

Synopsis
The cardiovascular response to asphyxia involves redistribution of cardiac output to maintain oxygen delivery to critical organs such as the adrenal gland, heart and brain, at the expense of other organs such as the gut, kidneys and skin. This results in reduced perfusion and localized hypoxia/ischemia in these organs, which if severe, can result in multi-organ failure. Liver injury, coagulopathy, bleeding, thrombocytopenia, renal dysfunction, pulmonary and gastrointestinal injury all result from hypoxia, under-perfusion or both. Current clinical therapies need to be considered together with therapeutic hypothermia and cardiovascular recovery.
Keywords: asphyxia, cardiovascular, multi-organ failure, kidneys, liver
Introduction
Birth asphyxia causes hypoxic-ischemic encephalopathy and multi-organ failure. The most studied organ affected by hypoxia is the cardiovascular system, and the resulting hemodynamic instability that occurs due to hypoxia, either in utero or during resuscitation and newborn transition, causing downstream effects on other organs. The clinical focus during the resuscitation of an asphyxiated infant is largely on the immediate changes in heart rate and systemic blood pressure that occur at delivery, based on the seminal findings of Dawes and associates [1–3]. The acute cardiorespiratory consequences of asphyxia require rapid intervention in the form of stimulation, ventilation and in the extreme cases cardiac resuscitation [4].
However, other cardiovascular consequences of asphyxia can also have long-term consequences to asphyxic newborns. One of the profound cardiovascular responses to asphyxia is the redistribution of cardiac output. Hypoxia diverts blood, partially through a primitive diving reflex, from less vital organs, such as the liver, kidney, and gut, to maintain oxygen delivery to critical organs such as the adrenal gland, heart and brain, at the expense of other organs [5–7]. Within these organs now deprived of blood flow, local vasoconstriction and redistribution of blood flow results in a decrease in oxygen delivery [7]. If prolonged, this can cause cellular injury and inadequate tissue function and can result in multi-organ dysfunction after birth. Further, the fetal cardiovascular response to asphyxia can also lead to redistribution of blood volume towards the placenta, leaving the asphyxic newborn volume depleted, and prone to circulatory shock.
All the clinical manifestations of organ failure must be managed in the setting of therapeutic hypothermia. While much of perinatology has been focused on the downstream consequences of asphyxia on the brain, and the devastating consequences of hypoxic ischemic encephalopathy (HIE), a growing appreciation of the consequences of asphyxia on other organs is emerging (Table 1) [8–11]. which further complicates the care of the asphyxiated infant.
Table 1.
Multi-organ failure after birth asphyxia in randomized therapeutic hypothermia trials
| Study | Selective Head CoolCap [8] | Whole-Body NICHD[9] | Whole-body TOBY trial[10] | Selective Head Chinese trial [11] | ||||
|---|---|---|---|---|---|---|---|---|
| Cooled | Control | Cooled | Control | Cooled | Control | Cooled | Control | |
| Increased Liver Enzymes | 38%* | 53% | 20% | 15% | NR | NR | 35% | 28% |
| Thrombocytopenia | 33% | 22% | NR | NR | 58% | 50% | 6% | 2% |
| Prolonged coagulation | 50% | 42% | 18% | 11% | 41% | 43% | NR | NR |
| Hemorrhage/Bleeding | NR | NR | 3% | 2% | NR | NR | 3% | 2% |
| Renal Failure | 65% | 70% | 22% | 26% | ND | ND | 23% | 22% |
P<0.05 verses control infants;
NR=not reported; ND= none requiring dialysis
Here we describe the circulatory responses to asphyxia and how they can lead to multi-organ dysfunction. We will focus on the physiologic derangements that occur in response to hypoxia, and the end organ damage caused by both the hypoxia and hypoperfusion caused by responsive shunting of blood. We will also briefly outline the current clinical strategies for minimizing multi-organ injury to attempt to reduce life-long morbidity.
Cardiovascular Response to Asphyxia
Irrespective of the cause of impaired gas exchange, there is a sequence of cardiovascular and respiratory changes that ensue in asphyxiated infants at birth. Shortly after the onset of asphyxia, the newborn undergoes a period of “primary apnea” which is also associated with a profound bradycardia [1, 2, 12, 13]. Blood pressure is usually maintained during primary apnea due to peripheral vasoconstriction and the redirection of blood from non-vital organs towards the heart, central nervous system and adrenal glands [14–20].
If asphyxia continues, after a period of gasping the fetus enters secondary apnea or terminal apnea [15, 21]. Secondary apnea is associated with a large decrease in blood pressure [12, 21] and without intervention, the newborn will eventually have cardiac arrest [12, 21]. The downstream consequences of asphyxia are detrimental to multiple organ systems. The underlying cause of multi-organ dysfunction is likely due to cardiovascular sequelae of asphyxia.
Heart Rate
The heart rate response to asphyxia has been well described in studies in fetal sheep. An immediate bradycardia is initiated with acute hypoxia and this is augmented by acidosis when the induced asphyxia is acute and severe [22]. The bradycardia associated with fetal asphyxia is mediated by the vagus nerve – supplying parasympathetic efferent innervation to the heart. Vagotomy will inhibit the bradycardic response to asphyxia and similar results are obtained using a pharmacological approach with atropine (a competitive acetylcholine muscarinic receptor antagonist) [23–26].
Debate has arisen previously over the afferent stimulus for the bradycardic response to asphyxia. Arterial chemo- and/or baroreceptors likely provide the afferent input into the cardiovascular control center, resulting in increased parasympathetic drive that mediates the heart rate response. Although peripheral chemoreceptors and baroreceptors are active and functional in the fetal sheep from 90 days gestational age, it is likely that the bradycardic response is primarily initiated by a chemoreflex for several reasons [27, 28]. Firstly, the increases in arterial blood pressure are gradual and occur after the more rapid decrease in heart rate [26]. Additionally, the bradycardic response occurs during brief episodes of fetal hypoxia without changes in blood pressure [29]. Finally, there is an absence of fetal heart rate decrease or arterial pressure increase to acute hypoxia in carotid body and aortic body denervated fetal lambs, suggesting the fetal bradycardic response is chemoreflex-mediated [30].
Similar bradycardic responses to asphyxia have been demonstrated in newborn animal model studies [3, 23, 31, 32]. These studies were conducted in animals of varying postnatal ages (from birth to 10 days postnatally) and the method of asphyxia was induced by placing the subjects head in a liquid-filled environment (water or normal saline) to prevent gaseous exchange. Recent studies have demonstrated that the heart rate response to asphyxia is altered if the asphyxia occurs with the fetus in utero compared to ex utero [33].
Arterial Blood Pressure and Peripheral Vasoconstriction
The knowledge of the distribution of fetal cardiac output is largely due to the seminal work of Rudolph and colleagues [34–36]. The normal organ distribution of fetal cardiac output is shown in Figure 1. Blood is oxygenated within the placenta and returns to the heart through the umbilical veins, bypasses the kidneys through the ductus venosus and enters the inferior vena cava. Inferior vena caval blood flow represents about two thirds of total venous return and is highly oxygenated; ~65% saturation. The oxygenated blood preferentially is directed through the foreman ovale to the left atrium. Left ventricular output in the fetus is only 33% of combined ventricular output and is distributed to the heart and upper body including the brain with only a quarter of left ventricular output flowing into the descending aorta. Right ventricular output makes up 66% of combined ventricular output in the fetus. The majority of right ventricular output bypasses the lungs via the ductus arteriosus whereupon it is distributed to the placenta, abdomen and lower body.
