantification bias.
For human sections, slides were scanned by the 20x lens of ScanScope AT Turbo (Leica Biosystems) and processed by the Aperio ImageScope software (Leica Biosystems).
Statistics
Continuous numerical data were presented as mean±standard error of the mean. Differences between two groups were assessed using the Student’s t-test and for groups of three or more with analysis of variance (ANOVA) with Bonferroni post-hoc test (unless stated otherwise). Groups were considered statistically different when at least a 95% confidence level was achieved (P<0.05). For all graphs statistical significance is indicated by * for P<0.05, ** for P<0.01 and *** for P<0.001.
Ethics
Our clinical investigation was conducted according to Declaration of Helsinki principles. Procedures were carried out with the University of Oxford Research Ethics Committee, Home Office approval and in accordance with the Animals (Scientific Procedures) Act of 1986 (UK).
Results
Gal-3 expression increased in periventricular inflammatory lesions in human MS
Gal-3 levels were previously shown to increase in human MS (Stancic et al. 2011) but since its expression in the corpus callosum, the SVZ and other periventricular regions was unknown, we carried out immunohistochemistry on controls and patients with MS (Table 1). Rabbit and rat anti-Gal-3 antibodies gave very similar results in human sections and, as before (Comte et al. 2011), both were negative in Gal-3−/− mice (data not shown). No primary control immunohistochemistry resulted in blank labeling. In controls (N=8), Gal-3 was expressed at low to undetectable levels around the lateral ventricles in the corpus callosum, the SVZ and ependymal cells (Fig. 1A, C–C‴). Gal-3 immunoreactivity could occasionally be detected associated with blood vessels in controls (Fig. 1C″). Gal-3 expression was significantly increased in periventricular regions in all human MS cases (N=7) (Fig. 1B, D–D‴). The cellular distribution of increased Gal-3 expression in the MS cases was associated with SVZ cells, ependymal cells and blood vessels (Fig. 1D) and was variable from case-to-case. Increases in Gal-3 expression were especially apparent in regions of demyelination delineated by the loss of proteolipid protein (PLP) immunoreactivity (Suppl. Fig. 1A–C).
Fig. 1. Periventricular regions express Gal-3+ in human MS.

(A,C) Gal-3 immunoreactivity was low surrounding the lateral ventricles (LV), and in the corpus callosum (CC) of controls. Boxed areas shown at higher magnification in C-C‴. Arrows show blood vessels. SVZ outlined in C and C‴. (B) Gal-3 expression was markedly increased in periventricular regions in MS cases. Boxed area shown at higher magnification in D-D‴. (E) GFAP immunoreactivity in the corpus callosum of a control human. (F) GFAP+ reactive astrocytes (ex. arrows) in the corpus callosum of an MS case. (G) CD68 immunoreactivity in a control human section. The SVZ is well delineated in this section by the lower cell density seen with haematoxylin counterstain in the hypocellular gap. Boxed areas shown in G′ and G″. (H) Many CD68+ cells (ex. arrow) were detected in the corpus callosum of a human with MS. Boxed areas shown in H′ and H″. All panels are of sections taken in the coronal plane. Scale bars: B=1 mm; D=500 μm; D‴=100 μm; F, H=100 μm; H″=10 μm.
We quantified this result and found that controls contained 685±194 Gal-3+ cells/cm2 in periventricular white matter. Within the lesions of MS cases this number increased dramatically to 17,689±5,438 cell/cm2, which was a statistically significant difference (p=0.035). We examined the lesions in more detail and found 9,848±2,661 Gal-3+ cells/cm2 in the core of the lesion, which was also significantly different, compared to controls (p=0.026). Periventricular white matter in MS cases outside of the lesion contained 3,918±1,905 Gal-3+ cells which was significantly more than in controls (p=0.009, Mann-Whitney U-Test). These results suggested that Gal-3 expression can be dramatically upregulated in MS lesions of periventricular regions and may influence SVZ neurogenic niche mediated repair of demyelination.
Inflammatory lesions in MS contain activated macrophages and microglia and reactive glial fibrillary acidic protein+ (GFAP+) astrocytes. We found many more strongly GFAP+ immunoreactive astrocytes in the corpus callosum of MS cases compared to controls (Fig. 1E–F). Immunoreactivity to the activated macrophage marker cluster of differentiation-68 (CD68) was also increased in MS cases (Fig. 1G–H′) in a pattern overlapping with increased Gal-3 expression (Suppl. Fig. 1D–E).
Gal-3 expression increased in the mouse forebrain after TMEV infection
Gal-3 expression after TMEV infection has not been reported, thus we examined its expression using several methods. ELISAs on dissected tissue showed the SVZ had higher constitutive levels of Gal-3 compared to the cerebral cortex in both SJL/J mice (TMEV-sensitive) and in C57BL/6 mice (TMEV-resistant) (Fig. 2A). Healthy SJL/J’s expressed more Gal-3 in the SVZ than C57BL/6 mice (Fig. 2A). However, 3 days post TMEV infection (dpi) Gal-3 expression was approximately 20x higher in C57BL/6 mice than in controls but only increased 3X in SJL/J’s (Fig. 2A). Gal-3 expression in the cerebral cortex after TMEV infection also increased to a greater extent in C57BL/6’s than in SJL/J’s but in both strains levels were consistently lower than those of SVZ tissue (Fig. 2A).
Fig. 2. TMEV infection in mice increased periventricular Gal-3 expression.

(A) Gal-3 protein levels in the cerebral cortex and the SVZ measured by ELISA. $$$ P<0.001 compared to controls, ***P<0.001 SJL/J compared to C57BL/6. (B–C) Gal-3 immunofluorescence was detected in the SVZ (arrow) in sham mice and was increased after TMEV. (D–G) Arrowhead points to a GFAP+ (green) and Gal-3+ (red) double-positive cell in the SVZ and the small arrows to double-positive cells in the striatum, 7 dpi (SJL/J mice). (H) Confocal image of striatal astrocyte expressing GFAP (green) and Gal-3 (red) (SJL/J mice). (I–J) Gal-3 was associated with blood vessels but seldom expressed by PECAM+ endothelial cells. (K–M) Iba1+ immunoreactivity in sham controls in the SVZ (ex. arrow), septum and corpus callosum (SJL/J mice). (N–P) Iba1+ immunoreactivity in the SVZ, septum and corpus callosum 7 dpi, (SJL/J mice). (Q–R) Iba1 (green) and Gal-3 immunoreactivity (red) in a sham mouse SVZ and 7 dpi. Arrows show rare double-positive cells (C57BL/6 mice). (S–T) Iba1+/Gal-3+ cell in the corpus callosum 7 dpi. Scale bars: C=500 μm; H=5 μm; M=15 μm; Q=10 μm; T=2 μm.
We next examined the cellular distribution of Gal-3 increases after TMEV. Similar to our previous work (Comte et al. 2011), Gal-3 was only immunohistochemically detectable in SVZ and ependymal cells of healthy controls but was not apparent in the parenchyma (Fig. 2B). As in the 129Sv strain (Comte et al. 2011), C57BL/6 and SJL/J mice expressed Gal-3 in SVZ astrocytes, ependymal cells, a subset of Mash1+ progenitors but was not found in Dcx+ neuroblasts (data not shown). Gal-3 expression increased in the SVZ at 14 dpi and also became immunodetectable in the dorsal septum and periventricular regions (Fig. 2C). The same SVZ cells expressed Gal-3 after TMEV infection as in controls. We found many cells that co-expressed Gal-3 and GFAP at 7 dpi (Fig. 2D–H). In the striatum 55.3±7.6% of GFAP+ cells were Gal-3+ and in the SVZ the percentage was 60.0±5.8%. Gal-3 was expressed de novo in GFAP+ cells of the striatum after TMEV infection (Fig. 2D–H). There was no detectable staining of Gal-3 on PECAM+ blood vessel endothelial cells (Fig. 2I–J) although Gal-3 was frequently expressed by perivascular cells surrounding PECAM+ vessels (Fig. 2J).
