

ABSTRACT
Rad/Rem/Rem2/Gem (RGK) proteins are Ras-like GTPases that potently inhibit all high-voltage-gated calcium (CaV1/CaV2) channels and are, thus, well-positioned to tune diverse physiological processes. Understanding how RGK proteins inhibit CaV channels is important for perspectives on their (patho)physiological roles and could advance their development and use as genetically-encoded CaV channel blockers. We previously reported that Rem can block surface CaV1.2 channels in 2 independent ways that engage distinct components of the channel complex: (1) by binding auxiliary β subunits (β-binding-dependent inhibition, or BBD); and (2) by binding the pore-forming α1C subunit N-terminus (α1C-binding-dependent inhibition, or ABD). By contrast, Gem uses only the BBD mechanism to block CaV1.2. Rem molecular determinants required for BBD CaV1.2 inhibition are the distal C-terminus and the guanine nucleotide binding G-domain which interact with the plasma membrane and CaVβ, respectively. However, Rem determinants for ABD CaV1.2 inhibition are unknown. Here, combining fluorescence resonance energy transfer, electrophysiology, systematic truncations, and Rem/Gem chimeras we found that the same Rem distal C-terminus and G-domain also mediate ABD CaV1.2 inhibition, but with different interaction partners. Rem distal C-terminus interacts with α1C N-terminus to anchor the G-domain which likely interacts with an as-yet-unidentified site. In contrast to some previous studies, neither the C-terminus of Rem nor Gem was sufficient to inhibit CaV1/CaV2 channels. The results reveal that similar molecular determinants on Rem are repurposed to initiate 2 independent mechanisms of CaV1.2 inhibition.
KEYWORDS: CaV1.2, L-type calcium channel, Gem, Rem, RGK protein
Introduction
High-voltage-activated (HVA) calcium channels (CaV1.1–1.4, CaV2.1–2.3) couple electrical excitation to physiological responses in excitable cells.1 These channels are hetero-oligomeric protein complexes comprising a pore-forming α1-subunit assembled with auxiliary proteins that include β/α2δ/γ subunits and calmodulin.2,3 There are 7 genes coding for HVA calcium channel α1-subunits, each with multiple splice variants. The transmembrane α1-subunit defines the channel subtype and contains the voltage sensor, the selectivity filter, and the water-filled pore that provides a passageway for Ca2+ ions to traverse the hydrophobic plasma membrane. The auxiliary subunits profoundly regulate the trafficking and gating properties of α1-subunits and are essential for the physiological function of CaV1/CaV2 channels. In particular, CaVβ (β1-β4) is crucial for forming functional CaV1/CaV2 channels as it is obligatory for α1 trafficking to the cell surface, increases the open probability (PO) of surface channels, and imposes isoform-dependent inactivation gating signatures.4,5 Modulation of specific CaV1/CaV2 channels by signaling proteins, Ca2+ ions, or small molecules is a powerful method to regulate diverse aspects of physiology including cardiac contractility, synaptic plasticity, insulin release, and gene expression.2,6-9 Molecules that block CaV1/CaV2 channels with high specificity and potency are sought after as therapeutics for various cardiovascular and neurological disorders including cardiac arrhythmias, pain, and neurodegenerative diseases.10-12
Rad, Rem, Rem2 and Gem/Kir (RGK) proteins are Ras-like monomeric G-proteins that potently and non-selectively inhibit all CaV1/CaV2 channels.13-16 Distinct RGK proteins are expressed in muscle, neurons, pancreas, and immune cells, and knockout mice studies suggest their inhibition of CaV channels is physiologically relevant in different systems.17-22 The potential of using RGK proteins as precisely targeted genetically-encoded CaV channel blockers for therapeutic applications has been explored in proof-of-concept experiments in heart in vivo and in vitro.23,24 A major limitation to the practical use of RGKs as CaV channel blockers is that they non-selectively inhibit CaV1/CaV2 channels. Development of selective RGK-derived CaV1/CaV2 channel inhibitors could accelerate their applied use as genetically-encoded CaV channel blockers.
All four RGK proteins interact directly with CaVβ.13,14,25 Recently, we examined the role of Rem/CaVβ interaction in Rem inhibition of recombinant CaV1.2 channels using a mutant β2a (β2aTM) which contains point mutations (D243A, D319A and D321A) that selectively abolish binding to RGK proteins,25 but retains the capacity to modulate CaV1/CaV2 channel trafficking and gating.26 We discovered that Rem utilizes 2 independent pathways to inhibit CaV1.2 channels—a β-binding-dependent (BBD) mode and a direct α1C-binding-dependent (ABD) mechanism, respectively. The BBD pathway likely explains the indiscriminate nature of RGK inhibition since all CaV1/CaV2 channels require assembly with β for their functional maturation.4,5 Understanding the molecular bases for the ABD Rem inhibition of CaV1.2 is of special interest because this mechanism could potentially be exploited to develop CaV1/CaV2 isoform-selective channel blockers. The ABD mechanism of CaV1.2 inhibition requires Rem binding to α1C N-terminus (α1CNT).26 However, the determinants on Rem itself necessary for binding α1CNT and initiating ABD CaV1.2 inhibition are unknown. Here, we report that the Rem distal C-terminus (RemDCT) and guanine nucleotide binding domain (G-domain) are both required for ABD CaV1.2 inhibition. RemDCT binds α1CNT anchoring the G-domain, which presumably engages with an as-yet-unidentified site either within the channel complex or elsewhere, to initiate CaV1.2 inhibition. Remarkably, these same Rem determinants—RemDCT and G-domain—are also utilized, but with different interaction partners (plasma membrane and CaVβ, respectively), for BBD CaV1.2 inhibition.26-29
Results
Rem and Gem differ in their capacity to use an α1C-binding-dependent mechanism to inhibit CaV1.2 channels
It is now well-established that RGK proteins strongly inhibit currents through CaV1/CaV2 channels.13-16,30-33 Here, we recapitulate this effect by examining the impact of Rem and Gem on CaV1.2 channels reconstituted by transient transfection of HEK293 cells with α1C + β2a subunits. As expected, control cells express large whole-cell L-type currents (ICa,L) which are deeply inhibited equally by either co-expressed Rem or Gem (Fig. 1A, B). To isolate the ABD component underlying RGK inhibition of ICa,L, we reconstituted CaV1.2 with a β2a triple mutant (β2aTM) that does not bind RGKs but retains modulatory actions on the channel complex.25,26 Channels reconstituted with α1C + β2aTM display robust currents which are differentially affected by Rem and Gem, respectively. Whereas Rem significantly inhibits α1C + β2aTM channels, Gem has no impact on ICa,L through these mutant CaV1.2 channels (Fig. 1C,D). These results confirm our recent finding that Gem uses a solely BBD mechanism to inhibit CaV1.2 channels, whereas Rem uses both BBD and ABD pathways to achieve ICa,L block.26
Figure 1.

Rem and Gem differ in their capacity to use a α1C-binding-dependent mechanism to inhibit CaV1.2 channels. (A) Exemplar Ba2+ currents from HEK293 cells expressing wild-type CaV1.2 (α1C + β2a) (left) in the presence of either Rem (middle) or Gem (right). (B) Population current density (Jpeak) vs. voltage relationships for wild-type CaV1.2 channels (▪, n = 6) co-expressed with either Rem ( ,n = 3) or Gem (
,n = 3) or Gem ( ,n = 4). (C) Exemplar Ba2+ currents from HEK293 cells expressing mutant CaV1.2 (α1C + β2aTM) (left) in the presence of either Rem (middle) or Gem (right). (D) Jpeak─voltage relationships for mutant CaV1.2 channels (▪, n = 9) co-expressed with Rem (,n = 7) or Gem (,n = 8). Data are means ± SEM.
,n = 4). (C) Exemplar Ba2+ currents from HEK293 cells expressing mutant CaV1.2 (α1C + β2aTM) (left) in the presence of either Rem (middle) or Gem (right). (D) Jpeak─voltage relationships for mutant CaV1.2 channels (▪, n = 9) co-expressed with Rem (,n = 7) or Gem (,n = 8). Data are means ± SEM.
Figure 2.
