

ABSTRACT
A leucine-to-proline missense mutation at residue 98 in the proline-serine-threonine phosphatase interacting protein 2 (Pstpip2) gene leads to autoinflammatory disease that is characterized by splenomegaly, necrosis, and spontaneous development of osteomyelitis in mice (Pstpip2cmo). Disease progression in these mice resembles that of chronic recurrent multifocal osteomyelitis in humans. Our group and others have shown that disease progression in Pstpip2cmo mice is mediated by the cytokine IL-1β, independently of inflammasomes or IL-1α. Our recent publication highlighted herein establishes that diet-induced changes in intestinal microbiota provide protection against the development of osteomyelitis in Pstpip2cmo mice. Moreover, the proteases caspase-1 and caspase-8 have redundant roles in cleaving IL-1β and promoting disease. This addendum reviews the current literature on the Pstpip2cmo murine disease model and the clinical significance of the role of PSTPIP2 in regulating autoinflammatory osteomyelitis, which is mediated by innate components of immune cells.
KEYWORDS: autoinflammatory, caspase, high-fat diet, IL-1beta, microbiota, neutrophil, osteomyelitis, Pstpip2
Introduction
Chronic recurrent multifocal osteomyelitis (CRMO) is an autoinflammatory disorder associated with periodic bone pain, fever, and the appearance of multiple bone lesions at any skeletal site. CRMO-like symptoms are observed in mice homozygous for a point mutation in the proline-serine-threonine phosphatase interacting protein 2 (Pstpip2) gene [referred to as Pstpip2cmo (chronic multifocal osteomyelitis) mice], which results in complete absence of the PSTPIP2 protein.1 Pstpip2cmo mice display bone deformities in the form of tail kinks and fibrosis of hind-footpad tissues.2 The footpads and popliteal lymph nodes of Pstpip2cmo mice contain inflammatory infiltrates of neutrophils, macrophages, and T and B cells.2 Bone marrow chimeras generated using the Pstpip2cmo model revealed that haematopoietic cells are drivers of the disease. Furthermore, Rag1−/−Pstpip2cmo mice develop osteomyelitis, demonstrating that disease initiation and progression occur independently of T and B cells.3 Together, these studies support that the innate immune arm is the inflammatory agent for disease initiation and progression. In fact, disease initiation in Pstpip2cmo mice is attributed to the hyperactivation of neutrophils, which secrete increased levels of interleukin 1β (IL-1β).4,5 Genetic studies by Cassel et al.4 and our group5 show that IL-1β is the key inflammatory cytokine that drives disease progression, and Pstpip2cmo mice deficient in IL-1β are protected from the disease. Interestingly, the closely related IL-1 cytokine IL-1α is not important for the disease phenotype.
IL-1β, a pleiotropic cytokine, is produced as a biologically inactive pro-form that needs to be cleaved before it is secreted. The cleavage of pro-IL-1β is mediated by caspase-1 after inflammasome complex formation; however, several other proteases can also cleave pro-IL-1β.6 Disease progression in Pstpip2cmo mice lacking caspse-1 or other important components of inflammasomes, such as the apoptosis-associated speck-like protein containing a CARD (ASC) and nod-like receptor family, pyrin domain containing 3 (NLRP3) develop disease similar to that observed in Pstpip2cmo mice.4,5 These observations indicate that PSTPIP2 negatively regulates IL-1β secretion in a caspase-1– and NLRP3 inflammasome–autonomous manner.
By using genetic crosses, we ruled out the involvement of various neutrophil-associated proteases in the processing and activation of pro-IL-1β in Pstpip2cmo mice and identified complex redundant roles for caspase-1 and caspase-8.7 Pstpip2cmo mice deficient for either caspase-1 or caspase-8 were not protected, however, deficiency in both caspases provided Pstpip2cmo mice with significant protection from disease induction.7 These outcomes were unexpected given our prior study that concluded bone disease in Pstpip2cmo mice developed independently of inflammasome activation.5 In this addendum, we highlight the complexity and challenge in deciphering the molecular mechanisms governing chronic multifocal myelitis in Pstpip2cmo mice as well as CRMO and related autoinflammatory diseases in humans.
Recent studies of intestinal microbiota, a complex community of microorganisms, show that autoinflammatory diseases are multifactorial. Our recent study highlights the crosstalk among the environment (diet), intestinal microbiota, and genetics (Pstpip2 mutation) in promoting IL-1β production and inducing osteomyelitic disease (Fig. 1). There is a growing body of evidence that identifies the correlation between diet, microbiota and increased susceptibility to common inflammatory diseases.8-10 We demonstrated that in Pstpip2cmo mice, a high-fat diet (HFD) induces changes in the microbiome, defined as the collective genome of the microbiota residing in the intestinal tissue, to protect the otherwise disease-susceptible mice from developing osteomyelitis. Our studies suggest a pathogenic role for Prevotella and a protective role for Lactobacillus genera in Pstpip2cmo mice. Furthermore, administration of broad-spectrum antibiotics reduced the levels of Prevotella and IL-1β and protected Pstpip2cmo mice from disease. In this addendum, we elaborate on the recently identified role for gut microbiota in regulating Pstpip2cmo-mediated disease and discuss the cell types, inflammasome complexes, and caspases involved in disease progression.