We asked if, similar to human MS, periventricular microglia were activated in TMEV infected mice. SVZ microglia expressed higher levels of Iba1 and often assumed highly ramified activated microglial morphology after TMEV infection (Fig. 2K,N). Far more Iba1+ activated microglia/macrophages were found in periventricular regions such as the septum and corpus callosum 7 dpi than in controls (Fig. 1L,M,O,P). This replicated our previous work showing TMEV-induced inflammation is especially prominent in the SVZ and periventricular regions (Goings et al. 2008). Activated microglia express Gal-3 following stroke and TBI (Lalancette-Hebert et al. 2012; Venkatesan et al. 2010). This was of interest since we had shown that SVZ microglia are constitutively semi-activated based on marker expression, morphology and proliferation (Goings et al. 2006) even though only a few of them express Gal-3 (Comte et al. 2011). We replicated this by showing that Iba1+ SVZ microglia were largely Gal-3 negative in sham controls (Fig. 2Q). Qualitative examination in this and our previous studies (Comte et al. 2011; Young et al. 2014) revealed only 1–3 Iba1+/Gal-3+ SVZ cells per section. We did not find any qualitative changes in this co-expression after TMEV, therefore we did not quantify them (Fig. 2R). Gal-3 was however, expressed in some highly ramified Iba1+ cells in the corpus callosum after TMEV infection (Fig. 2S–T). This is similar to our previous results and to published work in active human MS lesions showing Gal-3 expression in macrophages (Stancic et al. 2011) and provided validation of the TMEV model for further study.
We had previously shown that the TMEV virus is able to directly infect cells of the SVZ (Goings et al. 2008). Astrocytes were the major SVZ cell type that expressed Gal-3, thus we asked whether direct TMEV infection of these cells would stimulate Gal-3 secretion. A subset of GFAP+ cells expressed Gal-3 in cultured primary astrocytes from the SVZ (55.8±8.5%) and the cerebral cortex (54.4±4.6%) and they secreted low levels of Gal-3 (Suppl. Fig. 2A–I). Secreted Gal-3 concentrations were dramatically increased after TMEV infection in vitro (Suppl. Fig. 2I). Gal-3 secretion increases were more substantial from SVZ astrocytes than from cortical astrocytes (Suppl. Fig. 2I), as expected based on our in vivo ELISA results. We did not carry out MTS assays or caspase-3 staining on the astrocytes in vitro so we can not rule out the possibility that increased Gal-3 was associated with cell death. However, there were no morphological differences between the two cultures or signs of cell death such as blebbing, swelling, or vacuolation. These data indicated that TMEV-infection of astrocytes might stimulate them to release Gal-3 which could influence the local niche.
Increased chemokine expression after TMEV was blocked in Gal-3−/− mice
Chemokines and cytokines regulate hematopoietic and neural cell proliferation and migration and can influence the SVZ stem cell niche (Banisadr et al. 2011; Belmadani et al. 2006; Lane and Hosking 2010; Nansen et al. 2000; Tran et al. 2007; Tran et al. 2004). We next determined how loss of Gal-3 altered their expression levels in the SVZ and cerebral cortex after TMEV. We first carried out a chemokine/cytokine PCR microarray screen and found that many chemokine and cytokine mRNAs were increased 7 days after TMEV infection (Table 2 and Suppl. Table 2). The genes upregulated in the two strains were similar but in general C57BL/6 mice mounted a more robust chemokine response. Only 15 of the 23 genes that showed greater than 4 fold upregulation in C57BL/6 mice were similarly increased in SJL/J mice (Table 2, grey highlighting). Amongst the genes that were upregulated in C57BL/6 mice after TMEV were complement-3, CCL2, CCL11, CCL22, CXCL10, IFNγ and IL1β (N=3, p<0.05). Upregulation of chemokine receptors was also greater in C57BL/6 mice than in SJL/J’s. The only genes that were more upregulated in SJL/J than in C57BL/6 mice after TMEV were CCL5, CCL8, CXCL11, and interleukin 10 (IL10).
Table 2.
Chemokine and cytokine mRNA changes in the SVZ after TMEV.
| Gene Name | Gene symbol | C57BL/6 | SJL/J |
|---|---|---|---|
| Complement component 3 | C3 | 66.8±13.9 | 4.2±1.0 |
| Complement component 3a receptor 1 | C3ar1 | 13.1±1.1 | 5.7±0.6 |
| Chemokine (C-C) motif ligand 1 | Ccl1 | 11.8±0.9 | 2.9±0.7 |
| Chemokine (C-C) motif ligand 11 | Ccl11 | 20.4±3.4 | 1.1±0.8 |
| Chemokine (C-C) motif ligand 12 | Ccl12 | 63.9±18.2 | 45.5±5.1 |
| Chemokine (C-C) motif ligand 2 | Ccl2 | 277.6±47.4 | 43.5±1.5 |
| Chemokine (C-C) motif ligand 22 | Ccl22 | 14.1±4.4 | 2.2±0.3 |
| Chemokine (C-C) motif ligand 5 | Ccl5 | 257.8±67.8 | 639.4±206.3 |
| Chemokine (C-C) motif ligand 7 | Ccl7 | 354.3±29.9 | 99.5±49.0 |
| Chemokine (C-C) motif ligand 8 | Ccl8 | 29.2±11.0 | 148.8±14.8 |
| Chemokine (C-C)motif receptor 1 | Ccr1 | 6.9±2.3 | 2.3±0.1 |
| Chemokine (C-C)motif receptor 2 | Ccr2 | 17.3±6.3 | 6.3±1.6 |
| Chemokine (C-C)motif receptor 3 | Ccr3 | 6.4±0.7 | 3.0±0.4 |
| Chemokine (C-C)motif receptor 4 | Ccr4 | 4.0±1.4 | 2.0±0.1 |
| Chemokine (C-C)motif receptor 7 | Ccr7 | 4.2±0.7 | 1.6±0.0 |
| Chemokine (C-X-C motif) ligand 10 | Cxcl10 | 798.0±139.5 | 70.9±14.7 |
| Chemokine (C-X-C motif) ligand 11 | Cxcl11 | 46.8±3.7 | 61.3±9.2 |
| Chemokine (C-X-C motif) ligand 9 | Cxcl9 | 626.0±317.3 | 254.8±16.2 |
| Interferon gamma | Ifnγ | 25.8±6.3 | 6.4±0.2 |
| Interleukin 10 | Il10 | 5.8±1.1 | 13.9±0.9 |
| Interleukin 8 receptor acc. protein | Il18rap | 12.3±4.8 | 2.5±0.8 |
| Interleukin 1 beta | Il1β | 30.7±12.1 | 8.3±2.6 |
| Toll-like receptor 2 | Tlr2 | 13.6±1.8 | 8.6±8.6 |
Values are TMEV-induced fold increases±sd. Averages for triplicate samples of 7 dpi TMEV versus sham. Table shows genes that are present on SABiosciences RT-PCR array PAMM-077. Only genes which showed 4 or more fold increases in C57BL/6 mice are shown. Boxes shaded in grey are SJL genes that showed greater than 4 fold increase.
To confirm that TMEV altered levels of inflammatory molecules and to examine the role of Gal-3 in modulating them, protein levels of chemokines were measured using ELISAs. The chemokines CCL2, CCL5, CCL8 and CXCL10 were chosen as they showed the greatest changes at the mRNA level. Chemokine levels were higher in the SVZ than in the cerebral cortex of sham controls of both mouse strains (Fig. 3). Deletion of Gal-3 in sham controls did not significantly alter chemokine levels in the cerebral cortex of either strain (Fig. 3E–H, M–P). In both C57BL/6 and SJL/J mice, SVZ chemokine levels were significantly lower in Gal-3−/− compared to WT mice after TMEV infection (Fig. 3A–D, I–L), with the exception of CCL2 in C57BL/6 mice, which only had a small non-significant decrease. The greatest difference was seen for CCL5 and CCL8, Gal-3 deletion resulted in a dramatic decrease in their expression after TMEV infection for both C57BL/6 and SJL/J mice (Fig. 3B,C,J,K). Decreases in cortical CCL5 expression after TMEV were also seen in WT versus Gal-3−/− C57BL/6 mice (Fig. 3G). Intriguingly, CCL8 expression levels in the cerebral cortex changed in opposite directions in Gal-3−/− C57BL/6 compared to Gal-3−/− SJL/J mice (Fig. 3F,N). These chemokine differences in the SVZ suggested that the extent of immune cell infiltration into the SVZ after TMEV infection would be altered in Gal-3−/− mice.