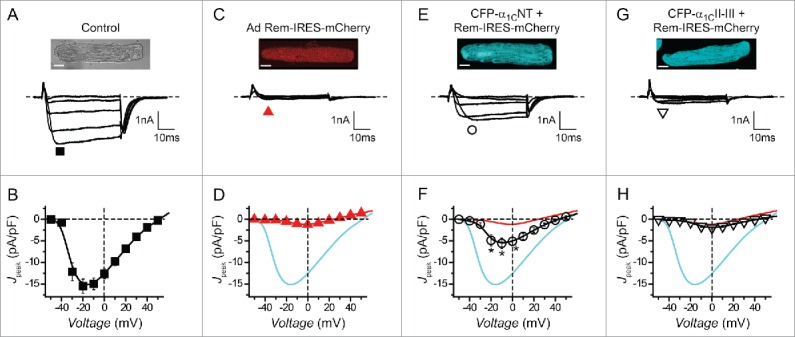
Cardiac myocytes possess a β-binding-independent mechanism to inhibit endogenous CaV1.2 channels. (A) Top, Gray scale image of rat ventricular myocyte. Scale bar, 10 μm. Bottom, representative whole-cell CaV1.2 channel currents from a cultured rat ventricular myocyte expressing CFP-α1CNT (▪, n = 8). (B) Population Jpeak─V relationship for control cardiomyocytes. (C-H) Data for cardiomyocytes expressing Rem-IRES-mCherry (,n = 8), CFP-α1CNT + Rem-IRES-mCherry (О, n = 10) and CFP-α1CII-III loop + Rem-IRES-mCherry (▿, n = 8), respectively; same format as A and B. Data for control (cyan line) and Rem-IRES-mCherry (red line) are reproduced for comparison. * P < 0.05 compared to either Rem-IRES-mCherry or control, one-way ANOVA.
α1C-binding-dependent Rem inhibition of ICa,L occurs in cardiac myocytes
The molecular determinants and mechanisms that distinct RGK proteins use to inhibit CaV channels can differ substantively in different cell types.30-32,34,35 Whether Rem inhibits endogenous CaV1.2 channels in their native context using the ABD mechanism is unknown. This information is crucial to gauge the potential physiological significance of this mode of channel inhibition and whether it can be exploited to design generally useful CaV1/CaV2 isoform-selective inhibitors. We previously showed that over-expressing an α1CNT peptide eliminates ABD CaV1.2 inhibition by competitively interfering with Rem binding to the full-length α1C N-terminus.26 We exploited this approach to determine whether Rem inhibits endogenous CaV1.2 channels in cardiac myocytes using the ABD mechanism.
Whole-cell patch clamp of cultured adult rat ventricular myocytes yielded large Ba2+ currents (Fig. 2, A and B; Ipeak = −15.5 ± 1.6 pA/pF, n = 8). Adenoviral-mediated over-expression of Rem-IRES-mCherry dramatically inhibited whole-cell current (Fig. 2, C and D; Ipeak = −1.2 ± 0.2 pA/pF, n = 8; P < 0.05 compared to control). Co-expressing CFP-α1CNT together with Rem-IRES-mCherry resulted in a partial rescue of current (Fig. 2, E and F; Ipeak = −5.6 ± 1.2 pA/pF, n = 8; P < 0.05 compared to Rem-IRES-mCherry alone), consistent with a significant contribution of the ABD mechanism to Rem inhibition of CaV1.2 in cardiac myocytes. This result was not due to the potentially trivial explanation that co-infecting myocytes with 2 adenoviruses led to reduced Rem expression because co-expressing CFP-α1C II-III loop did not appreciably rescue current blocked by Rem-IRES-mCherry (Fig. 2, G and H; Ipeak = −1.9 ± 0.3 pA/pF, n = 8). Patched cells were monitored for CFP and mCherry fluorescence ensuring that both proteins were expressed in the selected cardiomyocytes (Fig. S1).
These results demonstrate that ABD Rem inhibition of CaV1.2 occurs in a physiological context and provided strong motivation to probe the Rem molecular determinants underlying this mode of CaV1.2 inhibition.
Rem distal C-terminus interacts with α1CNT
How does Rem interact with α1CNT, and are the determinants for this interaction lacking in Gem? Initial expectations for answers to these questions were derived from comparing Rem and Gem primary sequences. Mouse Rem contains 297 amino acids and can be nominally divided into 3 parts based on comparison with the prototypical Ras: N-terminus (residues 1–77), G-domain (residues 78–246), and C-terminus (residues 247–297) (Fig. 3). Ras is principally composed of a G-domain, a structure comprised of a 6-stranded β-sheet surrounded by 5 α-helices with 5 conserved loops (G1-G5) that form the guanine-nucleotide binding site.36,37 The G-domains of all 4 RGK proteins are highly conserved, bind guanine nucleotides, and adopt a similar structural fold as the Ras G-domain.15,38 The N-terminus extensions of Rem and Gem are variable (< 30% homology); the C-termini extensions contain a variable proximal region (PCT; residues 247–257 in Rem and 244–256 in Gem, respectively) and a conserved distal region (DCT; 70% homology) (Fig. 3).
Figure 3.

Primary sequence alignment of Rem and Gem. Sequence alignment of murine Rem, human Gem and human H-Ras. Identical residues are shaded green; similar residues are shaded in cyan. PCT, proximal C-terminus; DCT, distal C-terminus.
We used a 3-cube fluorescence resonance energy transfer (FRET) assay39-41 to determine which regions of Rem associate with α1CNT and how these compared with determinants required for binding CaVβ (Fig. 4) We generated YFP-α1CNT and YFP-β3, respectively, and used these in 3-cube FRET experiments with CFP-tagged wild-type (wt) Rem and Rem-deletion mutants, respectively. As a negative control for these experiments, we first measured FRET between CFP-FRB and either YFP-α1CNT or YFP-β3, respectively. FRB is the rapamycin-binding domain from the kinase mTor,42,43 and is not expected to associate with either YFP-α1CNT or YFP-β3. HEK293 cells co-expressing CFP-FRB and either YFP-α1CNT or YFP-β3 displayed low FRET efficiencies (FRETeff) of 0.018 ± 0.005 and 0.031 ± 0.004, respectively (Fig. 4, B and C). By contrast, cells expressing CFP-Rem and either YFP-α1CNT or YFP-β3 displayed significantly elevated FRETeff of 0.147 ± 0.011 (n = 37) and 0.150 ± 0.007 (n = 42), respectively (Fig. 4, B and C). A truncated Rem lacking the final 32 amino acids of the C-terminus (CFP-Rem1–265) displayed no interaction with YFP-α1CNT (FRETeff = 0.037 ± 0.005, n = 33) (Fig. 4B), while the association with YFP-β3 was preserved (FRETeff = 0.125 ± 0.008, n = 40) (Fig. 4C). Conversely, CFP-Rem224–297 which lacks the Rem N-terminus and most of the G-domain showed robust binding to YFP-α1CNT (FRETeff = 0.439 ± 0.035, n = 20) but no interaction with YFP-β3 (FRETeff = 0.040 ± 0.004, n = 35) (Fig. 4, B and C). Consistent with these results, YFP-Rem, but not YFP-Rem265, interacted with full-length CFP-α1C as reported by an elevated FRET efficiency (Fig. S2).
Figure 4.
