

ABSTRACT
FGF13 (FHF2), the major fibroblast growth factor homologous factor (FHF) in rodent heart, directly binds to the C-terminus of the main cardiac sodium channel, NaV1.5. Knockdown of FGF13 in cardiomyocytes induces slowed ventricular conduction by altering NaV1.5 function. FGF13 has five splice variants, each of which possess the same core region and C terminus but differing in their respective N termini. Whether and how these alternatively spliced N termini impart isoform-specific regulation of NaV1.5, however, has not been reported. Here, we exploited a heterologous expression to explore the specific modulatory effects of FGF13 splice variants FGF13S, FGF13U and FGF13YV on NaV1.5 function. We found these three splice variants differentially modulated NaV1.5 current density. Although steady-state activation was unaltered by any of the FGF13 isoforms (compared to control cells expressing Nav1.5 but not expressing FGF13), open-state fast inactivation and closed-state fast inactivation were markedly slowed, steady-state availability was significantly shifted toward the depolarizing direction, and the window current was increased by each of FGF13 isoforms. Most strikingly, FGF13S hastened the rate of NaV1.5 entry into the slow inactivation state and induced a dramatic slowing of recovery from inactivation, which caused a large decrease in current after either low or high frequency stimulation. Overall, these data showed the diversity of the roles of the FGF13 N-termini in NaV1.5 channel modulation and suggested the importance of isoform-specific regulation.
KEYWORDS: activation, fibroblast growth factor homologous factors, FGF13, gating, inactivation, isoform, Nav1.5, N-termini, recovery from inactivation, voltage gated sodium channels
Introduction
Voltage gated sodium channels (VGSCs) underlie the rapid upstroke and propagation of the action potential in most excitable cells.1 VGSC channels are large macromolecular complexes composed of a pore-forming α subunit (NaV), auxiliary β subunits, and a growing list of channel interacting proteins (ChIPs).2-5 There are ten α subunits and four β subunits encoded in the human genome.6,7 The individual α subunits are expressed in different tissues and have different biophysical and pharmacological properties.8 Each α subunit is composed of four homologous domains (DI-DIV) that contain six transmembrane spanning segments.7 NaV1.5 is expressed in heart and is the major cardiac VGSC.
The importance of ChIPs on NaV1.5 function is highlighted by multiples studies showing that when ChIPs are mutated or when their binding site on NaV1.5 is mutated to affect interaction an increasingly wide range of cardiac rhythm disorders results, including Long-QT syndrome, Brugada syndrome, cardiac conduction disorders, idiopathic ventricular fibrillation, and sinus node dysfunction.9-13 Recently, fibroblast growth factor homologous factors (FHFs) have emerged as a novel class of VGSC ChIP.14-16 There are four FHF genes: FGF11-FGF14, each with different splice variants. Some of these splice variants, e.g., FGF12B, FGF13S, FGF13B, FGF14A, FGF14B, have been reported to bind to and modulate cardiac or neuronal VGSC.17-19 The relevance of FHFs on VGSC function is highlighted by various human disorders or mouse models. A human missense mutation in FGF14 or knockout in mice of Fgf14 causes ataxia and cognitive deficits.20,21 Conditional knockout of Fgf13 in mouse brain resulted in neuronal migration defects and altered learning and memory.22 Fgf13 knockdown in rat cardiomyocytes slowed cardiac conduction velocity by altering NaV1.5 function.23
This study focuses on FGF13. In rodent heart, Fgf13 is the main FHF expressed,23 so an understanding of how FGF13 proteins affect NaV1.5 channels provides essential information to understand the consequences of mouse models designed to study the effects of FHFs on cardiac VGSC currents. Moreover, FGF13, which encodes five isoforms, each with a distinct N-terminal sequence generated through alternative promoter usage and 5′ alternative splicing (Fig. 1), is expressed in human heart. So an understanding of the modulatory effects of FGF13 on NaV1.5 will provide context for unraveling certain human arrhythmias that result from variants in FGF13 or those within NaV1.5 that affect interaction with FGF13. The FGF-like core region, which forms the interaction domain for NaV1.5, and C-terminal sequence of the FGF13 isoforms are identical for all FGF13 isoforms. How specific isoforms of FGF13 isoforms differing only in their N-termini modulate NaV1.5 channel have not been explored. To understand the influence of FGF13 on cardiac VGSCs, we picked three specific FGF13 isoforms for detailed study: FGF13VY, which possess the longest N-terminus and is the isoform most highly expressed in rodent heart23; FGF13U, which possess the shortest N-terminus and thereby mimics a FGF13 composed of only its FGF-like core and C-terminus24; and FGF13S, which has a novel inactivation particle situated in the N-terminus, as reported in neurons.25 We found that the three splice variants differentially modulated NaV1.5 current density and gating properties including activation, fast and slow inactivation, steady-state inactivation, window current, and the frequency dependence of Nav1.5 inactivation. These data provide structural insights into FGF13 modulation on NaV1.5 currents and highlight the importance of FHF N-termini and their consequent isoform-specific regulation of VGSC.
