

Abstract
Decades of research have revealed numerous differences in brain structure size, connectivity and metabolism between males and females. Sex differences in neurobehavioral and cognitive function after various forms of central nervous system (CNS) injury are observed in clinical practice and animal research studies. Sources of sex differences include early life exposure to gonadal hormones, chromosome compliment and adult hormonal modulation. It is becoming increasingly apparent that mitochondrial metabolism and cell death signaling are also sexually dimorphic. Mitochondrial metabolic dysfunction is a common feature of CNS injury. Evidence suggests males predominantly utilize proteins while females predominantly use lipids as a fuel source within mitochondria and that these differences may significantly affect cellular survival following injury. These fundamental biochemical differences have a profound impact on energy production and many cellular processes in health and disease. This review will focus on the accumulated evidence revealing sex differences in mitochondrial function and cellular signaling pathways in the context of CNS injury mechanisms and the potential implications for neuroprotective therapy development.
Keywords: Sex differences, Mitochondria, Metabolism, Neuroprotection, Excitotoxicity, Mitophagy, Oxidative stress, Cell death
Introduction
Mitochondrial dysfunction is a common component of central nervous system (CNS) injury pathophysiology that has received increasing attention as a putative target for neuroprotective therapy development (Fiskum 2000; Robertson et al. 2006; Perez-Pinzon et al. 2012; Hagberg et al. 2014). Animal and human CNS injury studies indicate sexually dimorphic behavioral and histopathological outcomes yet animal and in vitro studies rarely include females for evaluation of injury and/or treatment responses. The National Institutes of Health of the US mandated the inclusion of women in clinical trials almost a decade ago, but this balanced approach was not adopted by basic science researchers. Recently, the NIH issued a statement that new guidelines will be established requiring the inclusion of both sexes or strong justification for exclusion of one sex in all future federally funded preclinical studies in an effort to increase the translational generalizability and replicability of basic scientific research (Clayton and Collins 2014).
Sex is determined in mammals by chromosome complement and the presence of a single gene on the Y chromosome, SRY, for sex-determining region of the Y chromosome, which codes for tdf, the testis determining factor (Fig. 1). Expression of this gene during a narrow window of early embryonic development directs the bipotential gonad to become a testis. In the absence of SRY the gonad will become an ovary. The testis begins steroidogenesis embryonically, exposing the developing male fetus to high concentrations of testosterone and its aromatized metabolite, estradiol. The fetal brain is rich in estrogen (ER) and androgen receptors (AR), as well as the enzyme aromatase that converts testosterone to estradiol. The ovary remains quiescent at this time, leaving the female relatively free from androgen and estrogen exposure, particularly in the brain. Androgens and estrogens program the developing brain by impacting numerous parameters including cell death and survival, neural and glial genesis, neuronal migration, myelination, synaptogenesis and synaptic pruning and neurochemical phenotype. In adulthood, gonadal and adrenal steroids act on and are influenced by this differentiated neural substrate in some but not all instances. Some adult brain sex differences are a byproduct of differences in gonadal steroids, meaning the sex difference goes away if steroids are either equalized or removed. Thus this is an instance of hormonal modulation as opposed to a primary sex difference per se. More recently it has become apparent there is also both a direct and a modulatory role for chromosome complement, with genes on the X or Y chromosome impacting brain and behavior in males and females. To-date there have been no clear connections drawn between a particular sex chromosome gene and a behavioral or phenotypic endpoint, but it is still early days for this research focus (review in (McCarthy 2008; McCarthy and Arnold 2011)).
Fig. 1.
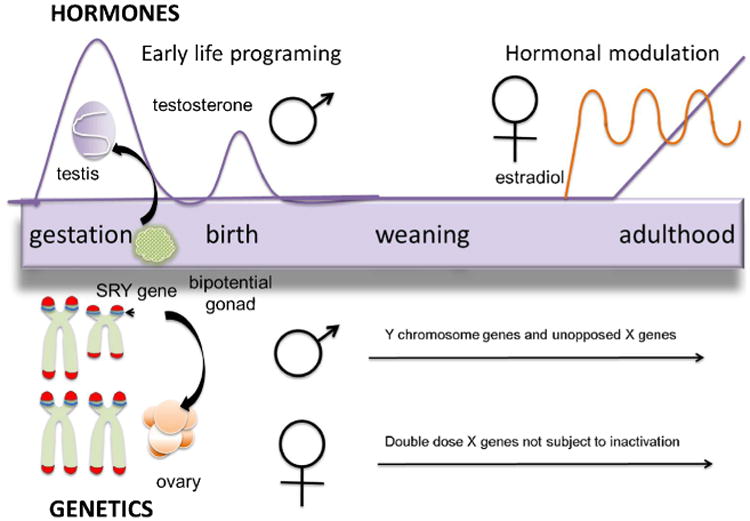
Sources of Sex Differences in the Brain. The bipotential gonad is directed towards testis development by the SRY gene on the Y chromosome. Testosterone production begins during fetal life and surges again at birth and then remains low until puberty. Steroids impact numerous endpoints in the developing brain, many of which will endure into adulthood. In females the ovary develops due to the lack of SRY and remains quiescent until puberty at which time estradiol production is cyclical. Genes on the X and Y chromosome are capable of influencing brain in behavior throughout life in manners still not well understood
Sex differences in metabolism have long been observed in humans (reviewed in (Widdowson 1976). Circumstantial evidence for sex differences comes from cases of severe nutrition deprivation observed in Germany following World War II. During the war, all people were given the same rations regardless of age or sex. In order to qualify for additional rations, individuals had to be considered severely malnourished and 60 % of those qualified individuals were male. Further studies of orphan children in 1947 indicated males were more severely malnourished than females. Subsequent studies of nutrition deprivation in pigs and rats revealed a significantly greater mortality in males, coinciding with a greater loss of total body protein, while females lost significantly more fat than males. Accordingly, Widdowson concludes “A large loss of protein from the body is metabolically a far more serious matter than loss of fat, and the greater catabolism of protein in males is probably an important reason why they withstand a deficiency of energy less well than females.” (Widdowson 1976). More recently, endurance exercise studies reveal that males rely almost exclusively on carbohydrates and amino acids (97 %) for fuel while females predominantly use fat (62 %) (Lamont 2005). Furthermore, starvation of mice results in significantly greater survival in females coinciding with increased plasma ketone bodies and associated estrogen mediated increases in mitochondrial uncoupling protein 1 in brown fat of females vs. males (Jikumaru et al. 2007). Sex differences in preferred fuel source are also apparent in cardiac and hepatic cells (Djouadi et al. 1998). The primary reliance on proteins and fatty acids for fuel in males and females, respectively, has been confirmed in cultured primary neurons and fibroblasts following nutrient deprivation (Du et al. 2009). Nutrient deprivation activates autophagy; the targeted degradation of cellular components for fuel. Given the evidence for sex differences in human metabolism and preferred cellular biofuels, there may also be sex differences in autophagy. These fundamental sex differences in metabolism under stressful conditions may also be influenced by intrinsic differences in genomic maintenance.
Only 13 of 92 mitochondrial proteins essential for oxidative phosphorylation are encoded by mitochondrial DNA. As a result, mitochondrial-nuclear communication for translation and mitochondrial import of respiratory chain subunit proteins is an essential process for maintaining adequate cellular energy supply. During stressful conditions, the mitochondrial-nuclear DNA coordination is especially critical for cellular survival. Therefore, mechanisms to prevent or repair DNA damage under stressful conditions would be predicted to improve cellular survival. Given the near exclusive maternal inheritance of mitochondrial DNA, there may be fundamental sex differences in mitochondrial metabolic regulation such that female mitochondria are better equipped to cope with stressful conditions and are relatively resilient to DNA damage and mutation to reduce the probability of producing inheritable metabolic disorders. Indeed, microarray analysis of embryonic day 10.5 mouse brains (previous to hormonal influence) identified over 50 differentially expressed transcripts between males and females (Dewing et al. 2003). Of note is the nearly 2 fold increase in expression of DNA replication and repair DNA polymerase delta 1 and GA binding protein subunit alpha (GABP) in female fetal brain vs. males (Dewing et al. 2003). Previous studies identify GABP as the nuclear DNA binding motif of the rat and human homologue nuclear respiratory factor-2 (NRF-2) (Virbasius and Virbasius 1993). NRF-2 upregulates the expression of complex II and IV electron transport chain subunits, ATP synthase β, mitochondrial biogenesis protein mitochondrial transcription activator A (TFAM) (Zhang and Wong-Riley 2000; Scarpulla 2002) and is activated by neuronal depolarization (Zhang and Wong-Riley 2000). These findings highlight the possibility that females are hardwired for favorable nuclear to mitochondrial coordination of critical mitochondrial respiratory subunits in response to stress compared to males. The few studies assessing sex differences in mitochondrial function following injury reveal substantial differences in mitochondrial membrane potential, enzyme activity and respiration between male and female rodents after cerebral neonatal hypoxic ischemia (Weis et al. 2012; Demarest et al. 2013). Therefore, the focus of this review is to examine the literature on sexually dimorphic phenomena relevant to metabolic and mitochondrial function in the context of well-established mechanisms of CNS injury.
Excitotoxicity and Calcium
Glutamate excitotoxicity is a widely accepted contributor to neurodegeneration following CNS injury. Under normal physiological conditions, activation of glutamate receptors increases intracellular sodium, calcium and downstream activation of second messengers, nitric oxide generation, mitochondrial calcium uptake, increased mitochondrial membrane potential (ΔΨ) and upregulation of rate-limiting TCA cycle dehydrogenases (Duchen 2000a, b). Excitotoxic neurodegeneration is predominantly mediated through extrasynaptic NR2B subunit containing N-Methyl-d-Aspartate receptors (NMDARs), however the exact receptor mediating excitotoxic cell death may differ with age and cellular composition of a given brain region (Chen et al. 1995; Portera-Cailliau et al. 1997; Hilton et al. 2006). Sex differences have been scarcely studied with regard to differential susceptibility to excitotoxic neurodegeneration. However, following severe traumatic brain injury (TBI) in humans, cerebral spinal fluid (CSF) glutamate content is significantly greater in males vs. females (Wagner et al. 2005). While female neuroprotection observed following injury is commonly attributed to female hormones, less attention has been paid to the influence of androgens mediating male susceptibility to brain injury (for review see (Quillinan et al. 2014)). For instance, in immature neurons, activation of ionotropic γ-aminobutyric acid receptors (GABAA) is excitatory and contributes to male vulnerability to excitotoxic injury in an androgen dependent manner (Nuñez et al. 2003; Nuñez and McCarthy 2008).
Sex differences in vulnerability to excitotoxic injury can also be influenced by chromosome complement. Sex stratified primary cortical neuronal cultures treated with exogenous glutamate/glycine, NMDA, AMPA or kainate demonstrate a significant decrease in cellular viability in male (XY) vs. female (XX) neurons. NMDAR antagonist MK-801 prevents decreases in cellular viability solely in XY neurons (Du et al. 2004) while 17β-estradiol treatment does not improve XY neuronal viability. Other studies exposing hippocampal (Weaver et al. 1997) and cortical neurons (Perrella and Bhavnani 2005) to glutamate demonstrate protection from excitotoxic cell death with exogenous administration of 17β-estradiol, but not in cerebellar granular neurons (Minano et al. 2007). However, these cultures were not sex-specific. Moreover, cultured mesencephalic dopamine neurons exposed to high concentrations of extracellular dopamine display male susceptibility to cell death, but only female neurons are rescued by NMDAR antagonist AP-5 (Lieb et al. 1995). In vivo CNS injury studies show some efficacy of NMDAR antagonists in preventing cell death but typically only use male animals or do not report which sex is used (Ford et al. 1989; Gaviria et al. 2000; Rao et al. 2001; Lapchak 2004; Han et al. 2009; Wang et al. 2014). Spinal cord excitotoxicity induced with high and low doses of kainic/quinolinic acid results in 80 % and 40 % mortality of male mice, respectively. Interestingly, all females survive both doses of either drug (Martin 2011). Further complicating the interpretation of sex differences in excitotoxic neurodegeneration is the observation that injection of naïve rats with NMDAR antagonist MK-801 results in significantly more neuronal cell death in females compared to males. However, similar treatment of several strains of mice does not result in the same severity of cell death nor a sex difference (Bender et al. 2010). These findings highlight sex, brain region and species dependent susceptibility to excitotoxic injury which require further investigation.