Figure 1.
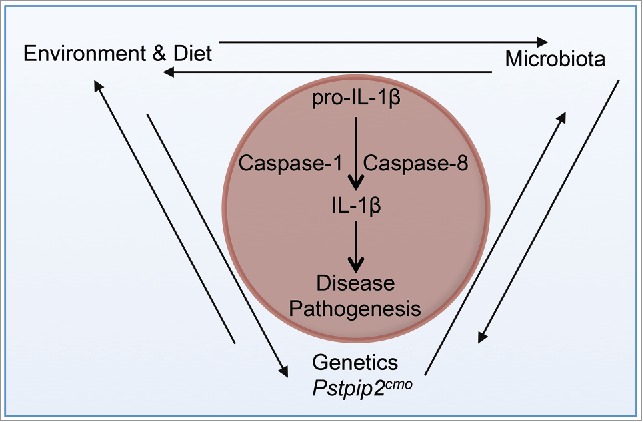
Schematic representing the crosstalk among genetics, environment (diet), and microbiota that induces bone disease in Pstpip2cmo mice. IL-1β is a critical cytokine that mediates bone disease in Pstpip2cmo mice. IL-1β processing is regulated by caspase-1 and caspase-8. Several factors regulate IL-1β in Pstpip2cmo mice. Pstpip2cmo mice have a dysregulated gut microbiota that promotes disease pathogenesis. Similarly, diet or environmental factors can influence disease progression in Pstpip2cmo mice by directly regulating immune cells or the gut microbiota. Thus, the disease pathogenesis in Pstpip2cmo mice is multifactorial because it requires a close coordination among genetics, diet, and the gut microbiota.
Diet-induced changes in microbiota inhibit osteomyelitis in Pstpip2cmo mice
The composition of gut microbiota has been implicated in modulating autoimmune and autoinflammatory diseases such as asthma, arthritis, colitis, diabetes, and lupus.8,11,12 Diet has a considerable effect on the composition of gut microbiota,13,14 and can thus have a measurable effect on the immune response. Western diets are under particular scrutiny because they are high in processed foods. Humanized gnotobiotic mice (i.e., mice born under germ-free conditions and then inoculated with human gut microbiota) switched from a diet low in fat and rich in plant polysaccharides to a Western diet display a dramatically altered gut microbiome as well as a change in the metatranscriptome of the microbiota, indicating that diet can alter not only composition of the gut microbiota, but also the function.13 Furthermore, a study comparing children from rural Africa and urban Europe identified Prevotella and Xylanibacter genera only in the African children. The presence of these bacteria is shaped by the children's diet and indicates the importance of intestinal microbiota that can digest the cellulose and xylan found in the diet of African children.15
To examine whether diet has a considerable effect on the autoinflammatory disease syndrome observed in Pstpip2cmo mice, disease onset was compared in mice given a HFD and mice given normal chow. Interestingly, the HFD completely protected Pstpip2cmo mice from osteomyelitis.7 Notably, the HFD did not significantly alter the body weight gain of Pstpip2cmo mice compared with mice given normal chow before disease onset. Thus, it could be argued that diet-induced metabolic changes do not have a major role in the protective outcome observed in Pstpip2cmo mice given a HFD.
Arguably, the relationship between obesity and the gut microbiota is a 2-way street. In addition to diet being an influence on the microbiome,16 germ-free mice colonized with microbiota from genetically predisposed or diet-induced obese mice exhibit significantly greater increase in body fat percentage compared to germ-free mice colonized with microbiota from non-obese mice.16,17 Pstpip2cmo mice reported in our studies are on a Balb/c background, and since Balb/c mice are resistant to diet-induced obesity18 mice on the HFD-fed mice did not gain weight.7 Furthermore, we observed gut microbiota dysbiosis in Pstpip2cmo mice prior to disease onset.7 Thus, we believe that the gut microbiota in Pstpip2cmo mice is shaped independently of alterations in adipose tissue function and accumulation. We also speculate that if Pstpip2cmo mice were sensitive to diet-induced obesity they would remain protected from disease unless a metabolic alteration led to significant changes in the gut microbiota. Lastly, specific dietary factors that conferred protection remain unknown, and future studies with Pstpip2cmo mice given modulated diets may help identify the dietary components that confer protection.
To investigate whether the protection provided by diet modulation was due to changes in the microbiota, our group performed metagenomic sequencing of 16S rRNA genes to identify the differences and similarities in commensal intestinal ecology in Pstpip2cmo mice given the HFD and those given normal chow. Notably, we found a greater ratio of Lactobacillus to Prevotella in protected Pstpip2cmo mice fed the HFD, whereas in the normal chow group, which developed disease, more Prevotella was present. Further, the footpads of Pstpip2cmo mice fed the HFD, contained lower levels of IL-1β compared to those on normal chow. Importantly, transplantation of fecal microbiota from protected HFD-fed Pstpip2cmo mice to pre-diseased Pstpip2cmo mice fed normal chow conferred protection from Prevotella outgrowth, decreased IL-1β production, and, subsequently, prevented disease onset,7 strongly indicating that diet-induced modulation of the gut microbiota can drive inflammatory bone disease.