Figure 1.
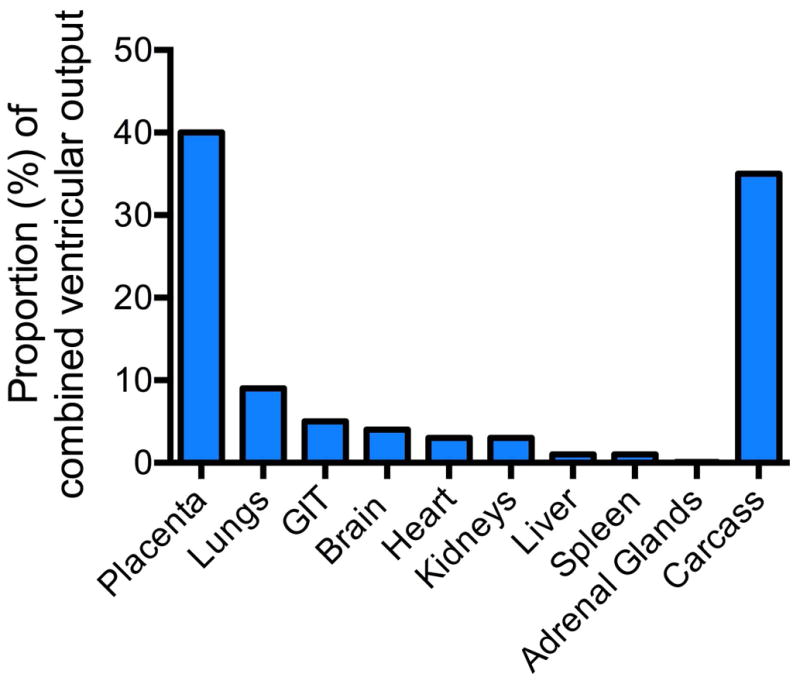
Distribution of cardiac output in the fetus; GIT = gastrointestinal tract. Data from 36, 37, 101]
In response to severe hypoxemia fetal cardiac output falls but umbilical flow remains constant [37]. In order to maintain arterial blood pressure and adequate oxygen supply to vital organs there is a re-direction of cardiac output to preferentially direct blood away from non-vital organs (the gut, kidney, lungs and skin) towards the heart, central nervous system and adrenal glands (Figure 2) [38–40]. This redistribution of cardiac output is the direct result of vasoconstriction in non-vital organs and vasodilation in vital organs such as the brain and heart. Increases in catecholamines and increased sympathetic activity via hypoxic-induced activation of the sympathetic-adrenergic system likely mediate these changes in peripheral vascular resistance [26, 41].
Figure 2.
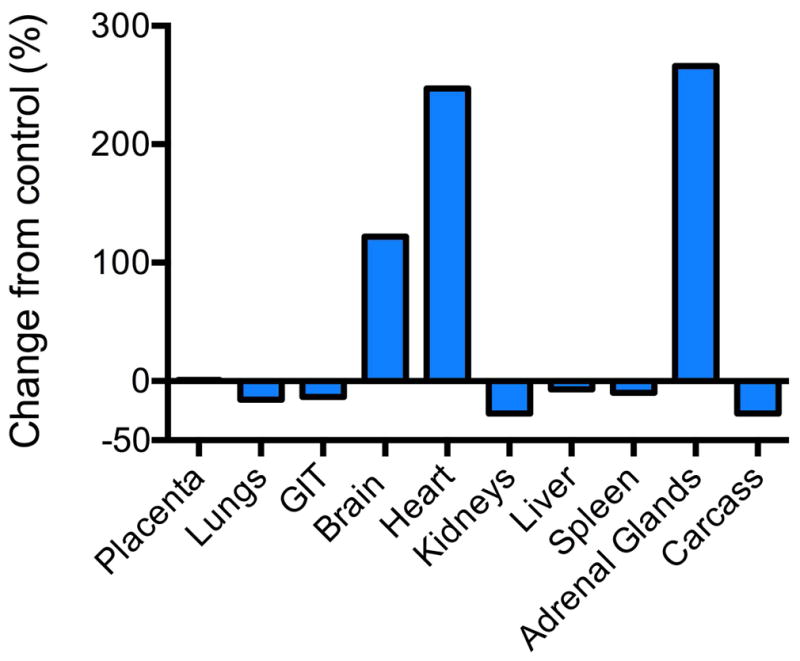
Change in cardiac output (%) from control values during reduction in uterine blood flow resulting in hypoxemia in fetal sheep. Illustrates the redistribution of cardiac output to the brain, adrenals and heart at the expense of other organs. Data from Jensen, A., C. Roman, and A.M. Rudolph, Effects of reducing uterine blood flow on fetal blood flow distribution and oxygen delivery. J Dev Physiol., 1991. 15(6): p. 309–23.
The carotid arterial chemoreceptor role in mediating peripheral vasoconstriction has been confirmed in studies in which carotid denervation abolished femoral vasoconstriction during hypoxia [26]. In addition, femoral vasoconstriction is inhibited using phentolamine (an α-adrenergic antagonist), confirming an adrenergic efferent mechanism mediating this pathway [26]. Pulmonary blood flow will decrease via pulmonary vasoconstriction in asphyxiated fetal and newborn studies [42, 43]. The increase in pulmonary vascular resistance was negated in sino-aortic denervated fetuses, suggesting similar chemo-reflexive mechanisms [44].
The high peripheral vasoconstriction also can result in the preferential redistribution of blood from the high resistance fetus, towards the low resistance placenta, resulting in a reduction in the blood volume to the newborn [45]. As asphyxia progresses, arterial blood pressure cannot be maintained in spite of peripheral vasoconstriction, because ventricular function begins to decline. As a result, during secondary apnea, blood pressure begins to fall, and will eventually lead to myocardial dysfunction and cardiac arrest.
Ventricular Output and Myocardial Dysfunction
The relative roles of heart rate and the Frank-Starling mechanism in effectively altering cardiac output in the newborn remain controversial. In adults, the Frank-Starling mechanism refers to the direct influence of end-diastolic volume on stroke volume; the intrinsic length-tension relationship of increasing cardiac muscle length by increasing end-diastolic volume will increase contractile force and therefore increase stroke volume. Some fetal studies have demonstrated that heart rate is the major determinant of fetal cardiac output rather than stroke volume [46–48]. These studies found that increasing preload as a means of increasing stroke volume via blood or saline infusion had little effect on cardiac output [47]. Rudolph and Heyman [48] further demonstrated that altering heart rate (from 160–180 bpm to 240–270 bpm) via left atrial pacing increased right ventricular output by just 12%. It has been suggested that the inability of the fetal heart to increase stroke volume is due to immaturity of the fetal myocardium [49]. However, structural immaturity cannot solely be responsible for limiting the cardiac response because left ventricular output is able to double immediately after birth [50]. Further, increasing heart rate (HR) via left atrial pacing in the fetus decreased right to left shunting through the foramen ovale [51]. As a result, increases in right ventricular output associated with left atrial pacing in the studies by Rudolph and Heyman [48] were likely to be exaggerated.