Regardless of the exact receptor and brain region specificity impacted by excitotoxicity, resulting cell death is calcium dependent (Choi 1985). Mitochondrial calcium buffering is an essential homeostatic process for maintenance of normal cell function. Mitochondrial calcium uptake in the context of excitotoxicity has been intensively studied in isolated mitochondria and primary neuronal cultures (for review see (Choi 1994; Nicholls 2004)). In general, a situation in which mitochondrial calcium is lower is associated with decreased cellular injury and too much calcium is associated with mitochondrial swelling and the opening of the mitochondrial permeability transition pore (mPTP) (Wang et al. 2001). Opening of the mPTP results in diffusion of molecules (<1,500 kD) from mitochondria to cytoplasm, ATP depletion and acute cell death. To our knowledge there have been no studies assessing putative sex differences in mPTP opening. However, studies of isolated mitochondria reveal rat brain (Kim et al. 2012) and mouse heart (Arieli et al. 2004) mitochondria have a sexually dimorphic capacity for calcium uptake with isolated male mitochondria having greater calcium uptake capacity than female mitochondria. This may be estrogen dependent as 17β-estradiol decreases calcium retention in brain mitochondria of both sexes (Kim et al. 2012) but overiectomy has no effect on calcium uptake in cardiac mitochondria (Arieli et al. 2004). Furthermore, brain mitochondria from cyclophilin D knockout mice have enhanced calcium uptake in both males and females but no sex difference. Cyclophilin D is a key regulator of mPTP opening where genetic knockout or pharmacological inhibition of cyclophilin D (e.g. by cyclosporine A) inhibits mPTP opening and cell death. Interestingly, survival analysis reveals that the increased lifespan normally observed in female vs. male wild-type mice is no longer apparent in cyclophiln D knockouts (Kim et al. 2012). These results beg the question - What is the physiological role of enhanced calcium uptake in mitochondria derived from males?
One possible explanation necessitating enhanced mitochondrial calcium uptake capacity by male mitochondria derives from secondary activation of the calcium-permeable transient receptor potential M2 (TRPM2) nonselective cation channels. TRPM2 channels are considered executioners of cell death following oxidative stress. They are activated by hydrogen peroxide and gated by intracellular adenine dinucleotide phosphate ribose (ADPr), (Fonfria et al. 2004) a breakdown product by poly(ADP)-ribose glycohydrolase (PARG) of poly(ADP-ribose) (PAR) polymers formed by poly(ADP-ribose) polymerase 1 (PARP-1). TRPM2 channels are present in both males and females at similar levels in cultured hippocampal neurons. However, electrophysiological evidence (Verma et al. 2012) and reductions in cell death by TRPM2 pharmacological or shRNA inhibition in an in vivo model of stroke, (Jia et al. 2011) or shRNA knockdown following in vitro oxygen glucose deprivation (OGD), (Verma et al. 2012) indicate that TRPM2 channels are only activated in males following injury. Contrarily, peroxide mediated in vitro toxicity shows no sex difference in cell death and TRPM2 inhibition is neuroprotective in both sexes (Verma et al. 2012) suggesting greater oxidative stress and/or PAR/ADPr generation in males following injury contributes to sex differences in TRPM2 mediated cell death.
As mentioned above, calcium propagates numerous cellular signaling cascades. Particularly relevant to CNS injury is the induction of nitric oxide synthase (NOS) and upregulation of TCA cycle enzymes. The TCA cycle enzyme α-ketoglutarate dehydrogenase (α-KGDH) is a potent generator and target of oxidative stress in the brain (Starkov et al. 2004; Starkov 2013) and regulatory mechanisms may limit ROS/RNS generation during times of cellular stress in a sex dependent manner. For example, TCA cycle enzyme regulation in myocardial ischemia suggests that increasing phosphorylation of α-KGDH and aldehyde dehydrogenase-2, reduces oxidative stress and confers cardioprotection in female heart as compared with male heart (Lagranha et al. 2010). These data suggest that female resilience to injury may be mediated by superior enzyme regulation and decreases in oxidative stress.
Oxidative/Nitrositive stress
Calcium induction of oxidative stress is well documented in brain cells (reviewed in (Duchen 2000a, b; Nicholls 2004; Peng and Jou 2010)). Oxidative and nitrositive stress (ROS/RNS) refers to the balance between the generation of free radicals and their detoxification via resident antioxidant systems. Mitochondria are a major source of cellular ROS/RNS generation. Under pathological conditions, high levels of ROS/RNS can damage proteins, lipids and nucleic acids that must be repaired in order to meet cellular energy demands and ensure cell survival. NOS induction by calcium influx is hypothesized to be a fundamental regulator of cellular energy demand. Nitric oxide (NO) synthesized by NOS is freely diffusible and competes with oxygen at complex IV to reversibly inhibit the rate of oxidative phosphorylation (Brown and Cooper 1994; Brown 1995, 2001, 2007). In this manner, slowing the flow of electrons could serve as a feedback mechanism to regulate the rate of oxidative phosphorylation in response to cellular energy demand under physiological and pathophysiological conditions. NO can also react with superoxide forming the highly reactive and damaging RNS, peroxinitrite (ONOO-) (Brown 2007). Interestingly, neuronal NOS (nNOS) induction is greater in male animals following cerebral ischemic injury (McCullough et al. 2005; Semenas et al. 2010). Pharmacological inhibition or genetic knockout of nNOS is neuroprotective in male mice but actually increases infarct volume in female mice (McCullough et al. 2005). This suggests NO production following injury has a beneficial role in females but whether this is due to vasodilation and restoration of cerebral blood flow or another mechanism is unclear.
Sex differences in NOS signaling in vitro have also been investigated. Organotypic hippocampal slices from males are more susceptible to cell death following NMDA exposure or OGD and produce more nitrate/nitrite vs. females (Li et al. 2005). nNOS inhibition prior to NMDA exposure or OGD prevents cell death in male slices while having no effect in female slices. Treatment with 17β-estradiol protects both male and female neurons in culture (Li et al. 2005). Treatment of cortical neurons with exogenous ONOO- results in cellular depletion of reduced glutathione (GSH) in XY neurons after 24 h, which is completely rescued by antioxidant N-acetyl-cysteine (NAC) but XX neurons have no detectible depletion of GSH and NAC treatment has no effect (Du et al. 2004). Male specific GSH depletion also occurs in an in vivo model of cardiac arrest asphyxia in postnatal day 17 rats (Du et al. 2004). Moreover, NAC treatment within 2 h of rat controlled cortical impact (CCI) TBI prevents GSH depletion, restores mitochondrial respiratory function and calcium buffering capacity; only male rats were used in this study (Xiong et al. 1999). In human infants and children suffering from severe TBI, GSH depletion in cerebrospinal fluid (CSF) is measured 5-7 days post-injury (Bayir et al. 2002) revealing the clinical importance of considering sex as a contributing variable to CNS injury.
The cellular origins of NOS may also be an important contributing variable. A mitochondrial localized NOS (mtNOS) has been proposed (Haynes et al. 2004; Giulivi 2007). While its undeniable existence is complicated by the lack of specific antibodies, mtNOS has been detected by immune electron microscopy (Bates et al. 1995). As such, it is tempting to speculate it plays an even larger role than nNOS in sexually dimorphic mitochondrial function. Regardless of cytosolic or mitochondrial origin, higher ONOO- generation under conditions of ROS/RNS damage mitochondrial and nuclear DNA thus activating PARP-1 for DNA repair (discussed in detail in cell death section). Additionally, protein nitration of critical antioxidant components may impair anti-oxidant defense systems. Following TBI in mice and humans, nitration of manganese superoxide dismutase (MnSOD/SOD2) impairs detoxification of mitochondrial superoxide (Bayir et al. 2007). Inhibition of neuronal NOS, but not endothelial or inducible NOS, attenuates MnSOD nitration following TBI. Interestingly, while total SOD activity remains unchanged over 72 h following TBI, mitochondrial MnSOD activity is significantly decreased over the same time period (Bayir et al. 2007). Indeed, over-expression of MnSOD in cells is associated with an increase in glutathione peroxidase (GPx) activity, decreases in lipid peroxidation and ONNO- formation ((Keller et al. 1998; Murakami et al. 1998) as cited by (Bayir et al. 2007)).
The antioxidant enzyme GPx is arguably the most important of this category of enzymes in both neurons and glia - providing the main detoxification pathway for hydrogen peroxide (H2O2) ((Dringen et al. 1999) as cited by (Nicholls 2004)). H2O2 is formed when MnSOD dismutates superoxide radicals formed by electron ‘leakage’ from the mitochondrial electron transport chain or by other mitochondrial activities. When catabolism of H2O2 by GPx (or catalase) is impaired, increasing lipid peroxidation can ensue, compromising cellular and mitochondrial membrane integrity and potentiating bioenergetic failure. Increased mitochondrial GPx activity in females vs. males was first observed in liver back in the 1960's (Pinto and Bartley 1969). More recently, Borras et al (2003) demonstrated increases in GPx and MnSOD activity in female derived liver mitochondria. Female derived mitochondria also produce about half the amount of H2O2 in liver, and perhaps more importantly, in synaptic and non-synaptic brain mitochondria vs. males. Mitochondria from overiectomized females have increased H2O2 production and decreased GSH vs. males, and these levels can be rescued by estrogen administration. These changes were most dramatic in synaptic mitochondria. Furthermore, female derived brain mitochondria have higher GSH content and four-fold less oxidative damage to mitochondrial DNA vs. males (Borras et al. 2003).
Mitochondrial DNA is oxidatively damaged with aging (Mecocci et al. 1993) and theorized as a main determining factor for lifespan (Harman 1956, 1972). In mammalian species, females generally live longer than males. Studies conducted as a part of The Biomarker of Aging Program from the NIH characterized longevity in several rodent strains and identified just two strains of mice where males live longer than females. Specifically, female C57BL6 mice have a 20 % shorter lifespan than males which is associated with decreases in antioxidant proteins and increases in oxidative stress in the hippocampus of 5 month-old mice. In older 24 month-old female C57BL6 mice, hippocampus, cortex and substantia nigra all have higher levels of oxidative stress vs. males. Chronic administration of a superoxide dismutase mimetic (SOD; from 12–24 months in drinking water) significantly increases longevity in female mice (p<0.05) compared to male mice (p=0.073) (Ali et al. 2006). Similar differences in longevity and associated increases in oxidative stress have been demonstrated in Drosophila (Ballard et al. 2007). Therefore, decreased ROS/RNS generation and enhanced detoxification by antioxidant systems in females vs. males may play an intimate part in sex differences observed across the lifespan.
The production and antioxidant detoxification of ROS/RNS plays a key role in normal cellular physiology and pathophysiology following CNS injury. Emerging evidence suggests ROS/RNS generation and antioxidant defense systems are sexually dimorphic. In humans, ROS damage measured by lipid peroxidation product F2-isoprostane is higher in male cord blood from preterm human twins (Minghetti et al. 2013) and following TBI in CSF of male adults vs. females (Bayir et al. 2004; Wagner et al. 2005). Another study finds higher F2-isoprostane in CSF of children and infants following severe TBI independent of sex (Varma et al. 2003). Further supporting the notion of sex-dependent oxidative damage are animal studies showing mitochondrial antioxidant enzymes paraoxynase-2 (PON2) (Costa et al. 2013; Giordano et al. 2013), thioredoxin (Trx) (Saeed et al. 2009; Chen et al. 2010), peroxiredoxin 6 (Prdx6) (Di et al. 2012), GPx (Borras et al. 2003) and glutaredoxin (GRx) (Diwakar et al. 2007; Saeed et al. 2009) at higher levels/activity in the female vs. male brain. Increased antioxidant enzymes in the female brain may be a result of the generally recognized neuroprotective effects conferred by female hormones. Accordingly, a rat model of global cerebral ischemia-reperfusion demonstrates metestrous (when estrogens are low) female susceptibility to lipid peroxidation, GSH depletion and decreased antioxidant enzyme activity of SOD and catalase vs. males (Mohagheghi et al. 2013b). This evidence suggests females may be relatively resilient to ROS/RNS mediated injury vs. males due to higher antioxidant enzyme defense systems and that males may particularly benefit from antioxidant treatments following CNS injury.