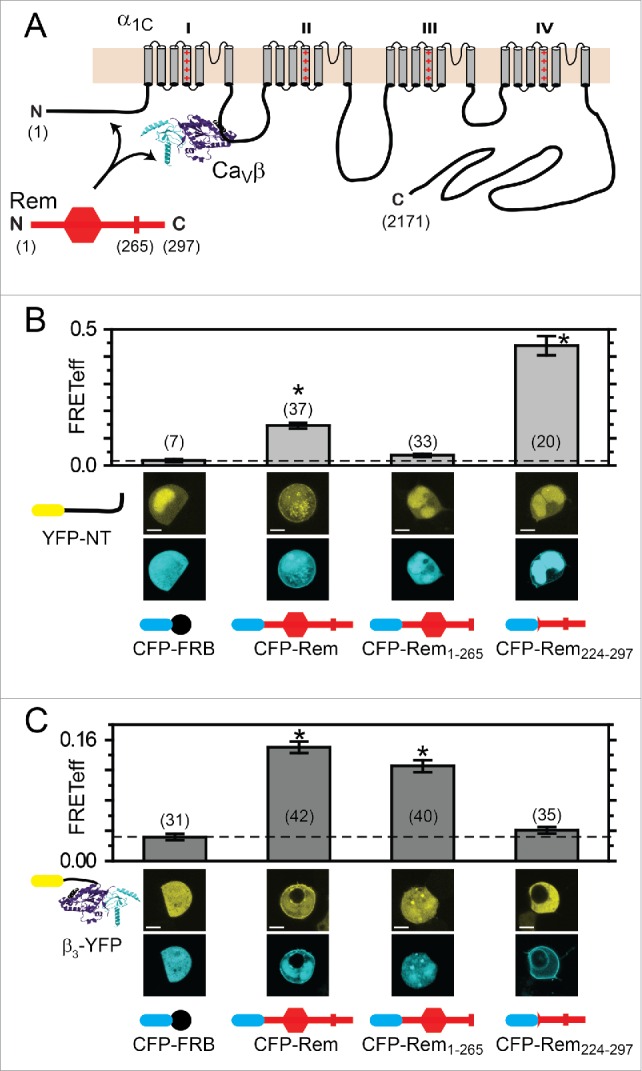
FRET detection of Rem determinants underlying interaction with α1C N-terminus and CaVβ. (A) Schematic of α1C, CaVβ, and Rem. Rem interacts independently with CaVβ and α1C N-terminus. (B) FRET detection of interactions between YFP-α1CNT and distinct CFP-tagged wt or truncated Rem constructs. *P < 0.05 compared with CFP-FRB using one-way ANOVA and Bonferroni test (C) FRET detection of interactions between YFP-CaVβ3 and distinct CFP-tagged wt or truncated Rem constructs. *P < 0.05 compared with CFP-FRB using one-way ANOVA and Bonferroni test. Data are means ± SEM.
Taken together with previous results, these data support the binary interpretation that separate determinants underlie Rem binding to α1CNT and CaVβ, respectively: the RemDCT is responsible for association with α1CNT but plays no role in binding CaVβ; Rem G-domain mediates interaction with CaVβ but does not contribute to α1CNT binding.
RemDCT determinants required for binding α1CNT and ABD CaV1.2 inhibition
To more precisely localize the residues within RemDCT responsible for binding α1CNT we generated 2 additional Rem deletion mutants (CFP-Rem1–285 and CFP-Rem1–275) and used FRET to assess their interaction with YFP-α1CNT (Fig. 5). CFP-Rem1–285 co-expressed with YFP-α1CNT yielded a robust FRET signal (FRETeff = 0.071 ± 0.01, n = 32) that was comparable to that obtained with wt CFP-Rem (FRETeff = 0.085 ± 0.009, n = 24, P = 1 compared to CFP-Rem1–285) (Fig. 5B). By contrast, CFP-Rem1–275 displayed a significantly reduced FRET signal when co-expressed with YFP-α1CNT (FRETeff = 0.040 ± 0.009, n = 13, P = 0.028 compared to CFP-Rem, one-way ANOVA), consistent with reduced binding between the 2 proteins (Fig. 5B). These results were bolstered by complementary co-immunoprecipitation experiments (Fig. 5C). We co-expressed CFP-tagged wt Rem or the deletion mutants without (lane 1) or with (lanes 2–5) YFP-α1CNT in HEK293 cells. Western blots of whole-cell lysates using anti-GFP antibody showed similar expression levels of all the Rem constructs, and confirmed the presence of co-expressed YFP-α1CNT (Fig. 5C, top). Immunoprecipitation using anti-Rem antibody led to comparable pull-down of all the Rem constructs. However, the amount of YFP-α1CNT that was co-immunoprecipitated differed among the various groups. A comparable amount of YFP-α1CNT was pulled down with CFP-Rem and CFP-Rem1–285, respectively. By comparison, a substantially lower quantity of YFP-α1CNT was co-immunoprecipitated with CFP-Rem1–275 and CFP-Rem1–265, respectively.
Figure 5.

Mapping Rem distal C-terminus determinants required for β-binding-dependent and α1C-binding-dependent inhibition of CaV1.2 (A) Sequence of Rem C-terminus with truncation sites indicated for Rem1–265, Rem1–275, and Rem1–285. (B) FRET detection of interactions between YFP-α1CNT and distinct CFP-tagged wt or truncated Rem constructs. *P < 0.05 compared with CFP-Rem using one-way ANOVA and Bonferroni test. Data are means ± SEM (C) Co-immunoprecipitation detection of interactions between YFP-α1CNT and CFP-tagged wt or truncated Rem constructs. Top panel shows Western blot of whole-cell lysates (input). (D) Bar chart showing mean peak current density from wild-type (black) and mutant (white) CaV1.2 channels ± wt or truncated Rem constructs. #P < 0.05 compared with α1C + β2a using one-way ANOVA and Bonferroni tests. *P < 0.05 compared with α1C + β2aTM using one-way ANOVA and Bonferroni tests. Data are means ± SEM.
We next comparatively evaluated how effectively the distinct Rem C-terminus deletion mutants inhibited CaV1.2 channels using the BBD and ABD mechanisms, respectively. Cells expressing either wt (α1C/β2a) or mutant (α1C/β2aTM) CaV1.2 channels were both inhibited by wt Rem but unaffected by Rem1–265, indicating that both the BBD and ABD mechanisms require RemDCT (Fig. 5D). Rem1–285 inhibited both α1C/β2a and α1C/β2aTM channels with a pattern indicating that both the BBD and ABD pathways were largely intact (Fig. 5D). By contrast, Rem1–275 significantly inhibited α1C/β2a but not α1C/β2aTM channels (Fig. 5D), indicating a selective loss of the ABD mode of inhibition. These data show that both the BBD and ABD modes of inhibition require RemDCT but not in an identical manner.
Rem C-terminus is not sufficient to inhibit CaV1.2
Could the interaction between Rem C-terminus and α1C be sufficient to inhibit CaV1.2? We explored this question by assessing the impact of a construct containing the last 75 amino acids of Rem (CFP-Rem224–297) on ICa,L from HEK293 cells expressing either wt (α1C/β2a) or mutant (α1C/β2aTM) CaV1.2 channels (Fig. 6). A similar extended C-terminus construct derived from Gem was previously reported to be sufficient to inhibit recombinant CaV2.1 channels.44 Surprisingly, CFP-Rem224–297 had no effect on either α1C/β2a or α1C/β2aTM channels (Fig. 6, A and B), indicating Rem C-terminus is necessary but not sufficient for either BBD or ABD CaV1.2 inhibition. This result implied an additional structural component in Rem is required for the observed ABD inhibition of CaV1.2.
Figure 6.
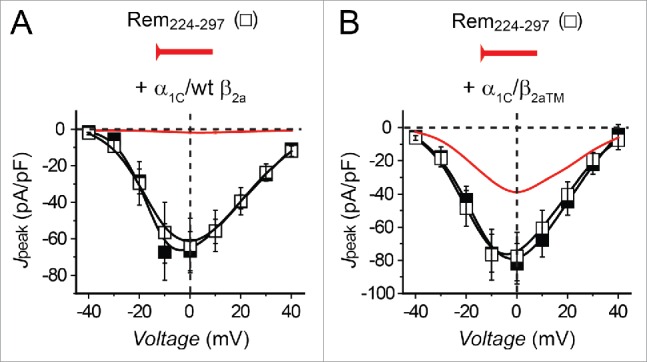
Rem C-terminus is not sufficient for CaV1.2 inhibition. (A) Jpeak─V relationships for α1C + β2a (▪, n = 4) and α1C + β2a + Rem224–297 (n = 8) channels. (B) Jpeak─V relationships for α1C + β2aTM (▪, n = 9) and α1C + β2aTM + Rem224–297 (□, n = 6) channels.