Interestingly, Kirkpatrick and colleagues [52] demonstrated that spontaneous changes in fetal HR (114–180 bpm) were not associated with significant changes in fetal left ventricular output. Further, Anderson et al. [53] found that changes in end-diastolic ventricular volume influenced ventricular stroke volume and left ventricular output in chronically instrumental fetal lambs. These studies suggest that the Frank-Starling mechanism is a major determinant of fetal cardiac output [52, 53].
Irrespective of the precise mechanisms effecting newborn cardiac output, ongoing severe asphyxia is associated with significantly reduced ventricular output and stroke volume [54, 55]. It is likely that the causes of eventual myocardial dysfunction and subsequent reduced cardiac output in the asphyxiated neonate are multifactorial; these include the low HR associated with asphyxia, acidosis[56], as well as reduced myocardial contractility due to poor perfusion and ischaemic cardiac injury. It is known that asphyxiated newborns are at increased risk of ischaemic cardiac injury due to decreased cardiac output and decreased coronary perfusion [57]. Asphyxiated newborns have increased troponin levels –a protein located on the actin filament of myocardium and that is an indicator of myocardial cell death and myocardial damage [58–60]. Increased serum troponin levels (taken within 24 hours of delivery) in asphyxiated human infants have been associated with increased myocardial damage and consequently lower left ventricular output and stroke volume[59].
Therefore, progressive asphyxia in the newborn without intervention results in ongoing circulatory deterioration eventually leading to myocardial dysfunction, circulatory shock, right and left ventricular failure, tricuspid regurgitation and hypotension and eventual cardiac arrest. Clinical management of cardiac dysfunction relies on maintaining adequate perfusion to organs, maintaining blood pressure, and assisting in cardiac contractility. Review of cardiac management and blood pressure control in the newborn are beyond the scope of this review but has been nicely summarized in a recent review article by Giesinger and McNamara[61].
Liver Injury, Coagulopathy, and Bleeding
Liver injury is likely due to hypoperfusion rather than hypoxia [62]. Transaminases (AST and ALT) often increase from initial measurements, but typically significantly improve by end of 72 hours of therapeutic hypothermia [63, 64]. Some correlation between the severity of the perinatal asphyxia and the elevation of liver transaminases has been seen [65]. Conversely, in a piglet model of HIE, there was a poor correlation between the serum transaminases and the degree of tissue damage on pathologic specimens, suggesting significant liver dysfunction can occur in the setting of normal transaminase levels [66]. In selective head cooling trial, 53% of non-cooled asphyxiated infants had elevated liver enzymes versus 38% in cooled group (p=0.02) [8]. There were no differences in coagulopathies (17%) and prolonged coagulation times (46%) between infants receiving therapeutic cooling and those that were not [33]. The overall rate of death or disability was higher in both groups of the Cool-Cap study than other HIE cooling trials, suggesting the infants were overall sicker than in the other studies which may explain the higher percent of hepatic dysfunction [9–11](Table 1). A head-cooling trial from China also showed no difference between groups with 30% demonstrating increased liver enzymes [11].
Coagulation dysfunction in hypoxic infants was reported as early as 1971 in a subset of infants with birth asphyxia, with the cause associated with a consumptive coagulopathy (increased fibrin degradation products) followed by disseminated intravascular coagulation [67, 68]. The effect on coagulation was of short duration and may only play a minor role overall morbidity of birth aphyxia. Levels of factor XIII are lower in infants with birth asphyxia. Plasma levels of thrombin-antithrombin complexes, D-dimer, fibrinogen, and fibrin degradation products are higher in infants with birth asphyxia [69]. It should be noted that levels of coagulation factors II, VII, IX, and X, protein C, protein S and antithrombin are also reduced in preterm compared with term plasma [70]. Prothrombin time (PT) and activated partial thromboplastin time (aPTT) are higher in preterm infants, but there is no correlation with increased risk of intraventricular hemorrhage (IVH) with higher coagulation times [70].
Coagulopathy requiring fresh frozen plasma (FFP) occurred in up to 50% of infants with HIE (Table 1) [8–11, 63]. In the National Institute of Child Health and Human Development (NICHD) whole body cooling trial, 17% of infants had hepatic dysfunction and 14% had disseminated intravascular coagulation (DIC) [9]. Although difficult to determine the cause of coagulation abnormalities in the setting of perinatal asphyxia, therapeutic cooling does not appear to increase coagulopathy. This is reassuring since hypothermia impairs coagulation in whole blood samples and accelerates microvascular coagulation in mice [71, 72]. In a retrospective analysis of bleeding in 76 infants with birth asphyxia undergoing therapeutic cooling, 54% had some form of active bleeding with an even distribution of intracranial, pulmonary, gastrointestinal and hematuria [73]. Infants with bleeding had a lower platelet count, fibrinogen level and higher maximum INR, and authors suggest levels associated with increased bleeding were minimum fibrinogen level of 1.54 g/L, mininum platelet count of 130,000 ×106/L, and international normalized ratio (INR) of 1.98 [73]. The bleeding was not severe enough to require surgery or neurosurgical intervention, and the rate of major bleeding was consistent with the lower rates in the randomized trials [73]. There is a wide range of clinical practice with regard to maintaining coagulation lab values in the normal range in asphyxiated children and no clinical guidelines have been determined. Fresh frozen plasma is often transfused in these infants to correct INR values or as part of fluid resuscitation for blood pressure control.
Thrombocytopenia
Thrombocytopenia, as defined by a platelet count of <100,000 ×106/L, is common in ill infants and infants with birth asphyxia (Table 1). It is difficult to gain consensus amongst neonatologists about the level of thrombocytopenia which is dangerous for ill neonates or infants with birth asphyxia. In a prospective, observational study of thrombocytopenia in all infants admitted to seven neonatal intensive care units (NICUs), 5% of all admissions had platelet count <60,000 ×106/L and 78% of these infants were less than 28 weeks gestational age [74]. Although a large percent of infants born less than 1500 grams had severe thrombocytopenia, only 9% developed a major hemorrhage [74]. In late preterm and term asphyxiated infants, a large retrospective study found that 31% had thrombocytopenia [75]. The nadir for the platelet count in these asphyxiated infants was on day 3, but normalization of thrombocytopenia took an average of 19 days [75]. There is a large variation in the rate of thrombocytopenia in the clinical trials of therapeutic hypothermia, with rate ranging from 6% to 55% of asphyxiated infants [8–11, 63].
Hypoxia is believed to have a direct effect on platelet formation for a clinical presentation termed thrombocytopenia of perinatal asphyxia [75]. Hypoxia in adult mice causes a decrease in the size and production of the megakaryocytes in the bone marrow [76]. Although the megakaryocytes appear to not be injured by hypoxia, the cells in the bone marrow surrounding them are affected and decrease the release of platelet promoting factors [77]. Increased destruction of platelets contributes importantly to role in thrombocytopenia of birth asphyxia, as is evident by the rapid decrease in platelet number after platelet transfusions in infants with birth asphyxia [78]. Thrombocytopenia was more common in infants with more chronic hypoxia (>24 hours), as classified by increased nucleated red blood cells count [73]. Infants with thrombocytopenia after asphyxia were more likely to die in one study, but the cause of death was not due to active bleeding or hemorrhagic consequences [73].
