

Abstract
AIM: To assess the role and mechanism of metformin in inducing apoptosis of pancreatic cancer cells.
METHODS: The human pancreatic cancer cell lines ASPC-1, BxPc-3, PANC-1 and SW1990 were exposed to metformin. The inhibition of cell proliferation and colony formation via apoptosis induction and S phase arrest in pancreatic cancer cell lines of metformin was tested.
RESULTS: In each pancreatic cancer cell line tested, metformin inhibited cell proliferation in a dose dependent manner in MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium assays). Flow cytometric analysis showed that metformin reduced the number of cells in G1 and increased the percentage of cells in S phase as well as the apoptotic fraction. Enzymelinked immunosorbent assay (ELISA) showed that metformin induced apoptosis in all pancreatic cancer cell lines. In Western blot studies, metformin induced poly-ADP-ribose polymerase (PARP) cleavage (an indicator of caspase activation) in all pancreatic cancer cell lines. The general caspase inhibitor (VAD-fmk) completely abolished metformin-induced PARP cleavage and apoptosis in ASPC-1 BxPc-3 and PANC-1, the caspase-8 specific inhibitor (IETD-fmk) and the caspase-9 specific inhibitor (LEHD-fmk) only partially abrogated metformin-induced apoptosis and PARP cleavage in BxPc-3 and PANC-1 cells. We also observed that metformin treatment dramatically reduced epidermal growth factor receptor (EGFR) and phosphorylated mitogen activated protein kinase (P-MAPK) in both a time- and dose-dependent manner in all cell lines tested.
CONCLUSION: Metformin significantly inhibits cell proliferation and apoptosis in all pancreatic cell lines. And the metformin-induced apoptosis is associated with PARP cleavage, activation of caspase-3, -8, and -9 in a time- and dose-dependent manner. Hence, both caspase-8 and -9-initiated apoptotic signaling pathways contribute to metformin-induced apoptosis in pancreatic cell lines.
Keywords: Metformin, Pancreatic cancer, Molecular classification, Apoptosis
INTRODUCTION
The incidence of pancreatic cancer has steadily increased year by year, however its prognosis is still dismal, despite of all efforts in early diagnosis and therapy. Even with a complete surgical resection, the 5-year survival rate is < 20%[1]. It is the fourth leading cause of cancer-related deaths in Western industrialized countries[2]. In 2006, it was estimated that more than 33 700 new cases of pancreatic cancer would be diagnosed in the United States, with virtually the same number of deaths (32 300) from this disease[3]. Conventional therapies associated with surgery and radiotherapy often in combination with chemotherapy show modest efficacy in local control and palliation and no real progress in patient survival[4-6]. Thus, novel approaches to human pancreatic carcinoma therapy are urgently needed.
Metformin (1,1-dimethylbiguanide hydrochloride) is the most widely prescribed anti-hyperglycemic agent in the world. It reduces blood glucose, is not associated with significant toxicity or hypoglycemia, increases insulin sensitivity and reduces serum insulin levels[7]. Population-based studies have shown that patients treated with metformin exhibit unexpected but beneficial reductions in both obesity and cancer of several subtypes[8]. We have studied the effects of metformin on pancreatic cancer and identified novel molecular mechanisms of metformin activity.
MATERIALS AND METHODS
Reagents
Metformin was purchased from Sigma Chemical Co., MO and dissolved in sterile water to make a 1M stock solution. Caspase-3 substrate, Ac-DEVD-pNA, caspase-8 substrate, Ac-IETD-pNA, and caspase-9 substrate, Ac-LEHD-pNA, were obtained from Alexis Biochemicals, San Diego, CA. Specific pan-caspase inhibitor, z-VAD-fmk, caspase-8 inhibitor, z-IETD-fmk, and caspase-9 inhibitor, z-LEHD-fmk, were obtained from BD Biosciences, San Jose, CA.
Antibodies for Western blot analysis were from following sources: caspase-8 mouse mAb (1C12), caspase-9 polyclonal antibody, and caspase-3 rabbit mAb (8G10), P-MAPK (Phospho-p44/42 MAP Kinase Thr202/Tyr204), Akt, and P-Akt (Phospho-Akt, Ser-473) (Cell Signaling Technology, Inc., Beverly, MA); MAPK (ERK2) (Santa Cruz, CA, USA); Poly (ADP-ribose) polymerase (PARP) mAb (C-2-10) (BIOMOL Research Laboratories Inc., Plymouth Meeting, PA); EGFR mouse mAb (clone F4), β-actin mouse mAb (clone AC-75) (Sigma Chemical Co.). All other reagents were purchased from Sigma Chemical Co. unless otherwise specified.