Because of the observed correlation between the HFD-induced alteration in the microbiome composition and reduced IL-1β production in Pstpip2cmo mice, we hypothesized that the intestinal microbiota could directly control IL-1β levels in the gut. Thus, we compared the expression of Il1b transcript in CD45+ cells isolated from colons of specific pathogen-free (SPF) mice and those isolated from germ-free mice. The germ-free mice expressed significantly lower levels of Il1b than did SPF mice.7 Broad-spectrum antibiotics administered in the drinking water of SPF Pstpip2cmo mice given normal chow significantly reduced the levels of Prevotella and Il1b in the gut and protected the mice from disease.7 We did not investigate the possibility that HFD could directly modulate Il1b expression in germ-free mice or mice treated with antibiotics, but speculate that induction of Il1b is dependent on the presence of microbiota and not the dietary fat content. Together, these data support our hypothesis that the gut microbiota promotes Il1b expression, thus driving disease progression in Pstpip2cmo mice.
To verify the direct relationship between intestinal microbiota and disease, Pstpip2cmo mice need to be re-derived under germ-free conditions and the level of Il1b measured. Likewise, the use of mono-associated, or germ-free mice colonized with a single strain of bacteria, would be an effective tool to specifically implicate Prevotella in driving and Lactobacillus in suppressing Il1b expression. There are many studies that have highlighted the immunomodulatory effect that Lactobacillus strains exert on the host immune system. For instance, macrophages that interact with Lactobacillus rhamnosus can discriminate between probiotic and pathogenic bacteria by interferon-mediated TLR gene regulation,19 and the interaction between Lactobacillus casei and gut-associated immune cells induces an increase in TLR2 expression.20 However, in our model we do not know if Lactobacillus is engaging directly with immune cells to suppress Il1b expression or if the outgrowth of Lactobacillus in the HFD-fed Pstpip2cmo mice merely competes with harmful Prevotella. Nevertheless, Lactobacilli are a promising, potentially therapeutic probiotic for CRMO patients.
Given the role of Prevotella in promoting Il1b overexpression in Pstpip2cmo mice, germ-free Pstpip2cmo mice should be completely protected from disease onset and have reduced Il1b expression, and colonization of these mice with Prevotella should rapidly induce disease. These outcomes would suggest that mutation in Pstpip2 predisposes neutrophils to hypersecretion of IL-1β, however their activation is dependent on gut microbiome dysbiosis. Diet and the composition of the intestinal microbiota control the basal levels of Il1b, and functional PSTPIP2 restrains an autoinflammatory response and prevent disease. Conversely, it is possible that a fine balance between the protective and pathogenic microbiome is necessary, and, in the complete absence of intestinal microbiota, genetic aberration in germ-free Pstpip2cmo mice may be sufficient to drive osteomyelitis.
The possibility that diet-induced changes in Prevotella and Lactobacillus or other commensal bacteria alter the disease state provides new directions for developing therapies for patients with CRMO. Instead of the currently prescribed corticosteroids, tumor necrosis factor (TNF-) α-blockers, and bisphosphonates for pain relief and to dampen inflammatory cytokines, probiotics can be included in future treatments to alter the ecology of the gut and provide long-term protection against disease progression and even symptom reversal. Furthermore, it is possible that these autoinflammatory diseases can be controlled by a diet that enriches healthy gut microbes and consequently calms inflammation.