There are large variations in indications for platelet transfusion between NICUs in US and Canada, with many units transfusing platelets in non-bleeding infants with platelet counts greater than 50,000 ×106/L [79]. The average response to a platelet transfusion is an increase of 52,000 ×106/L [74]. When preterm infants were randomized at platelet count of 50,000 to ether platelet transfusion to 150,000 ×106/L or left alone, there were no differences in bleeding or IVH. Since bleeding is often not severe in infants with thrombocytopenia (3% of infants in NICHD whole body cooling study [9]), the National Institutes of Health (NIH) conducted an expert conference to determine whether platelet transfusions were necessary for many of these infants. No consensus could be given since no clear randomized trial on platelet use has been performed [80]. British guidelines suggest keeping platelet counts greater than 20 to 30,000 ×106/L in stable newborns, and transfuse platelets if less than 50,000 ×106/L in sick infants with active bleeding or coagulation defects [81, 82]. The Platelets for Neonatal Transfusion - Study 2 (PlaNeT-2) is a randomized study of high and low platelet transfusion thresholds in newborn and is currently being conducted in Europe [83]. Until further studies are performed, each individual clinician must decide whether treating thrombocytopenia in a stable, non-bleeding asphyxiated infant is necessary.
Renal Dysfunction and Electrolyte Disturbances after Birth Asphyxia
There are poor definitions for acute kidney injury (AKI) in neonates due to the variation in serum creatinine levels at birth, which often reflect the maternal levels for the first 48 hours, and the large changes in glomerular filtration rates that occur at birth and cause variations in urine production [84, 85]. However, AKI after neonatal asphyxia occurs in as many as 56% of asphyxiated infants, with a combination of oliguric and non-oliguric renal failure complicating the clinical management. All nephrons are formed by 34 weeks gestational age but the GFR increases 6 fold from birth until 1 year of life [86]. The renal blood flow rate is low at 4% of cardiac output in infants at birth and increases slowly over the first six weeks to about 15% of cardiac output (adult levels are 20 to 25%) [85]. Some studies define AKI as creatinine as >1.5 mg/dL and decreasing urine output (<1 ml/kg/h), but oligouria only occurs in 50% of infants with AKI [87]. Infants can often maintain urine output >1 ml/kg/hr and still have renal dysfunction [84]. Oliguric renal failure is associated with higher mortality than nonoliguric failure [88], but there is no evidence that converting oliguric renal failure to nonoliguric renal failure improves prognosis [84].
Kidney injury is common in infants with birth asphyxia (Table 1). In hypothermia trials, 50% of asphyxiated infants had increased creatinine and 18 to 39% had urine output less than 0.5 ml/kg/hr for >24 hours [63]. In selective head cooling (CoolCap), 67% of the infants had abnormal renal function, while in the whole-body cooling study, only 19% had oliguria and 5% were anuric [9]. The percentage of infants with oliguria is similar to the 20% of infants in head-cooling trial from China which had increased blood urea nitrogen (BUN) and creatinine levels [11]. The differences in the incidence of AKI between studies may have been due to severity of HIE in CoolCap study. In a small study of 36 infants, AKI persisted in 17% of infants at 96 hours after birth asphyxia [89]. The degree of kidney injury is correlated with the clinical severity of the birth asphyxia and infants with AKI were more likely to have abnormal findings on the brain MRI [90, 91]. Asphyxiated infants with AKI are also ventilated on average for 4 days longer than infants without kidney disease [89]. Electrolyte abnormalities were seen in over 50% of infants, with hyponatremia, hypokalemia, and hypocalcemia the most prominent [8, 63].
Since the definition of AKI is not very refined in the newborn period, the assessment of biomarkers of kidney injury are often sub-optimal. Serum creatinine will not begin to rise until 25 to 50% of renal function is lost, thus significant injury can occur without changes in creatinine [92]. The change in serum creatinine levels over the first few days is predictive of renal injury, with normal values of >50% decline or serum creatinine <0.6 is found in 70% of infants after HIE [93]. A fractional excretion of sodium of greater than 3% is moderately specific after 48 hours for AKI [84]. Although Cystatin C (CysC) and neutrophil gelatinase-associated lipocalin (NGAL) are both specific markers of other pediatric kidney injury, they can be elevated by birth asphyxia and unable to determine whether an infant will develop AKI [93, 94]. A recent study demonstrated a NGAL level greater than 250 ng/ml was significantly associated with severe HIE and mortality [95]. Researchers are continuing to develop new assays for determining AKI in hopes earlier detection might alter the high rate of persistent renal failure in survivors of pediatric acute kidney disease.
The response to asphyxia-associated kidney disease is dependent on the clinical scenario – as some infants have initial oliguria followed by high output renal failure from acute tubular necrosis. Close monitoring of fluid balance, serum electroytes, and body weight can prevent situations of hypovolemia or fluid overload [85]. Fluid overload at time of renal replacement therapy is associated with increased mortality in multiple pediatric critical care settings [96]. Since renal dysfunction can occur with normal urine output, it is important to monitor drug levels and avoid nephrotoxic medications. Although prophylactic theophylline was shown in multiple randomized studies to improve AKI after birth asphyxia, it is not heavily used for this purpose in NICUs in the US since long-term follow-up is not available and studies were done prior to the introduction of therapeutic hypothermia [97]. The use of diuretics in AKI was not shown to be beneficial and could be harmful [85]. Rapid fluid and electrolyte shifts can occur after birth asphyxia, so care needs to be given to correction of hyponatremia, hypokalemia, and hypocalcemia often seen in HIE.
Lung and Gastrointestinal Complications
Most infants with significant birth asphyxia have injury to the lungs and many require mechanical ventilation. Since mechanical ventilation may be necessary because of either injury to the lung parenchyma, alterations in the blood vessels in the lung, or the respiratory drive in the brainstem, the respiratory management will vary significantly from one patient to the next. As with any critically ill infant, infants after birth asphyxia should have oxygenation maximized to decrease shunting away from vital organs. The lungs, if allowed to recover using minimal mechanical ventilation, often respond well and many infants with severe brain injury remain on room air. Pulmonary hypertension is also common in birth asphyxia and maybe due to the underlying cause of the birth asphyxia (in utero hypoxia with vascular hypertrophy) or in response to the concurrent hypoxia [8, 9, 63]. Whether pulmonary hypertension occurs during therapeutic hypothermia or during the rewarming process, it can be treated with nitric oxide, muscle relaxation, and medications for pain and anxiety. Occasionally an infant with birth asphyxia may require extracorporeal membrane oxygenation (ECMO) for pulmonary reasons, but many centers use severe birth asphyxia as a contraindication for ECMO. Pulmonary hemorrhage has been reported in severe asphyxia and with significant coagulopathy [73]. Overall, the lung can repair itself quickly. Prolonged ventilation is often necessary for neurologic reasons instead of intrinsic issues with the lungs.
The gut has multiple watershed regions that are prone to hypoxic injury from birth asphyxia. Necrotizing enterocolitis has been reported in these infants, but is a rare complication [98]. Current guidelines for therapeutic hypothermia have infants remain nothing by mouth (NPO) until completion of the hypothermia and this delay in feeding may allow for the gut mucosa to repair. Hypothermia might improve gut morbidities after HIE and has been considered as therapy in older infants with necrotizing enterocolitis [99]. The ability to use total parental nutrition in these infants has allowed clinicians to rest the gut mucosa for multiple days and allow for reconstituting the gut barrier lost in hypoxic events [100].