Figure 1.

Kinetics of Nav1.5 channel activation were not affected by FGF13 isoforms. (A) Diagram for FGF13 isoforms. Alternative splicing FGF13 isoforms are schematically shown with different color. (B) Representative families of currents recorded from tsA201 cells transiently co-transfected with Nav1.5 and either pIRES-GFP (Control) or one of the FGF13 isoforms (FGF13S, FGF13U and FGF13VY) subcloned into pIRES-GFP vector. (C) Summarized relative peak currents density for control or the FGF13 isoforms (**P < 0.01, compared to control). Numbers of cells tested are shown on the top of each column. (D) Steady-state conductance-voltage relationships of Nav1.5 channels in the absence and presence of FGF13 isoforms. Conductance was normalized to single Boltzmann fits, whose averages are represented by solid curves. (E) Normalized peak current-voltage relationships of Nav1.5 elicited from a holding potential of −120 mV to the indicated test voltages.
Results
Activation
As shown in Figure 1A, all FGF13 isoforms share the same core domain and C-terminus, but have different N-termini, which vary in length and amino acid sequence (Fig. 1A). In tsA201 cells, NaV1.5 was co-expressed with one of the three FGF13 isoforms chosen for study or IRES-GFP (control), and whole-cell currents were investigated by the voltage-clamp technique. To focus on the effects of the FGF13 isoforms, no β subunits were included in these studies. Exemplar traces are shown in Figure 1B. FGF13S did not altered NaV1.5 current density, but FGF13U and FGF13VY significantly decreased current density, compared to control (Fig. 1C). Current density can be regulated either by channel trafficking to the plasma membrane and/or by effects upon channel gating, and FHFs are known to exert cell-type specific regulation of channel trafficking.26-28 We therefore tested whether the reduced current density observed when FGF13VY was expressed resulted from effects upon channel trafficking to the membrane. By immunoblotting and a cell surface biotinylation assay, we found that FGF13VY expression not only reduced the amount of Nav1.5 expression lysates, but also significantly decreased amount of NaV1.5 at the cell surface in tsA201 cells (Supplemental Fig. 1). Thus, FGF13 isoforms can specifically affect trafficking of NaV1.5 to the plasma membrane. Having established this isoform-specific effect on current density, we then turned our attention to how FGF13 isoforms exert specific modulation on NaV1.5 channel gating.
We determined the voltage-dependence of channel activation by normalizing conductance and fitted the data with a Boltzmann function. The midpoint (V1/2) and the slope factor (k) were not altered when any of the three FGF13 isoforms were co-expressed compared to control (Fig. 1D and Table 1). Further, normalized peak current-voltage relationship of NaV1.5 elicited from a holding potential of −120 mV to the indicated test voltages were not different from control when any of the FGF13 isoforms were co-expressed (Fig. 1E).
Table 1.
Activation properties of Nav1.5 co-expression of pIRES-GFP or FGF13 isoforms.
| V1/2 (mV) | k | n | |
|---|---|---|---|
| Control | −36.3 ± 0.4 | 5.8 ± 0.2 | 9 |
| FGF13S | −39 ± 0.4 | 5.2 ± 0.2 | 9 |
| FGF13U | −36.7 ± 0.5 | 5.3 ± 0.2 | 6 |
| FGF13VY | −39.3 ± 0.45 | 5.6 ± 0.2 | 8 |
V1/2 is the membrane voltage of half-maximal activation and k is the slope factor.