Fig. 3. Loss of Gal-3 prevented TMEV-induced chemokine increases in the SVZ.

(A–D) Concentrations of CXCL10, CCL8, CCL5 and CCL2 in the SVZ measured by ELISA in shams and 7 dpi in WT and Gal-3−/− C57BL/6 and SJL/J mice (I–L). (E–H) Concentrations of CXCL10, CCL8, CCL5 and CCL2 in the cerebral cortex measured by ELISA in shams and 7 dpi in WT and Gal-3−/− C57BL/6 and SJL/J mice (M–P). *P<0.05 Gal-3−/− compared to WT, ** P<0.01, ***P<0.001, $ P<0.05 TMEV treated compared to shams, $$ P<0.01.
In order to demonstrate that the chemokines described above are chemoattractants for macrophages we conducted real time chemotaxis experiments with CCL2, CCL5, CCL8 and CXCL10 (all at 10 nM). The enriched macrophage population used for chemotaxis experiments were analysed using flow cytometry. Representative dot plots of forward (FSC) and side scatter (SSC) profiles and singlet populations (Suppl. Fig. 3B,C) were consistent with a population of predominantly mononuclear cells, which are 86% positive for the macrophage marker F4/80 (Suppl. Fig. 3D). CCL5 showed clear evidence of directed macrophage migration over a 4-hour period as compared to vehicle alone (Suppl. Fig. 3E). Suppl. Fig. 3F,G show analysis of pooled chemotaxis data (n>3) for CCL2 and CCL5. Slope and max-min measure initial rate of migration and magnitude of response, respectively. Altered migration compared to vehicle was not observed in response to CXCL10 or CCL8 (data not shown). In order to assess whether recombinant Gal-3 can act as a chemoattractant for murine macrophages we tested the ability of Gal-3 to mediate directed migration at concentrations up to 300 nM. In contrast to the reported effects of recombinant human Gal-3 on human monocytes/macrophages in modified Boyden Chamber assays (Sano et al. 2000) we observed no directed migration of murine macrophages to Gal-3 (Suppl. Fig. 3E). The recombinant Gal-3 used was shown to have biological activity in a separate neurosphere adhesion assay (Suppl. Fig. 3H).
Loss of Gal-3 decreased the number of immune cells post-TMEV in the SVZ
We hypothesized that the decreased chemokine levels in Gal-3−/− mice post TMEV infection may have reduced the number of activated immune cells in the SVZ. Amoeboid CD45+ cells probably represented a combination of activated microglia, infiltrating macrophages and T cells and were virtually absent in control sections (Goings et al. 2008; Goings et al. 2006). We found many amoeboid CD45+ cells in the dorsal SVZ, ventral SVZ, striatum and septum at 3, 7, and 14 dpi (Fig. 4A). In Gal-3−/− SJL/J mice, significantly fewer amoeboid CD45+ cells were found in all four regions at 7 dpi, the time of maximum inflammation in WT mice (Fig. 4B). Similarly, Gal-3−/− C57BL/6 mice had significantly fewer amoeboid CD45+ cells in these regions compared to WT C57BL/6 mice (Fig. 4C). In contrast to SJL/J’s, this inhibition was apparent at 3 and 14 dpi but not at 7 dpi (Fig. 4C). Thus, loss of Gal-3 inhibited the increase in immune cell numbers in both the TMEV-resistant and the TMEV-susceptible strain although the timing differed.
Fig. 4. Gal-3 loss reduced the number of CD45+ cells in the SVZ after TMEV infection.

(A) CD45 immunoreactivity (green) in WT C57BL/6 mice. Images are compressed confocal Z stacks. Arrowheads point to amoeboid CD45+ cells in the SVZ. Scale bar = 50 μm. (B) Number of CD45+ cells in the dorsal SVZ, ventral SVZ, striatum and septum at 3, 7 and 14 dpi in SJL/J mice. (C) Number of CD45+ cells in the dorsal SVZ, ventral SVZ, striatum and septum at 3, 7 and 14 dpi in C57BL/6 mice. *P<0.05 Gal-3−/− compared to WT, ** P<0.01, ***P<0.001.
To further explore the differences between the two mouse strains, we studied CD3+ cell migration into the SVZ. CD3+ cells (of any morphology) were not observed in forebrain sections in healthy mice (Suppl. Fig. 4C) however TMEV caused a high concentration of CD3+ cells to appear in the SVZ (Suppl. Fig. 4). These were frequently in the vicinity of periventricular blood vessels, and at the junction between the septum and the corpus callosum (Suppl. Fig. 4A). Many of the infiltrating CD3+ cells were associated with the midline azygous pericallosal artery (Suppl. Fig. 4B). CD3+ cells also appeared in the periventricular septum and striatum 7 dpi but were found in lower densities than in the SVZ (Suppl. Fig. 4C). Numbers of CD3+ cells in the SVZ of SJL/J mice increased through 14 dpi (Suppl. Fig. 4D). In TMEV resistant C57BL/6 mice, the maximum density of CD3+ cells in the SVZ peaked at 7 dpi and there was a large reduction by 14 dpi (Suppl. Fig. 3E). About half of CD3+ cells (40–60%) expressed Gal-3 at 7 and 14 dpi in the forebrain regions examined (Suppl. Fig. 3F). These results show that CD3+ (T cells) infiltrate the SVZ soon after TMEV infection and contribute to the inflammatory response. They also suggest that C57BL/6 T cell infiltration is more transitory compared to SJL/J’s.
Loss of Gal-3 restored SVZ proliferation after TMEV
Inflammation can decrease proliferation and neurogenesis in adult neurogenic niches (Ekdahl et al. 2003; Monje et al. 2003) thus we sought to determine if this was the case after TMEV and if loss of Gal-3 impacted it. Our previous study indicated that the total number of PHi3+ cells (SVZ neural cells combined with hematopoietic lineage cells in M phase of the cell cycle) was unchanged in the SVZ several weeks after TMEV (Goings et al. 2008). However, the Iba1+ cell activation and presence of high numbers of CD3+ T cells in the first two weeks after TMEV suggested neural proliferation in the SVZ might be decreased. We excluded hematopoietic lineage cells from this analysis by counting PHi3+/CD45− cells. Gal-3−/− 129Sv mice did not have altered basal levels of proliferation (Comte et al. 2011) and in these studies we found the same result in sham SJL/J and C57BL/6 mice, both with PHi3+/CD45− and BrdU+/CD45− cell counts (Fig. 5B–E).
Fig. 5. Decreased SVZ proliferation after TMEV infection was blocked in Gal-3−/− mice.

(A) Pulse-labeled BrdU+ cells in the SVZ of a sham WT SJL/J mice. (B–C) Quantification of Phi3+/CD45− and BrdU+/CD45− cells (proliferating cells of the neural lineage) in the SVZ after TMEV. (D–E) Quantification of Phi3+/CD45− and BrdU+/CD45− cells in the SVZ of C57BL/6 mice after TMEV. (F–G) Dcx immunohistochemistry in the SVZ of a sham C57BL/6 mouse and 7 dpi (G) showing disruption and loss of Dcx+ cells. DAPI nuclear counterstain (blue). (H–I) Quantification of number of Dcx+ cells in the dorsolateral SVZ of C57BL/6 and SJL/J WT and Gal-3−/− mice. *P<0.05 Gal-3−/− compared to WT, ** P<0.01, ***P<0.001, $ P<0.05 TMEV treated compared to shams, $$ P<0.01. (J–K) NeuN immunoreactivity in shams compared to 7 dpi. (L–M) Caspase-3 immunoreactivity in shams compared to 7 dpi in WT. Scale bars: A=30 μm; G=15 μm; M=200 μm.