Therapeutic Hypothermia and Multi-Organ Failure
In most of the clinical trials of therapeutic hypothermia to prevent death or poor neurologic outcome, there were no differences in markers of organ failure between the asphyxiated infants who received cooling or not (Table 1) [8–11]. A slight improvement in liver function tests was measured in selective head cooling, but not in other trials that included slightly less ill infants [8–11]. Proponents of selective head cooling argue that selective head cooling has less systemic effects, whereas whole body cooling centers may believe the over-all lower metabolic rate from hypothermia should be beneficial to all organs. In a single-center comparative study, there were no differences in end-organ damage over the first 72 hours of cooling between whole-body and selective head cooling [63] The rates of multi-organ system failure also did not differ between infants with HIE that had good or poor neurologic outcomes suggesting multi-organ failure is not a good predictor of overall outcome [64].
Conclusion
Multiorgan failure is common in infants born after acute or prolonged asphyxia, and likely results from downstream consequences of cardiovascular redistribution. The redistribution of blood flow and oxygen delivery results in regionalized hypoxia and consequently cellular death. Current clinical therapies of multi-organ dysfunction are now conducted on a background of therapeutic hypothermia. The efficacy of many of these therapies is yet to be truly demonstrated in this environment. Understanding the circulatory consequences of asphyxia may lead to improved cardiovascular stability after birth with potential benefit on multiple organ systems.
Best Practices Box.
What is the current practice?
Current newborn resuscitation guidelines recommend therapeutic hypothermia for severe birth asphyxia. Although neonatologists have focused on decreasing neurologic injury, the cardiovascular derangements from birth asphyxia lead to multiorgan failure. Therapeutic hypothermia has not changed the rate of multi-organ failure, and coagulopathy and kidney can lead to increased morbidity and mortalities.
What changes in current practice are likely to improve outcomes?
Improvements in management of blood pressure and perfusion will improve multi-organ failure
Although many clinicians have expanded the criteria for use of therapeutic hypothermia, clinical studies are warranted to study whether later use (>6h) or on infants with lower gestational age are beneficial.
Major Recommendations
Maintaining adequate circulatory support after birth asphyxia, based on blood pressure and echocardiographic data, will help decrease multi-organ failure.
Supportive measures to maintain adequate platelet counts and correction of coagulopathy should decrease bleeding events, but this has not been shown within clinical studies.
Renal dysfunction should be closely monitored in infants with birth asphyxia and medication doses adjusted
Rating for the Strength of the Evidence: Moderate
Summary Statement.
Birth asphyxia leads to cardiovascular changes in heart rate, cardiac output, and vasoconstriction that lead to multi-organ failure. In the setting of therapeutic hypothermia, clinicians need to provide symptomatic support to other affected organ systems as the organs recover from the hypoperfusion event.
Key Points.
The cardiovascular consequences of asphyxia likely underlie downstream multi-organ injury.
The major mechanism is due to redistribution of blood flow and oxygen delivery to vital organs, resulting in poor perfusion and hypoxia in other organs.
Current clinical management needs to consider the cardiovascular effects of therapeutic hypothermia on the asphyxia mediated organ dysfunction and recovery.
Footnotes
Disclosure statement: The authors have nothing to disclose
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Graeme R. Polglase, The Ritchie Centre, Hudson Institute of Medical Research, 27–31 Wright Street, Clayton, Victoria, 3168, Australia.
Tracey Ong, Email: tong1@student.monash.edu, The Ritchie Centre, Hudson Institute of Medical Research, 27–31 Wright Street, Clayton, Victoria, 3168, Australia.
Noah H Hillman, Email: hillman@slu.edu, Noah Hillman: Saint Louis University, Department of Pediatrics, 1100 S. Grand Blvd, St. Louis, MO 63124.
References
- 1.Dawes GS. Foetal and neonatal physiology: a comparative study of the changes at birth. Year Book Medical Publishers; 1968. [Google Scholar]
- 2.Cross KW. Resuscitation of the asphyxiated infant. British Medical Bulletin. 1966;22(1):73–78. doi: 10.1093/oxfordjournals.bmb.a070442. [DOI] [PubMed] [Google Scholar]
- 3.Dawes GS, et al. Some observations on foetal and new-born rhesus monkeys. The Journal of physiology. 1960;152:271–298. doi: 10.1113/jphysiol.1960.sp006487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wyllie J, et al. Part 7: Neonatal resuscitation: 2015 International Consensus on Cardiopulmonary Resuscitation and Emergency Cardiovascular Care Science with Treatment Recommendations. Resuscitation. 2015;95:e169–201. doi: 10.1016/j.resuscitation.2015.07.045. Epub 2015 Oct 15. [DOI] [PubMed] [Google Scholar]
- 5.Peeters LL, et al. Blood flow to fetal organs as a function of arterial oxygen content. Am J Obstet Gynecol. 1979;135(5):637–46. doi: 10.1016/s0002-9378(16)32989-1. [DOI] [PubMed] [Google Scholar]
- 6.Sheldon RE, et al. Redistribution of cardiac output and oxygen delivery in the hypoxemic fetal lamb. Am J Obstet Gynecol. 1979;135(8):1071–8. doi: 10.1016/0002-9378(79)90739-7. [DOI] [PubMed] [Google Scholar]
- 7.Jensen A, Garnier Y, Berger R. Dynamics of fetal circulatory responses to hypoxia and asphyxia. Eur J Obstet Gynecol Reprod Biol. 1999;84(2):155–72. doi: 10.1016/s0301-2115(98)00325-x. [DOI] [PubMed] [Google Scholar]
- 8.Gluckman PD, et al. Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: multicentre randomised trial. Lancet. 2005;365(9460):663–70. doi: 10.1016/S0140-6736(05)17946-X. [DOI] [PubMed] [Google Scholar]
- 9.Shankaran S, et al. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N Engl J Med. 2005;353(15):1574–84. doi: 10.1056/NEJMcps050929. [DOI] [PubMed] [Google Scholar]
- 10.Azzopardi DV, et al. Moderate hypothermia to treat perinatal asphyxial encephalopathy. N Engl J Med. 2009;361(14):1349–58. doi: 10.1056/NEJMoa0900854. [DOI] [PubMed] [Google Scholar]
- 11.Zhou WH, et al. Selective head cooling with mild systemic hypothermia after neonatal hypoxic-ischemic encephalopathy: a multicenter randomized controlled trial in China. J Pediatr. 2010;157(3):367–72. 372e1–3. doi: 10.1016/j.jpeds.2010.03.030. [DOI] [PubMed] [Google Scholar]