Rem G-domain is required for ABD CaV1.2 inhibition
We used a chimeric method to probe which additional Rem structural component(s) is required for BBD CaV1.2 inhibition. The approach exploited the functional difference between Rem and Gem with regards to the prevalence of the 2 distinct modes of CaV1.2 inhibition: whereas Rem diminishes ICa,L using both BBD and ABD mechanisms, Gem utilizes only the BBD pathway (Fig. 1). The inability of Gem to reconstitute ABD CaV1.2 inhibition could be due to a failure to bind α1C-NT. Alternatively, Gem could potentially bind α1C-NT well but lack the additional component required to transduce the functional effect. FRET experiments in cells co-expressing CFP-Gem and YFP-α1CNT indicated no interaction between the 2 proteins (Fig. 7A). Could simply donating the capacity to bind α1C-NT to Gem be sufficient to reconstitute ABD CaV1.2 block? To address this we examined the functional properties of a chimeric protein, Gem-r, in which the C-terminus of Gem was replaced with the corresponding region from Rem. Cells co-expressing YFP-α1CNT and CFP-Gem-r displayed robust FRET indicating successful transplantation of the capacity to directly bind α1C (Fig. 7A). Functionally, CFP-Gem-r potently inhibits wt α1C/β2a but has no effect on ICa,L recorded from mutant α1C/β2aTM channels (Fig. 7B), indicating this chimera displays only BBD CaV1.2 inhibition. Hence, simply targeting Gem to α1C-NT is not sufficient to reconstitute ABD inhibition of CaV1.2. The results further suggested that additional component(s) present in Rem N-terminus and/or G-domain but lacking in Gem were necessary for ABD inhibition of CaV1.2.
Figure 7.
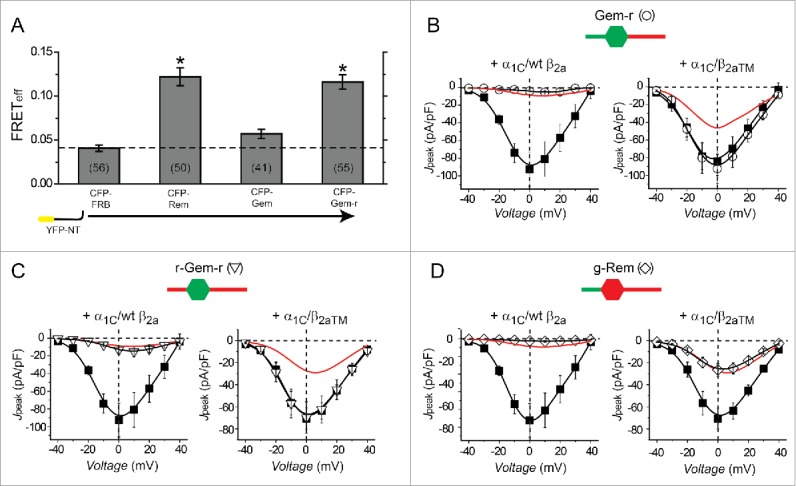
Chimeric Rem/Gem analyses of determinants required for α1C-binding-dependent CaV1.2 inhibition. (A) FRET detection of interactions between YFP-α1CNT and CFP-tagged Rem/Gem constructs. *P < 0.05 compared with CFP-FRB using one-way ANOVA and Bonferroni tests. Data are means ± SEM. (B) Left, Jpeak─V relationships for α1C + β2a (▪, n = 6) and α1C + β2a + Gem-r (О, n = 4) channels. Data for α1C + β2a + Rem is reproduced for comparison (red line). Right, Jpeak─V relationships for α1C + β2aTM (▪, n = 9) and α1C + β2aTM + Gem-r (О, n = 8) channels. Data for α1C + β2aTM + Rem is reproduced for comparison (red line). (C) Left, Jpeak─V relationships for α1C + β2a (▪, n = 6) (data are same as in (B)) and α1C + β2a + r-Gem-r (▿, n = 7) channels. Data for α1C + β2a + Rem is reproduced for comparison (red line). Right, Jpeak─V relationships for α1C + β2aTM (▪, n = 9) and α1C + β2aTM + r-Gem-r (▿, n = 10) channels. Data for α1C + β2aTM + Rem is reproduced for comparison (red line). (D) Left, Jpeak─V relationships for α1C + β2a (▪, n = 6) (data are same as in (B)) and α1C + β2a + g-Rem (◊, n = 9) channels. Data for α1C + β2a + Rem is reproduced for comparison (red line). Right, Jpeak─V relationships for α1C + β2aTM (▪, n = 9) (data are same as in (C)) and α1C + β2aTM + g-Rem (◊, n = 8) channels. Data for α1C + β2aTM + Rem is reproduced for comparison (red line). Data are means ± SEM.
We generated 2 additional chimeras to test this assumption: r-Gem-r contains the N- and C-terminus extensions of Rem appended to Gem G-domain; and g-Rem which consists of Gem N-terminus attached to Rem G-domain and C-terminus. Both r-Gem-r and g-Rem retained the capacity to potently inhibit wt α1C/β2a channels (Fig. 7, C and D). By contrast, the 2 chimeras showed a sharp dichotomy in their impact on mutant α1C/β2aTM channels—r-Gem-r was without effect while g-Rem inhibited these channels to the same extent as wt Rem. Taken together, the results indicate that the ABD mode of CaV1.2 inhibition minimally requires both the RemDCT and G-domain.
Discussion
This study provides new insights into molecular determinants underlying Rem-mediated inhibition of CaV1.2 channels. The data demonstrate that RemDCT binds α1CNT to initiate ABD Rem inhibition of CaV1.2 channels. However, Rem C-terminus is not by itself sufficient to reconstitute ABD inhibition. Chimeric Rem/Gem analyses indicated that Rem (but not Gem) G-domain is also necessary for the ABD mechanism of CaV1.2 inhibition.
The finding that RemDCT and G-domain are the essential motifs required for ABD inhibition of CaV1.2 was unexpected for 2 main reasons. First, previous work has established that these same 2 regions also underlie BBD Rem inhibition of CaV1.2.26-29,32 To activate BBD inhibition, RemDCT binds the plasma membrane while the G-domain interacts with CaVβ in the channel complex. This configuration essentially uses Rem to cross-link CaVβ and by association, the intracellular α1C I-II loop, to the plasma membrane (Fig. 8). This is hypothesized to induce a conformational change that effectively closes the channel pore. We previously exploited these insights into the BBD inhibition mechanism to develop a general approach─ termed channel inactivation induced by membrane-tethering an associated protein (ChIMP)—for generating novel genetically-encoded CaV1/CaV2 channel blockers.27 For the ABD inhibition pathway, this study shows that RemDCT and G-domain are also utilized, but do so by interacting with different binding partners. In this case, RemDCT interacts with α1CNT. Precisely how the Rem G-domain participates in ABD inhibition of CaV1.2 is not clear. We can deduce it plays an active role in the process because the homologous Gem G-domain cannot substitute for its function. The most likely scenario is that Rem G-domain selectively binds to another site within the cell, effectively cross-linking α1CNT to an intracellular anchor to initiate ABD CaV1.2 inhibition (Fig. 8). Candidate regions for the putative Rem G-domain interaction site include somewhere on the α1C subunit itself or the cytoskeleton. RGK proteins are known to interact with and regulate the cytoskeleton.15,45 Identification of the presumed interaction site for Rem G-domain that is necessary for ABD CaV1.2 inhibition is an important goal for future studies.
Figure 8.

Rem distal C-terminus and G-domain underlie both β-binding-dependent and α1C-binding-dependent CaV1.2 inhibition. Cartoon showing Rem determinants and putative interaction sites responsible for β-binding-dependent and α1C-binding-dependent CaV1.2 inhibition. Both types of inhibition rely on the Rem distal C-terminus (DCT) and G-domain (GD). For β-binding-dependent inhibition, RemDCT and GD bind the plasma membrane and CaVβ, respectively. For α1C-binding-dependent inhibition RemDCT binds α1C N-terminus while GD interacts with a secondary site either on the channel itself or elsewhere.
The second reason why the finding that RemDCT and G-domain underlie ABD inhibition was surprising is that these 2 regions are the most highly conserved among RGK proteins. Nevertheless, Gem and Rem2 lack the capacity for ABD CaV1.2 inhibition.26 The distal C-termini of all 4 RGKs anchor the respective proteins to the plasma membrane.46 Similarly, the G-domains of all RGKs bind CaVβ. The dual capability of all RGKs to bind the plasma membrane and CaVβs provides a simple explanation for the rather unique feature that RGKs potently and non-selectively inhibit all CaV1/CaV2 channels, i.e. they accomplish this through the BBD pathway. The unique capability of Rem to bind α1CNT and initiate ABD CaV1.2 inhibition reveals functional specialization among RGK C-termini and G-domains despite their high sequence homology.