We did find significantly fewer proliferating PHi3+/CD45− and BrdU/CD45− SVZ cells in WT SJL/J and C57BL/6 mice after TMEV infection (Fig. 5B–E). Interestingly, SJL/J Gal-3−/− mice had significantly more mitotic SVZ cells than WT controls at 14 dpi (Fig. 5B–C). In C57BL/6 Gal-3−/− mice the number of mitotic cells was also increased compared to C57BL/6 WT at 7 and 14 dpi (Fig. 5D–E). These results suggested that loss of Gal-3 protected against the TMEV-induced decline in SVZ neural cell proliferation.
We next used doublecortin (Dcx) immunohistochemistry to study the SVZ neuroblast response (Brown et al. 2003; Yang et al. 2004). We had previously observed that at 24 dpi and later time points, the tight chains of Dcx+ SVZ neuroblasts were disrupted in TMEV infected mice (Goings et al. 2008). Here we found a qualitatively similar effect at 3 through 14 dpi (Fig. 5F–G). The total number of Dcx+ cells in the SVZ was decreased significantly after TMEV in C57BL/6 WT mice but this was not altered in C57BL/6 Gal-3−/− mice (Fig. 5H). In contrast, the loss of Gal-3 significantly increased the number of SVZ neuroblasts in SJL/J’s (Fig. 5I). We were concerned that the changes in the number of Dcx+ cells in the SVZ could be due to precocious differentiation or excessive cell death in the SVZ. We therefore carried out neuronal nuclei (NeuN) and Caspase-3 immunohistochemistry to detect mature neurons and apoptotic cells, respectively. We did not detect NeuN+ cells in the SVZ in either shams or at 7 dpi (SJL/J’s) (Fig. 5J–K). Additionally, we found very few Caspase-3+ cells in the SVZ and there was no difference in the number of Caspase-3+ cells between shams and 7 dpi (Fig. 5L–M).
Loss of Gal-3 increased Dcx+ cell numbers in the corpus callosum
SVZ progenitor cells migrate towards injuries and demyelinating lesions and contribute to neuroreplacement and neuroprotection (Jin et al. 2010; Pluchino et al. 2003; Pluchino et al. 2005). Since we showed that loss of Gal-3 can restore SVZ proliferation, we asked if this was associated with increased numbers of Dcx+ cells in the corpus callosum (white matter) or striatum (grey matter) (Fig. 6A). The number of Dcx+ cells found in the corpus callosum was increased in C57BL/6 Gal-3−/− mice compared to WT mice at 3, 7 and 14 dpi (Fig. 6B–C). The large majority of these cells had a distinctive bipolar migratory morphology, with leading processes and trailing edges (Fig. 6B–C) (Martinez-Molina et al. 2011; Nam et al. 2007). Increased numbers of Dcx+ cells were found in the striatum after TMEV, but interestingly this effect was blocked in C57BL/6 Gal-3−/− mice (Fig. 6D–F). The data suggest that loss of Gal-3 redirected SVZ emigration from the grey matter (striatum) to the white matter (corpus callosum) in C57BL/6 mice.
Fig. 6. The number of Dcx+ cells in the corpus callosum was greater in Gal-3−/− mice after TMEV infection.

(A) Forebrain schematic showing location of analysis for SVZ neuroblast emigration into the corpus callosum (cc) and striatum (str). (B–C) Dcx immunofluorescence in the corpus callosum and SVZ of C57BL/6 WT and knockout mice. Arrowheads point to Dcx+ cells in the corpus callosum. (D–E) fewer Dcx+ cells were found in the striatum of C57BL/6 Gal-3−/− mice after TMEV. Arrowhead points to Dcx+ cell in the striatum. Scale bar: F=20 μm. (F) The number of Dcx+ cells increased in the corpus callosum in C57BL/6 Gal-3−/− mice compared to WT. The number of Dcx+ cells decreased in the striatum in C57BL/6 Gal-3−/− mice compared to WT. (G) The number of Dcx+ cells increased in the corpus callosum in SJL/J Gal-3−/− mice compared to WT SJL/J. The number of Dcx+ cells in the striatum in SJL/J Gal-3−/− mice was not significantly different compared to WT SJL/J’s.*P<0.05 Gal-3−/− compared to WT, ** P<0.01, ***P<0.001, $ P<0.05 TMEV treated compared to shams, $$ P<0.01.
Similar to C57BL/6 Gal-3−/− mice, SJL/J Gal-3−/− mice had increased numbers of Dcx+ cells in the corpus callosum, the differences being significant at 7 and 14 days after TMEV (Fig. 6G). Unlike the C57BL/6 Gal-3−/− mice, SJL/J Gal-3−/− did not exhibit significantly different numbers of Dcx+ cells in the striatum after TMEV (Fig. 6G). These data show that loss of Gal-3 increases the number Dcx+ cells in the corpus callosum after TMEV.
Discussion
We show in this study that Gal-3 expression increased in periventricular regions in human MS and after murine TMEV infection. CCL2, CCL5, CCL8 and CXCL10 chemokine expression in the SVZ was dramatically augmented after TMEV infection, but this was inhibited in Gal-3 knockout mice. TMEV infection induced the appearance of large numbers of immune cells in the SVZ but this was also blocked in Gal-3−/− mice. We hypothesize that this was due to the lack of chemokine expression increases since we showed that CCL2 and CCL5 are chemoattractants to macrophages. Thus loss of Gal-3 seemed to block the SVZ’s inflammatory response and this was associated with a partial restoration of SVZ proliferation after TMEV infection as well as increased numbers of Dcx+ cells in the corpus callosum.
In a previous study, TMEV-IDD caused random inflammation throughout the CNS except for the SVZ where inflammation was seen in periventricular regions early and in every section of each animal (Goings et al. 2008). In that work, we found increased numbers of CD45+ cells in the SVZ at 14 dpi but were uncertain which immune cells were activated and how soon this occurred (Goings et al. 2008). Here, CD3+ T cells were found in the SVZ and periventricular regions already 3 dpi and peaked around 7 dpi. Many Iba1+ microglia and macrophages were also found in and around the SVZ after TMEV and overlapped with peak T cell infiltration. Both strains showed a peak of SVZ inflammation at 7 dpi but it resolved more quickly in C57BL/6 mice, which may contribute to this strain’s TMEV-resistance. Interestingly, Gal-3 was expressed at higher levels in healthy SJL/J’s than in C57BL/6’s but it was increased more dramatically after TMEV infection in C57BL/6’s. The temporal and spatial pattern of TMEV-induced immune cell activation in mice resembled that of increased Gal-3 expression, and was similar to what was seen in human MS sections. Thus the lateral ventricle region seems to be targeted both in human MS and TMEV infection can model rapid SVZ inflammation.
At this stage we do not know for certain if astrocytes in this work were directly infected by TMEV. Our Goings et al. 2008 study indicated that the SVZ seemed to be preferentially targeted by the BeAn TMEV virus and immunohistochemistry showed good overlap between the virus and SVZ neuroblasts. In this study we have infected a culture of astrocytes and showed increased Gal-3 release. The majority of cells in the astrocyte cultures were indeed astrocytes, the large majority of DAPI+ nuclei were in GFAP+ cells. Nevertheless it is difficult to completely remove microglia from these cultures and we cannot exclude that some of the effects were due to microglial contaminants. Although it is clear that Gal-3 expression increased after TMEV infection, the exact cell source and signaling pathways involved were not examined. A particularly interesting feature of this study and our previous work was that both Gal-3 and chemokines were expressed at higher levels in the SVZ and periventricular regions, both constitutively and after TMEV injections. Gal-3 is selectively expressed in the SVZ in healthy animals (Comte et al. 2011) and immunohistochemistry showed that Gal-3 was increased in the SVZ after TMEV injections. Furthermore ELISAs on dissected SVZ tissue showed large increases in chemokines after TMEV injections. These chemokine increases were blocked in Gal-3−/− mice suggesting that Gal-3 mediates their expression. Gal-3 could act intra- or extracellularly in the SVZ to regulate chemokine expression, however we do not know which scenario predominated in this study.