- 12.Wyllie J. Applied physiology of newborn resuscitation. Current Paediatrics. 2006;16(6):379–385. [Google Scholar]
- 13.Gupta JM, Tizard JP. The sequence of events in neonatal apnoea. Lancet. 1967;2(7506):55–59. [PubMed] [Google Scholar]
- 14.Low JA. Intrapartum fetal asphyxia: Definition, diagnosis, and classification. American Journal of Obstetrics and Gynecology. 1997;176(5):957–959. doi: 10.1016/s0002-9378(97)70385-5. [DOI] [PubMed] [Google Scholar]
- 15.Wall SN, et al. Reducing Intrapartum-Related Neonatal Deaths in Low- and Middle-Income Countries-What Works? Seminars in Perinatology. 2010;34(6):395–407. doi: 10.1053/j.semperi.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 16.Sheldon RE, Peeters LLH, Jones MD., Jr Redistribution of cardiac output and oxygen delivery in the hypoxemic fetal lamb. American Journal of Obstetrics and Gynecology. 1979;135(8):1071–1078. doi: 10.1016/0002-9378(79)90739-7. [DOI] [PubMed] [Google Scholar]
- 17.Peeters LLH, Sheldon RE, Jones MD., Jr Blood flow to fetal organs as a function of arterial oxygen content. American Journal of Obstetrics and Gynecology. 1979;135(5):637–646. doi: 10.1016/s0002-9378(16)32989-1. [DOI] [PubMed] [Google Scholar]
- 18.Jensen A, Roman C, Rudolph AM. Effects of reducing uterine blood flow on fetal blood flow distribution and oxygen delivery. Journal of Developmental Physiology. 1991;15(6):309–323. [PubMed] [Google Scholar]
- 19.Itskovitz J, LaGamma EF, Rudolph AM. Effects of cord compression on fetal blood flow distribution and O2 delivery. The American journal of physiology. 1987;252(1 Pt 2):H100–109. doi: 10.1152/ajpheart.1987.252.1.H100. [DOI] [PubMed] [Google Scholar]
- 20.Zaichkin J, Weiner GM. Neonatal Resuscitation Program (NRP) 2011: New science, new strategies. Advances in Neonatal Care. 2011;11(1):43–51. doi: 10.1097/ANC.0b013e31820e429f. [DOI] [PubMed] [Google Scholar]
- 21.Lakshminrusimha S, Carrion V. Perinatal Physiology and Principles of Neonatal Resuscitation. Clinical Pediatric Emergency Medicine. 2008;9(3):131–139. [Google Scholar]
- 22.Bennet L, et al. The cardiovascular and cerebrovascular responses of the immature fetal sheep to acute umbilical cord occlusion. Journal of Physiology. 1999;517(1):247–257. doi: 10.1111/j.1469-7793.1999.0247z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adamsons K, et al. The treatment of acidosis with alkali and glucose during asphyxia in foetal rhesus monkeys. The Journal of Physiology. 1963;169(3):679–689. doi: 10.1113/jphysiol.1963.sp007288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thakor AS, Giussani DA. Effects of acute acidemia on the fetal cardiovascular defense to acute hypoxemia. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 2009;296(1):R90–R99. doi: 10.1152/ajpregu.90689.2008. [DOI] [PubMed] [Google Scholar]
- 25.Reeves JT, Daoud FS, Eastin C. Effects of vagotomy on arterial pressure and blood gases in the fetal calf. Am J Physiol. 1971;221:349–355. doi: 10.1152/ajplegacy.1971.221.1.349. [DOI] [PubMed] [Google Scholar]
- 26.Giussani DA, et al. Afferent and efferent components of the cardiovascular reflex responses to acute hypoxia in term fetal sheep. Journal of Physiology. 1993;461:431–449. doi: 10.1113/jphysiol.1993.sp019521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blanco C, et al. Carotid baroreceptors in fetal and newborn sheep. Pediatric research. 1988;24(3):342–346. doi: 10.1203/00006450-198809000-00014. [DOI] [PubMed] [Google Scholar]
- 28.Blanco C, et al. The response to hypoxia of arterial chemoreceptors in fetal sheep and new-born lambs. The Journal of Physiology. 1984;351(1):25–37. doi: 10.1113/jphysiol.1984.sp015229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parer J, et al. Increased fetal heart rate variability with acute hypoxia in chronically instrumented sheep. European Journal of Obstetrics & Gynecology and Reproductive Biology. 1980;10(6):393–399. doi: 10.1016/0028-2243(80)90025-8. [DOI] [PubMed] [Google Scholar]
- 30.Itskovitz J, et al. Cardiovascular responses to hypoxemia in sinoaortic-denervated fetal sheep. Pediatric research. 1991;30(4):381–387. doi: 10.1203/00006450-199110000-00016. [DOI] [PubMed] [Google Scholar]
- 31.Born G, Dawes G, Mott JC. Oxygen lack and autonomic nervous control of the foetal circulation in the lamb. The Journal of physiology. 1956;134(1):149–166. doi: 10.1113/jphysiol.1956.sp005631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dawes G, et al. The treatment of asphyxiated, mature foetal lambs and rhesus monkeys with intravenous glucose and sodium carbonate. The Journal of physiology. 1963;169(1):167–184. doi: 10.1113/jphysiol.1963.sp007248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sobotka KS, et al. Circulatory responses to asphyxia differ if the asphyxia occurs in utero or ex utero in near-term lambs. PLoS One. 2014;9(11):e112264. doi: 10.1371/journal.pone.0112264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rudolph AM, Heymann MA. The circulation of the fetus in utero. Methods for studying distribution of blood flow, cardiac output and organ blood flow. Circ Res. 1967;21(2):163–84. doi: 10.1161/01.res.21.2.163. [DOI] [PubMed] [Google Scholar]
- 35.Jensen A, Roman C, Rudolph AM. Effects of reducing uterine blood flow on fetal blood flow distribution and oxygen delivery. J Dev Physiol. 1991;15(6):309–23. [PubMed] [Google Scholar]
- 36.Rudolph AM. Distribution and regulation of blood flow in the fetal and neonatal lamb. Circ Res. 1985;57(6):811–21. doi: 10.1161/01.res.57.6.811. [DOI] [PubMed] [Google Scholar]
- 37.Cohn HE, et al. Cardiovascular responses to hypoxemia and acidemia in fetal lambs. Am J Obstet Gynecol. 1974;120(6):817–24. doi: 10.1016/0002-9378(74)90587-0. [DOI] [PubMed] [Google Scholar]
- 38.Peeters L, et al. Blood flow to fetal organs as a function of arterial oxygen content. American journal of obstetrics and gynecology. 1979;135(5):637–646. doi: 10.1016/s0002-9378(16)32989-1. [DOI] [PubMed] [Google Scholar]
- 39.Cohn H, et al. Cardiovascular responses to hypoxemia and acidemia in fetal lambs. Am J Obstet Gynecol. 1974;120(6):817–824. doi: 10.1016/0002-9378(74)90587-0. [DOI] [PubMed] [Google Scholar]
- 40.Rudolph AM. Distribution and regulation of blood flow in the fetal and neonatal lamb. Circulation research. 1985;57(6):811–821. doi: 10.1161/01.res.57.6.811. [DOI] [PubMed] [Google Scholar]
- 41.Mulder AL, et al. Alpha-adrenergic contribution to the cardiovascular response to acute hypoxemia in the chick embryo. Am J Physiol Regul Integr Comp Physiol. 2001;281(6):R2004–10. doi: 10.1152/ajpregu.2001.281.6.R2004. [DOI] [PubMed] [Google Scholar]