Deepened understanding of how ABD inhibition arises could potentially be exploited to design novel genetically-encoded CaV channel blockers, in the same manner as we previously accomplished with the BBD mode of inhibition.27 The particular advantage of leveraging the ABD pathway in this manner is the likelihood that this approach could yield isoform-selective genetically-encoded CaV1/CaV2 blockers. One potential merit of such blockers is that they can be genetically targeted to precise cell populations and sub-cellular localizations, affording a degree of spatial selectivity that is difficult to achieve with small molecules.8,23,24,47 The prospect of engineering channel isoform selectivity into genetically encoded CaV channel blockers is intriguing and could potentially help address difficulties in developing selective small molecule blockers for specific CaV channel isoforms.48,49
What is the mechanism underlying ABD CaV1.2 inhibition by Rem? We previously showed that Rem inhibits recombinant wt α1C/β2a channels using at least 3 distinct mechanisms: (1) by reducing the number of channels at the cell surface; or by reducing the open probability (Po) of surface channels in 2 distinguishable ways─ (2) without an impact on voltage sensor movement (no effect on gating charge); (3) by partially immobilizing α1C voltage sensors (reduced gating charge).32 In mutant α1C/β2aTM channels, 2 of the mechanistic signatures of Rem inhibition (decreased channel surface density and reduced Po without an impact on voltage sensors) but not the third (reduced Po by voltage sensor immobilization) were eliminated. These previous results suggest that ABD inhibition by Rem is due to a reduced Po of surface channels mediated by a partial immobilization of α1C voltage sensor(s). Given the continuity between α1CNT and the domain I (DI) S1-S4 voltage sensor it is tempting to speculate based on the “cross-linking model” we propose wherein RemDCT binds α1CNT while the G-domain binds to a second site, that Rem impedes movement of at least the α1C DI voltage sensor. Voltage clamp fluorimetry experiments could be used to directly test how and which of the 4 α1C voltage sensors are affected by Rem.50
Our findings add to a growing list of molecules regulating the gating of CaV1 and CaV2 channels by targeting the N-termini of pore-forming α1-subunits. These include reports that the α1B N-terminus acts as a gated module that enables voltage-dependent G-protein βγ subunit inhibition of CaV2.2 channels;51 a role for α1C N-terminus in protein kinase C modulation of CaV1.2 channels;52,53 that the N-termini of CaV1.2 and CaV1.3 channels contains a Ca2+-CaM binding site (termed NSCaTE for N-terminal spatial Ca2+ transforming element) that controls local vs. global spatial Ca2+ selectivity for CaM regulation of CaV channels;54,55 and that deleting segments of α1C N-terminus increases ICa,L by enhancing channel Po.52,56
Some aspects of our findings contrast with previous reports. While we found that RemDCT is necessary for CaV1.2 inhibition, a Rem224–297 peptide that contained this whole region was not sufficient to block ICa,L. This result is in agreement with previous observations that a peptide comprising the Rem distal C-terminus alone (Rem266–297) did not inhibit CaV1.2 channels reconstituted in tsA201 cells,29 and that Rem2 C-terminus had no impact on endogenous CaV2.2 channels in SCG neurons.30 By contrast, it was previously reported that Gem223–296 (which lacks the Gem G-domain) was sufficient to bind auxiliary CaVβ and strongly inhibit recombinant CaV2.1 channels reconstituted in Xenopus oocytes.44 Further, a different study showed that a 12 amino acid peptide derived from Gem C-terminus is also sufficient to inhibit CaV2.1 channels in excised patches from Xenopus oocytes.57 These seemingly conflicting results could be due to differences in experimental techniques, cell systems, or simply the result of an emerging pattern of distinct mechanistic differences among RGK proteins with regard to the CaV channels inhibition. However, we found that CFP-Gem223–296 did not bind CaVβ or inhibit either CaV1.2 or CaV2.1 channels reconstituted in HEK293 cells (Fig. S3).
Recently, Beqollari et al. (2014) found that while both Rad and Rem overexpressed in adult mice flexor digitorum brevis fibers potently inhibited endogenous CaV1.1 currents, only Rad also reduced CaV1.1 Vage sensor movement.58 This is in contrast to cultured skeletal myotubes where Rem inhibited endogenous CaV1.1 current concomitantly with a reduction in gating charge.59 Chimeric Rad/Rem analyses indicated that the N-terminus of Rad was necessary for the reduced CaV1.1 Vage-sensor movement in adult skeletal muscle fibers. Overall, taken together with these and other previous reports, our results add to a growing awareness that RGK inhibition of CaV channels is underlain by a rich variety of determinants and mechanisms that are specific for the RGK subtype, CaV1/CaV2 channel isoforms, and the cell context.15
Materials and methods
Molecular biology
To generate cyan fluorescent protein (CFP)-tagged RGK constructs [mouse Rem (NM_009047); human Gem (NM_181702)] we first cloned CFP into pcDNA4.1 (Invitrogen) using KpnI and BamHI sites. Subsequently, Rem and Gem cDNA were amplified using polymerase chain reaction (PCR) and cloned downstream of CFP using BamHI and EcoRI sites. To generate yellow fluorescent protein (YFP)-tagged CaVβ2a, we PCR amplified and cloned YFP into pAd CMV vector using BamHI and XbaI sites. CaVβ2a was amplified by PCR and cloned upstream of YFP using NheI and BamHI sites. To generate YFP-tagged CaVβ3 we first cloned YFP into pcDNA3 using KpnI and BamHI sites. Subsequently, CaVβ3 was amplified downstream of YFP using BamHI and Xba sites. Point mutations in CaVβ were generated using QuikChange Site-Directed Mutagenesis Kit (Stratagene). PCR amplification and cloning was used to generate truncated Rem275 and Rem285 using BamHI and EcoRI sites. Chimeric RGK proteins were generated using overlap-extension PCR amplification and cloned into pcDNA4.1 (Invitrogen). All constructs were verified by sequencing.
Cell culture and transfection
HEK293 cells were maintained in DMEM supplemented with 10% FBS and 100 μg ml−1 penicillin-streptomycin. For electrophysiology experiments, HEK293 cells cultured in 35-mm tissue culture dishes were transiently transfected with α1C (4 μg), β2a (3 μg), T-antigen (2 μg) and the appropriate Rem, Gem or chimeric RGK construct (3 μg) using the calcium phosphate precipitation method. Cells were washed with PBS 4–6 h after transfection and maintained in supplemented DMEM. For confocal microscopy and FRET imaging experiments, transfected HEK293 cells were replated onto 35-mm fibronectin-coated No. 0 glass bottom culture dishes (MatTek). For electrophysiology experiments, cells were replated onto fibronectin-coated glass coverslips 24–48 h after transfection.
Generation of adenoviruses
Rem-IRES-mCherry adenoviral vectors were generated using the Adeno-X CMV vector kit (Clontech). CFP-tagged adenoviral vectors were generated using the AdEasy XL Adenoviral Vector System (Agilent Technologies) as previously described.31 The cDNA sequence comprising α1CNT (residues 1–153) and II-III intracellular loop (residues 800–942) were amplified using PCR and spliced in-frame downstream of CFP using overlap extension PCR. The whole cDNA cassette comprising either CFP-α1CNT or CFP-α1C II-III loop was cloned into pShuttle for construction of adenoviral vectors using the AdEasy system.
Adult rat ventricular myocyte culture and infection
Primary cultures of adult rat heart ventricular myocytes were prepared as previously described,60,61 and in accordance with the guidelines of the Columbia University Animal Care and Use Committee. Briefly, male Sprague-Dawley rats (Harlan) were euthanized with an overdose of halothane. Hearts were excised and ventricular myocytes isolated by enzymatic digestion with 1.7 mg Liberase-TM enzyme mix (Roche) using a Langendorff perfusion apparatus. Myocytes were cultured on laminin-coated glass coverslips (for electrophysiology experiments) or MatTek dishes (for confocal imaging experiments) in Medium 199 (Life Technologies) supplemented with (in mM): 5 carnitine, 5 creatine, 5 taurine, 0.5% penicillin-streptomycin-glutamine (Life Technologies), and 5% (vol/vol) FBS (Life Technologies). Cells were infected with 10–20 μL of viral stock in a final volume of 1–2 mL.