Fast inactivation
We then measured the time constants of open-state fast inactivation by fitting the decay of current with a single exponential function. Cells were held at −120 mV to remove inactivation and then test pulses from −80 to +40 mV were applied in 5-mV increments. The normalized traces in Figure 2A and 2B emphasized the rate of decay of VGSC currents recorded at −20 mV (peak of the I-V curve) from cells expressing NaV1.5 in either the presence or absence of FGF13 isoforms. Compared to currents from cells not expressing FGF13, currents from cells expressing any one of the three FGF13 isoforms showed a slowed rate of decay. The time constants for open-state fast inactivation, measured between −60 mV and +40 mV are shown in Figure 2C. FGF13S significantly slowed the fast inactivation at all test potentials between +10 mV and −40 mV. However, FGF13U or FGF13VY only slowed the fast inactivation at test potentials more negative than −20 mV. We also examined the persistent current, measured at the end of a 100 ms test pulse to −20 mV and shown as percentage of peak current. There were no significant difference between control and FGF13 isoforms (Fig. 2D).
Figure 2.
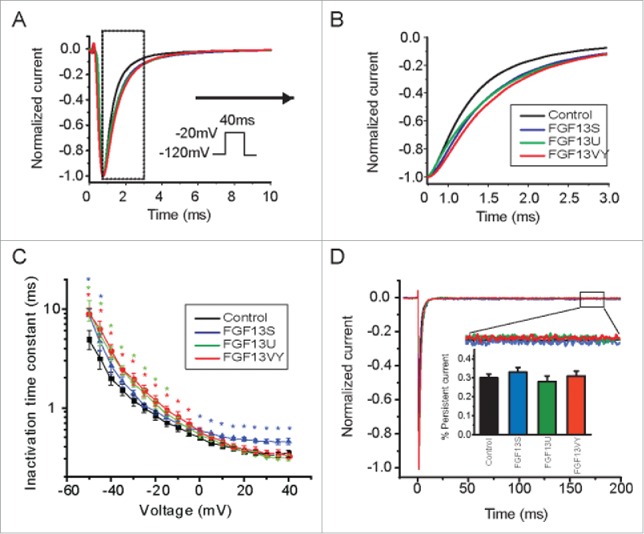
FGF13 isoforms affect fast inactivation. (A,B) Currents were recorded from tsA201 cells transiently co-transfected with Nav1.5 and either pIRES-GFP or FGF13 isoforms (FGF13S, FGF13U and FGF13VY). Comparison normalized sample current traces recorded at −20 mV illustrating slowed inactivation of FGF13 isoforms co-expression groups as compared with control group. (C) Fast inactivation time constants were obtained by fitting the fast component of current decay with single exponential equation among each groups. Solid line are exponential fits to the mean time constants (Color stars indicate respective groups, *P < 0.05, compared to control). (D) Representative TTX-subtracted, whole-cell currents were recorded. Persistent current was measured in response to a 200-ms voltage step to −20 mV from a holding potential of −120 mV, and each trace was normalized to the peak sodium current. Insert (box) shows magnification of the persistent currents. Summarized data showed there were no different for persistent currents in each groups (insert).
Closed state inactivation
To measure closed-state fast inactivation, cells were held at −120 mV, prepulsed to −70 mV or −90 mV for increasing amounts of time (1-200 ms) and then stepped to −20 mV and the fraction of current inactivated during the prepulse was determined. Compared to control, FGF13 isoforms markedly decreased the development of NaV1.5 close-state fast inactivation (Fig. 3 and Table 2).
Figure 3.

Development of close-state inactivation. (A, B) Development of inactivation at −70 mV and −90 mV. FGF13 isoforms co-expression altered the voltage-dependence of development of close-state inactivation.
Table 2.