Cells and cell culture
The human pancreatic cancer cell lines SW1990 ASPC-1 BxPc-3 PANC-1 were obtained from the American Type Culture Collection (ATCC, Rockville, MD) and maintained in RPMI 1640 (Life Technologies). All cell lines were cultured at a 37°C humidified atmosphere containing 95% air and 5% CO2 and were split twice a week.
Cell proliferation assay
The CellTiter96™ AQ non-radioactive cell proliferation kit (Promega Corp., Madison, WI) was used to determine the cell viability. In brief, cells were plated onto 96-well plates with complete medium for 24 h incubation in a 37°C humidified atmosphere containing 95% air and 5% CO2. Cells were then grown in either 0.1 mL medium with 5% FBS as control, or 0.1 mL of the same medium containing a series of doses of metformin and incubated for another 72 h. After reading all wells at 490 nm with a micro-plate reader, the percentages of surviving cells from each group relative to controls, defined as 100% survival, were determined by reduction of MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt).
Clonogenic assay
In brief, cells were seeded into 6-well plates in triplicates at a density of 100-500 cells/well in 2 mL of medium containing 10% fetal bovine serum (FBS). After 24 h incubation, cells were then cultured with medium with 5% FBS as control, or the same medium containing a series of doses of metformin for 14 d in a 37°C humidified atmosphere containing 95% air and 5% CO2. The cell clones were stained for 15 min with a solution containing 0.5% crystal violet and 25% methanol, followed by three rinses with tap water to remove excess dye. The colony numbers were counted with gel documentation system EAGLE EYETM II (Stratagene, La Jolla, CA).
Flow cytometric analysis
Flow cytometric analyses were performed to define the cell cycle distribution for metformin treated and untreated cells. In brief, cells grown in 100-mm culture dishes were harvested by trypsinization and fixed with 70% ethanol. Cells were stained for total DNA content with a solution containing 50 μg/mL propidium iodide and 100 μg/mL RNase I in phosphate buffered saline (PBS) for 30 min at 37°C. Cell cycle distribution was then analyzed at the Flow Cytometry Core Facility of UCDHSC with a FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA).
Caspase enzymatic activity assay
Caspase enzymatic activities were measured using a modified colorimetric assay kit from Clontech Laboratories, Inc. (Palo Alto, CA). The assay was based on spectrophotometric detection of the chromophore p-nitroanilide (pNA), which is cleaved from caspase-specific substrates by activated caspases (DEVD-pNA by activated caspase-3, IETD-pNA by activated caspase-8, and LEHD-pNA by activated caspase-9).
Quantification of apoptosis
An apoptosis ELISA kit (Roche Diagnostics Corp.) was used to quantitatively measure cytoplasmic histone-associated DNA fragments (mononucleosomes and oligonucleosomes). This photometric enzyme immunoassay was performed according to the manufacturer’s instructions.
Western blotting analysis
Protein expression levels were determined by Western blot analysis. Briefly, cells were lysed in a buffer containing 50 mmol/L Tris, pH 7.4, 50 mmol/L NaCl, 0.5% NP-40, 50 mmol/L NaF, 1 mmol/L Na3VO4, 1 mmol/L phenylmethylsulfonyl fluoride, 25 g/mL leupeptin, and 25 g/mL aprotinin. The lysates were centrifuged at full speed in a microcentrifuge for 20 min and the supernatants were collected for protein concentration determination by the Coomassie Plus protein assay reagent (Pierce Chemical Co., Rockford, IL). Equal amounts of cell lysates were boiled in Laemmli SDS-sample buffer, resolved by SDS-PAGE, and Western blot analysis with specific antibodies as described in the Figure legends.