Chemokines are central in beckoning cells to inflammation and could serve as primary chemoattractants to immune cells post-TMEV infection. We showed that chemokines were expressed at higher levels in the SVZ than in the cerebral cortex of healthy controls and that their expression increased markedly after TMEV. We hypothesize that immune cells migrate into the SVZ much earlier and more robustly than into nearby regions due to its relatively larger chemokine increases. Loss of Gal-3 inhibited CCL2, CCL5, CCL8 and CXCL10 upregulation in TMEV infected mice. Since all four chemokines are chemoattractant to T cells in the CNS (Huang et al. 2001; Karpus et al. 1995; Muzio et al. 2010; Tsunoda et al. 2004), it is likely they contributed to the diminished immune cell infiltration in Gal-3−/− mice. We also provided direct evidence that CCL2 and CCL5 were chemoattractants to mouse macrophages in an vitro assay and thus believe these two chemokines specifically influenced macrophage migration into the SVZ in vivo. CCL2 was previously shown to be highly up-regulated by microglia and astrocytes after TMEV and anti-CCL2 treatment reduced numbers of CNS-infiltrating T-cells and macrophages (Banisadr et al. 2011; Belmadani et al. 2006; Karpus et al. 2006). In our study we showed that both T cells and macrophage cell numbers are dramatically increased in the SVZ after TMEV infections. We also found that Gal-3−/− mice have reduced numbers of CD45+ cells in the SVZ after TMEV but do not know which cell type this corresponds to.
Recent studies from several laboratories have demonstrated that chemokines play a key role in regulating the migration of stem cells and SVZ neuroblasts (Belmadani et al. 2006; Tran et al. 2007; Tran et al. 2004) and SVZ progenitor cells express numerous types of chemokine receptors (Banisadr et al. 2011). CCL2 (MCP-1) is a chemoattractant for SVZ neuroblasts in vitro as well as after injury (Gordon et al. 2009; Gordon et al. 2012; Yan et al. 2007), whilst the function of CCL5 in the SVZ is unknown. After TMEV injections, CCL2 is highly up-regulated by microglia and astrocytes and CCR2 receptors are expressed by migrating neural progenitors (Banisadr et al. 2011; Belmadani et al. 2006; Tran et al. 2007). Future functional studies will test to what extent CCL2 and CCL5 are necessary for SVZ neuroblast migration after TMEV.
Although we focused on four chemokines at the protein level, our RT-PCR results suggested many chemokine, cytokine, interleukin ligand and receptor expression levels markedly changed after TMEV. They are likely to have multiple distinct effects in this model and interestingly, there were differences in how TMEV induced them in C57BL/6 versus SJL/J mice. It is possible that the greater increase in Gal-3 expression in the resistant strain induced expression of molecules that helped clear the disease. Gal-3 itself has been proposed as a chemoattractant to macrophages (Sano et al. 2000), however we did not reproduce this result in vitro and SJL/J’s contained far more T cells in the SVZ after TMEV than C57BL/6’s, despite their more modest Gal-3 increase. Data from both strains indicated that the SVZ seems to be a hot-spot of infiltration in TMEV and that Gal-3 regulated chemokine expression (Papaspyridonos et al. 2008).
Inflammation and various MS models can decrease SVZ proliferation and adult neurogenesis (Ekdahl et al. 2003; Monje et al. 2003; Tepavcevic et al. 2011; Wang et al. 2010). We showed that TMEV infection decreased the number of Phi3+ and BrdU+ CD45-negative neural lineage cells as well as the number of Dcx+ SVZ neuroblasts. Gal-3 loss did not impact homeostatic SVZ proliferation in 129Sv mice (Comte et al. 2011), a result replicated here in SJL/J and C57BL/6 shams. This suggests that the decreased proliferation after TMEV was probably not caused by increased Gal-3 but may have been influenced by either the changes in chemokine levels and/or the infiltration of immune cells. The reduced proliferation in C57BL/6 Gal-3−/− mice at 7 dpi followed the earlier reduction in amoeboid CD45+ cells at 3 dpi. SJL/J Gal-3−/− mice had reduced amoeboid CD45+ cell numbers at 7 dpi and more normal levels of SVZ proliferation at 14 dpi. The results suggest reduced SVZ immune cell infiltration in Gal-3−/− mice allowed maintenance of more normal proliferation levels.
We found that Gal-3−/− mice had significantly more Dcx+ cells in the corpus callosum after TMEV. Since Dcx is expressed by newborn migratory SVZ neuroblasts, the simplest explanation is that they migrated from the neurogenic niche and the cells had distinctive migratory morphology. However, Dcx is expressed in some non-neurogenic regions (Luzzati et al. 2008) and TMEV may have induced aberrant Dcx expression. Since SVZ progenitors have been found to be neuroprotective, the increased emigration may contribute to neuroprotection. This will ultimately have to be tested in long-term studies that determine cellular and behavioural correlates of increased SVZ progenitor emigration to the corpus callosum. In particular it will be important to show if they are neuroprotective, survive and contribute remyelinating cells.
We postulate that the increased SVZ neuroblast emigration was in part due to the rescued SVZ proliferation in Gal-3/− mice, which produced more neuroblasts that could emigrate after TMEV than in WT mice. We demonstrated before that Gal-3 was necessary for rostral migration in the SVZ; Gal-3−/− had slower and more local exploratory cell migration patterns (Comte et al. 2011) and local migration may have contributed to the greater rates of emigration after TMEV. We do not know if the decreased rostral migration of SVZ neuroblasts we showed in (Comte et al. 2011) is associated with decreased olfactory functions. However, human MS has been associated with reduced olfaction and we found that demyelinating human pathologies, including MS, involve olfactory bulb and tract demyelination (DeLuca et al. 2014).
Overall this study provides evidence that Gal-3 has pivotal functions in TMEV-induced chemokine expression, T cell infiltration, SVZ progenitor proliferation and neuroblast numbers. This is important because SVZ progenitor cells can be neuroprotective in several types of injury (Jin et al. 2010; Pluchino et al. 2003; Pluchino et al. 2005) and because white matter surrounding the SVZ frequently has demyelinated lesions in human MS (Dawson 1916). Gal-3 periventricular distribution was unknown and we showed that it increased in all MS cases examined and corresponded with the location of astrocytosis and microgliosis in active periventricular lesions. Gal-3 is constitutively produced by astrocytes and ependymal cells in the SVZ (Comte et al. 2011) and in this study striatal and septal astrocytes began to express Gal-3 after TMEV infection. As well, direct infection of cultured astrocytes with TMEV stimulated the secretion of high levels of Gal-3. Taken together our work suggests that the SVZ, a source of reparative endogenous progenitors is specifically targeted by TMEV, which may limit its beneficial role. It also suggests that Gal-3 regulates this process and since it is druggable, may in the future provide a therapeutic target for treating MS.
Supplementary Material
Suppl. Fig. 1. Gal-3 increased in area of demyelination.
(A) Gal-3 expression was increased in an active lesion (arrow) in the CC of an MS case, haematoxylin counterstain. (B) Higher magnification view of lesion in A. (C) PLP immunoreactivity loss in same lesion as B, adjacent section. (D, E) Gal-3 and CD68 immunoreactivity in adjacent sections at high magnification in same lesion as A–C. Asterisks show blood vessels. Scale bars: A=5 mm; B–C=2mm; D–E=100 μm.
Suppl. Fig. 2. TMEV infection increased astrocyte Gal-3 expression.
(A–D) Typical Gal-3 expression by GFAP+ cerebral cortex and (E–H) SVZ-derived primary astrocyte cultures. (I) ELISA measurement of Gal-3 secreted from primary astrocytes in controls and after TMEV multiplicity of infection (MOI) 1 and 10.
Suppl. Fig. 3. Macrophage Chemotaxis Assay
(A) Outline of murine monocyte/macrophage elicitation, enrichment by adhesion to bacterial petri dishes before analysis. (B–D) flow cytometry showing relative purity of macrophages. (E) Representative trace from a CIM-16 plate where each line represents the mean of 3–4 wells per chemoattractant. (F–G) Chemotaxis data from n=2–6 independent experiments quantified as Slope or Max-min analysis. Statistical analysis in V and W was performed using one way ANOVA and Dunnett’s multiple comparisons post test relative to vehicle alone, ***p<0.001. (H) Addition of exogenous rGal-3 caused neurospheres to clump together.