Electrophysiology
Whole-cell recordings on HEK293 cells were conducted 48–72 h after transfection at room temperature using an EPC-8 or EPC-10 patch clamp amplifier controlled by PULSE software (HEKA). Micropipettes were fashioned from 1.5 mm thin-walled glass with filament (World Precision Instruments). Series resistance was typically 1.7–2.5 MΩ. Internal solution contained (in mM): 135 cesium methanesulphonate (MeSO3), 5 cesium chloride, 5 EGTA, 1 MgCl2, 10 HEPES and 4 MgATP added fresh (pH 7.3). External solution contained (in mM): 140 tetraethylammonium-MeSO3, 5 BaCl2 and 10 HEPES (pH 7.3). Whole-cell I-V curves were generated from a family of step depolarizations (−50 to +70 mV from a holding potential of −90 mV). Currents were sampled at 25 kHz and filtered at 10 kHz. Traces were acquired at a repetition interval of 6 s. Leak and capacitive currents were subtracted using a P/8 protocol.
Whole-cell recordings of cultured rat ventricular myocytes were conducted at room temperature. Patch pipettes used typically had 1–2 MΩ series resistance when filled with internal solution containing (in mM): 150 cesium-methanesulfonate, 10 EGTA, 5 CsCl, 1MgCl2, 10 HEPES and 4 MgATP added fresh (pH 7.3). Cells were perfused with normal Tyrode external solution during formation of gigaohm seal. Tyrode external solution contained (in mM): 138 NaCl, 4KCl, 2 CaCl2, 1 MgCl2, 0.33 NaH2PO4, 10 HEPES (pH 7.4). After successful break-in to the whole-cell configuration the perfusing medium was switched to an external recording solution containing (in mM): 155 N-methy-D-glucamine-aspartate, 10 4-aminopyridine, 1 MgCl2, 5 BaCl2, 10 HEPES (pH 7.4). Currents were sampled at 50 KHz and filtered at 5 KHz and leak and capacitive currents were subtracted using a P/8 protocol.
Immunoprecipitation and Western blotting
Confluent cultures of HEK293 cells plated in 60-mm tissue culture dishes were harvested 48 h after transfection. Cells were washed in PBS and resuspended in 0.5 mL cold lysis buffer (50 mmol/L Tris-HCl, 150 mmol/L NaCl, 1% NP-40) containing protease inhibitor cocktail for 30 minutes. Cell lysates were centrifuged at 10,000 × g for 15 minutes at 4°C, and the supernatant precleared by incubation with 30 μL protein G beads slurry for 1 h. The mixture was centrifuged and the resulting supernatant incubated with 4 μg anti-Rem (SC58472, Santa Cruz) antibody and 30 μL protein G slurry for 1 h on a rotator. The mixture was again centrifuged, and the pellet washed 4 times with lysis buffer. 50 μL Laemmli sample buffer was added to the bead pellet and the mixture vortexed and heated (95°C for 10 minutes). The sample was centrifuged and the supernatant loaded onto a gel for subsequent SDS-PAGE and Western blot analyses. For immunoblots, primary antibodies to GFP (Invitrogen, A6455) were detected by horse-radish peroxidase-conjugated secondary antibodies (goat-anti rabbit obtained from Thermo Scientific, 32260) and enhanced chemiluminescence (Thermo Scientific, 34080).
Confocal imaging
Static images of HEK 293 and cultured rat ventricular myocytes cells expressing CFP- and YFP-tagged proteins were imaged using a Leica TCS SP2 AOBS MP Confocal microscope with a 40 × oil objective (HCX PL APO 1.25–.75 NA). 458/514 nm argon laser line was used for excitation of fluorescent protein-tagged constructs.
FRET imaging
Three-cube FRET assay with CFP- (donor) and YFP-tagged (acceptor) molecules was used to probe specific protein-protein interactions in live cells as previously described.39-41,62 Fluorescence images were acquired using a 40x oil objective (NA 1.3) on a Nikon Eclipse Ti-U inverted microscope fitted with an electron-multiplying CCD camera (QuantEM:512SC, Photometrics). Excitation wavelengths of 440 nm (CFP and FRET cubes) and 500 nm (YFP cube) were applied using a random access monochromator with a 75 W Xenon Arc lamp housing (PTI DeltaRam X, Photon Technology International). FRET efficiency was measured by acquiring 3 separate signals for each donor-acceptor pair condition: the donor channel (DD) which excites and detects donor emission, the acceptor channel (AA) which excites and detects acceptor emission, and the FRET channel (DA) which excites the donor and detects acceptor emission. The filter cubes used were (dichroic, emission): DD (455DCLP, D480/30M); AA (525DRLP, 530EFLP); DA (455DRLP, 535DF25). Cross-talk parameters were determined by imaging cells expressing either donor (CFP) or acceptor (YFP) fluorescent proteins alone. To avoid outlying data points only donor/acceptor ratios from 0.1 to 6 and signal/noise ratios with a minimum of 2 were used for FRET efficiency calculations. FRET efficiency and relative donor and acceptor concentrations were calculated as described.40,41,62
Data and statistical analyses
Data were analyzed off-line using PulseFit (HEKA), Microsoft Excel and Origin software. Data were plotted and statistical analyses were performed in Origin using built-in functions. Statistically significant differences between means (P < 0.05) were determined using one-way ANOVA followed by Bonferroni post hoc analyses for comparisons involving more than 2 groups. Data are represented as means ± SEM.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Ming Chen for technical assistance and members of the Colecraft lab for helpful discussions.
Funding
This research was supported by a grant from the National Institutes of Health (R01 GM107585 to HMC); an AHA predoctoral fellowship (13PRE13970018 to AAP), a UNCF-Merck graduate dissertation fellowship (CU11-2479 to AAP), and an American Heart Association Established Investigator Award (HMC).
References
- [1].Hille B. Ion Channels of Excitable Membranes. Sunderland: Sinauer Associates, Inc., 2001 [Google Scholar]
- [2].Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol 2000; 16:521-55; PMID:11031246; http://dx.doi.org/ 10.1146/annurev.cellbio.16.1.521 [DOI] [PubMed] [Google Scholar]
- [3].Simms BA, Zamponi GW. Neuronal voltage-gated calcium channels: structure, function, and dysfunction. Neuron 2014; 82:24-45; PMID:24698266; http://dx.doi.org/ 10.1016/j.neuron.2014.03.016 [DOI] [PubMed] [Google Scholar]
- [4].Buraei Z, Yang J. The {beta} Subunit of Voltage-Gated Ca2+ Channels. Physiol Rev 2010; 90:1461-506; PMID:20959621; http://dx.doi.org/ 10.1152/physrev.00057.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dolphin AC. Beta subunits of voltage-gated calcium channels. J Bioenerg Biomembr 2003; 35:599-620; PMID:15000522; http://dx.doi.org/ 10.1023/B:JOBB.0000008026.37790.5a [DOI] [PubMed] [Google Scholar]
- [6].Neely A, Hidalgo P. Structure-function of proteins interacting with the alpha1 pore-forming subunit of high-voltage-activated calcium channels. Frontiers in physiology 2014; 5:209; PMID:24917826; http://dx.doi.org/ 10.3389/fphys.2014.00209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ben-Johny M, Yue DT. Calmodulin regulation (calmodulation) of voltage-gated calcium channels. J Gen Physiol 2014; 143:679-92; PMID:24863929; http://dx.doi.org/ 10.1085/jgp.201311153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Xu X, Colecraft HM. Engineering proteins for custom inhibition of Ca(V) channels. Physiology (Bethesda) 2009; 24:210-8; PMID:19675352; http://dx.doi.org/ 10.1152/physiol.00010.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Striessnig J. Pharmacology, structure and function of cardiac L-type Ca(2+) channels. Cell Physiol Biochem 1999; 9:242-69; PMID:10575201; http://dx.doi.org/ 10.1159/000016320 [DOI] [PubMed] [Google Scholar]