Redundant roles for caspase-1 and caspase-8 in cleaving IL-1β
The excessive production of IL-1 and signaling through the IL-1 receptor (IL-1R) can contribute to multiple autoinflammatory diseases, many of which respond to treatment with IL-1R blocking agents.21-23 IL-1 production is primarily regulated by the inflammasome, a multimeric protein complex that is assembled in response to various inflammatory triggers such as danger signals, microbial toxins, and crystalline substances. A prototypical inflammasome complex consists of a cytoplasmic sensor (NLR, AIM2, or Pyrin), the common adaptor molecule ASC, and the cysteine protease caspase-1. Cleavage of caspase-1 by the inflammasome results in its activation, which in turn cleaves pro-IL-1β. Our group was one of the first to show that autoinflammatory disease in Pstpip2cmo mice is specifically triggered by IL-1β, independently of IL-1α.5 Surprisingly, IL-1β production in Pstpip2cmo mice was independent of NLRP3, ASC, or even caspase-1, because Pstpip2cmo mice bred with Nlrp3−/−, Asc−/−, or Casp1−/− mice were not protected from the onset of osteomyelitis. Because neutrophils are the sentinel cells required for disease progression in Pstpip2cmo mice, we hypothesized that proteases abundant within neutrophils might be involved in IL-1β processing independently of caspase-1. However, generation of Pstpip2cmo mice deficient in neutrophil proteinase 3 or elastase did not confer protection.7
Recent studies have also identified caspase-8 as a direct protease for IL-1β.24,25 Recombinant caspase-8 directly cleaves pro-IL-1β after the in vitro stimulation of macrophages by TLR3 or TLR4 agonists.26 Caspase-8–deficient mice are embryonic lethal and can be rescued by breeding with Ripk3−/− mice.27 To test whether caspase-8 is a direct protease for IL-1β in Pstpip2cmo mice, Ripk3−/−Casp8−/−Pstpip2cmo mice were generated. Again, caspase-8 deficiency did not protect Pstpip2cmo mice from disease onset. We hypothesized that caspase-1 and caspase-8 play redundant roles in IL-1β processing, thus masking the effect of deficiency in either caspase-1 or capase-8. To test whether both caspase-1 and caspase-8 are important in IL-1β processing and subsequent disease progression in Pstpip2cmo mice, Ripk3−/−Casp8−/−Casp1−/−Pstpip2cmo mice (QKO mice) were generated. The QKO mice showed complete protection from osteomyelitic disease, which demonstrated the redundant roles for caspase-1 and caspase-8 in Pstpip2cmo mice.7
Our results with caspase-1 and caspase-8 reveal several possibilities for further examination. We established that caspase-1 and caspase-8 form independent complexes to uniquely process IL-1β (Fig. 2A,B), such that a deficiency in either protease does not confer even partial protection. Moreover, it is well established that caspase-1 is activated in the inflammasome complex (Fig. 2A). In future studies, we will identify the upstream molecules involved in formation of the caspase-1 complex. We will use Ripk3−/−Casp8−/−Pstpip2cmo mice to breed with Asc−/− mice. We expect that ASC deficiency in Ripk3−/−Casp8−/−Pstpip2cmo mice will provide complete protection, thereby phenocopying the QKO mice. Upstream cytoplasmic sensors NLRP3, NLRC4, NLRP1b, and AIM2 will also be tested to identify which inflammasome is required to activate caspase-1.
Figure 2.
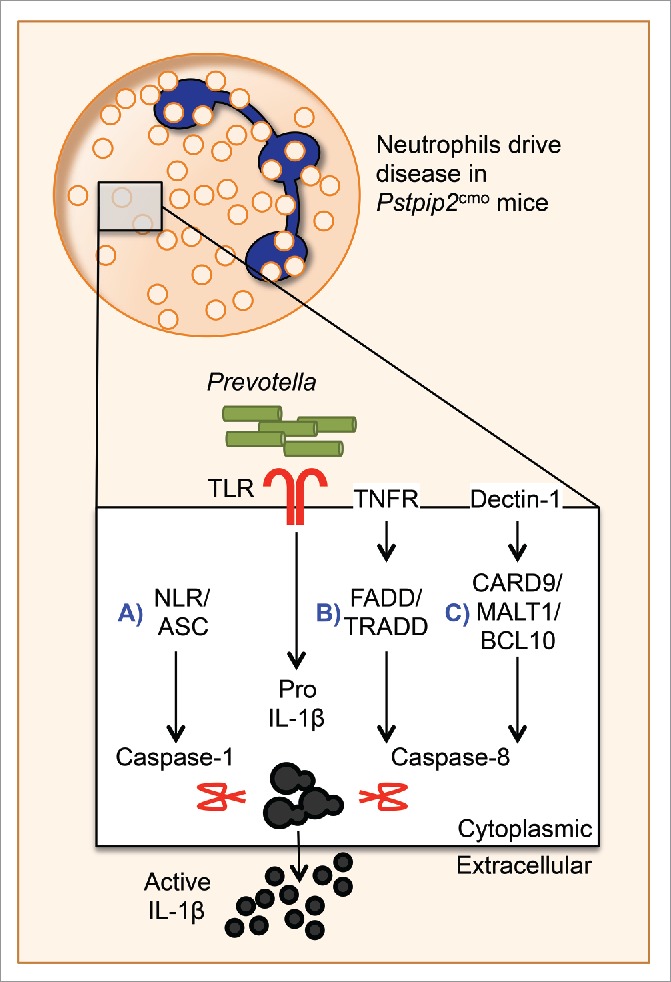
Caspase-1 and caspase-8 play redundant roles in neutrophils to induce disease in Pstpip2cmo mice. Here, we propose potential complexes that activate caspase-1 and caspase-8 to promote IL-1β cleavage in neutrophils and promote disease. Pstpip2cmo mice harbor pathogenic Prevotella, which promotes the upregulation of pro-IL-1β through yet-unknown toll-like receptors. (A) Caspase-1 processes pro-IL-1β in the inflammasome complex containing NLRP3 and ASC. Caspase-8 also functions in redundant complexes to process pro-IL-1β downstream of (B) TNFR1 and (C) Dectin-1.