Suppl. Fig. 4. CD3+ cells infiltrated the SVZ and forebrain post-TMEV infection.
(A) Low magnification image of CD3+ immunofluorescence in the SVZ surrounding the lateral ventricles (lv). Note the two prominent blood vessels (white arrowheads) in the SVZ and the azygos pericallosal artery (azPA), the midline vessel just dorsal to the corpus callosum (red arrow). These blood vessels are lined with CD3+ cells. (B) Large numbers of CD3+ cells migrated into the CNS through the azPA. Scale bars: A=200 μm; B=100 μm.
(C) Quantification of CD3+ cells in CNS subregions of TMEV infected SJL/J mice. Sham mice only had 1–2 CD3+ cells per section, therefore the bars are not visible. (D–E) Number of CD3+ cells in the SVZ in SJL/J and C57BL/6 mice. (F) Approximately 40–60% of CD3+ cells expressed Gal-3, 7 and 14 dpi in SJL/J mice.
Suppl. Table 1. Fold changes in gene expression by RT-PCR array in SVZ tissue and astrocyte cultures after TMEV infection.
Main Points.
Gal-3 is increased in MS.
Gal-3 is necessary for chemokine increases in MS model.
Loss of Gal-3 reduces number of immune cells in SVZ.
Acknowledgments
We would like to thank members of the Szele lab for critical reading of the manuscript. We would also like to thank Margaret Esiri, Steven Chance and Daniel Anthony for helpful comments on the project and the manuscript. RJ supported by a Wellcome Trust Studentship. FS supported by BBSRC project and NIH RO1 grants.
Footnotes
The authors declare no competing financial interest.
References
- Banisadr G, Frederick TJ, Freitag C, Ren DJ, Jung HS, Miller SD, Miller RJ. The role of CXCR4 signaling in the migration of transplanted oligodendrocyte progenitors into the cerebral white matter. Neurobiology of Disease. 2011;44:19–27. doi: 10.1016/j.nbd.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmadani A, Tran PB, Ren D, Miller RJ. Chemokines regulate the migration of neural progenitors to sites of neuroinflammation. J Neurosci. 2006;26:3182–91. doi: 10.1523/JNEUROSCI.0156-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bo L, Geurts JJG, Mork SJ, van der Valk P. Grey matter pathology in multiple sclerosis. Acta Neurologica Scandinavica. 2006;113:48–50. doi: 10.1111/j.1600-0404.2006.00615.x. [DOI] [PubMed] [Google Scholar]
- Brown JP, Couillard-Despres S, Cooper-Kuhn CM, Winkler J, Aigner L, Kuhn HG. Transient expression of doublecortin during adult neurogenesis. J Comp Neurol. 2003;467:1–10. doi: 10.1002/cne.10874. [DOI] [PubMed] [Google Scholar]
- Bruck W. The pathology of multiple sclerosis is the result of focal inflammatory demyelination with axonal damage. Journal of Neurology. 2005;252:V3–V9. doi: 10.1007/s00415-005-5002-7. [DOI] [PubMed] [Google Scholar]
- Comte I, Kim Y, Young CC, van der Harg JM, Hockberger P, Bolam PJ, Poirier F, Szele FG. Galectin-3 maintains cell motility from the subventricular zone to the olfactory bulb. J Cell Sci. 2011;124:2438–47. doi: 10.1242/jcs.079954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connick P, Kolappan M, Crawley C, Webber DJ, Patani R, Michell AW, Du MQ, Luan SL, Altmann DR, Thompson AJ, et al. Autologous mesenchymal stem cells for the treatment of secondary progressive multiple sclerosis: an open-label phase 2a proof-of-concept study. Lancet Neurol. 2012;11:150–6. doi: 10.1016/S1474-4422(11)70305-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson JD. The histology of disseminated sclerosis. Royal Soc Edin. 1916;50:517–740. [Google Scholar]
- DeLuca GC, Ebers GC, Esiri MM. Axonal loss in multiple sclerosis: a pathological survey of the corticospinal and sensory tracts. Brain. 2004;127:1009–1018. doi: 10.1093/brain/awh118. [DOI] [PubMed] [Google Scholar]
- DeLuca GC, Joseph A, George J, Yates RL, Hamard M, Hofer M, Esiri MM. Olfactory Pathology in Central Nervous System Demyelinating Diseases. Brain Pathol. 2014 doi: 10.1111/bpa.12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Sanchez M, Williams K, DeLuca GC, Esiri MM. Protein co-expression with axonal injury in multiple sclerosis plaques. Acta Neuropathol. 2006;111:289–99. doi: 10.1007/s00401-006-0045-0. [DOI] [PubMed] [Google Scholar]
- Ekdahl CT, Claasen J-H, Bonde S, Kokaia Z, Lindvall O. Inflammation is detrimental for neurogenesis in adult brain. PNAS. 2003;100:13632–13637. doi: 10.1073/pnas.2234031100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goings GE, Greisman A, James RE, Abram LK, Begolka WS, Miller SD, Szele FG. Hematopoietic cell activation in the subventricular zone after Theiler’s virus infection. J Neuroinflammation. 2008;5:44–66. doi: 10.1186/1742-2094-5-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goings GE, Kozlowski DA, Szele FG. Differential activation of microglia in neurogenic versus non-neurogenic regions of the forebrain. Glia. 2006;54:329–42. doi: 10.1002/glia.20381. [DOI] [PubMed] [Google Scholar]
- Gordon RJ, McGregor AL, Connor B. Chemokines direct neural progenitor cell migration following striatal cell loss. Mol Cell Neurosci. 2009 doi: 10.1016/j.mcn.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Gordon RJ, Mehrabi NF, Maucksch C, Connor B. Chemokines influence the migration and fate of neural precursor cells from the young adult and middle-aged rat subventricular zone. Exp Neurol. 2012;233:587–94. doi: 10.1016/j.expneurol.2011.11.029. [DOI] [PubMed] [Google Scholar]
- Hoyos HC, Rinaldi M, Mendez-Huergo SP, Marder M, Rabinovich GA, Pasquini JM, Pasquini LA. Galectin-3 controls the response of microglial cells to limit cuprizone-induced demyelination. Neurobiol Dis. 2014;62:441–55. doi: 10.1016/j.nbd.2013.10.023. [DOI] [PubMed] [Google Scholar]
- Huang DR, Wang J, Kivisakk P, Rollins BJ, Ransohoff RM. Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J Exp Med. 2001;193:713–26. doi: 10.1084/jem.193.6.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal AJ, Regan-Komito D, Christou I, White GE, McNeill E, Kenyon A, Taylor L, Kapellos TS, Fisher EA, Channon KM, et al. A real time chemotaxis assay unveils unique migratory profiles amongst different primary murine macrophages. PLoS One. 2013;8:e58744. doi: 10.1371/journal.pone.0058744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonska B, Aguirre A, Raymond M, Szabo G, Kitabatake Y, Sailor KA, Ming GL, Song H, Gallo V. Chordin-induced lineage plasticity of adult SVZ neuroblasts after demyelination. Nat Neurosci. 2010;13:541–50. doi: 10.1038/nn.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon SB, Yoon HJ, Chang CY, Koh HS, Jeon SH, Park EJ. Galectin-3 exerts cytokine-like regulatory actions through the JAK-STAT pathway. J Immunol. 2010;185:7037–46. doi: 10.4049/jimmunol.1000154. [DOI] [PubMed] [Google Scholar]
- Jiang HR, Al Rasebi Z, Mensah-Brown E, Shahin A, Xu D, Goodyear CS, Fukada SY, Liu FT, Liew FY, Lukic ML. Galectin-3 deficiency reduces the severity of experimental autoimmune encephalomyelitis. J Immunol. 2009;182:1167–73. doi: 10.4049/jimmunol.182.2.1167. [DOI] [PubMed] [Google Scholar]