- [10].Surmeier DJ, Schumacker PT. Calcium, bioenergetics, and neuronal vulnerability in Parkinson's disease. J Biol Chem 2013; 288:10736-41; PMID:23086948; http://dx.doi.org/ 10.1074/jbc.R112.410530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Triggle DJ. Calcium channel antagonists: clinical uses–past, present and future. Biochem Pharmacol 2007; 74:1-9; PMID:17276408; http://dx.doi.org/ 10.1016/j.bcp.2007.01.016 [DOI] [PubMed] [Google Scholar]
- [12].Kochegarov AA. Pharmacological modulators of voltage-gated calcium channels and their therapeutical application. Cell Calcium 2003; 33:145-62; PMID:12600802; http://dx.doi.org/ 10.1016/S0143-4160(02)00239-7 [DOI] [PubMed] [Google Scholar]
- [13].Beguin P, Nagashima K, Gonoi T, Shibasaki T, Takahashi K, Kashima Y, Ozaki N, Geering K, Iwanaga T, Seino S. Regulation of Ca2+ channel expression at the cell surface by the small G-protein kir/Gem. Nature 2001; 411:701-6; PMID:11395774; http://dx.doi.org/ 10.1038/35079621 [DOI] [PubMed] [Google Scholar]
- [14].Finlin BS, Crump SM, Satin J, Andres DA. Regulation of voltage-gated calcium channel activity by the Rem and Rad GTPases. Proc Natl Acad Sci U S A 2003; 100:14469-74; PMID:14623965; http://dx.doi.org/ 10.1073/pnas.2437756100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yang T, Colecraft HM. Regulation of voltage-dependent calcium channels by RGK proteins. Biochim Biophys Acta 2013; 1828:1644-54; PMID:23063948; http://dx.doi.org/ 10.1016/j.bbamem.2012.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Flynn R, Zamponi GW. Regulation of calcium channels by RGK proteins. Channels (Austin) 2010; 4:434-9; PMID:20953143; http://dx.doi.org/ 10.4161/chan.4.6.12865 [DOI] [PubMed] [Google Scholar]
- [17].Maguire J, Santoro T, Jensen P, Siebenlist U, Yewdell J, Kelly K. Gem: an induced, immediate early protein belonging to the Ras family. Science 1994; 265:241-4; PMID:7912851; http://dx.doi.org/ 10.1126/science.7912851 [DOI] [PubMed] [Google Scholar]
- [18].Reynet C, Kahn CR. Rad: a member of the Ras family overexpressed in muscle of type II diabetic humans. Science 1993; 262:1441-4; PMID:8248782; http://dx.doi.org/ 10.1126/science.8248782 [DOI] [PubMed] [Google Scholar]
- [19].Finlin BS, Andres DA. Rem is a new member of the Rad- and Gem/Kir Ras-related GTP-binding protein family repressed by lipopolysaccharide stimulation. J Biol Chem 1997; 272:21982-8; PMID:9268335; http://dx.doi.org/ 10.1074/jbc.272.35.21982 [DOI] [PubMed] [Google Scholar]
- [20].Chang L, Zhang J, Tseng YH, Xie CQ, Ilany J, Bruning JC, Sun Z, Zhu X, Cui T, Youker KA, et al.. Rad GTPase deficiency leads to cardiac hypertrophy. Circulation 2007; 116:2976-83; PMID:18056528; http://dx.doi.org/ 10.1161/CIRCULATIONAHA.107.707257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Magyar J, Kiper CE, Sievert G, Cai W, Shi GX, Crump SM, Li L, Niederer S, Smith N, Andres DA, et al.. Rem-GTPase regulates cardiac myocyte L-type calcium current. Channels (Austin) 2012; 6:166-73; PMID:22854599; http://dx.doi.org/ 10.4161/chan.20192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gunton JE, Sisavanh M, Stokes RA, Satin J, Satin LS, Zhang M, Liu SM, Cai W, Cheng K, Cooney GJ, et al.. Mice Deficient in GEM GTPase Show Abnormal Glucose Homeostasis Due to Defects in Beta-Cell Calcium Handling. PLoS One 2012; 7:e39462; PMID:22761801; http://dx.doi.org/ 10.1371/journal.pone.0039462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Murata M, Cingolani E, McDonald AD, Donahue JK, Marban E. Creation of a genetic calcium channel blocker by targeted gem gene transfer in the heart. Circ Res 2004; 95:398-405; PMID:15242970; http://dx.doi.org/ 10.1161/01.RES.0000138449.85324.c5 [DOI] [PubMed] [Google Scholar]
- [24].Makarewich CA, Correll RN, Gao H, Zhang H, Yang B, Berretta RM, Rizzo V, Molkentin JD, Houser SR. A caveolae-targeted L-type Ca(2)+ channel antagonist inhibits hypertrophic signaling without reducing cardiac contractility. Circ Res 2012; 110:669-74; PMID:22302787; http://dx.doi.org/ 10.1161/CIRCRESAHA.111.264028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Beguin P, Ng YJ, Krause C, Mahalakshmi RN, Ng MY, Hunziker W. RGK small GTP-binding proteins interact with the nucleotide kinase domain of Ca2+-channel beta-subunits via an uncommon effector binding domain. J Biol Chem 2007; 282:11509-20; PMID:17303572; http://dx.doi.org/ 10.1074/jbc.M606423200 [DOI] [PubMed] [Google Scholar]
- [26].Yang T, Puckerin A, Colecraft HM. Distinct RGK GTPases Differentially Use alpha(1)- and Auxiliary beta-Binding-Dependent Mechanisms to Inhibit Ca(V)1.2/Ca(V)2.2 Channels. PLoS One 2012; 7:e37079; PMID:22590648; http://dx.doi.org/ 10.1371/journal.pone.0037079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yang T, He LL, Chen M, Fang K, Colecraft HM. Bio-inspired voltage-dependent calcium channel blockers. Nat Commun 2013; 4:2540; PMID:24096474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yang T, Suhail Y, Dalton S, Kernan T, Colecraft HM. Genetically encoded molecules for inducibly inactivating CaV channels. Nat Chem Biol 2007; 3:795-804; PMID:17952065; http://dx.doi.org/ 10.1038/nchembio.2007.42 [DOI] [PubMed] [Google Scholar]
- [29].Correll RN, Pang C, Finlin BS, Dailey AM, Satin J, Andres DA. Plasma membrane targeting is essential for Rem-mediated Ca2+ channel inhibition. J Biol Chem 2007; 282:28431-40; PMID:17686775; http://dx.doi.org/ 10.1074/jbc.M706176200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chen H, Puhl HL 3rd, Niu SL, Mitchell DC, Ikeda SR. Expression of Rem2, an RGK family small GTPase, reduces N-type calcium current without affecting channel surface density. J Neurosci 2005; 25:9762-72; PMID:16237180; http://dx.doi.org/ 10.1523/JNEUROSCI.3111-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Xu X, Marx SO, Colecraft HM. Molecular mechanisms, and selective pharmacological rescue, of Rem-inhibited CaV1.2 channels in heart. Circ Res 2010; 107:620-30; PMID:20616312; http://dx.doi.org/ 10.1161/CIRCRESAHA.110.224717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yang T, Xu X, Kernan T, Wu V, Colecraft HM. Rem, a member of the RGK GTPases, inhibits recombinant CaV1.2 channels using multiple mechanisms that require distinct conformations of the GTPase. J Physiol 2010; 588:1665-81; PMID:20308247; http://dx.doi.org/ 10.1113/jphysiol.2010.187203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wang G, Zhu X, Xie W, Han P, Li K, Sun Z, Wang Y, Chen C, Song R, Cao C, et al.. Rad as a novel regulator of excitation-contraction coupling and beta-adrenergic signaling in heart. Circ Res 2010; 106:317-27; PMID:19926875; http://dx.doi.org/ 10.1161/CIRCRESAHA.109.208272 [DOI] [PubMed] [Google Scholar]
- [34].Finlin BS, Mosley AL, Crump SM, Correll RN, Ozcan S, Satin J, Andres DA. Regulation of L-type Ca2+ channel activity and insulin secretion by the Rem2 GTPase. J Biol Chem 2005; 280:41864-71; PMID:15728182; http://dx.doi.org/ 10.1074/jbc.M414261200 [DOI] [PubMed] [Google Scholar]