As mentioned previously, specific brain regions appear to be particularly susceptible to injury. In particular, dopaminergic areas are very sensitive to ROS/RNS mediated damage. Antioxidant enzyme PON2 is highly enriched in astrocytes compared to neurons and highest in dopaminergic brain regions (e.g. striatum) (Giordano et al. 2013), possibly playing a role in the aforementioned female resilience of mesencephalic cultures to excitotoxicity (Lieb et al. 1995; Alano et al. 2010). In the in vitro and in vivo 6-hydroxydopamine model of Parkinson's disease (PD), male mice have more lactate dehydrogenase release, apoptotic and necrotic cell death vs. females. XY neurons also display decreases in mRNA and protein levels of mitochondrial respiratory chain subunits coinciding with decreases in ATP and higher ROS generation vs. XX neurons (Misiak and Beyer 2010). Similar ROS and mitochondrial changes are observed in sex-dependent vulnerability of cultured astrocytes in the 1-methyl-4-phenylpyridinium (MPTP) model of PD (Sundar et al. 2011). These molecular mechanisms of ROS/RNS production and antioxidant detoxification may underlie the higher incidence of early onset (age 50–59) PD in human males (Pringsheim et al. 2014) and contribute to their poorer quality of life due to increased burden of symptoms vs. females (Lubomski et al. 2014). Dopaminergic regions of the brain (i.e. midbrain & striatum) are also susceptible to injury following TBI (Yan et al. 2007; Hutson et al. 2011; Shin et al. 2012; van Bregt et al. 2012) but sex dependent vulnerability of these regions following TBI is unknown.
Antioxidant defense systems are a necessary vital component for proper mitochondrial function. For example, the antioxidant enzyme GRx is essential to maintenance of mitochondrial complex I activity where reductions in GRx result in loss of complex I activity. Following ROS/RNS induction by β-N-oxalyl amino-l-alanine lumbosacral spinal cord injection or MPTP exposure of motor neurons, complex I activity in males is severely inhibited while activity is unaffected in females (Kenchappa et al. 2002, 2004; Kenchappa and Ravindranath 2003; Diwakar et al. 2007). In overiectomized and estrogen receptor antagonized (ICI 182,780) females, complex I is also inhibited. In the context of CNS injury, the higher antioxidant capacity of females may provide neuroprotection but evaluation of putative sex differences in mitochondrial function following TBI have yet to be assessed. Therefore, novel approaches to neuroprotection utilizing antioxidant treatments are most likely to benefit males vs. females (reviewed in (Fernandez-Gajardo et al. 2014)) and should be evaluated in a sex-specific manner.
Oxidative stress promotes loss of mitochondrial ΔΨ and mitochondrial fission. This is thought to ‘select’ mitochondria for degradation by mitophagy. Indeed, oxidative stress is sufficient to induce autophagy/mitophagy ((Scherz-Shouval et al. 2007) as cited by (Au et al. 2010)) and is necessary for the normal progression of autophagic recycling ((Zhu et al. 2007) as cited by (Au et al. 2010)). The following section reviews autophagy in the context of brain injury and sex differences.
Autophagy/Mitophagy
Autophagy/mitophagy generally refers to the cellular process of targeted lysosomal degradation of macromolecules for metabolic recycling into amino acid and fatty acid constituents (Smith et al. 2011). This process is necessary for normal cellular protein and lipid turnover and augmented following excitotoxic, ischemic and traumatic CNS injury (Diskin et al. 2005; Guo et al. 2014; Lin et al. 2014; Ginet et al. 2014; Zheng et al. 2014; Zhou et al. 2014). Recent studies reveal that during starvation induced autophagy, mitochondria supply membranes for autophagasome formation via association with autophagy mediating protein Atg5 and subsequent association with autophagasome protein microtubule-associated-protein-1 light chain 3 (LC3) (Hailey et al. 2010) thus suggesting a critical role for mitochondria in the induction of autophagy. While mitochondrial degradation via mitophagy is a focus of this section, the term autophagy by definition includes mitophagy and will be used interchangeably hereafter as it is the most commonly used terminology.
There is a significant signaling role for the mitochondrial specific diphosphatylglycerol lipid cardiolipin in coordination of mitophagy progression (Kirkland et al. 2002; Chu et al. 2013, 2014). Cardiolipin peroxidation causes a conformational flip from the normal position on the inner mitochondrial membrane to the outer mitochondrial membrane in a phospholipid scrambalase-3 dependent manner. This promotes oxidized cardiolipin association with the lapidated form of autophagasome protein microtubule-associated-protein-1 light chain 3 (LC3-II) (Chu et al. 2013). LC3-II is considered a reliable biochemical marker for preautophagasomal membrane formation and is widely used to detect changes in autophagy.
Sex differences in autophagy are observed using in vitro and in vivo models of cardiac ischemia, (Chen et al. 2013) cerebral neonatal hypoxia-ischemia, (Weis et al. 2014) and iron-induced brain injury (Chen et al. 2012). In one of the most informative studies pertaining to sex differences in autophagy, Du et al. (2009) demonstrates fundamental differences following nutrient deprivation of neuronal cultures. They observe a decrease in XY cellular viability and associated increases in LC3-II protein levels compared to XX cells, an observation supported by time-lapsed microscopy confirmation of lysosomal fusion. Pharmacological or siRNA mediated inhibition of autophagy initiating protein Atg7 attenuates loss of cell viability and cell death of XY neurons to levels of XX neurons. Furthermore, XX neurons display phospholipase A2 mediated increases in lipid droplet formation following nutrient deprivation which is not apparent in XY neurons. Treatment of cultures with L-carnitine, a necessary co-factor for import of free fatty acids into the mitochondrial matrix for β-oxidation, improves XY neuronal viability and attenuates cell death after nutrient deprivation but has no effect in XX neurons (Du et al. 2009). Thus, autophagy may play a detrimental role in XY cells under stressful conditions and the relative resistance of XX cells to nutrient deprivation may be attributed to an enhanced capacity to synthesize and utilize free fatty acids as alternative biofuels. Related to these results, we observe a male susceptibility to brain mitochondrial respiratory impairment following cerebral neonatal hypoxic-ischemia and in vivo administration of acetyl-L-carnitine post-injury partially prevents respiratory impairment in male mitochondria while having no effect on female mitochondrial respiration (Demarest et al. 2013). These data support the aforementioned hypothesis that human females preferentially utilize lipids while males utilize proteins as primary biofuels at the subcellular level and imply that under stressful conditions, males may need to cannibalize cellular components via autophagy to obtain the requisite protein fuel.
In support of the notion that too much autophagy is detrimental following stressful conditions, Atg7 deficient mice subjected to the Rice-Vanucci model of cerebral neonatal hypoxia ischemia have less hippocampal pyramidal neuron death vs. wild-type (Koike et al. 2008). On the contrary, neuroprotection is not observed in older Atg7 deficient mice (Koike et al. 2008). Notably, sex was not specified in this study and may be a contributing factor to the discrepancies between ages. In another study using male animals subject to closed head TBI, rapamycin, an activator of mTOR mediated autophagy induction, is neuroprotective both histologically and behaviorally vs. vehicle treated controls (Erlich et al. 2007). Further clouding our sex-specific understanding of the involvement of autophagy following injury, female mice subjected to cerebral neonatal hypoxia ischemia have increased cortical LC3B-II levels compared to males (Weis et al. 2014). The authors interpret this increase as a marker of failed autophagy progression. Autophagy is undoubtedly involved in response to various CNS injuries and evidence suggests it may be sex-dependent but the exact beneficial/detrimental role is incompletely understood. Similarly unresolved in functional significance in response to a CNS injury is the observed synthesis of new mitochondria or mitochondrial biogenesis.
Mitochondrial Quality Control
Mitochondrial quality control (MQC) is essential for maintaining adequate cellular energy supply. Main processes contributing to MQC maintenance include mitochondrial biogenesis, dynamics (fission and fusion balance) and the aforementioned recycling of damaged/aged organelles via autophagy (reviewed in (Michel et al. 2012)). Targeting MQC processes in CNS injury where mitochondrial dysfunction plays a role in pathogenesis has been advocated (for review see (Anne et al. 2013)).
In animal models of neonatal (Yin et al. 2008) and adult ischemic stroke, mitochondrial biogenesis is increased in the ischemic hemisphere (Xie et al. 2014). It is hypothesized that this is an endogenous compensatory response attempting to restore ATP levels by increasing mitochondrial number. In vitro OGD of sex specific cerebral granule neurons (CGNs) shows sex-dependent responses with XY cells maintaining cellular ATP, mitochondrial ΔΨ and less cell death vs. XX CGNs. These results are attributed to mitochondrial biogenesis shown by increases in mtDNA, mitochondrial biogenesis proteins peroxisome proliferator-activated receptor-γ coactivator 1alpha (PGC1α), TFAM, nuclear respiratory factor 1 (NRF-1), mitochondrial heath shock protein 60 (HSP60) and cytochrome c oxidase (COXIV) in XY while XX CGNs lack or have suppressed responses (Sharma et al. 2014).
The functional consequences of enhanced mitochondrial biogenesis following brain injury are unknown. Nonetheless, promoters of mitochondrial biogenesis as therapeutics have been investigated. Gemfibrozil (an activator of mitochondrial biogenesis) treatment of rats following global cerebral ischemia induces TFAM and NRF-1 in pre-treated metestrous females but suppresses levels in males (Mohagheghi et al. 2013a). Correspondingly, treatment with gemfibrozil inhibits caspase-dependent apoptosis in females by upregulating antioxidant defenses but promotes caspase-dependent and caspase-independent cell death in male hippocampus (Mohagheghi et al. 2013b) (cell death signaling discussed in the next section). Selenium pretreatment of murine hippocampal neurons or mice is neuroprotective in models of ischemic stroke by reducing oxidative damage and promoting mitochondrial biogenesis via PGC-1α and NRF-1 induction but was not tested in female cells/animals (Mehta et al. 2012). On the opposing side of mitochondrial biogenesis, enhanced rates of mitochondrial fragmentation or fission have been reported to contribute to cell damage following CNS injury (Cao et al. 2013; Slupe et al. 2013; Kashani et al. 2014).
Mitochondrial fission and fusion processes are generally thought to segregate functional and dysfunctional mitochondria in order to maintain a healthy population of efficient energy producing organelles within a given cell (for review see (van der Bliek et al. 2013)). The overall concept is that dysfunctional mitochondria tend toward fission while healthy functional mitochondria undergo fusion and maintain an elongated tubular morphology thereby protecting mitochondria from autophagic degradation (Rambold et al. 2011). During injury, upregulation of fission proteins (e.g. FIS1, DRP-1) is associated with damage while fusion proteins (Mitofusins 1 and 2, OPA1) are considered beneficial (Jahani-Asl et al. 2011). DRP-1 is upregulated in disease processes and inhibition by siRNA preserves mitochondrial function and attenuates cell death in a hippocampal cell model of excitotoxicity (Zhang et al. 2014). Moreover, DRP-1 levels are decreased or cleared by induction of autophagy in primary rat striatal neurons (Purnell and Fox 2013). Very few studies have investigated the role of mitochondrial dynamics in a sex-specific manner. One study of cultured astrocytes indicates sex differences in mitochondrial dynamics and cell survival in response to estradiol or progesterone treatment. Specifically, treatment with progesterone or estradiol increases cell number, fusion and fission protein expression in female astrocytes and decreases cell number by inducing apoptotic cell death in male astrocytes (Andrabi et al. 2008). Expression of apoptotic proteins BCL-2 and BAX are upregulated after progesterone treatment in female astrocytes but decreased in male astrocytes indicating opposing effects of progesterone on critical apoptosis mediating proteins (Arnold et al. 2008). The detrimental effect of progesterone on male derived astrocytes may be a contributing factor to the recent discontinuation of the progesterone phase III clinical trial for treatment of TBI based on lack of evidence for neuroprotection in 875 enrolled participants (www.ninds.nih.gov/research/tbi). Indeed, at least one preclinical TBI study reported a reduction of anti-apoptotic proteins (BCL-2, AKT) and the astrocyte marker GFAP in male rats administered progesterone (Djebaili et al. 2005). Given the putative clinical implications of furthering our understanding of sex-specific regulation and functional consequences of mitochondrial biogenesis and dynamics, enhancing future research efforts in this area is warranted.
Cell Death Pathways
We now know cell death is not simply apoptotic or necrotic as once proposed, but rather a continuum encompassing apoptosis, necrosis, programmed necrosis, necroptosis and parthanatos. Each of these modes of cell death has unique defining features but one thing in common, mitochondrial involvement (reviewed in (Fatokun et al. 2014)). Prevention of cell death and resulting cognitive behavioral deficits is the essence of neuroprotection. The general dogma is that males are more susceptible to CNS injury primarily due to the lack of the inherently neuroprotective effects of female hormones which can converge on mitochondria (reviewed in (Nilsen and Brinton 2004; Simpkins et al. 2005; Simpkins and Dykens 2008; Arnold and Beyer 2009)). While hormones clearly play a role in sex differences, other intrinsic hormone-independent cell signaling mechanisms are likely involved since sex differences can be modeled in cell culture absent of circulating hormones (Du et al. 2004; Li et al. 2005) and are present during embryonic development prior to hormone involvement (Dewing et al. 2003).