- 42.Campbell A, et al. Pulmonary vasoconstriction and changes in heart rate during asphyxia in immature foetal lambs. The Journal of physiology. 1967;192(1):93. doi: 10.1113/jphysiol.1967.sp008290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cassin S, Dawes G, Ross B. Pulmonary blood flow and vascular resistance in immature foetal lambs. The Journal of physiology. 1964;171(1):80–89. doi: 10.1113/jphysiol.1964.sp007362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moore P, Hanson M. The role of peripheral chemoreceptors in the rapid response of the pulmonary vasculature of the late gestation sheep fetus to changes in PaO2. Journal of developmental physiology. 1991;16(3):133–138. [PubMed] [Google Scholar]
- 45.Van Bel F, Walther FJ. Myocardial dysfunction and cerebral blood flow velocity following birth asphyxia. Acta Paediatr Scand. 1990;79(8–9):756–62. doi: 10.1111/j.1651-2227.1990.tb11551.x. [DOI] [PubMed] [Google Scholar]
- 46.Kenny J, et al. Effects of heart rate on ventricular size, stroke volume, and output in the normal human fetus: a prospective Doppler echocardiographic study. Circulation. 1987;76(1):52–58. doi: 10.1161/01.cir.76.1.52. [DOI] [PubMed] [Google Scholar]
- 47.Gilbert RD. Control of fetal cardiac output during changes in blood volume. Am J Physiol. 1980;238(1):H80–H86. doi: 10.1152/ajpheart.1980.238.1.H80. [DOI] [PubMed] [Google Scholar]
- 48.Rudolph AM, Heyman M. Fetal and neonatal circulation and respiration. Annual review of physiology. 1974;36(1):187–207. doi: 10.1146/annurev.ph.36.030174.001155. [DOI] [PubMed] [Google Scholar]
- 49.Friedman WF. The intrinsic physiologic properties of the developing heart. Progress in cardiovascular diseases. 1972;15(1):87–111. doi: 10.1016/0033-0620(72)90006-0. [DOI] [PubMed] [Google Scholar]
- 50.Teitel D, Rudolph A. Perinatal oxygen delivery and cardiac function. Advances in pediatrics. 1984;32:321–347. [PubMed] [Google Scholar]
- 51.Pitlick PT, Kirkpatrick SE, Friedman WF. Distribution of fetal cardiac output: importance of pacemaker location. Am J Physiol. 1976;231(1):204–8. doi: 10.1152/ajplegacy.1976.231.1.204. [DOI] [PubMed] [Google Scholar]
- 52.Kirkpatrick SE, et al. Frank-Starling relationship as an important determinant of fetal cardiac output. Am J Physiol. 1976;231(2):495–500. doi: 10.1152/ajplegacy.1976.231.2.495. [DOI] [PubMed] [Google Scholar]
- 53.Anderson P, et al. The effect of heart rate on in utero left ventricular output in the fetal sheep. The Journal of physiology. 1986;372(1):557–573. doi: 10.1113/jphysiol.1986.sp016025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Walther FJ, et al. Cardiac output in newborn infants with transient myocardial dysfunction. The Journal of pediatrics. 1985;107(5):781–785. doi: 10.1016/s0022-3476(85)80417-0. [DOI] [PubMed] [Google Scholar]
- 55.Van Bel F, Walther FJ. Myocardial dysfunction and cerebral blood flow velocity following birth asphyxia. Acta Paediatrica Scandinavica. 1990;79(8–9):756–762. doi: 10.1111/j.1651-2227.1990.tb11551.x. [DOI] [PubMed] [Google Scholar]
- 56.Fisher DJ. Left ventricular oxygen consumption and function in hypoxemia in conscious lambs. Am J Physiol. 1983;244(5):H664–71. doi: 10.1152/ajpheart.1983.244.5.H664. [DOI] [PubMed] [Google Scholar]
- 57.Sehgal A, Wong F, Mehta S. Reduced cardiac output and its correlation with coronary blood flow and troponin in asphyxiated infants treated with therapeutic hypothermia. European journal of pediatrics. 2012;171(10):1511–1517. doi: 10.1007/s00431-012-1764-y. [DOI] [PubMed] [Google Scholar]
- 58.Costa S, et al. Is serum troponin T a useful marker of myocardial damage in newborn infants with perinatal asphyxia? Acta Paediatrica. 2007;96(2):181–184. doi: 10.1111/j.1651-2227.2007.00104.x. [DOI] [PubMed] [Google Scholar]
- 59.Wei Y, et al. Left ventricular systolic function of newborns with asphyxia evaluated by tissue Doppler imaging. Pediatric cardiology. 2009;30(6):741–746. doi: 10.1007/s00246-009-9421-6. [DOI] [PubMed] [Google Scholar]
- 60.Clark S, et al. Cardiac troponin T in cord blood. Archives of Disease in Childhood-Fetal and Neonatal Edition. 2001;84(1):F34–F37. doi: 10.1136/fn.84.1.F34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Giesinger RE, McNamara PJ. Hemodynamic instability in the critically ill neonate: An approach to cardiovascular support based on disease pathophysiology. Semin Perinatol. 2016;40(3):174–88. doi: 10.1053/j.semperi.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 62.Beath SV. Hepatic function and physiology in the newborn. Semin Neonatol. 2003;8(5):337–46. doi: 10.1016/S1084-2756(03)00066-6. [DOI] [PubMed] [Google Scholar]
- 63.Sarkar S, et al. Effects of therapeutic hypothermia on multiorgan dysfunction in asphyxiated newborns: whole-body cooling versus selective head cooling. J Perinatol. 2009;29(8):558–63. doi: 10.1038/jp.2009.37. [DOI] [PubMed] [Google Scholar]
- 64.Shah P, et al. Multiorgan dysfunction in infants with post-asphyxial hypoxic-ischaemic encephalopathy. Arch Dis Child Fetal Neonatal Ed. 2004;89(2):F152–5. doi: 10.1136/adc.2002.023093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Islam MT, et al. Status of liver enzymes in babies with perinatal asphyxia. Mymensingh Med J. 2011;20(3):446–9. [PubMed] [Google Scholar]
- 66.Karlsson M, et al. Liver enzymes cannot be used to predict liver damage after global hypoxia-ischemia in a neonatal pig model. Neonatology. 2009;96(4):211–8. doi: 10.1159/000215591. [DOI] [PubMed] [Google Scholar]
- 67.Chessells JM, Wigglesworth JS. Coagulation studies in severe birth asphyxia. Arch Dis Child. 1971;46(247):253–6. doi: 10.1136/adc.46.247.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chadd MA, et al. Coagulation defects in hypoxic full-term newborn infants. Br Med J. 1971;4(5786):516–8. doi: 10.1136/bmj.4.5786.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Suzuki S, Morishita S. Hypercoagulability and DIC in high-risk infants. Semin Thromb Hemost. 1998;24(5):463–6. doi: 10.1055/s-2007-996040. [DOI] [PubMed] [Google Scholar]
- 70.Neary E, et al. Coagulation indices in very preterm infants from cord blood and postnatal samples. J Thromb Haemost. 2015;13(11):2021–30. doi: 10.1111/jth.13130. [DOI] [PubMed] [Google Scholar]
- 71.Dirkmann D, et al. Hypothermia and acidosis synergistically impair coagulation in human whole blood. Anesth Analg. 2008;106(6):1627–32. doi: 10.1213/ane.0b013e31817340ad. [DOI] [PubMed] [Google Scholar]
- 72.Lindenblatt N, et al. Sustained hypothermia accelerates microvascular thrombus formation in mice. Am J Physiol Heart Circ Physiol. 2005;289(6):H2680–7. doi: 10.1152/ajpheart.00425.2005. [DOI] [PubMed] [Google Scholar]