The mechanism of caspase-8 activation in the osteoarthritic model of Pstpip2cmo mice also remains unknown. Caspase-8 acts downstream of various death receptors, including CD95 (Fas), TRAIL receptor, and TNFR1 (Fig. 2B), which recruit the protease via its adaptor protein FADD.6 The recruited caspase-8 molecules homodimerize, undergo autocleavage, and process downstream targets.6 Although caspase-8 activation through this pathway usually induces apoptosis, it is not known whether caspase-8 activation in these complexes in the absence of functional PSTPIP2 promotes pyroptosis, IL-1β cleavage, and inflammation. Breeding Pstpip2cmoCasp1−/− mice with mice deficient in CD95, TRAIL receptor, TNFR, or FADD will sequentially test the hypothesis that caspase-8 acts downstream of these signaling molecules to cleave pro-IL-1β.
Caspase-8–mediated cleavage of pro-IL-1β has also been described in response to fungi and mycobacteria.24 Dectin-1–induced signaling through Syk promotes the assembly of a complex comprising adaptor proteins CARD9, Bcl-10, and MALT1. The CARD9–Bcl-10–MALT1 complex further recruits caspase-8 and activates it to promote pro-IL-1β processing (Fig. 2C). This complex, termed the noncanonical inflammasome, works independently of caspase-1. To test whether a similar caspase-8 complex is formed in Pstpip2cmo mice, Pstpip2cmoCasp1−/− mice will be bred with mice deficient in CARD9, Bcl-10, or MALT1. Results from these crosses will reveal the requirement of potential molecules in the caspase-8 complex that drive IL-1β processing in Pstpip2cmo mice.
Our studies have identified a complex crosstalk and redundancy between caspase-1 and caspase-8 in IL-1β processing (Fig. 2). Our future studies and use of novel genetic strategies will reveal the redundant complexes that promote IL-1β processing and trigger autoinflammatory disease.
Hyperactivated neutrophils secrete high levels of IL-1β in the absence of functional PSTPIP2
IL-1β is a critical cytokine that is dysregulated in Pstpip2cmo mice and required for disease initiation and progression. Thus, Pstpip2cmo mice deficient in IL-1β are completely protected from the onset of autoinflammatory bone disease. In vitro and in vivo, innate immune cells, specifically macrophages, dendritic cells, and monocytes, are the major source of IL-1β. Immunohistochemical staining of inflamed footpad sections showed that the lesions contain neutrophils and macrophages.7 Given that macrophages are the major producer of IL-1β, we hypothesized that Pstpip2cmo macrophages have dysregulated IL-1β production. However, in vitro studies on macrophages from wild type and Pstpip2cmo mice found no major difference in their ability to produce IL-1β after exposure to several stimuli.4,7 Interestingly, neutrophils from Pstpip2cmo mice secreted significantly higher levels of IL-1β than those from wild type mice. These studies demonstrate a negative regulatory role for PSTPIP2 in neutrophils and implicate neutrophils as the innate immune cells that potentially mediate autoinflammatory bone disease.
Our group showed that depletion of neutrophils by using Ly6G antibodies conferred complete protection in Pstpip2cmo mice.7 Not surprisingly, neutrophils from Pstpip2cmo mice treated with a caspase-1 inhibitor continued to secrete IL-1β, whereas cells were sensitive to the pan-serine protease inhibitor diisopropylfluorophosphate.4 Neutrophil proteinases and elastase were dispensable for disease initiation, whereas caspase-1 and caspase-8 played redundant roles in IL-1β processing and secretion.7 Although it is now well accepted that neutrophil-mediated hypersecretion of IL-1β is necessary for disease initiation, it remains unknown how or whether other inflammatory cells and cytokines contribute to disease persistence and severity. We ruled out the role for adaptive T and B cells in Pstpip2cmo-mediated disease, because Pstpip2cmo mice on a Rag1-deficient background still develop disease.3 Whether other cells such as macrophages, dendritic cells, and innate lymphoid cells play a role in responding to neutrophil-produced IL-1β to perpetuate the disease needs to be determined in Pstpip2cmo mice specifically deficient for or depleted of these innate immune cell types.
Diseased hind paws of Pstpip2cmo mice are characterized by increased induction of pathogenic factors associated with the recruitment and expansion of granulocytes and macrophages. The inflammatory environment has high levels of TNF-α, G-CSF, KC (also known as CXCL1), MIP-1α, and MCP-1 in addition to IL-1β.5 Our studies with Pstpip2cmoIl1b−/− mice show that these inflammatory cytokines are induced secondary to IL-1β. However, how IL-1β induces these cytokines and which immune cells respond to IL-1β to produce these additional inflammatory cytokines are unknown. The effects of these cytokines and how they aid disease progression in Pstpip2cmo mice is also unknown. The recruitment of innate immune cells (e.g., macrophages and monocytes) to inflamed bones has undetermined consequences in Pstpip2cmo mice. Determining their role in disease progression could help in developing novel therapies for patients with CRMO.