Sex differences in cell death pathways is one of the most heavily studied areas of sexually dimorphic neurotrauma and neuroprotection. Since TBI research rarely includes females, the majority of our knowledge comes from animal stroke models. Primary cultured neurons also show sex differences in cell death pathways (Du et al. 2004; Li et al. 2005). It has been proposed that male cell death is a product of caspase-independent cell death while female cell death is caspase dependent with bioenergetic failure (decreased ATP) and neuronal NOS induction being the common preceding features observed in both sexes (reviewed in (Turtzo and McCullough 2010; Hill and Fitch 2012; Zuo et al. 2013)). Briefly, caspase independent cell death is mediated by PARP-1 activation and mitochondrial to nuclear translocation apoptosis inducing factor (AIF) while caspase-dependent cell death (reviewed in (Hyman and Yuan 2012)) is mediated by cytochrome c mitochondrial to cytosol translocation, apoptosome formation, caspase cleavage and activation of complement. Both pathways result in DNA fragmentation and cell death. In the caspase-independent pathway mitochondrial to nuclear translocation of AIF occurs in combination with endonuclease G (EndoG) mediated DNA fragmentation as shown by AIF/Endo G nuclear colocalization following transient focal ischemia in mice (Lee et al. 2005). In caspase dependent cell death DNA fragmentation is initiated by caspase-3 cleavage and release of caspase activated DNase (CAD) from inhibitor of caspase activated DNase (ICAD) (Enari et al. 1998). The different endonucleases contributing to DNA fragmentation downstream of cell death signaling pathways offer unique neuroprotective targets for sex specific treatments. However, sex dependent specificity of endonucleases needs to be confirmed.
It is important to appreciate that these pathways are not exclusive to either sex but overlap considerably (Fig. 2). For example, cytochrome c release and active caspase 3 are observed in males as well as females following injury (Clark et al. 2000; Djebaili et al. 2005). The sex ‘specific’ distinction comes primarily from neuroprotection studies demonstrating that caspase inhibitors are solely neuroprotective in females, while having little to no effect in males. Females also have an endogenous caspase inhibitor, X-linked inhibitor of apoptosis (XIAP) expressed at higher levels than males independent of estradiol or overiectomy in adult mice. However, protein levels in cytosolic and mitochondrial fractions are lower in females vs. males. This may be due to miR-23a regulation of XIAP mRNA (Siegel et al. 2011). Similarly, mRNA expression of Smac/DIABLO, an endogenous inhibitor of XIAP, is higher in females vs. males but protein levels are lower. Following stroke Smac/DIABLO mRNA and protein is decreased more in females than males. This suggests females have a genetic advantage in inhibition of caspase dependent cell death signaling. Accordingly inhibition of XIAP with embelin in adult male and female mice after stroke exacerbates lesion volume in females but has no effect in males (Siegel et al. 2011). Furthermore, treatment of female rat pups with embelin following rat cerebral neonatal hypoxic ischemia injury exacerbates anatomical damage and behavioral deficits vs. vehicle treated controls (Hill et al. 2011). These results imply that under conditions eliciting the same degree of caspase activation in both sexes, females may be more resilient to caspase dependent cell death vs. males.
Fig. 2.
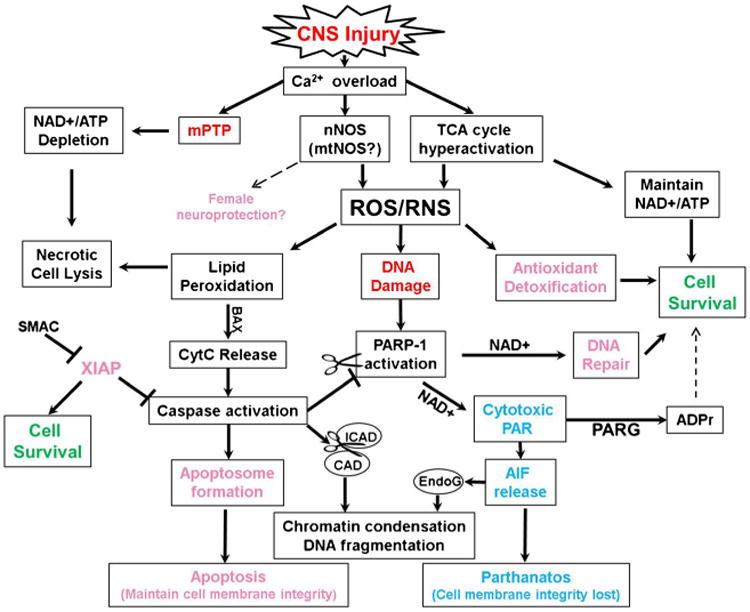
Sex Dependent Cell Death Proclivity and Interactions. Male predominant pathways are in blue, females in pink, and interacting pathways in black. Abbreviations: mitochondrial permeability transition pore (mPTP); neuronal/mitochondrial nitric oxide synthase (nNOS/mtNOS); tricarboxylic acid cycle (TCA cycle); nicotinamide adenine dinucleotide (NAD+); Reactive oxygen species (ROS); reactive nitrogen species (RNS); adenosine triphosphate (ATP);cytochrome c (CytC); poly(ADP-ribose) polymerase 1 (PARP-1); poly(ADP-ribose) (PAR); poly(ADP-ribose) glycohydrolase (PARG); ADP-ribose (ADPr); apoptosis inducing factor (AIF); X-linking inhibition of apoptosis (XIAP); Second mitochondria-derived activator of caspases (SMAC). Inhibitor of caspase activated DNase (ICAD); Caspase activated DNase (CAD); Endonuclease G (EndoG)
Female resilience to injury may also be afforded by PARP-1. Genetic studies have demonstrated that, following experimental stroke, male PARP-1 homozygous knockout mice have a substantially diminished infarct size while female PARP-1 knockout mice have increased lesion volume (McCullough et al. 2005; Yuan et al. 2009). Instead of sex-specificity, it is helpful to think of a cell death continuum where each sex has a predominant or ‘preferred’ cell death pathway but there is still overlap between male and female cell death signaling (Fig. 2). Intriguingly, following TBI in human infants and children, poly(ADP-ribose) (PAR), the product of PARP-1, is detected at higher levels in the CSF of males vs. females and positively correlated with age (Fink et al. 2008). Excitotoxicity, oxidative/nitrositive stress, autophagy and cell death all converge on PARP-1 (Huang and Shen 2009; Fatokun et al. 2014). PARP-1 is activated by ROS/RNS mediated DNA damage and consumes nicotinamide adenine dinucleotide (NAD+) to form cytotoxic PAR. Cell death studies evaluating the involvement of PAR have coined the term parthanatos, PAR for poly(ADP-ribose) and the Greek word for death, thanatos (for review see (Andrabi et al. 2008; Fatokun et al. 2014)). Accumulation of PAR polymers serves as a cell death signal by stimulating mitochondria to release AIF, (Wang et al. 2009, 2011) but not cytochrome c, possibly via the mPTP (Baek et al. 2013). PAR can be catabolized by PARG into ADPr, suppressing cytotoxic levels of PAR, however, ADPr can activate the aforementioned TRPM2 calcium channel in males. TBI studies report mixed results on behavioral improvement with PARP-1 inhibition. For instance, Clark et al. (2007) reports improvement on the spatial learning task, the Morris water maze (MWM) but no histopathological improvement following CCI in male mice with PARP-1 inhibitor INO-1001 while Stoica et al. (2014) report inhibition of microglial activation and limited neuronal preservation without MWM improvement after PARP-1 inhibitor, PJ34, treatment. PARP-1 inhibition presumably inhibits NAD+depletion following injury.
NAD+depletion is necessary and sufficient for neuronal cell death (parthanatos) (Alano et al. 2010) and contributes to astrocyte cell death (Alano and Ying 2004). NAD+metabolism is altered during the pathophysiology of acute brain injury (reviewed in (Owens et al. 2013)). Male mice have twice as much baseline NAD+vs. females. Following middle cerebral artery occlusion, NAD+depletion occurs in males and overiectomized females but not in intact females (Siegel and McCullough 2013). In PARP-1 knockout mice, stroke induced NAD+depletion is prevented in males but aggravated in females. Treatment with nicotinamide, an NAD+precursor, reduces infarct volume in both sexes of PARP-1 knockout mice but has no effect in wild type females. So why do males have twice as much NAD+compared to females? Since males have higher baseline ROS/RNS production and lower antioxidant defense systems compared to females, excess NAD+may be necessary for PARP-1 to routinely repair oxidatively damaged DNA.
Recent studies suggest a mitochondrial localization of PARP-1 and that NAD+consumption specifically by intramitochondrial PARP-1 contributes to oxidative stress induced cell death (Du et al. 2003). PAR polymerized proteins are detected in mitochondrial subcellular fractions from liver (Masmoudi and Mandel 1987) and brain (Masmoudi et al. 1988; Du et al. 2003). PAR coimmunoprecipitates with AIF in HeLa cells and increases following NMDA exposure (Wang et al. 2011). Whether the intramitochondrial PAR observed is a product of nuclear or mitochondrial localized PARP-1 and whether cytosolic or mitochondrial NAD+depletion is the cause of cell death is controversial. Pankotai et al. (2009) observed that a component of α-KGDH complex (DLDH) also consumes NAD+and addition of DLDH to isolated mitochondria results in PAR formation demonstrating PARP-like activity of DLDH. Thus, intramitochondrial PARP-like proteins may contribute to the observed PAR formation within mitochondria. On the other hand, PARP-1 itself coimmunoprecipitates with inner mitochondrial transmembrane protein, mitofilin, suggesting intramitochondrial localization. Moreover, PARP-1 coimmunoprecipitates with mitochondrial DNA and α-DNA ligase III in HeLa cells (Rossi et al. 2009). These results suggest that if PARP-1 is indeed present in the mitochondrial matrix, it may be involved in mitochondrial DNA repair. As such, it is tempting to speculate that increased male production of ROS/RNS and consequential DNA damage induces PARP-1 activity more in males vs. females, thus necessitating higher basal NAD +levels for homeostatic maintenance.
Summary and future directions
There exists substantial evidence for fundamental sex differences in brain metabolism. A model adapted from the collective evidence presented in this manuscript portraying sex differences in pathophysiological cell signaling is depicted in Fig. 3 and highlights of each section are summarized in Table 1. Thus far these differences have been largely ignored and necessitate full consideration when designing future experiments. Appropriate inclusion of both sexes in future studies will help fill significant knowledge gaps. Some unanswered questions for future experimental evaluation include the role of mitochondrial quality control, the mPTP, and AIF and caspase dependent endonucleases in sexually dimorphic cell death signaling. Evaluation of putative sex specific brain regional vulnerability following TBI may also provide insight into the cognitive and behavioral consequences of CNS injury observed in humans. Furthermore, segregating cell culture studies by genotype and including a female sex comparison in CNS injury studies will evoke generalizability of experimental findings and appropriate translational relevance. While it may be unrealistic that all hormonal states (e.g. estrus vs. proestrus) are accounted for in every experiment, there are experimental strategies that can be followed to distinguish between hormonal, environmental and chromosomal influences on sex differences (McCarthy et al. 2012). Future appreciation of sexually dimorphic mitochondrial metabolism, cell signaling and pathophysiology will ultimately aid in the generation of novel efficacious neuroprotective treatment strategies for both men and women suffering from CNS injury.
Fig. 3.