- Jin K, Wang X, Xie L, Mao XO, Greenberg DA. Transgenic ablation of doublecortin-expressing cells suppresses adult neurogenesis and worsens stroke outcome in mice. Proc Natl Acad Sci U S A. 2010;107:7993–7998. doi: 10.1073/pnas.1000154107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpus WJ, Kennedy KJ, Fife BT, Bennett JL, Dal Canto MC, Kunkel SL, Lukacs NW. Anti-CCL2 treatment inhibits Theiler’s murine encephalomyelitis virus-induced demyelinating disease. J Neurovirol. 2006;12:251–61. doi: 10.1080/13550280600873819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpus WJ, Lukacs NW, McRae BL, Strieter RM, Kunkel SL, Miller SD. An important role for the chemokine macrophage inflammatory protein-1 alpha in the pathogenesis of the T cell-mediated autoimmune disease, experimental autoimmune encephalomyelitis. J Immunol. 1995;155:5003–10. [PubMed] [Google Scholar]
- Kokaia Z, Martino G, Schwartz M, Lindvall O. Cross-talk between neural stem cells and immune cells: the key to better brain repair? Nat Neurosci. 2012;15:1078–87. doi: 10.1038/nn.3163. [DOI] [PubMed] [Google Scholar]
- Lalancette-Hebert M, Swarup V, Beaulieu JM, Bohacek I, Abdelhamid E, Weng YC, Sato S, Kriz J. Galectin-3 is required for resident microglia activation and proliferation in response to ischemic injury. J Neurosci. 2012;32:10383–95. doi: 10.1523/JNEUROSCI.1498-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane TE, Hosking MP. The pathogenesis of murine coronavirus infection of the central nervous system. Crit Rev Immunol. 2010;30:119–30. doi: 10.1615/critrevimmunol.v30.i2.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu FT, Rabinovich GA. Galectins as modulators of tumour progression. Nat Rev Cancer. 2005;5:29–41. doi: 10.1038/nrc1527. [DOI] [PubMed] [Google Scholar]
- Luzzati F, Bonfanti L, Fasolo A, Peretto P. DCX and PSA-NCAM Expression Identifies a Population of Neurons Preferentially Distributed in Associative Areas of Different Pallial Derivatives and Vertebrate Species. Cereb Cortex. 2008 doi: 10.1093/cercor/bhn145. [DOI] [PubMed] [Google Scholar]
- Martinez-Molina N, Kim Y, Hockberger P, Szele FG. Rostral migratory stream neuroblasts turn and change directions in stereotypic patterns. Cell Adh Migr. 2011;5:83–95. doi: 10.4161/cam.5.1.13788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DP, Richards MH, Miller SD. Mouse models of multiple sclerosis: experimental autoimmune encephalomyelitis and Theiler’s virus-induced demyelinating disease. Methods Mol Biol. 2012;900:381–401. doi: 10.1007/978-1-60761-720-4_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecha M, Carrillo-Salinas FJ, Mestre L, Feliu A, Guaza C. Viral models of multiple sclerosis: neurodegeneration and demyelination in mice infected with Theiler’s virus. Prog Neurobiol. 2013a;101–102:46–64. doi: 10.1016/j.pneurobio.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecha M, Feliu A, Carrillo-Salinas FJ, Mestre L, Guaza C. Mobilization of progenitors in the subventricular zone to undergo oligodendrogenesis in the theiler s virus model of multiple sclerosis: Implications for remyelination at lesions sites. Exp Neurol. 2013b doi: 10.1016/j.expneurol.2013.10.011. [DOI] [PubMed] [Google Scholar]
- Menn B, Garcia-Verdugo JM, Yaschine C, Gonzalez-Perez O, Rowitch D, Alvarez-Buylla A. Origin of oligodendrocytes in the subventricular zone of the adult brain. J Neurosci. 2006;26:7907–18. doi: 10.1523/JNEUROSCI.1299-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monje ML, Toda H, Palmer TD. Inflammatory Blockade Restores Adult Hippocampal Neurogenesis. Science. 2003;302:1760–1765. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]
- Muzio L, Cavasinni F, Marinaro C, Bergamaschi A, Bergami A, Porcheri C, Cerri F, Dina G, Quattrini A, Comi G, et al. Cxcl10 enhances blood cells migration in the sub-ventricular zone of mice affected by experimental autoimmune encephalomyelitis. Mol Cell Neurosci. 2010;43:268–80. doi: 10.1016/j.mcn.2009.11.008. [DOI] [PubMed] [Google Scholar]
- Nait-Oumesmar B, Decker L, Lachapelle F, Avellana-Adalid V, Bachelin C, Van Evercooren AB. Progenitor cells of the adult mouse subventricular zone proliferate, migrate and differentiate into oligodendrocytes after demyelination. Eur J Neurosci. 1999;11:4357–66. doi: 10.1046/j.1460-9568.1999.00873.x. [DOI] [PubMed] [Google Scholar]
- Nait-Oumesmar B, Picard-Riera N, Kerninon C, Decker L, Seilhean D, Hoglinger GU, Hirsch EC, Reynolds R, Baron-Van Evercooren A. Activation of the subventricular zone in multiple sclerosis: evidence for early glial progenitors. Proc Natl Acad Sci U S A. 2007;104:4694–9. doi: 10.1073/pnas.0606835104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam SC, Kim Y, Dryanovski D, Walker A, Goings G, Woolfrey K, Kang SS, Chu C, Chenn A, Erdelyi F, et al. Dynamic features of postnatal subventricular zone cell motility: A two-photon time-lapse study. J Comp Neurol. 2007;505:190–208. doi: 10.1002/cne.21473. [DOI] [PubMed] [Google Scholar]
- Nansen A, Marker O, Bartholdy C, Thomsen AR. CCR2+ and CCR5+ CD8+ T cells increase during viral infection and migrate to sites of infection. Eur J Immunol. 2000;30:1797–806. doi: 10.1002/1521-4141(200007)30:7<1797::AID-IMMU1797>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Papaspyridonos M, McNeill E, de Bono JP, Smith A, Burnand KG, Channon KM, Greaves DR. Galectin-3 is an amplifier of inflammation in atherosclerotic plaque progression through macrophage activation and monocyte chemoattraction. Arterioscler Thromb Vasc Biol. 2008;28:433–40. doi: 10.1161/ATVBAHA.107.159160. [DOI] [PubMed] [Google Scholar]
- Patrikios P, Stadelmann C, Kutzelnigg A, Rauschka H, Schmidbauer M, Laursen H, Sorensen PS, Bruck W, Lucchinetti C, Lassmann H. Remyelination is extensive in a subset of multiple sclerosis patients. Brain. 2006;129:3165–72. doi: 10.1093/brain/awl217. [DOI] [PubMed] [Google Scholar]
- Picard-Riera N, Decker L, Delarasse C, Goude K, Nait-Oumesmar B, Liblau R, Pham-Dinh D, Evercooren AB. Experimental autoimmune encephalomyelitis mobilizes neural progenitors from the subventricular zone to undergo oligodendrogenesis in adult mice. Proc Natl Acad Sci U S A. 2002;99:13211–6. doi: 10.1073/pnas.192314199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluchino S, Gritti A, Blezer E, Amadio S, Brambilla E, Borsellino G, Cossetti C, Del Carro U, Comi G, t Hart B, et al. Human neural stem cells ameliorate autoimmune encephalomyelitis in non-human primates. Ann Neurol. 2009;66:343–54. doi: 10.1002/ana.21745. [DOI] [PubMed] [Google Scholar]
- Pluchino S, Quattrini A, Brambilla E, Gritti A, Salani G, Dina G, Galli R, Del Carro U, Amadio S, Bergami A, et al. Injection of adult neurospheres induces recovery in a chronic model of multiple sclerosis. Nature. 2003;422:688–94. doi: 10.1038/nature01552. [DOI] [PubMed] [Google Scholar]
- Pluchino S, Zanotti L, Rossi B, Brambilla E, Ottoboni L, Salani G, Martinello M, Cattalini A, Bergami A, Furlan R, et al. Neurosphere-derived multipotent precursors promote neuroprotection by an immunomodulatory mechanism. Nature. 2005;436:266–71. doi: 10.1038/nature03889. [DOI] [PubMed] [Google Scholar]