Development of inactivation properties of Nav1.5 co-expression of pIRES-GFP or FGF13 isoforms at −70mV and −90mV.
| Offset (%) | tau | n | |
|---|---|---|---|
| −70mV Control | 0.3 ± 0.2 | 31.9 ± 1.1 | 4 |
| −70mV FGF13S | 33.4 ± 0.1** | 84.4 ± 11** | 4 |
| −70mV FGF13U | 40.9 ± 0.2** | 95.4 ± 7.6** | 4 |
| −70mV FGF13VY | 26.3 ± 0.2 ** | 73.1 ± 7.3** | 4 |
| −90mV Control | 42.7 ± 3 | 69.3 ± 11.8 | 4 |
| −90mV FGF13S | 83 ± 8.7** | 164.9 ± 12.3** | 4 |
| −90mV FGF13U | 90.8 ± 5.4** | 153.3 ± 12.9** | 4 |
| −90mV FGF13VY | 73.2 ± 10.2 ** | 268.1 ± 28.4** | 4 |
Offset is the proportion of current that did not inactivate.
P < 0.01 compared to control at the same voltage.
Steady state inactivation
To measure the voltage dependence of steady-state fast inactivation in NaV1.5, we plotted the peak current amplitudes as a function of the prepulse potential (protocol as shown in Fig. 4A, inset) and fitted the data with a Boltzmann function as shown in Figure 4. FGF13 isoforms markedly shifted the V1/2 of steady-state fast inactivation to the depolarizing direction compared to control (see also Table 3). Those changes are consistent with the effect observed in closed-state inactivation (Fig. 3). Window current was measured by calculating the area under the normalized activation curve and steady-state fast inactivation curve. Compared to control, FGF13 isoforms increased the relative window current by 463.5%, 879.5% and 968.4%, for FGF13S, FGF13U and FGF13VY, respectively (Fig. 4B).
Figure 4.
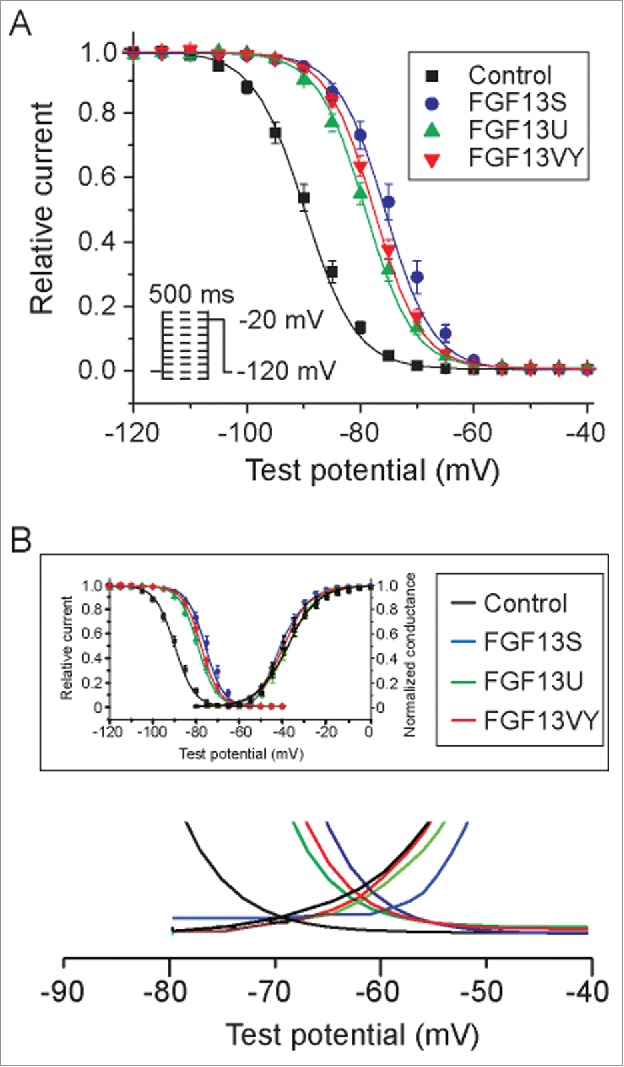
Voltage dependence of steady-state inactivation. (A) Inactivation was elicited by a 500 ms conditioning pulse from a holding potential of −120 mV to voltages between −130 and −10 mV in 5 mV increments prior to a test pulse at −20 mV (inset). Cells co-expressing Nav1.5 and FGF13 isoforms (FGF13S, FGF13U, FGF13VY) displayed an availability curve that was significantly depolarized shift comparing to cells expressing Nav1.5 and IRES-GFP (Control). (B) FGF13 isoforms increase window current.