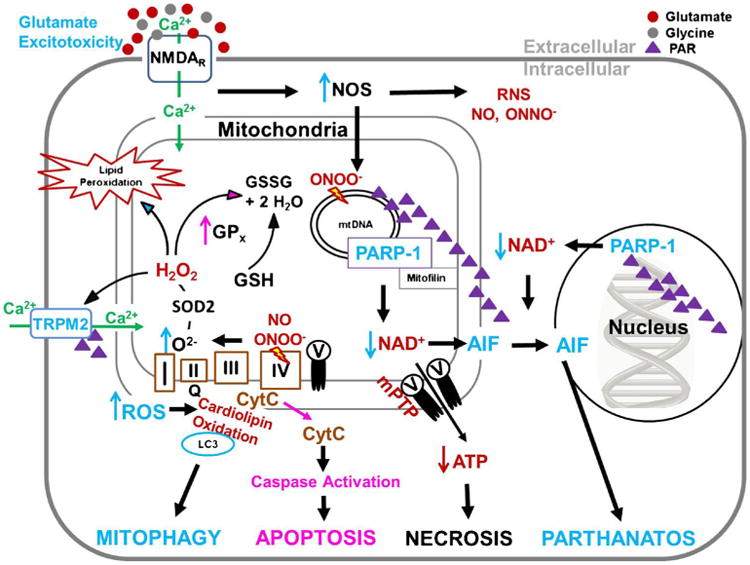
Summary of Sexually Dimorphic Cell signaling following CNS Injury. Male predominant pathways are represented in blue and female predominant pathways in pink. Following CNS injury, extracellular glutamate activates extrasynaptic NMDARs triggering Ca2+ influx. Intracellular Ca2+ activates NOS, to a higher degree in male cells. Mitochondrial Ca2+ uptake increases oxidative phosphorylation, ROS generation, subsequent DNA damage and mitochondrial swelling. ROS mediated damage is greater in male cells likely due to poorer antioxidant defense systems vs. females, (i.e. glutathioned peroxidase; GPx), which activates PARP-1 consumption of NAD+to generate PAR and ADPr. In surviving cells, ROS oxidation of cardiolipin, likely more common in males, causes a conformational flip to the outer membrane where oxidized cardiolipin binds LC3 to promote mitophagy, removing damaged organelles. In male cells, the combination of peroxide and ADPr activates TRPM2 cation channels further exacerbating Ca2+ influx and oxidative damage. In severe injury, Ca2+ overload can lead to activation of the mitochondrial permeability transition pore (mPTP), acute loss of ATP and necrosis or mitochondrial release of cytochrome c and AIF. Cytochrome c release results in caspase-dependent apoptosis in female cells while the combination of NAD depletion and AIF release leads to caspase-independent parthanatos in male cells
Table 1. Highlights: Sex differences in mitochondrial (dys)function.
Excitotoxicity and Calcium
|
Acknowledgments
Supported by NIHP01HD016596-27.
Contributor Information
Tyler G. Demarest, Email: tdemarest@anes.umm.edu, Program in Neuroscience, University of Maryland School of Medicine, Baltimore, MD, USA; Department of Anesthesiology, University of Maryland School of Medicine, Baltimore, MD, USA.
Margaret M. McCarthy, Program in Neuroscience, University of Maryland School of Medicine, Baltimore, MD, USA; Department of Pharmacology, University of Maryland School of Medicine, Baltimore, MD, USA
References
- Alano CC, Ying W, Swanson RA. Poly(ADP-ribose) polymerase-1-mediated cell death in astrocytes requires NAD+depletion and mitochondrial permeability transition. J Biol Chem. 2004;279:18895–18902. doi: 10.1074/jbc.M313329200. [DOI] [PubMed] [Google Scholar]
- Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA. NAD+depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J Neurosci. 2010;30:2967–2978. doi: 10.1523/JNEUROSCI.5552-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali SS, Xiong C, Lucero J, Behrens MM, Dugan LL, Quick KL. Gender differences in free radical homeostasis during aging: shorter-lived female C57BL6 mice have increased oxidative stress. Aging Cell. 2006;5:565–574. doi: 10.1111/j.1474-9726.2006.00252.x. [DOI] [PubMed] [Google Scholar]
- Andrabi SA, Dawson TM, Dawson VL. Mitochondrial and nuclear cross talk in cell death: parthanatos. Ann N Y Acad Sci. 2008;1147:233–241. doi: 10.1196/annals.1427.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anne SR, Leak RK, Gao Y, Chen J. The dynamics of the mitochondrial organelle as a potential therapeutic target. J Cereb Blood Flow Metab. 2013;33:22–32. doi: 10.1038/jcbfm.2012.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arieli Y, Gursahani H, Eaton MM, Hernandez LA, Schaefer S. Gender modulation of Ca(2+) uptake in cardiac mitochondria. J Mol Cell Cardiol. 2004;37:507–513. doi: 10.1016/j.yjmcc.2004.04.023. [DOI] [PubMed] [Google Scholar]
- Arnold S, Beyer C. Neuroprotection by estrogen in the brain: the mitochondrial compartment as presumed therapeutic target. J Neurochem. 2009;110:1–11. doi: 10.1111/j.1471-4159.2009.06133.x. [DOI] [PubMed] [Google Scholar]
- Arnold S, de Araujo GW, Beyer C. Gender-specific regulation of mitochondrial fusion and fission gene transcription and viability of cortical astrocytes by steroid hormones. J Mol Endocrinol. 2008;41:289–300. doi: 10.1677/JME-08-0085. [DOI] [PubMed] [Google Scholar]
- Au AK, Bayir H, Kochanek PM, Clark RS. Evaluation of autophagy using mouse models of brain injury. Biochim Biophys Acta. 2010;1802:918–923. doi: 10.1016/j.bbadis.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek SH, Bae ON, Kim EK, Yu SW. Induction of mitochondrial dysfunction by poly(ADP-ribose) polymer: implication for neuronal cell death. Mol Cell. 2013;36:258–266. doi: 10.1007/s10059-013-0172-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard JW, Melvin RG, Miller JT, Katewa SD. Sex differences in survival and mitochondrial bioenergetics during aging in Drosophila. Aging Cell. 2007;6:699–708. doi: 10.1111/j.1474-9726.2007.00331.x. [DOI] [PubMed] [Google Scholar]
- Bates TE, Loesch A, Burnstock G, Clark JB. Immunocytochemical evidence for a mitochondrially located nitric oxide synthase in brain and liver. Biochem Biophys Res Commun. 1995;213:896–900. doi: 10.1006/bbrc.1995.2213. [DOI] [PubMed] [Google Scholar]
- Bayir H, Kagan VE, Tyurina YY, Tyurin V, Ruppel RA, Adelson PD, Graham SH, Janesko K, Clark RS, Kochanek PM. Assessment of antioxidant reserves and oxidative stress in cerebrospinal fluid after severe traumatic brain injury in infants and children. Pediatr Res. 2002;51:571–578. doi: 10.1203/00006450-200205000-00005. [DOI] [PubMed] [Google Scholar]
- Bayir H, Marion DW, Puccio AM, Wisniewski SR, Janesko KL, Clark RS, Kochanek PM. Marked gender effect on lipid peroxidation after severe traumatic brain injury in adult patients. J Neurotrauma. 2004;21:1–8. doi: 10.1089/089771504772695896. [DOI] [PubMed] [Google Scholar]
- Bayir H, Kagan VE, Clark RS, Janesko-Feldman K, Rafikov R, Huang Z, Zhang X, Vagni V, Billiar TR, Kochanek PM. Neuronal NOS-mediated nitration and inactivation of manganese superoxide dismutase in brain after experimental and human brain injury. J Neurochem. 2007;101:168–181. doi: 10.1111/j.1471-4159.2006.04353.x. [DOI] [PubMed] [Google Scholar]
- Bender C, de OS, Bueno A, de OJ, Lorenzo A. Comparative analyses of the neurodegeneration induced by the non-competitive NMDA-receptor-antagonist drug MK801 in mice and rats. Neurotoxicol Teratol. 2010;32:542–550. doi: 10.1016/j.ntt.2010.05.002. [DOI] [PubMed] [Google Scholar]
- Borras C, Sastre J, Garcia-Sala D, Lloret A, Pallardo FV, Vina J. Mitochondria from females exhibit higher antioxidant gene expression and lower oxidative damage than males. Free Radic Biol Med. 2003;34:546–552. doi: 10.1016/s0891-5849(02)01356-4. [DOI] [PubMed] [Google Scholar]
- Brown GC. Nitric oxide regulates mitochondrial respiration and cell functions by inhibiting cytochrome oxidase. FEBS Lett. 1995;369:136–139. doi: 10.1016/0014-5793(95)00763-y. [DOI] [PubMed] [Google Scholar]
- Brown GC. Regulation of mitochondrial respiration by nitric oxide inhibition of cytochrome c oxidase. Biochim Biophys Acta. 2001;1504:46–57. doi: 10.1016/s0005-2728(00)00238-3. [DOI] [PubMed] [Google Scholar]
- Brown GC. Nitric oxide and mitochondria. Front Biosci. 2007;12:1024–1033. doi: 10.2741/2122. [DOI] [PubMed] [Google Scholar]
- Brown GC, Cooper CE. Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett. 1994;356:295–298. doi: 10.1016/0014-5793(94)01290-3. [DOI] [PubMed] [Google Scholar]
- Cao Y, Lv G, Wang YS, Fan ZK, Bi YL, Zhao L, Guo ZP. Mitochondrial fusion and fission after spinal sacord injury in rats. Brain Res. 2013;1522:59–66. doi: 10.1016/j.brainres.2013.05.033. [DOI] [PubMed] [Google Scholar]
- Chen Q, Harris C, Brown CS, Howe A, Surmeier DJ, Reiner A. Glutamate-mediated excitotoxic death of cultured striatal neurons is mediated by non-NMDA receptors. Exp Neurol. 1995;136:212–224. doi: 10.1006/exnr.1995.1098. [DOI] [PubMed] [Google Scholar]
- Chen TY, Tsai KL, Lee TY, Chiueh CC, Lee WS, Hsu C. Sex-specific role of thioredoxin in neuroprotection against iron-induced brain injury conferred by estradiol. Stroke. 2010;41:160–165. doi: 10.1161/STROKEAHA.109.562850. [DOI] [PubMed] [Google Scholar]
- Chen CW, Chen TY, Tsai KL, Lin CL, Yokoyama KK, Lee WS, Chiueh CC, Hsu C. Inhibition of autophagy as a therapeutic strategy of iron-induced brain injury after hemorrhage. Autophagy. 2012;8:1510–1520. doi: 10.4161/auto.21289. [DOI] [PubMed] [Google Scholar]
- Chen C, Hu LX, Dong T, Wang GQ, Wang LH, Zhou XP, Jiang Y, Murao K, Lu SQ, Chen JW, Zhang GX. Apoptosis and autophagy contribute to gender difference in cardiac ischemia-reperfusion induced injury in rats. Life Sci. 2013;93:265–270. doi: 10.1016/j.lfs.2013.06.019. [DOI] [PubMed] [Google Scholar]
- Choi DW. Glutamate neurotoxicity in cortical cell culture is calcium dependent. Neurosci Lett. 1985;58:293–297. doi: 10.1016/0304-3940(85)90069-2. [DOI] [PubMed] [Google Scholar]
- Choi DW. Calcium and excitotoxic neuronal injury. Ann N Y Acad Sci. 1994;747:162–171. doi: 10.1111/j.1749-6632.1994.tb44407.x. [DOI] [PubMed] [Google Scholar]
- Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, Tyurin VA, Yanamala N, Shrivastava IH, Mohammadyani D, Qiang Wang KZ, Zhu J, Klein-Seetharaman J, Balasubramanian K, Amoscato AA, Borisenko G, Huang Z, Gusdon AM, Cheikhi A, Steer EK, Wang R, Baty C, Watkins S, Bahar I, Bayir H, Kagan VE. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013;15:1197–1205. doi: 10.1038/ncb2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CT, Bayir H, Kagan VE. LC3 binds externalized cardiolipin on injured mitochondria to signal mitophagy in neurons: implications for Parkinson disease. Autophagy. 2014;10:376–378. doi: 10.4161/auto.27191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RS, Kochanek PM, Watkins SC, Chen M, Dixon CE, Seidberg NA, Melick J, Loeffert JE, Nathaniel PD, Jin KL, Graham SH. Caspase-3 mediated neuronal death after traumatic brain injury in rats. J Neurochem. 2000;74:740–753. doi: 10.1046/j.1471-4159.2000.740740.x. [DOI] [PubMed] [Google Scholar]
- Clark RS, Vagni VA, Nathaniel PD, Jenkins LW, Dixon CE, Szabo C. Local administration of the poly(ADP-ribose) polymerase inhibitor INO-1001 prevents NAD+depletion and improves water maze performance after traumatic brain injury in mice. J Neurotrauma. 2007;24:1399–1405. doi: 10.1089/neu.2007.0305. [DOI] [PubMed] [Google Scholar]
- Clayton JA, Collins FS. Policy: NIH to balance sex in cell and animal studies. Nature. 2014;509:282–283. doi: 10.1038/509282a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa LG, de Laat R, Dao K, Pellacani C, Cole TB, Furlong CE. Paraoxonase-2 (PON2) in brain and its potential role in neuroprotection. Neurotoxicology. 2013;43:3–9. doi: 10.1016/j.neuro.2013.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demarest TG, Waddell J, Schuh RA, McKenna MC, Fiskum G. Sexually dimorphic impairment of brain mitochondrial respiration following neonatal hypoxic-ischemia [abstract] J Neurotrauma. 2013;30:A4. [Google Scholar]
- Dewing P, Shi T, Horvath S, Vilain E. Sexually dimorphic gene expression in mouse brain precedes gonadal differentiation. Brain Res Mol Brain Res. 2003;118:82–90. doi: 10.1016/s0169-328x(03)00339-5. [DOI] [PubMed] [Google Scholar]