RESULTS
Metformin inhibits cell proliferation/survival in pancreatic cancer cells
In each pancreatic cancer cell line tested, metformin inhibited cell proliferation in a dose-dependent manner in MTS assays (Figure 1A). The MTS IC50 values for metformin activity were less than 5 mmol/L (in three of the four lines ASPC 1, BxPc 3, and PANC 1); while for SW1990 cells it was 10 mmol/L. To study the long- term effects of metformin on pancreatic cancer cells, we used clonogenicity assays. Metformin resulted in significantly fewer colonies at concentrations well below what was inhibitory using the 72 h MTS assay described above. The lowest inhibitory concentrations for the 4 cell lines ASPC 1, BxPc 3, and PANC 1, and SW1990 were 1 mmol/L, 0.01 mmol/L, 1 mmol/L, and 0.05 mmol/L, respectively (Figure 1B).
Figure 1.
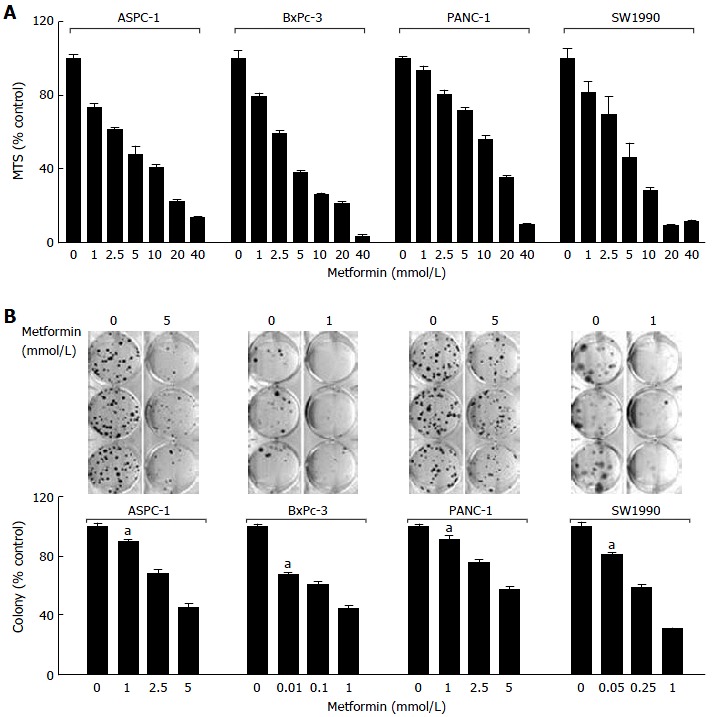
Metformin inhibits proliferation/survival in the basal-like subtype of pancreatic cancer cells. A: ASPC-1, BxPc-3, PANC-1 and SW1990 were plated onto 96-well plates with either complete medium or medium containing a series doses of metformin; B: ASPC-1, BxPc-3, PANC-1 and SW1990 were grown in triplicates in the absence or presence of metformin at different concentrations for 2-3 wk.
Metformin blocks cell cycle progression and induces apoptosis in pancreatic cancer cells
To study the effects of metformin on cell cycle distribution and progression, we used flow cytometric methods on metformin treated and untreated cells. Metformin reduced the number of cells in G1 and increased the percentage of cells in S phase as well as the apoptotic fraction (Figure 2). The percentages of apoptotic cells in all four pancreatic cancer cell lines were increased significantly in metformin-treated as compared with untreated cells (Figure 2). These data suggest that the effects of metformin on pancreatic cancer cells were distinct and, therefore, should occur through different molecular mechanisms.
Figure 2.