Table 3.
Inactivation properties of Nav1.5 co-expression of pIRES-GFP or FGF13 isoforms.
| V1/2 (mV) | k | n | |
|---|---|---|---|
| Control | −89.8 ± 0.3 | 4.5 ± 0.1 | 11 |
| FGF13S | −75.6 ± 0.4* | 4.2 ± 0.2 | 10 |
| FGF13U | −79.1 ± 0.3* | 4.2 ± 0.1 | 6 |
| FGF13VY | −77.4 ± 0.3* | 4.1 ± 0.1 | 7 |
V1/2 is the membrane voltage of half-maximal activation and k is the slope factor.
P < 0.05, compared to control.
Slow inactivation
We also examined whether FGF13 isoforms affected slower forms of inactivation that might be critical for channel availability on a time scale from several seconds to minutes. To measure slow inactivation, cells were depolarized to −20 mV for durations ranging from 500 ms to 10 s, allowed to recover from fast inactivation at −120 mV for 40 ms, and subjected to a 50 ms test pulse to −20 mV. Compared with control, only FGF13S showed a significantly faster onset of slow inactivation (Fig. 5, Table 4).
Figure 5.

FGF13S markedly hasten entry into inactivation state. Entry of Nav1.5 currents into inactivated state was assayed by varying the during of conditioning pulse to −20 mV followed by a return to −120 mV for 40 ms, then followed by a 50 ms test pulse to −20 mV (insert).
Table 4.
Entry into inactivation state properties of Nav1.5 co-expression of pIRES-GFP or FGF13 isoforms.
| Offset (%) | tau | n | |
|---|---|---|---|
| Control | 31.9 ± 7.6 | 5851.6 ± 149.3 | 6 |
| FGF13S | 18.9 ± 5.5* | 4477.2 ± 152.6* | 5 |
| FGF13U | 23.5 ± 9.2 | 5401.8 ± 162 | 4 |
| FGF13VY | 30.7 ± 10 | 5606.5 ± 189.5 | 4 |
Offset is the proportion of current that did not inactivate.
P<0.05 compared to control.
Recovery from inactivation and response to repetitive stimulation
A previous study reported that FGF13S, but not FGF13U slowed the recovery of NaV1.6 from inactivation.29 To examine the effects of FGF13 isoforms on NaV1.5 recovery from inactivation, we used a two-pulse protocol, as shown in Fig. 6. Cells were held at −120 mV before a test depolarization to −20 mV for 30 ms followed by a variable (1-1000 ms) interval were the membrane potential was returned to −120 mV for recovery and then a second test pulse for 30 ms to −20 mV. The peak current obtained during the second test pulse after a specific recovery interval was then normalized to the first and the data were fitted with double exponential functions. As shown in Figure 6, FGF13S cause a pronounced slower recovery from inactivation of NaV1.5 compared to control, but FGF13U and FGF13VY were without effect (Table 5). We then tested whether the FGF13 isoforms differentially affected NaV1.5 in response to repetitive stimulation. Cells were held at −120 mV before a series of 20 successive test potentials at −20 mV for 40 ms delivered over a range of frequencies (Fig. 7C, insert). Consistent with the effects upon recovery from inactivation, co-expression of FGF13S caused a larger decrement in peak current at higher stimulation frequencies (2 Hz, 5 Hz and 10 Hz) than that at lower stimulation frequencies (0.5 Hz and 1 Hz). There were no significant differences in peak currents at all frequencies tested for other FGF13 isoforms (Fig. 7).
Figure 6.