- Di DF, Casalena G, Jia J, Sultana R, Barone E, Cai J, Pierce WM, Cini C, Mancuso C, Perluigi M, Davis CM, Alkayed NJ, Butterfield DA. Sex differences in brain proteomes of neuron-specific STAT3-null mice after cerebral ischemia/reperfusion. J Neurochem. 2012;121:680–692. doi: 10.1111/j.1471-4159.2012.07721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diskin T, Tal-Or P, Erlich S, Mizrachy L, Alexandrovich A, Shohami E, Pinkas-Kramarski R. Closed head injury induces upregulation of Beclin 1 at the cortical site of injury. J Neurotrauma. 2005;22:750–762. doi: 10.1089/neu.2005.22.750. [DOI] [PubMed] [Google Scholar]
- Diwakar L, Kenchappa RS, Annepu J, Ravindranath V. Downregulation of glutaredoxin but not glutathione loss leads to mitochondrial dysfunction in female mice CNS: implications in excitotoxicity. Neurochem Int. 2007;51:37–46. doi: 10.1016/j.neuint.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Djebaili M, Guo Q, Pettus EH, Hoffman SW, Stein DG. The neurosteroids progesterone and allopregnanolone reduce cell death, gliosis, and functional deficits after traumatic brain injury in rats. J Neurotrauma. 2005;22:106–118. doi: 10.1089/neu.2005.22.106. [DOI] [PubMed] [Google Scholar]
- Djouadi F, Weinheimer CJ, Saffitz JE, Pitchford C, Bastin J, Gonzalez FJ, Kelly DP. A gender-related defect in lipid metabolism and glucose homeostasis in peroxisome proliferator- activated receptor alpha- deficient mice. J Clin Invest. 1998;102:1083–1091. doi: 10.1172/JCI3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dringen R, Kussmaul L, Gutterer JM, Hirrlinger J, Hamprecht B. The glutathione system of peroxide detoxification is less efficient in neurons than in astroglial cells. J Neurochem. 1999;72:2523–2530. doi: 10.1046/j.1471-4159.1999.0722523.x. [DOI] [PubMed] [Google Scholar]
- Du L, Zhang X, Han YY, Burke NA, Kochanek PM, Watkins SC, Graham SH, Carcillo JA, Szabo C, Clark RS. Intra-mitochondrial poly(ADP-ribosylation) contributes to NAD+depletion and cell death induced by oxidative stress. J Biol Chem. 2003;278:18426–18433. doi: 10.1074/jbc.M301295200. [DOI] [PubMed] [Google Scholar]
- Du L, Bayir H, Lai Y, Zhang X, Kochanek PM, Watkins SC, Graham SH, Clark RS. Innate gender-based proclivity in response to cytotoxicity and programmed cell death pathway. J Biol Chem. 2004;279:38563–38570. doi: 10.1074/jbc.M405461200. [DOI] [PubMed] [Google Scholar]
- Du L, Hickey RW, Bayir H, Watkins SC, Tyurin VA, Guo F, Kochanek PM, Jenkins LW, Ren J, Gibson G, Chu CT, Kagan VE, Clark RS. Starving neurons show sex difference in autophagy. J Biol Chem. 2009;284:2383–2396. doi: 10.1074/jbc.M804396200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR. Mitochondria and Ca(2+) in cell physiology and pathophysiology. Cell Calcium. 2000a;28:339–348. doi: 10.1054/ceca.2000.0170. [DOI] [PubMed] [Google Scholar]
- Duchen MR. Mitochondria and calcium: from cell signalling to cell death. J Physiol. 2000b;1:57–68. doi: 10.1111/j.1469-7793.2000.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391:43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- Erlich S, Alexandrovich A, Shohami E, Pinkas-Kramarski R. Rapamycin is a neuroprotective treatment for traumatic brain injury. Neurobiol Dis. 2007;26:86–93. doi: 10.1016/j.nbd.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Fatokun AA, Dawson VL, Dawson TM. Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br J Pharmacol. 2014;171:2000–2016. doi: 10.1111/bph.12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Gajardo R, Matamala JM, Carrasco R, Gutierrez R, Melo R, Rodrigo R. Novel therapeutic strategies for traumatic brain injury: acute antioxidant reinforcement. CNS Drugs. 2014;28:229–248. doi: 10.1007/s40263-013-0138-y. [DOI] [PubMed] [Google Scholar]
- Fink EL, Lai Y, Zhang X, Janesko-Feldman K, Adelson PD, Szabo C, Berger RP, Sarnaik AA, Kochanek PM, Clark RS. Quantification of poly(ADP-ribose)-modified proteins in cerebrospinal fluid from infants and children after traumatic brain injury. J Cereb Blood Flow Metab. 2008;28:1523–1529. doi: 10.1038/jcbfm.2008.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiskum G. Mitochondrial participation in ischemic and traumatic neural cell death. J Neurotrauma. 2000;17:843–855. doi: 10.1089/neu.2000.17.843. [DOI] [PubMed] [Google Scholar]
- Fonfria E, Marshall IC, Benham CD, Boyfield I, Brown JD, Hill K, Hughes JP, Skaper SD, McNulty S. TRPM2 channel opening in response to oxidative stress is dependent on activation of poly(ADP-ribose) polymerase. Br J Pharmacol. 2004;143:186–192. doi: 10.1038/sj.bjp.0705914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford LM, Sanberg PR, Norman AB, Fogelson MH. MK-801 prevents hippocampal neurodegeneration in neonatal hypoxic-ischemic rats. Arch Neurol. 1989;46:1090–1096. doi: 10.1001/archneur.1989.00520460072016. [DOI] [PubMed] [Google Scholar]
- Gaviria M, Privat A, d'Arbigny P, Kamenka J, Haton H, Ohanna F. Neuroprotective effects of a novel NMDA antagonist, Gacyclidine, after experimental contusive spinal cord injury in adult rats. Brain Res. 2000;874:200–209. doi: 10.1016/s0006-8993(00)02581-6. [DOI] [PubMed] [Google Scholar]
- Ginet V, Spiehlmann A, Rummel C, Rudinskiy N, Grishchuk Y, Luthi-Carter R, Clarke PG, Truttmann AC, Puyal J. Involvement of autophagy in hypoxic-excitotoxic neuronal death. Autophagy. 2014;10:846–860. doi: 10.4161/auto.28264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano G, Tait L, Furlong CE, Cole TB, Kavanagh TJ, Costa LG. Gender differences in brain susceptibility to oxidative stress are mediated by levels of paraoxonase-2 expression. Free Radic Biol Med. 2013;58:98–108. doi: 10.1016/j.freeradbiomed.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulivi C. Mitochondria as generators and targets of nitric oxide. Novartis Found Symp. 2007;287:92–100. [PubMed] [Google Scholar]
- Guo Z, Cao G, Yang H, Zhou H, Li L, Cao Z, Yu B, Kou J. A combination of four active compounds alleviates cerebral ischemia-reperfusion injury in correlation with inhibition of autophagy and modulation of AMPK/mTOR and JNK pathways. J Neurosci Res. 2014;92(10):1295–1306. doi: 10.1002/jnr.23400. [DOI] [PubMed] [Google Scholar]
- Hagberg H, Mallard C, Rousset CI, Thornton C. Mitochondria: hub of injury responses in the developing brain. Lancet Neurol. 2014;13:217–232. doi: 10.1016/S1474-4422(13)70261-8. [DOI] [PubMed] [Google Scholar]
- Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–667. doi: 10.1016/j.cell.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han RZ, Hu JJ, Weng YC, Li DF, Huang Y. NMDA receptor antagonist MK-801 reduces neuronal damage and preserves learning and memory in a rat model of traumatic brain injury. Neurosci Bull. 2009;25:367–375. doi: 10.1007/s12264-009-0608-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc. 1972;20:145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- Haynes V, Elfering S, Traaseth N, Giulivi C. Mitochondrial nitric-oxide synthase: enzyme expression, characterization, and regulation. J Bioenerg Biomembr. 2004;36:341–346. doi: 10.1023/B:JOBB.0000041765.27145.08. [DOI] [PubMed] [Google Scholar]
- Hill CA, Fitch RH. Sex differences in mechanisms and outcome of neonatal hypoxia-ischemia in rodent models: implications for sex-specific neuroprotection in clinical neonatal practice. Neurol Res Int. 2012;2012:1–9. doi: 10.1155/2012/867531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill CA, Alexander ML, McCullough LD, Fitch RH. Inhibition of X-linked inhibitor of apoptosis with embelin differentially affects male versus female behavioral outcome following neonatal hypoxia-ischemia in rats. Dev Neurosci. 2011;33:494–504. doi: 10.1159/000331651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton GD, Nunez JL, Bambrick L, Thompson SM, McCarthy MM. Glutamate-mediated excitotoxicity in neonatal hippocampal neurons is mediated by mGluR-induced release of Ca++ from intracellular stores and is prevented by estradiol. Eur J Neurosci. 2006;24:3008–3016. doi: 10.1111/j.1460-9568.2006.05189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q, Shen HM. To die or to live: the dual role of poly(ADP-ribose) polymerase-1 in autophagy and necrosis under oxidative stress and DNA damage. Autophagy. 2009;5:273–276. doi: 10.4161/auto.5.2.7640. [DOI] [PubMed] [Google Scholar]
- Hutson CB, Lazo CR, Mortazavi F, Giza CC, Hovda D, Chesselet MF. Traumatic brain injury in adult rats causes progressive nigrostriatal dopaminergic cell loss and enhanced vulnerability to the pesticide paraquat. J Neurotrauma. 2011;28:1783–1801. doi: 10.1089/neu.2010.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman BT, Yuan J. Apoptotic and non-apoptotic roles of caspases in neuronal physiology and pathophysiology. Nat Rev Neurosci. 2012;13:395–406. doi: 10.1038/nrn3228. [DOI] [PubMed] [Google Scholar]
- Jahani-Asl A, Pilon-Larose K, Xu W, MacLaurin JG, Park DS, McBride HM, Slack RS. The mitochondrial inner membrane GTPase, optic atrophy 1 (Opa1), restores mitochondrial morphology and promotes neuronal survival following excitotoxicity. J Biol Chem. 2011;286:4772–4782. doi: 10.1074/jbc.M110.167155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J, Verma S, Nakayama S, Quillinan N, Grafe MR, Hurn PD, Herson PS. Sex differences in neuroprotection provided by inhibition of TRPM2 channels following experimental stroke. J Cereb Blood Flow Metab. 2011;31:2160–2168. doi: 10.1038/jcbfm.2011.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jikumaru M, Hiramoto K, Honma T, Sato EF, Sekiyama A, Inoue M. Effect of starvation on the survival of male and female mice. Physiol Chem Phys Med NMR. 2007;39:247–257. [PubMed] [Google Scholar]
- Kashani IR, Rajabi Z, Akbari M, Hassanzadeh G, Mohseni A, Eramsadati MK, Rafiee K, Beyer C, Kipp M, Zendedel A. Protective effects of melatonin against mitochondrial injury in a mouse model of multiple sclerosis. Exp Brain Res. 2014;232(9):2835–2846. doi: 10.1007/s00221-014-3946-5. [DOI] [PubMed] [Google Scholar]
- Keller JN, Kindy MS, Holtsberg FW, St Clair DK, Yen HC, Germeyer A, Steiner SM, Bruce-Keller AJ, Hutchins JB, Mattson MP. Mitochondrial manganese superoxide dismutase prevents neural apoptosis and reduces ischemic brain injury: suppression of peroxynitrite production, lipid peroxidation, and mitochondrial dysfunction. J Neurosci. 1998;18:687–697. doi: 10.1523/JNEUROSCI.18-02-00687.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenchappa RS, Ravindranath V. Glutaredoxin is essential for maintenance of brain mitochondrial complex I: studies with MPTP. FASEB J. 2003;17:717–719. doi: 10.1096/fj.02-0771fje. [DOI] [PubMed] [Google Scholar]