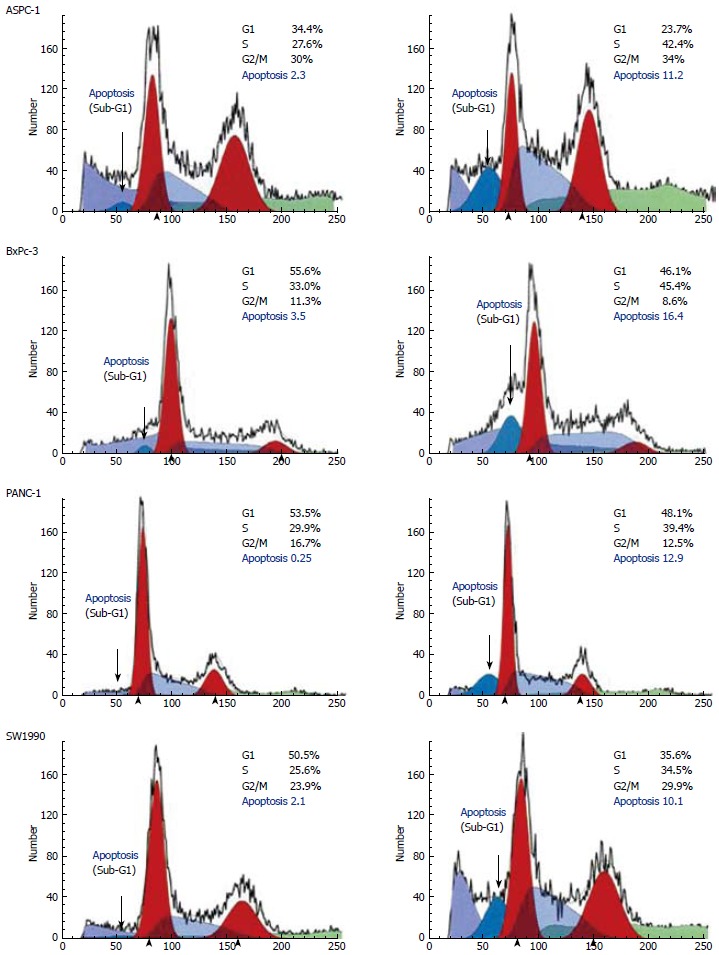
Metformin induces apoptosis and blocks cell cycle progression in the pancreatic cancer cells.
Metformin selectively induces apoptosis via caspases activation in pancreatic cancer cells
ELISA specific assays for apoptosis were used to quantitatively evaluate metformin associated apoptosis in pancreatic cancer cell lines. Metformin induced apoptosis in all pancreatic cancer cell lines (Figure 3A). In Western blot studies, metformin also induced PARP cleavage (an indicator of caspase activation) pancreatic cancer cell lines (Figure 3A). Consistent with these data, protein levels of the pro-caspases-8, -9, and -3 were each reduced by metformin in the pancreatic cell lines (Figure 3A). Using these three assays (Western blot analyses of PARP cleavage, an apoptosis specific ELISA and caspase activity assays) the effects of metformin over a range of treatment intervals and drug concentrations were evaluated.
Figure 3.
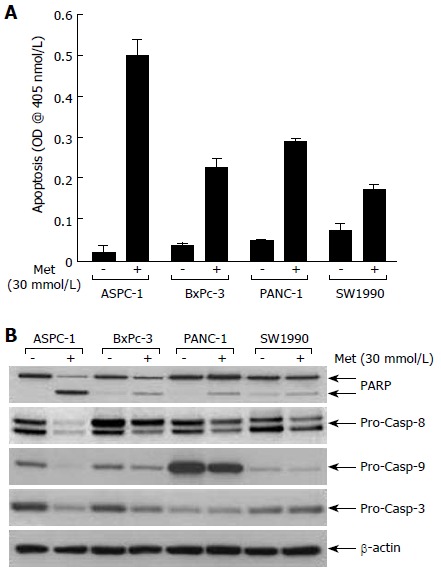
Metformin increases PARP cleavage, reduces levels of pro-caspase-8, -9, -3, and induces apoptosis in pancreatic cancer cells.
Activation of both caspase-8 and caspase-9 contributes to metformin-induced apoptosis
There are two well characterized caspase cascades in apoptosis: one initiated by cell surface death receptors (the so-called extrinsic pathway via caspase-8) and the other triggered by changes in mitochondrial integrity (caspase-9 activation, known as the intrinsic pathway)[9-11]. To further define the effects of metformin, we used specific caspase inhibitors to determine which might block metformin-induced apoptosis. The general caspase inhibitor, VAD-fmk completely abolished metformin-induced PARP cleavage and apoptosis in three cell lines (Figure 4). The caspase-8 specific inhibitor (IETD-fmk) and the caspase-9 specific inhibitor (LEHD-fmk) only partially abrogated metformin-induced apoptosis and PARP cleavage in BxPc-3 and PANC-1 cells (Figure 4B and C), although IETD-fmk had a much greater effect than LEHD-fmk in ASPC-1 cells (Figure 4A).
Figure 4.