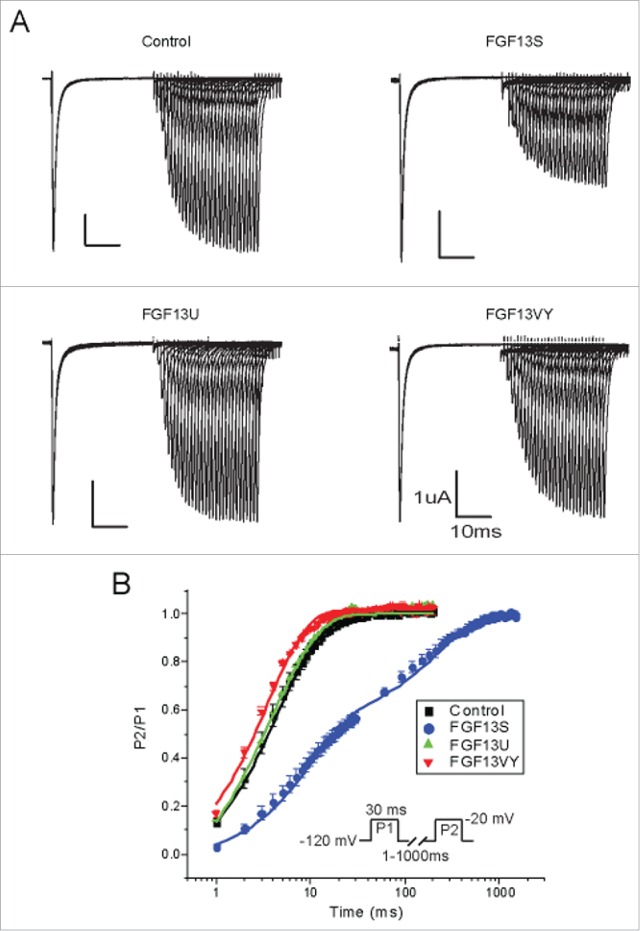
FGF13S dramatically slow the recovery from fast inactivation of Nav1.5. (A) Recovery from fast inactivation was determined from a holding potential of −120 mV, using two 30 ms pulses separated by a recovery time from 1 to 1000 ms at −120 mV (B) Mean data was fitted by double exponential equation.
Table 5.
Recovery from fast inactivation properties of Nav1.5 co-expression of pIRES-GFP or FGF13 isoforms.
| tau1 | tau2 | n | |
|---|---|---|---|
| Control | 4.3 ± 0.3 | 20.6 ± 4.5 | 7 |
| FGF13S | 9.7 ± 0.07* | 226.4 ± 7** | 6 |
| FGF13U | 5.4 ± 0.9 | 20.4 ± 1.6 | 3 |
| FGF13VY | 3.5 ± 0.1 | 15.3 ± 0.1 | 6 |
P < 0.05,
P < 0.01 compared to control.
Figure 7.

FGF13S markedly decrease the response to different frequency trains. Cells were held at −120 mV for 40 ms for 20 episodes at a variety of frequencies (0.5, 1, 2, 5 and 10 Hz). (A-D). Averages are summarized at variety of frequencies for cells expression of Nav1.5 with GFP (A) (n = 8), FGF13S (B) (n = 5), FGF13U (C) (n = 7), FGF13VY (D) (n = 6). (E) The level of inactivation of the Nav1.5 current was increased by co-expression of FGF13S at frequencies of 2, 5 and 10 Hz (*P<0.05, compared to GFP group).
Discussion
In this study we focused on the functional effects of FGF13 isoforms on gating properties of cardiac NaV1.5 channels expressed in mammalian cells. We found that individual FGF13 isoforms did not affect channel activation but had multiple effects on channel inactivation. Overall, we observed that co-expression of all three FGF13 isoforms examined decreased channel availability (depolarized the V1/2 of steady-state inactivation) and markedly slowed both the open-state and close-state fast inactivation. Among the FGF13 isoforms, the FGF13S splice variant was the most potent. Because the FGF13 splice variants share an identical core domain, which contains the major NaV1.5 interaction site, our data suggest that the alternatively spliced FGF13 N-termini exert the isoform-dependent effects on NaV1.5 gating properties.
Fast inactivation of sodium channel is a critical process that occurs within milliseconds of channel opening.2 A critical structure in open-state fast inactivation is the intracellular loop connecting domains III and IV, which serves as a hinged lid that occludes the inner pore.30 A triplet of hydrophobic residues (IFM) near the center of the III-IV linker may comprise a latch that holds the lid in a closed position over the inner pore. The docking site for the lid consists of multiple regions, including the cytoplasmic linkers connecting segments 4 and 5 in domains III and IV and the cytoplasmic end of the S6 segment in domain IV.31 The cardiac NaV1.5 channel exhibits faster close-state inactivation than skeletal muscle and neuronal sodium channels at potentials close to the normal cardiac resting potential. Inherited mutations associated with congenital arrhythmias have been shown to modify the close-state inactivation.32 Here, we showed that the development of closed-state inactivation at voltages of −90 mV or −70 mV (near the normal cardiomyocyte resting potential) was slowed when FGF13 isoforms were co-expressed. This suggests that mutations in FHFs or in the NaV1.5 channel at the site to which FHFs bind may lead to changes in closed-state inactivation and consequently to arrhythmogenesis.