- Kenchappa RS, Diwakar L, Boyd MR, Ravindranath V. Thioltransferase (glutaredoxin) mediates recovery of motor neurons from excitotoxic mitochondrial injury. J Neurosci. 2002;22:8402–8410. doi: 10.1523/JNEUROSCI.22-19-08402.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenchappa RS, Diwakar L, Annepu J, Ravindranath V. Estrogen and neuroprotection: higher constitutive expression of glutaredoxin in female mice offers protection against MPTP-mediated neurodegeneration. FASEB J. 2004;18:1102–1104. doi: 10.1096/fj.03-1075fje. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Magrane J, Starkov AA, Manfredi G. The mitochondrial calcium regulator cyclophilin D is an essential component of oestrogen-mediated neuroprotection in amyotrophic lateral sclerosis. Brain. 2012;135:2865–2874. doi: 10.1093/brain/aws208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland RA, Adibhatla RM, Hatcher JF, Franklin JL. Loss of cardiolipin and mitochondria during programmed neuronal death: evidence of a role for lipid peroxidation and autophagy. Neuroscience. 2002;115:587–602. doi: 10.1016/s0306-4522(02)00512-2. [DOI] [PubMed] [Google Scholar]
- Koike M, Shibata M, Tadakoshi M, Gotoh K, Komatsu M, Waguri S, Kawahara N, Kuida K, Nagata S, Kominami E, Tanaka K, Uchiyama Y. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008;172:454–469. doi: 10.2353/ajpath.2008.070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagranha CJ, Deschamps A, Aponte A, Steenbergen C, Murphy E. Sex differences in the phosphorylation of mitochondrial proteins result in reduced production of reactive oxygen species and cardioprotection in females. Circ Res. 2010;106:1681–1691. doi: 10.1161/CIRCRESAHA.109.213645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamont LS. Gender differences in amino acid use during endurance exercise. Nutr Rev. 2005;63:419–422. doi: 10.1301/nr.2005.dec.419-422. [DOI] [PubMed] [Google Scholar]
- Lapchak PA. The neuroactive steroid 3-alpha-ol-5-beta-pregnan-20-one hemisuccinate, a selective NMDA receptor antagonist improves behavioral performance following spinal cord ischemia. Brain Res. 2004;997:152–158. doi: 10.1016/j.brainres.2003.10.047. [DOI] [PubMed] [Google Scholar]
- Lee BI, Lee DJ, Cho KJ, Kim GW. Early nuclear translocation of endonuclease G and subsequent DNA fragmentation after transient focal cerebral ischemia in mice. Neurosci Lett. 2005;386:23–27. doi: 10.1016/j.neulet.2005.05.058. [DOI] [PubMed] [Google Scholar]
- Li H, Pin S, Zeng Z, Wang MM, Andreasson KA, McCullough LD. Sex differences in cell death. Ann Neurol. 2005;58:317–321. doi: 10.1002/ana.20538. [DOI] [PubMed] [Google Scholar]
- Lieb K, Andrae J, Reisert I, Pilgrim C. Neurotoxicity of dopamine and protective effects of the NMDA receptor antagonist AP-5 differ between male and female dopaminergic neurons. Exp Neurol. 1995;134:222–229. doi: 10.1006/exnr.1995.1052. [DOI] [PubMed] [Google Scholar]
- Lin CJ, Chen TH, Yang LY, Shih CM. Resveratrol protects astrocytes against traumatic brain injury through inhibiting apoptotic and autophagic cell death. Cell Death Differ. 2014;5:e1147. doi: 10.1038/cddis.2014.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubomski M, Louise RR, Lee W, Bertram KL, Williams DR. Sex differences in Parkinson's disease. J Clin Neurosci. 2014;21(9):1503–1506. doi: 10.1016/j.jocn.2013.12.016. [DOI] [PubMed] [Google Scholar]
- Martin LJ. An approach to experimental synaptic pathology using green fluorescent protein-transgenic mice and gene knockout mice to show mitochondrial permeability transition pore-driven excitotoxicity in interneurons and motoneurons. Toxicol Pathol. 2011;39:220–233. doi: 10.1177/0192623310389475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masmoudi A, Mandel P. ADP-ribosyl transferase and NAD glycohydrolase activities in rat liver mitochondria. Biochemistry. 1987;26:1965–1969. doi: 10.1021/bi00381a027. [DOI] [PubMed] [Google Scholar]
- Masmoudi A, Islam F, Mandel P. ADP-ribosylation of highly purified rat brain mitochondria. J Neurochem. 1988;51:188–193. doi: 10.1111/j.1471-4159.1988.tb04854.x. [DOI] [PubMed] [Google Scholar]
- McCarthy MM. Estradiol and the developing brain. Physiol Rev. 2008;88:91–124. doi: 10.1152/physrev.00010.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MM, Arnold AP. Reframing sexual differentiation of the brain. Nat Neurosci. 2011;14:677–683. doi: 10.1038/nn.2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MM, Arnold AP, Ball GF, Blaustein JD, De Vries GJ. Sex differences in the brain: the not so inconvenient truth. J Neurosci. 2012;32:2241–2247. doi: 10.1523/JNEUROSCI.5372-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough LD, Zeng Z, Blizzard KK, Debchoudhury I, Hurn PD. Ischemic nitric oxide and poly (ADP-ribose) polymerase-1 in cerebral ischemia: male toxicity, female protection. J Cereb Blood Flow Metab. 2005;25:502–512. doi: 10.1038/sj.jcbfm.9600059. [DOI] [PubMed] [Google Scholar]
- Mecocci P, MacGarvey U, Kaufman AE, Koontz D, Shoffner JM, Wallace DC, Beal MF. Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain. Ann Neurol. 1993;34:609–616. doi: 10.1002/ana.410340416. [DOI] [PubMed] [Google Scholar]
- Mehta SL, Kumari S, Mendelev N, Li PA. Selenium preserves mitochondrial function, stimulates mitochondrial biogenesis, and reduces infarct volume after focal cerebral ischemia. BMC Neurosci. 2012;13:79. doi: 10.1186/1471-2202-13-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel S, Wanet A, De PA, Rommelaere G, Arnould T, Renard P. Crosstalk between mitochondrial (dys)function and mitochondrial abundance. J Cell Physiol. 2012;227:2297–2310. doi: 10.1002/jcp.23021. [DOI] [PubMed] [Google Scholar]
- Minano A, Cerbon MA, Xifro X, Malagelada C, Aguilera J, Rodriguez-Alvarez J. 17beta-estradiol does not protect cerebellar granule cells from excitotoxicity or apoptosis. J Neurochem. 2007;102:354–364. doi: 10.1111/j.1471-4159.2007.04475.x. [DOI] [PubMed] [Google Scholar]
- Minghetti L, Greco A, Zanardo V, Suppiej A. Early-life sex-dependent vulnerability to oxidative stress: the natural twining model. J Matern Fetal Neonatal Med. 2013;26:259–262. doi: 10.3109/14767058.2012.733751. [DOI] [PubMed] [Google Scholar]
- Misiak M, Beyer C, Arnold S. Gender-specific role of mitochondria in the vulnerability of 6-hydroxydopamine-treated mesencephalic neurons. Biochim Biophys Acta. 2010;1797:1178–1188. doi: 10.1016/j.bbabio.2010.04.009. [DOI] [PubMed] [Google Scholar]
- Mohagheghi F, Ahmadiani A, Rahmani B, Moradi F, Romond N, Khalaj L. Gemfibrozil pretreatment resulted in a sexually dimorphic outcome in the rat models of global cerebral ischemia-reperfusion via modulation of mitochondrial pro-survival and apoptotic cell death factors as well as MAPKs. J Mol Neurosci. 2013a;50:379–393. doi: 10.1007/s12031-012-9932-0. [DOI] [PubMed] [Google Scholar]
- Mohagheghi F, Khalaj L, Ahmadiani A, Rahmani B. Gemfibrozil pretreatment affecting antioxidant defense system and inflammatory, but not Nrf-2 signaling pathways resulted in female neuroprotection and male neurotoxicity in the rat models of global cerebral ischemia-reperfusion. Neurotox Res. 2013b;23:225–237. doi: 10.1007/s12640-012-9338-3. [DOI] [PubMed] [Google Scholar]
- Murakami K, Kondo T, Kawase M, Li Y, Sato S, Chen SF, Chan PH. Mitochondrial susceptibility to oxidative stress exacerbates cerebral infarction that follows permanent focal cerebral ischemia in mutant mice with manganese superoxide dismutase deficiency. J Neurosci. 1998;18:205–213. doi: 10.1523/JNEUROSCI.18-01-00205.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG. Mitochondrial dysfunction and glutamate excitotoxicity studied in primary neuronal cultures. Curr Mol Med. 2004;4:149–177. doi: 10.2174/1566524043479239. [DOI] [PubMed] [Google Scholar]
- Nilsen J, Brinton RD. Mitochondria as therapeutic targets of estrogen action in the central nervous system. Curr Drug Targets CNS Neurol Disord. 2004;3:297–313. doi: 10.2174/1568007043337193. [DOI] [PubMed] [Google Scholar]
- Nuñez JL, McCarthy MM. Androgens predispose males to GABAA-mediated excitotoxicity in the developing hippocampus. Exp Neurol. 2008;210:699–708. doi: 10.1016/j.expneurol.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuñez JL, Alt JJ, McCarthy MM. A new model for prenatal brain damage I GABAA receptor activation induces cell death in developing rat hippocampus. Exp Neurol. 2003;181:258–269. doi: 10.3201/eid0906.030118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens K, Park JH, Schuh R, Kristian T. Mitochondrial dysfunction and NAD(+) metabolism alterations in the pathophysiology of acute brain injury. Transl Stroke Res. 2013;4:618–634. doi: 10.1007/s12975-013-0278-x. [DOI] [PubMed] [Google Scholar]
- Pankotai E, Lacza Z, Muranyi M, Szabo C. Intra-mitochondrial poly(ADP-ribosyl)ation: potential role for alpha-ketoglutarate dehydrogenase. Mitochondrion. 2009;9:159–164. doi: 10.1016/j.mito.2009.01.013. [DOI] [PubMed] [Google Scholar]
- Peng TI, Jou MJ. Oxidative stress caused by mitochondrial calcium overload. Ann N Y Acad Sci. 2010;1201:183–188. doi: 10.1111/j.1749-6632.2010.05634.x. [DOI] [PubMed] [Google Scholar]
- Perez-Pinzon MA, Stetler RA, Fiskum G. Novel mitochondrial targets for neuroprotection. J Cereb Blood Flow Metab. 2012;32:1362–1376. doi: 10.1038/jcbfm.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrella J, Bhavnani BR. Protection of cortical cells by equine estrogens against glutamate-induced excitotoxicity is mediated through a calcium independent mechanism. BMC Neurosci. 2005;6:34. doi: 10.1186/1471-2202-6-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto RE, Bartley W. The nature of the sex-linked differences in glutathione peroxidase activity and aerobic oxidation of glutathione in male and female rat liver. Biochem J. 1969;115:449–456. doi: 10.1042/bj1150449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portera-Cailliau C, Price DL, Martin LJ. Non-NMDA and NMDA receptor-mediated excitotoxic neuronal deaths in adult brain are morphologically distinct: further evidence for an apoptosis-necrosis continuum. J Comp Neurol. 1997;378:88–104. [PubMed] [Google Scholar]
- Pringsheim T, Jette N, Frolkis A, Steeves TD. The prevalence of Parkinson's disease: A systematic review and meta-analysis. Mov Disord. 2014 doi: 10.1002/mds.25945. [DOI] [PubMed] [Google Scholar]
- Purnell PR, Fox HS. Autophagy-mediated turnover of dynamin-related protein 1. BMC Neurosci. 2013;14:86. doi: 10.1186/1471-2202-14-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quillinan N, Deng G, Grewal H, Herson PS. Androgens and stroke: Good, bad or indifferent? Exp Neurol. 2014 doi: 10.1016/j.expneurol.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci U S A. 2011;108:10190–10195. doi: 10.1073/pnas.1107402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao VL, Dogan A, Todd KG, Bowen KK, Dempsey RJ. Neuroprotection by memantine, a non-competitive NMDA receptor antagonist after traumatic brain injury in rats. Brain Res. 2001;911:96–100. doi: 10.1016/s0006-8993(01)02617-8. [DOI] [PubMed] [Google Scholar]
- Robertson CL, Soane L, Siegel ZT, Fiskum G. The potential role of mitochondria in pediatric traumatic brain injury. Dev Neurosci. 2006;28:432–446. doi: 10.1159/000094169. [DOI] [PubMed] [Google Scholar]