- Sano H, Hsu DK, Yu L, Apgar JR, Kuwabara I, Yamanaka T, Hirashima M, Liu FT. Human galectin-3 is a novel chemoattractant for monocytes and macrophages. J Immunol. 2000;165:2156–64. doi: 10.4049/jimmunol.165.4.2156. [DOI] [PubMed] [Google Scholar]
- Stancic M, van Horssen J, Thijssen VL, Gabius HJ, van der Valk P, Hoekstra D, Baron W. Increased expression of distinct galectins in multiple sclerosis lesions. Neuropathol Appl Neurobiol. 2011;37:654–71. doi: 10.1111/j.1365-2990.2011.01184.x. [DOI] [PubMed] [Google Scholar]
- Sun M, Fu H, Cheng H, Cao Q, Zhao Y, Mou X, Zhang X, Liu X, Ke Y. A dynamic real-time method for monitoring epithelial barrier function in vitro. Anal Biochem. 2012;425:96–103. doi: 10.1016/j.ab.2012.03.010. [DOI] [PubMed] [Google Scholar]
- Tepavcevic V, Lazarini F, Alfaro-Cervello C, Kerninon C, Yoshikawa K, Garcia-Verdugo JM, Lledo PM, Nait-Oumesmar B, Baron-Van Evercooren A. Inflammation-induced subventricular zone dysfunction leads to olfactory deficits in a targeted mouse model of multiple sclerosis. J Clin Invest. 2011;121:4722–34. doi: 10.1172/JCI59145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran PB, Banisadr G, Ren D, Chenn A, Miller RJ. Chemokine receptor expression by neural progenitor cells in neurogenic regions of mouse brain. The Journal of Comparative Neurology. 2007;500:1007–1034. doi: 10.1002/cne.21229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran PB, Ren D, Veldhouse TJ, Miller RJ. Chemokine receptors are expressed widely by embryonic and adult neural progenitor cells. J Neurosci Res. 2004;76:20–34. doi: 10.1002/jnr.20001. [DOI] [PubMed] [Google Scholar]
- Tsunoda I, Kuang LQ, Libbey JE, Fujinami RS. Axonal injury heralds virus-induced demyelination. Am J Pathol. 2003;162:1259–69. doi: 10.1016/S0002-9440(10)63922-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunoda I, Lane TE, Blackett J, Fujinami RS. Distinct roles for IP-10/CXCL10 in three animal models, Theiler’s virus infection, EAE, and MHV infection, for multiple sclerosis: implication of differing roles for IP-10. Mult Scler. 2004;10:26–34. doi: 10.1191/1352458504ms982oa. [DOI] [PubMed] [Google Scholar]
- Venkatesan C, Chrzaszcz M, Choi N, Wainwright MS. Chronic upregulation of activated microglia immunoreactive for galectin-3/Mac-2 and nerve growth factor following diffuse axonal injury. J Neuroinflammation. 2010;7:32. doi: 10.1186/1742-2094-7-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther M, Kuklinski S, Pesheva P, Guntinas-Lichius O, Angelov DN, Neiss WF, Asou H, Probstmeier R. Galectin-3 is upregulated in microglial cells in response to ischemic brain lesions, but not to facial nerve axotomy. J Neurosci Res. 2000;61:430–5. doi: 10.1002/1097-4547(20000815)61:4<430::AID-JNR9>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Wang T, Lee MH, Johnson T, Allie R, Hu L, Calabresi PA, Nath A. Activated T-cells inhibit neurogenesis by releasing granzyme B: rescue by Kv1.3 blockers. J Neurosci. 2010;30:5020–7. doi: 10.1523/JNEUROSCI.0311-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Ji BX, Su L, Dong HQ, Sun WL, Wan SG, Liu YO, Zhang P, Liu CY. Clinical outcome of autologous peripheral blood stem cell transplantation in opticospinal and conventional forms of secondary progressive multiple sclerosis in a Chinese population. Annals of Hematology. 2011;90:343–8. doi: 10.1007/s00277-010-1071-5. [DOI] [PubMed] [Google Scholar]
- Yamout B, Hourani R, Salti H, Barada W, El-Hajj T, Al-Kutoubi A, Herlopian A, Baz EK, Mahfouz R, Khalil-Hamdan R, et al. Bone marrow mesenchymal stem cell transplantation in patients with multiple sclerosis: a pilot study. J Neuroimmunol. 2010;227:185–9. doi: 10.1016/j.jneuroim.2010.07.013. [DOI] [PubMed] [Google Scholar]
- Yan YP, Sailor KA, Lang BT, Park SW, Vemuganti R, Dempsey RJ. Monocyte chemoattractant protein-1 plays a critical role in neuroblast migration after focal cerebral ischemia. J Cereb Blood Flow Metab. 2007;27:1213–24. doi: 10.1038/sj.jcbfm.9600432. [DOI] [PubMed] [Google Scholar]
- Yang HK, Sundholm-Peters NL, Goings GE, Walker AS, Hyland K, Szele FG. Distribution of doublecortin expressing cells near the lateral ventricles in the adult mouse brain. J Neurosci Res. 2004;76:282–95. doi: 10.1002/jnr.20071. [DOI] [PubMed] [Google Scholar]
- Young CC, Al-Dalahmah O, Lewis NJ, Brooks KJ, Jenkins MM, Poirier F, Buchan AM, Szele FG. Blocked angiogenesis in Galectin-3 null mice does not alter cellular and behavioral recovery after middle cerebral artery occlusion stroke. Neurobiol Dis. 2014;63:155–64. doi: 10.1016/j.nbd.2013.11.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Suppl. Fig. 1. Gal-3 increased in area of demyelination.
(A) Gal-3 expression was increased in an active lesion (arrow) in the CC of an MS case, haematoxylin counterstain. (B) Higher magnification view of lesion in A. (C) PLP immunoreactivity loss in same lesion as B, adjacent section. (D, E) Gal-3 and CD68 immunoreactivity in adjacent sections at high magnification in same lesion as A–C. Asterisks show blood vessels. Scale bars: A=5 mm; B–C=2mm; D–E=100 μm.
Suppl. Fig. 2. TMEV infection increased astrocyte Gal-3 expression.
(A–D) Typical Gal-3 expression by GFAP+ cerebral cortex and (E–H) SVZ-derived primary astrocyte cultures. (I) ELISA measurement of Gal-3 secreted from primary astrocytes in controls and after TMEV multiplicity of infection (MOI) 1 and 10.
Suppl. Fig. 3. Macrophage Chemotaxis Assay
(A) Outline of murine monocyte/macrophage elicitation, enrichment by adhesion to bacterial petri dishes before analysis. (B–D) flow cytometry showing relative purity of macrophages. (E) Representative trace from a CIM-16 plate where each line represents the mean of 3–4 wells per chemoattractant. (F–G) Chemotaxis data from n=2–6 independent experiments quantified as Slope or Max-min analysis. Statistical analysis in V and W was performed using one way ANOVA and Dunnett’s multiple comparisons post test relative to vehicle alone, ***p<0.001. (H) Addition of exogenous rGal-3 caused neurospheres to clump together.
Suppl. Fig. 4. CD3+ cells infiltrated the SVZ and forebrain post-TMEV infection.
(A) Low magnification image of CD3+ immunofluorescence in the SVZ surrounding the lateral ventricles (lv). Note the two prominent blood vessels (white arrowheads) in the SVZ and the azygos pericallosal artery (azPA), the midline vessel just dorsal to the corpus callosum (red arrow). These blood vessels are lined with CD3+ cells. (B) Large numbers of CD3+ cells migrated into the CNS through the azPA. Scale bars: A=200 μm; B=100 μm.
(C) Quantification of CD3+ cells in CNS subregions of TMEV infected SJL/J mice. Sham mice only had 1–2 CD3+ cells per section, therefore the bars are not visible. (D–E) Number of CD3+ cells in the SVZ in SJL/J and C57BL/6 mice. (F) Approximately 40–60% of CD3+ cells expressed Gal-3, 7 and 14 dpi in SJL/J mice.
Suppl. Table 1. Fold changes in gene expression by RT-PCR array in SVZ tissue and astrocyte cultures after TMEV infection.