The potency of the FGF13S isoform may have significant physiological and pathophysiological significance. Although FGF12 appears to be the main FHF in human heart, in which FGF13S expression appears relatively limited,33 any upregulation of FGF13S, such as during a disease state, would have important consequences on NaV1.5 currents and subsequently upon arrhythmogenesis.
The molecular mechanism underlying the specific effects of FGF13S on NaV1.5 gating remains unclear. One possibility is that the N terminus of FGF13S might directly interact with some component of the NaV1.5 inactivation machinery (e.g., the III-IV intracellular linker) to promote inactivation or to prevent recovery from inactivation once the process is initiated. Further studies will be needed to clarify the structural basis underlying the specific interaction between the FGF13S N-terminus and NaV1.5 channels. In addition, calmodulin is known to interact with sodium channel and FHFs in a ternary complex.34 However, it is not known whether calmodulin contribute to the different modulation of FGF13 isoforms on Nav1.5 gating.
Besides modulation of Nav1.5 channel gating, we found that FGF13VY affected Nav1.5 current density in a heterologous expression system by reducing overall protein expression of Nav1.5 and by downregulating the amount of NaV1.5 at the cell surface. Together, these data suggest FGF13VY is an important regulator of VGSCs in heart through multiple mechanisms.
Materials and methods
Plasmids and virus
6xHis (His6) tagged human FGF13 isoforms (FGF13S, FGF13U and FGF13VY) were subcloned into pIRES2-AcGFP1 (Clontech).
Cell culture and transfections
tsA201 cells were maintained in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 4 mM L-glutamine, 10% fetal bovine serum (Gibco), 100 U ml−1 penicillin, and 100 μg ml−1 streptomycin and incubated at 37° with 5% CO2. tsA201 cells were transfected at 80%-90% confluency using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. The total amount of DNA for all transfections was kept constant. All experiments were done 48-72 hours post-transfection.
Electrophysiology
Transfected cells were identified by GFP fluorescence. Na+ currents (INa) were recorded using the whole-cell patch-clamp technique at room temperature (20-22 ºC) 48–72 h after transfection. Electrode resistance ranged from 1-2 MΩ before series resistance compensation of 80%. Currents were filtered at 5 kHz. In tsA201 cells expressing NaV1.5 and FGF13, INa was recorded in solution containing (in mmol/L): NaCl 130, KCl 4, CaCl2 1.8, MgCl2 1, HEPES 10, glucose 10, pH 7.35 (adjusted with NaOH). The intracellular solution contained (in mmol/L): CsF 110, EGTA 10, NaF 10, CsCl 20, HEPES 10, pH 7.35 (adjusted with CsOH). Currents were elicited by a 40 ms pulse from a holding potential of −120 mV to test potentials between 100 mV and +60 mV in 10 mV increments. To determine the voltage-dependence of steady-state activation, the sodium conductance (G) was calculated by dividing the peak current for each voltage step by the driving force (Vm-Vrev) then normalized to the peak conductance (Gmax). Data were fitted with the Boltzmann relationship, G/Gmax=1/{1+exp[(V1/2-Vm)/k]} in which V1/2 is the voltage at which half of NaV1.5 channels is activated, k is the slope factor and Vm is the membrane potential. Standard two-pulse protocols were used to generate the steady-state inactivation curves: from the holding potential −120 mV, cells were stepped to 500-ms preconditioning potentials varying between 130 mV and −10 mV (prepulse), followed by a 20 ms test pulse to −20 mV. Currents (I) were normalized to Imax and fit to a Boltzmann function of the form I/Imax =1/{1+exp[(Vm-V1/2)/k]} in which V1/2 is the voltage at which half of NaV1.5 channels is inactivated, k is the slope factor and Vm is the membrane potential. Data analysis was performed using Clampfit 10.2 software (Axon Instruments) and Origin 8 (Originlab Corporation). Development of closed state inactivation at −70mV was measured by holding cells at −120 mV, prepulsing to −70 mV for increasing amounts of time (1-200 ms), and then stepping to −20 mV to determine the fraction of current inactivated during the pre-pulse. For recovery from inactivation, two-pulse protocol was used. Cells were held at −120 mV and two depolarizations to −20 mV for 30 ms were applied, with an increasing interval of time (1-1000 ms) between them for recovery. Curves were fitted with a double rising exponential function.