Additive potential of diet and anti-IL-1β therapy in autoinflammatory disease
In humans, Majeed syndrome and DIRA (deficient in IL-1R antagonist) are forms of CRMO that are caused by single gene mutations in LPIN2 (encodes a phosphatidate phosphatase) and IL-1RN (encodes IL-1 receptor antagonist protein), respectively.29-32 Despite identifying associated genetic mutations, the molecular mechanisms that influence the development of a sterile autoinflammatory environment in CRMO are not understood. Because there are no diagnostic tests or inflammatory markers for CRMO,33 patients with Majeed syndrome are often treated with anti-TNF-α, NSAIDs, and corticosteroids, which provide only partial, temporary relief.30 IL-1 inhibition with anakinra, a recombinant version of the IL-1 receptor antagonist protein, effectively treats both the Majeed syndrome and DIRA.31,34 Anakinra blocks the binding of IL-1α and IL-1β to IL-1R, which is expressed in nearly all tissues. Interestingly, anakinra is also used for patients with rheumatological, autoinflammatory, and other inflammatory conditions (Cavalli et al. Supplemental Table S1),35 but the etiology of these disorders is not well understood. It is critical to understand the pathogenesis of autoinflammatory conditions to improve their diagnosis and treatment. Our recent studies in autoinflammatory disease models indicate specific roles for IL-1α36 and IL-1β.5 Given the specific and independent functions of IL-1α,37 a more targeted therapy against IL-1β can benefit patients with osteomyelitis or CRMO.5,7
IL-1β is not detected in healthy tissues, and transcription is the rate-limiting step in IL-1β production. We have identified that the microbiome is a potential modifier of Il1b transcriptional levels and that the microbiome is shaped by diet.7 IL-1β has been implicated as the key disease-associated cytokine in patients with CRMO, and studies in Pstpip2cmo mice indicate that diet can modulate the levels of IL-1β produced by neutrophils. Thus, diet might be a novel and additional form of therapy to modulate IL-1β levels and dampen inflammation in patients with CRMO, thereby potentially ameliorating disease. More importantly, in conditions wherein genetic mutations such as IL-1RA deficiency predispose patients to autoinflammatory disease, it may be possible to further improve therapies and provide long-term disease regression through dietary restrictions, adding a probiotic regimen or administering a fecal microbiota transplant.
Concluding remarks
Our recent work reveals a role for diet-induced alterations in the microbiome in a CRMO-like autoinflammatory disease in Pstpip2cmo mice. The enrichment of Lactobacillus species in the gut reduced IL-1β secretion and leukocyte infiltration in footpads of disease-prone Pstpip2cmo mice. IL-1β hypersecretion by neutrophils in Pstpip2cmo mice was dependent on both caspase-1 and caspase-8. Our studies support the effectiveness of using agents that block IL-1β in patients with CRMO and identify new avenues for anti-inflammatory therapy, such as protease inhibition and diet modulation.
The mechanism by which caspase-8 acts as a redundant factor in cleaving IL-1β remains to be determined. We propose that in Pstpip2cmo mice, caspase-8 can be activated either downstream of its traditional death receptors or in the CARD9–Bcl-10–MALT1 complex downstream of Syk. We also discuss the possibility that although neutrophils and IL-1β are required for disease initiation, other innate immune agents such as chemokines and cytokines could perpetuate the inflammatory environment to contribute to disease progression.
A deeper understanding of the multiple factors that contribute to autoinflammatory diseases will aid efforts to prevent and treat these complex disorders. Although mutations in PSTPIP2 have not yet been associated with disease in humans, the murine Pstpip2cmo model has provided critical molecular mechanistic insights into IL-1β biology and autoinflammatory bone diseases in general. Future studies on these mice will reveal more about the relation among diet, inflammation, and disease, which will undoubtedly accelerate the identification of novel therapeutic targets to combat these debilitating bone diseases.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Grosse J, Chitu V, Marquardt A, Hanke P, Schmittwolf C, Zeitlmann L, Schropp P, Barth B, Yu P, Paffenholz R, et al.. Mutation of mouse Mayp/Pstpip2 causes a macrophage autoinflammatory disease. Blood 2006; 107:3350-8; PMID:16397132; http://dx.doi.org/ 10.1182/blood-2005-09-3556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ferguson PJ, Bing X, Vasef MA, Ochoa LA, Mahgoub A, Waldschmidt TJ, Tygrett LT, Schlueter AJ, El-Shanti H. A missense mutation in pstpip2 is associated with the murine autoinflammatory disorder chronic multifocal osteomyelitis. Bone 2006; 38:41-7; PMID:16122996; http://dx.doi.org/ 10.1016/j.bone.2005.