- Rossi MN, Carbone M, Mostocotto C, Mancone C, Tripodi M, Maione R, Amati P. Mitochondrial localization of PARP-1 requires interaction with mitofilin and is involved in the maintenance of mitochondrial DNA integrity. J Biol Chem. 2009;284:31616–31624. doi: 10.1074/jbc.M109.025882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed U, Karunakaran S, Meka DP, Koumar RC, Ramakrishnan S, Joshi SD, Nidadavolu P, Ravindranath V. Redox activated MAP kinase death signaling cascade initiated by ASK1 is not activated in female mice following MPTP: novel mechanism of neuroprotection. Neurotox Res. 2009;16:116–126. doi: 10.1007/s12640-009-9058-5. [DOI] [PubMed] [Google Scholar]
- Scarpulla RC. Nuclear activators and coactivators in mammalian mitochondrial biogenesis. Biochim Biophys Acta. 2002;1576:1–14. doi: 10.1016/s0167-4781(02)00343-3. [DOI] [PubMed] [Google Scholar]
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenas E, Nozari A, Sharma HS, Basu S, Rubertsson S, Wiklund L. Sex differences in cerebral injury after severe haemorrhage and ventricular fibrillation in pigs. Acta Anaesthesiol Scand. 2010;54:343–353. doi: 10.1111/j.1399-6576.2009.02125.x. [DOI] [PubMed] [Google Scholar]
- Sharma J, Johnston MV, Hossain MA. Sex differences in mitochondrial biogenesis determine neuronal death and survival in response to oxygen glucose deprivation and reoxygenation. BMC Neurosci. 2014;15:9. doi: 10.1186/1471-2202-15-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SS, Bray ER, Dixon CE. Effects of nicotine administration on striatal dopamine signaling after traumatic brain injury in rats. J Neurotrauma. 2012;29:843–850. doi: 10.1089/neu.2011.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel CS, McCullough LD. NAD+and nicotinamide: sex differences in cerebral ischemia. Neuroscience. 2013;237:223–231. doi: 10.1016/j.neuroscience.2013.01.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel C, Li J, Liu F, Benashski SE, McCullough LD. miR-23a regulation of X-linked inhibitor of apoptosis (XIAP) contributes to sex differences in the response to cerebral ischemia. Proc Natl Acad Sci U S A. 2011;108:11662–11667. doi: 10.1073/pnas.1102635108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpkins JW, Dykens JA. Mitochondrial mechanisms of estrogen neuroprotection. Brain Res Rev. 2008;57:421–430. doi: 10.1016/j.brainresrev.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Simpkins JW, Wang J, Wang X, Perez E, Prokai L, Dykens JA. Mitochondria play a central role in estrogen-induced neuroprotection. Curr Drug Targets CNS Neurol Disord. 2005;4:69–83. doi: 10.2174/1568007053005073. [DOI] [PubMed] [Google Scholar]
- Slupe AM, Merrill RA, Flippo KH, Lobas MA, Houtman JC, Strack S. A calcineurin docking motif (LXVP) in dynamin-related protein 1 contributes to mitochondrial fragmentation and ischemic neuronal injury. J Biol Chem. 2013;288:12353–12365. doi: 10.1074/jbc.M113.459677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CM, Chen Y, Sullivan ML, Kochanek PM, Clark RS. Autophagy in acute brain injury: feast, famine, or folly? Neurobiol Dis. 2011;43:52–59. doi: 10.1016/j.nbd.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkov AA. An update on the role of mitochondrial alpha-ketoglutarate dehydrogenase in oxidative stress. Mol Cell Neurosci. 2013;55:13–16. doi: 10.1016/j.mcn.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkov AA, Fiskum G, Chinopoulos C, Lorenzo BJ, Browne SE, Patel MS, Beal MF. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J Neurosci. 2004;24:7779–7788. doi: 10.1523/JNEUROSCI.1899-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoica BA, Loane DJ, Zhao Z, Kabadi SV, Hanscom M, Byrnes KR, Faden AI. PARP-1 inhibition attenuates neuronal loss, microglia activation and neurological deficits after traumatic brain injury. J Neurotrauma. 2014;31:758–772. doi: 10.1089/neu.2013.3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundar BS, Barbara VM, Roemgens A, Beyer C, Arnold S. Sex-and brain region-specific role of cytochrome c oxidase in 1-methyl-4-phenylpyridinium-mediated astrocyte vulnerability. J Neurosci Res. 2011;89:2068–2082. doi: 10.1002/jnr.22669. [DOI] [PubMed] [Google Scholar]
- Turtzo LC, McCullough LD. Sex-specific responses to stroke. Future Neurol. 2010;5:47–59. doi: 10.2217/fnl.09.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bregt DR, Thomas TC, Hinzman JM, Cao T, Liu M, Bing G, Gerhardt GA, Pauly JR, Lifshitz J. Substantia nigra vulnerability after a single moderate diffuse brain injury in the rat. Exp Neurol. 2012;234:8–19. doi: 10.1016/j.expneurol.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Bliek AM, Shen Q, Kawajiri S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb Perspect Biol. 2013 doi: 10.1101/cshperspect.a011072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma S, Janesko KL, Wisniewski SR, Bayir H, Adelson PD, Thomas NJ, Kochanek PM. F2-isoprostane and neuron-specific enolase in cerebrospinal fluid after severe traumatic brain injury in infants and children. J Neurotrauma. 2003;20:781–786. doi: 10.1089/089771503767870005. [DOI] [PubMed] [Google Scholar]
- Verma S, Quillinan N, Yang YF, Nakayama S, Cheng J, Kelley MH, Herson PS. TRPM2 channel activation following in vitro ischemia contributes to male hippocampal cell death. Neurosci Lett. 2012;530:41–46. doi: 10.1016/j.neulet.2012.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virbasius JV, Virbasius CA, Scarpulla RC. Identity of GABP with NRF-2, a multisubunit activator of cytochrome oxidase expression, reveals a cellular role for an ETS domain activator of viral promoters. Genes Dev. 1993;7:380–392. doi: 10.1101/gad.7.3.380. [DOI] [PubMed] [Google Scholar]
- Wagner AK, Fabio A, Puccio AM, Hirschberg R, Li W, Zafonte RD, Marion DW. Gender associations with cerebrospinal fluid glutamate and lactate/pyruvate levels after severe traumatic brain injury. Crit Care Med. 2005;33:407–413. doi: 10.1097/01.ccm.0000153931.23488.dd. [DOI] [PubMed] [Google Scholar]
- Wang L, Cherednichenko G, Hernandez L, Halow J, Camacho SA, Figueredo V, Schaefer S. Preconditioning limits mitochondrial Ca(2+) during ischemia in rat hearts: role of K(ATP) channels. Am J Physiol Heart Circ Physiol. 2001;280:H2321–H2328. doi: 10.1152/ajpheart.2001.280.5.H2321. [DOI] [PubMed] [Google Scholar]
- Wang Y, Dawson VL, Dawson TM. Poly(ADP-ribose) signals to mitochondrial AIF: a key event in parthanatos. Exp Neurol. 2009;218:193–202. doi: 10.1016/j.expneurol.2009.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kim NS, Haince JF, Kang HC, David KK, Andrabi SA, Poirier GG, Dawson VL, Dawson TM. Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos) Sci Signal. 2011;4:20. doi: 10.1126/scisignal.2000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Huang XJ, Van KC, Went GT, Nguyen JT, Lyeth BG. Amantadine improves cognitive outcome and increases neuronal survival after fluid percussion traumatic brain injury in rats. J Neurotrauma. 2014;31:370–377. doi: 10.1089/neu.2013.2917. [DOI] [PubMed] [Google Scholar]
- Weaver CE, Jr, Park-Chung M, Gibbs TT, Farb DH. 17beta-Estradiol protects against NMDA-induced excitotoxicity by direct inhibition of NMDA receptors. Brain Res. 1997;761:338–341. doi: 10.1016/s0006-8993(97)00449-6. [DOI] [PubMed] [Google Scholar]
- Weis SN, Pettenuzzo LF, Krolow R, Valentim LM, Mota CS, Dalmaz C, Wyse AT, Netto CA. Neonatal hypoxia-ischemia induces sex-related changes in rat brain mitochondria. Mitochondrion. 2012;12:271–279. doi: 10.1016/j.mito.2011.10.002. [DOI] [PubMed] [Google Scholar]
- Weis SN, Toniazzo AP, Ander BP, Zhan X, Careaga M, Ashwood P, Wyse AT, Netto CA, Sharp FR. Autophagy in the brain of neonates following hypoxia-ischemia shows sex- and region-specific effects. Neuroscience. 2014;256:201–209. doi: 10.1016/j.neuroscience.2013.10.046. [DOI] [PubMed] [Google Scholar]
- Widdowson EM. The response of the sexes to nutritional stress. Proc Nutr Soc. 1976;35:175–180. doi: 10.1079/pns19760030. [DOI] [PubMed] [Google Scholar]
- Xie Y, Li J, Fan G, Qi S, Li B. Reperfusion promotes mitochondrial biogenesis following focal cerebral ischemia in rats. PLoS One. 2014;9:e92443. doi: 10.1371/journal.pone.0092443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Peterson PL, Lee CP. Effect of N-acetylcysteine on mitochondrial function following traumatic brain injury in rats. J Neurotrauma. 1999;16:1067–1082. doi: 10.1089/neu.1999.16.1067. [DOI] [PubMed] [Google Scholar]
- Yan HQ, Ma X, Chen X, Li Y, Shao L, Dixon CE. Delayed increase of tyrosine hydroxylase expression in rat nigrostriatal system after traumatic brain injury. Brain Res. 2007;1134:171–179. doi: 10.1016/j.brainres.2006.11.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin W, Signore AP, Iwai M, Cao G, Gao Y, Chen J. Rapidly increased neuronal mitochondrial biogenesis after hypoxic-ischemic brain injury. Stroke. 2008;39:3057–3063. doi: 10.1161/STROKEAHA.108.520114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M, Siegel C, Zeng Z, Li J, Liu F, McCullough LD. Sex differences in the response to activation of the poly (ADP-ribose) polymerase pathway after experimental stroke. Exp Neurol. 2009;217:210–218. doi: 10.1016/j.expneurol.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Wong-Riley MT. Depolarizing stimulation upregulates GA-binding protein in neurons: a transcription factor involved in the bigenomic expression of cytochrome oxidase subunits. Eur J Neurosci. 2000;12:1013–1023. doi: 10.1046/j.1460-9568.2000.00997.x. [DOI] [PubMed] [Google Scholar]
- Zhang C, Yuan XR, Li HY, Zhao ZJ, Liao YW, Wang XY, Su J, Sang SS, Liu Q. Downregualtion of dynamin-related protein 1 attenuates glutamate-induced excitotoxicity via regulating mitochondrial function in a calcium dependent manner in HT22 cells. Biochem Biophys Res Commun. 2014;443:138–143. doi: 10.1016/j.bbrc.2013.11.072. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Hou J, Liu J, Yao M, Li L, Zhang B, Zhu H, Wang Z. Inhibition of autophagy contributes to melatonin-mediated neuroprotection against transient focal cerebral ischemia in rats. J Pharmacol Sci. 2014;124:354–364. doi: 10.1254/jphs.13220fp. [DOI] [PubMed] [Google Scholar]
- Zhou H, Wang J, Jiang J, Stavrovskaya IG, Li M, Li W, Wu Q, Zhang X, Luo C, Zhou S, Sirianni AC, Sarkar S, Kristal BS, Friedlander RM, Wang X. N-acetyl-serotonin offers neuroprotection through inhibiting mitochondrial death pathways and autophagic activation in experimental models of ischemic injury. J Neurosci. 2014;34:2967–2978. doi: 10.1523/JNEUROSCI.1948-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JH, Horbinski C, Guo F, Watkins S, Uchiyama Y, Chu CT. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am J Pathol. 2007;170:75–86. doi: 10.2353/ajpath.2007.060524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo W, Zhang W, Chen NH. Sexual dimorphism in cerebral ischemia injury. Eur J Pharmacol. 2013;711:73–79. doi: 10.1016/j.ejphar.2013.04.024. [DOI] [PubMed] [Google Scholar]