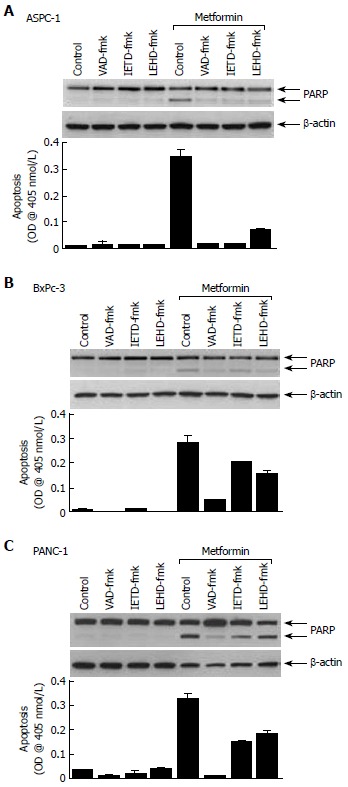
Activation of both caspase-8 and caspase-9 contributes to metformin-induced apoptosis in pancreatic cancer cells. A: ASPC-1; B: BxPc-3; C: PANC-1.
Metformin reduces epidermal growth factor receptor (EGFR) and inhibits Akt and MAPK signaling
Pancreatic carcinomas were well known for EGFR overexpression, and they appear to utilize EGFR as an important pro-carcinogenic, pro-growth receptor. Using Western blot analyses, we observed that metformin treatment dramatically reduced EGFR and P-MAPK, in both a time- and dose-dependent manner in three cell lines tested (Figure 5). Of interest, short-term (8 h) treatment with metformin transiently raised P-MAPK levels. Early induction of MAPK signaling may reflect metformin’s interaction with the insulin receptor[12]. P-Akt levels were also significantly lowered by metformin treatment in one cell line (ASPC-1; Figure 5A), although there were only minor changes of P-Akt in BxPc-3 cells (Figure 5B). P-Akt was undetectable and unchanged by metformin in PANC-1 cells (Figure 5C). This data suggests that apoptosis induced by metformin may be mechanistically driven by a reduced EGFR, with subsequent inactivation of downstream signaling involving MAPK and to a lesser extent Akt.
Figure 5.
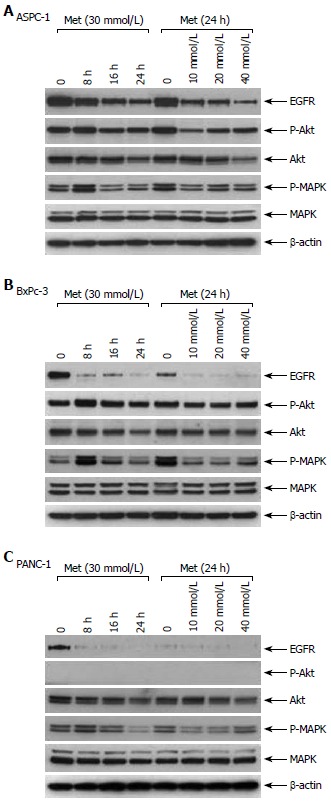
Metformin lowers EGFR expression levels and inhibits downstream signaling in pancreatic cancer cells. A: ASPC-1; B: BxPc-3; C: PANC-1.
DISCUSSION
The development, continued growth and metastasis of pancreatic cancer are driven by multiple genetic and epigenetic changes, including inactivation of tumor suppressor genes and activation of proto-oncogenes[13]. Since the last decade, molecular biology and technology have contributed significantly to the development of therapeutic agents in medicine, and especially in oncology. The major areas include inhibition of tumor growth, inhibition of metastatic invasion, and inhibition of intercellular signal transduction and compensation of gene expression[14]. But it still needs a long time to treat pancreatic cancer patients. Up to now, only two combinations, Gem plus erlotinib and Gem plus capecitabine have achieved a slightly longer survival of the patients[15].
Metformin has a long track record of human use, with limited toxicity and it is relatively inexpensive. It might, therefore, be of great clinical benefit for pancreatic cancer treatment. According to our data, concomitant metformin therapy can enhance the response of patients to DNA damaging agents (chemotherapy and radiation therapy) because of their extended arrest in the S phase. Metformin should also enhance RTK inhibitor and anti-EGFR treatment response, because of its action on EGFR and P-MAPK effects. Finally, metformin might enhance treatment with apoptosis-inducing agents that inhibit PARP, for it induces PARP cleavage. Its usage in pancreatic cancer patients may also have additional benefits, including weight control, stabilization of pre-diabetic syndromes, regulation of glucose/insulin and adipogenesis pathways.