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chitu V, Ferguson PJ, de Bruijn R, Schlueter AJ, Ochoa LA, Waldschmidt TJ, Yeung YG, Stanley ER. Primed innate immunity leads to autoinflammatory disease in PSTPIP2-deficient cmo mice. Blood 2009; 114:2497-505; PMID:19608749; http://dx.doi.org/ 10.1182/blood-2009-02-204925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Cassel SL, Janczy JR, Bing X, Wilson SP, Olivier AK, Otero JE, Iwakura Y, Shayakhmetov DM, Bassuk AG, Abu-Amer Y, et al.. Inflammasome-independent IL-1beta mediates autoinflammatory disease in Pstpip2-deficient mice. Proc Natl Acad Sci U S A 2014; 111:1072-7; PMID:24395802; http://dx.doi.org/ 10.1073/pnas.1318685111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lukens JR, Gross JM, Calabrese C, Iwakura Y, Lamkanfi M, Vogel P, Kanneganti TD. Critical role for inflammasome-independent IL-1beta production in osteomyelitis. Proc Natl Acad Sci U S A 2014; 111:1066-71; PMID:24395792; http://dx.doi.org/ 10.1073/pnas.1318688111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gurung P, Kanneganti TD. Novel roles for caspase-8 in IL-1beta and inflammasome regulation. Am J Pathol 2015; 185:17-25; PMID:25451151; http://dx.doi.org/ 10.1016/j.ajpath.2014.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lukens JR, Gurung P, Vogel P, Johnson GR, Carter RA, McGoldrick DJ, Bandi SR, Calabrese CR, Vande Walle L, Lamkanfi M, et al.. Dietary modulation of the microbiome affects autoinflammatory disease. Nature 2014; 516:246-9; PMID:25274309; http://dx.doi.org/ 10.1038/nature13788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Maslowski KM, Mackay CR. Diet, gut microbiota and immune responses. Nat Immunol 2011; 12:5-9; PMID:21169997; http://dx.doi.org/ 10.1038/ni0111-5 [DOI] [PubMed] [Google Scholar]
- [9].Agus A, Denizot J, Thevenot J, Martinez-Medina M, Massier S, Sauvanet P, Bernalier-Donadille A, Denis S, Hofman P, Bonnet R, et al.. Western diet induces a shift in microbiota composition enhancing susceptibility to Adherent-Invasive E. coli infection and intestinal inflammation. Sci Rep 2016; 6:19032; PMID:26742586; http://dx.doi.org/ 10.1038/srep19032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Asakura H, Suzuki K, Kitahora T, Morizane T. Is there a link between food and intestinal microbes and the occurrence of Crohn's disease and ulcerative colitis? J Gastroenterol Hepatol 2008; 23:1794-801; PMID:19120872; http://dx.doi.org/ 10.1111/j.1440-1746.2008.05681.x [DOI] [PubMed] [Google Scholar]
- [11].Chervonsky AV. Influence of microbial environment on autoimmunity. Nat Immunol 2010; 11:28-35; PMID:20016507; http://dx.doi.org/ 10.1038/ni.1801 [DOI] [PubMed] [Google Scholar]
- [12].Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med 2002; 347:911-20; PMID:12239261; http://dx.doi.org/ 10.1056/NEJMra020100 [DOI] [PubMed] [Google Scholar]
- [13].Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med 2009; 1:6ra14; PMID:20368178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, et al.. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014; 505:559-63; PMID:24336217; http://dx.doi.org/ 10.1038/nature12820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, Collini S, Pieraccini G, Lionetti P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A 2010; 107:14691-6; PMID:20679230; http://dx.doi.org/ 10.1073/pnas.1005963107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Turnbaugh PJ, Backhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 2008; 3:213-23; PMID:18407065; http://dx.doi.org/ 10.1016/j.chom.2008.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006; 444:1027-31; PMID:17183312; http://dx.doi.org/ 10.1038/nature05414 [DOI] [PubMed] [Google Scholar]
- [18].Montgomery MK, Hallahan NL, Brown SH, Liu M, Mitchell TW, Cooney GJ, Turner N. Mouse strain-dependent variation in obesity and glucose homeostasis in response to high-fat feeding. Diabetologia 2013; 56:1129-39; PMID:23423668; http://dx.doi.org/ 10.1007/s00125-013-2846-8 [DOI] [PubMed] [Google Scholar]
- [19].Miettinen M, Veckman V, Latvala S, Sareneva T, Matikainen S, Julkunen I. Live Lactobacillus rhamnosus and Streptococcus pyogenes differentially regulate Toll-like receptor (TLR) gene expression in human primary macrophages. J Leukoc Biol 2008; 84:1092-100; PMID:18625909; http://dx.doi.org/ 10.1189/jlb.1206737 [DOI] [PubMed] [Google Scholar]
- [20].Aragon F, Carino S, Perdigon G, de Moreno de LeBlanc A. The administration of milk fermented by the probiotic Lactobacillus casei CRL 431 exerts an immunomodulatory effect against a breast tumour in a mouse model. Immunobiology 2014; 219:457-64; PMID:24646876; http://dx.doi.org/ 10.1016/j.imbio.2014.02.005 [DOI] [PubMed] [Google Scholar]