- 73.Forman KR, et al. Coagulopathy in newborns with hypoxic ischemic encephalopathy (HIE) treated with therapeutic hypothermia: a retrospective case-control study. BMC Pediatr. 2014;14:277. doi: 10.1186/1471-2431-14-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stanworth SJ, et al. Prospective, observational study of outcomes in neonates with severe thrombocytopenia. Pediatrics. 2009;124(5):e826–34. doi: 10.1542/peds.2009-0332. [DOI] [PubMed] [Google Scholar]
- 75.Christensen RD, Baer VL, Yaish HM. Thrombocytopenia in late preterm and term neonates after perinatal asphyxia. Transfusion. 2015;55(1):187–96. doi: 10.1111/trf.12777. [DOI] [PubMed] [Google Scholar]
- 76.McDonald TP, et al. Effects of hypoxia on megakaryocyte size and number of C3H and BALB/c mice. Proc Soc Exp Biol Med. 1992;199(3):287–90. doi: 10.3181/00379727-199-43358. [DOI] [PubMed] [Google Scholar]
- 77.Saxonhouse MA, et al. Effects of hypoxia on megakaryocyte progenitors obtained from the umbilical cord blood of term and preterm neonates. Biol Neonate. 2006;89(2):104–8. doi: 10.1159/000088561. [DOI] [PubMed] [Google Scholar]
- 78.Bauman ME, Cheung PY, Massicotte MP. Hemostasis and platelet dysfunction in asphyxiated neonates. J Pediatr. 2011;158(2 Suppl):e35–9. doi: 10.1016/j.jpeds.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 79.Josephson CD, et al. Platelet transfusion practices among neonatologists in the United States and Canada: results of a survey. Pediatrics. 2009;123(1):278–85. doi: 10.1542/peds.2007-2850. [DOI] [PubMed] [Google Scholar]
- 80.Josephson CD, et al. A multidisciplinary “think tank”: the top 10 clinical trial opportunities in transfusion medicine from the National Heart, Lung, and Blood Institute-sponsored 2009 state-of-the-science symposium. Transfusion. 2011;51(4):828–41. doi: 10.1111/j.1537-2995.2010.02898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Carr R, Kelly AM, Williamson LM. Neonatal thrombocytopenia and platelet transfusion - a UK perspective. Neonatology. 2015;107(1):1–7. doi: 10.1159/000365163. [DOI] [PubMed] [Google Scholar]
- 82.Gibson BE, et al. Transfusion guidelines for neonates and older children. Br J Haematol. 2004;124(4):433–53. doi: 10.1111/j.1365-2141.2004.04815.x. [DOI] [PubMed] [Google Scholar]
- 83.Curley A, et al. Platelets for neonatal transfusion - study 2: a randomised controlled trial to compare two different platelet count thresholds for prophylactic platelet transfusion to preterm neonates. Neonatology. 2014;106(2):102–6. doi: 10.1159/000358481. [DOI] [PubMed] [Google Scholar]
- 84.Durkan AM, Alexander RT. Acute kidney injury post neonatal asphyxia. J Pediatr. 2011;158(2 Suppl):e29–33. doi: 10.1016/j.jpeds.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 85.Selewski DT, et al. Neonatal Acute Kidney Injury. Pediatrics. 2015;136(2):e463–73. doi: 10.1542/peds.2014-3819. [DOI] [PubMed] [Google Scholar]
- 86.Vanpee M, et al. Renal function in very low birth weight infants: normal maturity reached during early childhood. J Pediatr. 1992;121(5 Pt 1):784–8. doi: 10.1016/s0022-3476(05)81916-x. [DOI] [PubMed] [Google Scholar]
- 87.Karlowicz MG, Adelman RD. Nonoliguric and oliguric acute renal failure in asphyxiated term neonates. Pediatr Nephrol. 1995;9(6):718–22. doi: 10.1007/BF00868721. [DOI] [PubMed] [Google Scholar]
- 88.Agras PI, et al. Acute renal failure in the neonatal period. Ren Fail. 2004;26(3):305–9. doi: 10.1081/jdi-200026749. [DOI] [PubMed] [Google Scholar]
- 89.Kaur S, et al. Evaluation of glomerular and tubular renal function in neonates with birth asphyxia. Ann Trop Paediatr. 2011;31(2):129–34. doi: 10.1179/146532811X12925735813922. [DOI] [PubMed] [Google Scholar]
- 90.Aggarwal A, et al. Evaluation of renal functions in asphyxiated newborns. J Trop Pediatr. 2005;51(5):295–9. doi: 10.1093/tropej/fmi017. [DOI] [PubMed] [Google Scholar]
- 91.Sarkar S, et al. Relationship between acute kidney injury and brain MRI findings in asphyxiated newborns after therapeutic hypothermia. Pediatr Res. 2014;75(3):431–5. doi: 10.1038/pr.2013.230. [DOI] [PubMed] [Google Scholar]
- 92.Askenazi DJ, Ambalavanan N, Goldstein SL. Acute kidney injury in critically ill newborns: what do we know? What do we need to learn? Pediatr Nephrol. 2009;24(2):265–74. doi: 10.1007/s00467-008-1060-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gupta C, Massaro AN, Ray PE. A new approach to define acute kidney injury in term newborns with hypoxic ischemic encephalopathy. Pediatr Nephrol. 2016 doi: 10.1007/s00467-016-3317-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sweetman DU, Molloy EJ. Biomarkers of acute kidney injury in neonatal encephalopathy. Eur J Pediatr. 2013;172(3):305–16. doi: 10.1007/s00431-012-1890-6. [DOI] [PubMed] [Google Scholar]
- 95.Essajee F, Were F, Admani B. Urine neutrophil gelatinase-associated lipocalin in asphyxiated neonates: a prospective cohort study. Pediatr Nephrol. 2015;30(7):1189–96. doi: 10.1007/s00467-014-3035-9. [DOI] [PubMed] [Google Scholar]
- 96.Sutherland SM, et al. Fluid overload and mortality in children receiving continuous renal replacement therapy: the prospective pediatric continuous renal replacement therapy registry. Am J Kidney Dis. 2010;55(2):316–25. doi: 10.1053/j.ajkd.2009.10.048. [DOI] [PubMed] [Google Scholar]
- 97.Al-Wassia H, Alshaikh B, Sauve R. Prophylactic theophylline for the prevention of severe renal dysfunction in term and post-term neonates with perinatal asphyxia: a systematic review and meta-analysis of randomized controlled trials. J Perinatol. 2013;33(4):271–7. doi: 10.1038/jp.2012.97. [DOI] [PubMed] [Google Scholar]
- 98.Goldberg RN, Thomas DW, Sinatra FR. Necrotizing enterocolitis in the asphyxiated full-term infant. Am J Perinatol. 1983;1(1):40–2. doi: 10.1055/s-2007-1000050. [DOI] [PubMed] [Google Scholar]
- 99.Thornton KM, et al. Effects of whole body therapeutic hypothermia on gastrointestinal morbidity and feeding tolerance in infants with hypoxic ischemic encephalopathy. Int J Pediatr. 2014;2014:643689. doi: 10.1155/2014/643689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Grenz A, Clambey E, Eltzschig HK. Hypoxia signaling during intestinal ischemia and inflammation. Curr Opin Crit Care. 2012;18(2):178–85. doi: 10.1097/MCC.0b013e3283514bd0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Peeters LL, et al. Blood flow to fetal organs as a function of arterial oxygen content. Am J Obstet Gynecol. 1979;135(5):637–46. doi: 10.1016/s0002-9378(16)32989-1. [DOI] [PubMed] [Google Scholar]