- [35].Jhun BS, J OU, Wang W, Ha CH, Zhao J, Kim JY, Wong C, Dirksen RT, Lopes CM, Jin ZG. Adrenergic signaling controls RGK-dependent trafficking of cardiac voltage-gated L-type Ca2+ channels through PKD1. Circ Res 2012; 110:59-70; PMID:22076634; http://dx.doi.org/ 10.1161/CIRCRESAHA.111.254672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Colicelli J. Human RAS superfamily proteins and related GTPases. Sci STKE 2004; 2004:RE13; PMID:15367757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sprang SR. G protein mechanisms: insights from structural analysis. Annu Rev Biochem 1997; 66:639-78; PMID:9242920; http://dx.doi.org/ 10.1146/annurev.biochem.66.1.639 [DOI] [PubMed] [Google Scholar]
- [38].Sasson Y, Navon-Perry L, Huppert D, Hirsch JA. RGK family G-domain:GTP analog complex structures and nucleotide-binding properties. J Mol Biol 2011; 413:372-89; PMID:21903096; http://dx.doi.org/ 10.1016/j.jmb.2011.08.017 [DOI] [PubMed] [Google Scholar]
- [39].Erickson MG, Alseikhan BA, Peterson BZ, Yue DT. Preassociation of calmodulin with voltage-gated Ca(2+) channels revealed by FRET in single living cells. Neuron 2001; 31:973-85; PMID:11580897; http://dx.doi.org/ 10.1016/S0896-6273(01)00438-X [DOI] [PubMed] [Google Scholar]
- [40].Chen H, Puhl HL 3rd, Ikeda SR. Estimating protein-protein interaction affinity in living cells using quantitative Forster resonance energy transfer measurements. J Biomed Opt 2007; 12:054011; PMID:17994899; http://dx.doi.org/ 10.1117/1.2799171 [DOI] [PubMed] [Google Scholar]
- [41].Chen H, Puhl HL 3rd, Koushik SV, Vogel SS, Ikeda SR. Measurement of FRET efficiency and ratio of donor to acceptor concentration in living cells. Biophys J 2006; 91:L39-41; PMID:16815904; http://dx.doi.org/ 10.1529/biophysj.106.088773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Crabtree GR, Schreiber SL. Three-part inventions: intracellular signaling and induced proximity. Trends Biochem Sci 1996; 21:418-22; PMID:8987395; http://dx.doi.org/ 10.1016/S0968-0004(96)20027-1 [DOI] [PubMed] [Google Scholar]
- [43].Inoue T, Heo WD, Grimley JS, Wandless TJ, Meyer T. An inducible translocation strategy to rapidly activate and inhibit small GTPase signaling pathways. Nat Methods 2005; 2:415-8; PMID:15908919; http://dx.doi.org/ 10.1038/nmeth763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Leyris JP, Gondeau C, Charnet A, Delattre C, Rousset M, Cens T, Charnet P. RGK GTPase-dependent CaV2.1 Ca2+ channel inhibition is independent of CaVbeta-subunit-induced current potentiation. FASEB J 2009; 23:2627-38; PMID:19332647; http://dx.doi.org/ 10.1096/fj.08-122135 [DOI] [PubMed] [Google Scholar]
- [45].Correll RN, Pang C, Niedowicz DM, Finlin BS, Andres DA. The RGK family of GTP-binding proteins: regulators of voltage-dependent calcium channels and cytoskeleton remodeling. Cell Signal 2008; 20:292-300; PMID:18042346; http://dx.doi.org/ 10.1016/j.cellsig.2007.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Heo WD, Inoue T, Park WS, Kim ML, Park BO, Wandless TJ, Meyer T. PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science 2006; 314:1458-61; PMID:17095657; http://dx.doi.org/ 10.1126/science.1134389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ibanez-Tallon I, Nitabach MN. Tethering toxins and peptide ligands for modulation of neuronal function. Curr Opin Neurobiol 2012; 22:72-8; PMID:22119144; http://dx.doi.org/ 10.1016/j.conb.2011.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Huang H, Ng CY, Yu D, Zhai J, Lam Y, Soong TW. Modest CaV1.342-selective inhibition by compound 8 is beta-subunit dependent. Nat Commun 2014; 5:4481; PMID:25057870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kang S, Cooper G, Dunne SF, Dusel B, Luan CH, Surmeier DJ, Silverman RB. CaV1.3-selective L-type calcium channel antagonists as potential new therapeutics for Parkinson's disease. Nat Commun 2012; 3:1146; PMID:23093183; http://dx.doi.org/ 10.1038/ncomms2149 [DOI] [PubMed] [Google Scholar]
- [50].Pantazis A, Savalli N, Sigg D, Neely A, Olcese R. Functional heterogeneity of the four voltage sensors of a human L-type calcium channel. Proc Natl Acad Sci U S A 2014; 111:18381-6; PMID:25489110; http://dx.doi.org/ 10.1073/pnas.1411127112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Agler HL, Evans J, Tay LH, Anderson MJ, Colecraft HM, Yue DT. G protein-gated inhibitory module of N-type (ca(v)2.2) ca2+ channels. Neuron 2005; 46:891-904; PMID:15953418; http://dx.doi.org/ 10.1016/j.neuron.2005.05.011 [DOI] [PubMed] [Google Scholar]
- [52].Shistik E, Ivanina T, Blumenstein Y, Dascal N. Crucial role of N terminus in function of cardiac L-type Ca2+ channel and its modulation by protein kinase C. J Biol Chem 1998; 273:17901-9; PMID:9651396; http://dx.doi.org/ 10.1074/jbc.273.28.17901 [DOI] [PubMed] [Google Scholar]
- [53].McHugh D, Sharp EM, Scheuer T, Catterall WA. Inhibition of cardiac L-type calcium channels by protein kinase C phosphorylation of two sites in the N-terminal domain. Proc Natl Acad Sci U S A 2000; 97:12334-8; PMID:11035786; http://dx.doi.org/ 10.1073/pnas.210384297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Dick IE, Tadross MR, Liang H, Tay LH, Yang W, Yue DT. A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels. Nature 2008; 451:830-4; PMID:18235447; http://dx.doi.org/ 10.1038/nature06529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Tadross MR, Dick IE, Yue DT. Mechanism of local and global Ca2+ sensing by calmodulin in complex with a Ca2+ channel. Cell 2008; 133:1228-40; PMID:18585356; http://dx.doi.org/ 10.1016/j.cell.2008.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wei X, Neely A, Olcese R, Lang W, Stefani E, Birnbaumer L. Increase in Ca2+ channel expression by deletions at the amino terminus of the cardiac alpha 1C subunit. Receptors Channels 1996; 4:205-15; PMID:9065969 [PubMed] [Google Scholar]
- [57].Fan M, Zhang WK, Buraei Z, Yang J. Molecular Determinants of Gem Protein Inhibition of P/Q-type Ca2+ Channels. J Biol Chem 2012; 287:22749-58; PMID:22589533; http://dx.doi.org/ 10.1074/jbc.M111.291872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Beqollari D, Romberg CF, Meza U, Papadopoulos S, Bannister RA. Differential effects of RGK proteins on L-type channel function in adult mouse skeletal muscle. Biophys J 2014; 106:1950-7; PMID:24806927; http://dx.doi.org/ 10.1016/j.bpj.2014.03.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Bannister RA, Colecraft HM, Beam KG. Rem inhibits skeletal muscle EC coupling by reducing the number of functional L-type Ca2+ channels. Biophys J 2008; 94:2631-8; PMID:18192376; http://dx.doi.org/ 10.1529/biophysj.107.116467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Colecraft HM, Alseikhan B, Takahashi SX, Chaudhuri D, Mittman S, Yegnasubramanian V, Alvania RS, Johns DC, Marbán E, Yue DT. Novel functional properties of Ca2+ channel beta subunits revealed by their expression in adult rat heart cells. J Physiol 2002; 541:435-52; PMID:12042350; http://dx.doi.org/ 10.1113/jphysiol.2002.018515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Subramanyam P, Chang DD, Fang K, Xie W, Marks AR, Colecraft HM. Manipulating L-type calcium channels in cardiomyocytes using split-intein protein transsplicing. Proc Natl Acad Sci U S A 2013; 110:15461-6; PMID:24003157; http://dx.doi.org/ 10.1073/pnas.1308161110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Chang DD, Colecraft HM. Rad and Rem are non-canonical G-proteins with respect to the regulatory role of guanine nucleotide binding in Ca(V)1.2 channel regulation. J Physiol 2015; 593:5075-90; PMID:26426338; http://dx.doi.org/ 10.1113/JP270889 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.