- [21].Lukens JR, Dixit VD, Kanneganti TD. Inflammasome activation in obesity-related inflammatory diseases and autoimmunity. Discov Med 2011; 12:65-74; PMID:21794210 [PMC free article] [PubMed] [Google Scholar]
- [22].Ferguson PJ, Laxer RM. New discoveries in CRMO: IL-1beta, the neutrophil, and the microbiome implicated in disease pathogenesis in Pstpip2-deficient mice. Semin Immunopathol 2015; 37:407-12; PMID:25894861; http://dx.doi.org/ 10.1007/s00281-015-0488-2 [DOI] [PubMed] [Google Scholar]
- [23].Yang CA, Chiang BL. Inflammasomes and human autoimmunity: A comprehensive review. J Autoimmun 2015; 61:1-8; PMID:26005048; http://dx.doi.org/ 10.1016/j.jaut.2015.05.001 [DOI] [PubMed] [Google Scholar]
- [24].Gringhuis SI, Kaptein TM, Wevers BA, Theelen B, van der Vlist M, Boekhout T, Geijtenbeek TB. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1beta via a noncanonical caspase-8 inflammasome. Nat Immunol 2012; 13:246-54; PMID:22267217; http://dx.doi.org/ 10.1038/ni.2222 [DOI] [PubMed] [Google Scholar]
- [25].Gurung P, Anand PK, Malireddi RK, Vande Walle L, Van Opdenbosch N, Dillon CP, Weinlich R, Green DR, Lamkanfi M, Kanneganti TD. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J Immunol 2014; 192:1835-46; PMID:24453255; http://dx.doi.org/ 10.4049/jimmunol.1302839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Maelfait J, Vercammen E, Janssens S, Schotte P, Haegman M, Magez S, Beyaert R. Stimulation of Toll-like receptor 3 and 4 induces interleukin-1beta maturation by caspase-8. J Exp Med 2008; 205:1967-73; PMID:18725521; http://dx.doi.org/ 10.1084/jem.20071632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011; 471:363-7; PMID:21368763; http://dx.doi.org/ 10.1038/nature09852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Karki R, Man SM, Malireddi RK, Gurung P, Vogel P, Lamkanfi M, Kanneganti TD. Concerted activation of the AIM2 and NLRP3 inflammasomes orchestrates host protection against Aspergillus infection. Cell Host Microbe 2015; 17:357-68; PMID:25704009; http://dx.doi.org/ 10.1016/j.chom.2015.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ferguson PJ, Chen S, Tayeh MK, Ochoa L, Leal SM, Pelet A, Munnich A, Lyonnet S, Majeed HA, El-Shanti H. Homozygous mutations in LPIN2 are responsible for the syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia (Majeed syndrome). J Med Genet 2005; 42:551-7; PMID:15994876; http://dx.doi.org/ 10.1136/jmg.2005.030759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Al-Mosawi ZS, Al-Saad KK, Ijadi-Maghsoodi R, El-Shanti HI, Ferguson PJ. A splice site mutation confirms the role of LPIN2 in Majeed syndrome. Arthritis Rheum 2007; 56:960-4; PMID:17330256; http://dx.doi.org/ 10.1002/art.22431 [DOI] [PubMed] [Google Scholar]
- [31].Aksentijevich I, Masters SL, Ferguson PJ, Dancey P, Frenkel J, van Royen-Kerkhoff A, Laxer R, Tedgard U, Cowen EW, Pham TH, et al.. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med 2009; 360:2426-37; PMID:19494218; http://dx.doi.org/ 10.1056/NEJMoa0807865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Reddy S, Jia S, Geoffrey R, Lorier R, Suchi M, Broeckel U, Hessner MJ, Verbsky J. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N Engl J Med 2009; 360:2438-44; PMID:19494219; http://dx.doi.org/ 10.1056/NEJMoa0809568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Jansson A, Renner ED, Ramser J, Mayer A, Haban M, Meindl A, Grote V, Diebold J, Jansson V, Schneider K, et al.. Classification of non-bacterial osteitis: retrospective study of clinical, immunological and genetic aspects in 89 patients. Rheumatology (Oxford) 2007; 46:154-60; PMID:16782988; http://dx.doi.org/ 10.1093/rheumatology/kel190 [DOI] [PubMed] [Google Scholar]
- [34].Herlin T, Fiirgaard B, Bjerre M, Kerndrup G, Hasle H, Bing X, Ferguson PJ. Efficacy of anti-IL-1 treatment in Majeed syndrome. Ann Rheum Dis 2013; 72:410-3; PMID:23087183; http://dx.doi.org/ 10.1136/annrheumdis-2012-201818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Cavalli G, Dinarello CA. Treating rheumatological diseases and co-morbidities with interleukin-1 blocking therapies. Rheumatology (Oxford) 2015; PMID:26209330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lukens JR, Vogel P, Johnson GR, Kelliher MA, Iwakura Y, Lamkanfi M, Kanneganti TD. RIP1-driven autoinflammation targets IL-1alpha independently of inflammasomes and RIP3. Nature 2013; 498:224-7; PMID:23708968; http://dx.doi.org/ 10.1038/nature12174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Rider P, Carmi Y, Voronov E, Apte RN. Interleukin-1alpha. Semin Immunol 2013; 25:430-8; PMID:24183701; http://dx.doi.org/ 10.1016/j.smim.2013.10.005 [DOI] [PubMed] [Google Scholar]