Two recent studies have indicated that EGFR was detected in more than 95% of patients with pancreatic cancer[16,17]. Also, co-expression of EGFR and its ligands is a common occurrence in pancreatic cancer and has been shown to function as an autocrine[13]. Real advancement towards individualized pancreatic cancer treatment will require understanding the molecular mechanisms underlying pancreatic cancer biology[18,19]. Early studies of basal carcinomas focused on EGFR as a molecular target, although EGFR inhibitors have not shown great efficacy in clinical trials[11,20]. The data we present here indicates that metformin may serve as a valuable treatment option for pancreatic cancer patients.
Metformin is widely used as a first-line treatment for pre-diabetic syndromes and type II diabetes[21]. Of interest, some studies have shown that women treated with metformin have a lower overall incidence of cancer, including pancreatic cancer[8]. Metformin may therefore be useful as a preventative agent in selected patients to reduce, either directly through cell growth inhibition or indirectly through obesity and diabetes control. Further studies of metformin may provide more insights into its efficacy.
In summary, we have demonstrated that the anti-diabetic drug, metformin, selectively induces apoptosis through activation of the caspase cascade, abrogation of EGFR and downstream signaling in the basal subtype of pancreatic cancer cells. Metformin may be a low toxicity, novel therapeutic strategy for the difficult-to-treat pancreatic cancer patients.
ACKNOWLEDGMENTS
We thank all the staff in the Pancreatic Cancer Study Group.
COMMENTS
Background
Pancreatic carcinoma is one of the tumors with a high incidence rate. It is less sensitive to standard adjuvant chemotherapy. Metformin (1,1-dimethylbiguanide hydrochloride) is the most widely prescribed anti-hyperglycemic agent in the world. Population-based studies have shown that patients treated with metformin exhibit unexpected but beneficial reductions in both obesity and cancer. But no study about the therapeutic effect of metformin in pancreatic caner has been reported.
Research frontiers
There are many studies about the treatment of pancreatic cancer. Target therapy is the most popular part of pancreatic cancer research as well as the use of traditional medicine. Metformin is one of the traditional drugs which are thought to have anti-cancer effects.
Innovations and breakthroughs
This study showed that metformin significantly inhibited cell proliferation and induced apoptosis in all pancreatic cell lines, and the metformin-induced apoptosis was associated with poly-ADP-ribose polymerase leavage, activation of caspase-3, -8, and -9 in a time- and dose-dependent manner.
Applications
Metformin is a common drug which has been used for many years. This study has suggested that it had a potential therapeutic effect for pancreatic cancer. Although further in vivo studies are needed, metformin may be a low toxicity, novel therapeutic drug for the difficult-to-treat pancreatic cancer patients.
Peer review
This is a very interesting study. Metformin, the commonly used anti-diabetic drug has been proven to have anti-cancer effect in pancreatic carcinoma in in vitro studies with pancreatic cancer cell line. The study was strictly done with standard molecular techniques. The result is of clinical implication in the treatment of pancreatic cancer.
Footnotes
Peer reviewers: Jia-Yu Xu, Professor, Shanghai Second Medical University, Rui Jin Hospital, 197 Rui Jin Er Road, Shanghai 200025, China; Michael Steer, Professor, Department of Surgery, Tufts-Nemc, 860 Washington St, Boston, Ma 02111, United States
Supported by The National Natural Science Foundation of China, No. 30700360
S- Editor Zhong XY L- Editor Ma JY E- Editor Lin YP
References
- 1.Wagner M, Redaelli C, Lietz M, Seiler CA, Friess H, Buchler MW. Curative resection is the single most important factor determining outcome in patients with pancreatic adenocarcinoma. Br J Surg. 2004;91:586–594. doi: 10.1002/bjs.4484. [DOI] [PubMed] [Google Scholar]
- 2.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 3.Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, Thun MJ. Cancer statistics, 2006. CA Cancer J Clin. 2006;56:106–130. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- 4.Azria D, Ychou M, Jacot W, Thezenas S, Lemanski C, Senesse P, Prost P, Delard R, Masson B, Dubois JB. Treatment of unresectable, locally advanced pancreatic adenocarcinoma with combined radiochemotherapy with 5-fluorouracil and cisplatin. Pancreas. 2002;25:360–365. doi: 10.1097/00006676-200211000-00007. [DOI] [PubMed] [Google Scholar]
- 5.Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet. 2004;363:1049–1057. doi: 10.1016/S0140-6736(04)15841-8. [DOI] [PubMed] [Google Scholar]
- 6.Czito BG, Willett CG, Bendell JC, Morse MA, Tyler DS, Fernando NH, Mantyh CR, Blobe GC, Honeycutt W, Yu D, et al. Increased toxicity with gefitinib, capecitabine, and radiation therapy in pancreatic and rectal cancer: phase I trial results. J Clin Oncol. 2006;24:656–662. doi: 10.1200/JCO.2005.04.1749. [DOI] [PubMed] [Google Scholar]
- 7.Hundal RS, Inzucchi SE. Metformin: new understandings, new uses. Drugs. 2003;63:1879–1894. doi: 10.2165/00003495-200363180-00001. [DOI] [PubMed] [Google Scholar]
- 8.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304–1305. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 10.Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 11.Green MR. Targeting targeted therapy. N Engl J Med. 2004;350:2191–2193. doi: 10.1056/NEJMe048101. [DOI] [PubMed] [Google Scholar]
- 12.Holland W, Morrison T, Chang Y, Wiernsperger N, Stith BJ. Metformin (Glucophage) inhibits tyrosine phosphatase activity to stimulate the insulin receptor tyrosine kinase. Biochem Pharmacol. 2004;67:2081–2091. doi: 10.1016/j.bcp.2004.02.016. [DOI] [PubMed] [Google Scholar]
- 13.Papageorgio C, Perry MC. Epidermal growth factor receptor-targeted therapy for pancreatic cancer. Cancer Invest. 2007;25:647–657. doi: 10.1080/07357900701522653. [DOI] [PubMed] [Google Scholar]
- 14.Dhar A, Mehta S, Banerjee S, Dhar K, Dhar G, Sengupta K, Ray G, Banerjee SK, Campbell DR. Epidermal growth factor receptor: is a novel therapeutic target for pancreatic cancer? Front Biosci. 2005;10:1763–1767. doi: 10.2741/1659. [DOI] [PubMed] [Google Scholar]
- 15.Boeck S, Hinke A, Wilkowski R, Heinemann V. Importance of performance status for treatment outcome in advanced pancreatic cancer. World J Gastroenterol. 2007;13:224–227. doi: 10.3748/wjg.v13.i2.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiong HQ, Rosenberg A, LoBuglio A, Schmidt W, Wolff RA, Deutsch J, Needle M, Abbruzzese JL. Cetuximab, a monoclonal antibody targeting the epidermal growth factor receptor, in combination with gemcitabine for advanced pancreatic cancer: a multicenter phase II Trial. J Clin Oncol. 2004;22:2610–2616. doi: 10.1200/JCO.2004.12.040. [DOI] [PubMed] [Google Scholar]
- 17.Bloomston M, Bhardwaj A, Ellison EC, Frankel WL. Epidermal growth factor receptor expression in pancreatic carcinoma using tissue microarray technique. Dig Surg. 2006;23:74–79. doi: 10.1159/000093497. [DOI] [PubMed] [Google Scholar]
- 18.Baselga J, Arteaga CL. Critical update and emerging trends in epidermal growth factor receptor targeting in cancer. J Clin Oncol. 2005;23:2445–2459. doi: 10.1200/JCO.2005.11.890. [DOI] [PubMed] [Google Scholar]
- 19.Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med. 2005;353:172–187. doi: 10.1056/NEJMra044389. [DOI] [PubMed] [Google Scholar]
- 20.Spector NL, Xia W, Burris H 3rd, Hurwitz H, Dees EC, Dowlati A, O'Neil B, Overmoyer B, Marcom PK, Blackwell KL, Smith DA, Koch KM, Stead A, Mangum S, Ellis MJ, Liu L, Man AK, Bremer TM, Harris J, Bacus S. Study of the biologic effects of lapatinib, a reversible inhibitor of ErbB1 and ErbB2 tyrosine kinases, on tumor growth and survival pathways in patients with advanced malignancies. J Clin Oncol. 2005;23:2502–2512. doi: 10.1200/JCO.2005.12.157. [DOI] [PubMed] [Google Scholar]
- 21.Kirpichnikov D, McFarlane SI, Sowers JR. Metformin: an update. Ann Intern Med. 2002;137:25–33. doi: 10.7326/0003-4819-137-1-200207020-00009. [DOI] [PubMed] [Google Scholar]