Statistical analyses
Results are presented as means ± standard error; the statistical significance of differences between groups was assessed using either a two-tailed Student's t test or one-way ANOVA and was set at P < 0.05.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Bingcai Guan for expert technical assistance in patch clamp recording.
Funding
This work was supported by the National Natural Science Foundation of China (No. 31171097), Natural Science Foundation of Hebei Province (No.C2014206419) to C.W.; Research Project of Science and Technology of Higher Education of Hebei Province (No.ZD2015007 and No.ZD2016002) to C.W and Y.J.; National Heart, Lung, and Blood Institute (NHLBI), R01HL71665 and R01HL112928 to G.S.P.; NHLBI F30 HL131217 (D.S.S.); the Duke Medical Scientist Training Program T32 GM007171 (D.S.S.); National Natural Science Foundation of China (No.31270882), and the National Basic Research Program of China (2013CB531302) to H.Z.
References
- [1].Catterall WA. The molecular basis of neuronal excitability. Science 1984; 223:653-61; PMID:6320365; http://dx.doi.org/ 10.1126/science.6320365 [DOI] [PubMed] [Google Scholar]
- [2].Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 2000; 26:13-25; PMID:10798388; http://dx.doi.org/ 10.1016/S0896-6273(00)81133-2 [DOI] [PubMed] [Google Scholar]
- [3].Goldin AL. Evolution of voltage-gated Na(+) channels. J Exp Biol 2002; 205:575-84; PMID:11907047 [DOI] [PubMed] [Google Scholar]
- [4].Amin AS, Asghari-Roodsari A, Tan HL. Cardiac sodium channelopathies. Pflugers Arch 2010; 460:223-37; PMID:20091048; http://dx.doi.org/ 10.1007/s00424-009-0761-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Shy D, Gillet L, Abriel H. Cardiac sodium channel NaV1.5 distribution in myocytes via interacting proteins: the multiple pool model. Biochim Biophys Acta 2013; 1833:886-94; PMID:23123192; http://dx.doi.org/ 10.1016/j.bbamcr.2012.10.026 [DOI] [PubMed] [Google Scholar]
- [6].Catterall WA, Goldin AL, Waxman SG. International union of pharmacology. XLVII. nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev 2005; 57:397-409; PMID:16382098; http://dx.doi.org/ 10.1124/pr.57.4.4 [DOI] [PubMed] [Google Scholar]
- [7].Payandeh J, Gamal El-Din TM, Scheuer T, Zheng N, Catterall WA. Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 2012; 486:135-9; PMID:22678296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Goldin AL, Barchi RL, Caldwell JH, Hofmann F, Howe JR, Hunter JC, Kallen RG, Mandel G, Meisler MH, Netter YB, et al.. Nomenclature of voltage-gated sodium channels. Neuron 2000; 28:365-8; PMID:11144347; http://dx.doi.org/ 10.1016/S0896-6273(00)00116-1 [DOI] [PubMed] [Google Scholar]
- [9].Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA, Keating MT. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 1995; 80:805-11; PMID:7889574; http://dx.doi.org/ 10.1016/0092-8674(95)90359-3 [DOI] [PubMed] [Google Scholar]
- [10].Tan HL, Bezzina CR, Smits JP, Verkerk AO, Wilde AA. Genetic control of sodium channel function. Cardiovasc Res 2003; 57:961-73; PMID:12650874; http://dx.doi.org/ 10.1016/S0008-6363(02)00714-9 [DOI] [PubMed] [Google Scholar]
- [11].Akai J, Makita N, Sakurada H, Shirai N, Ueda K, Kitabatake A, Nakazawa K, Kimura A, Hiraoka M. A novel SCN5A mutation associated with idiopathic ventricular fibrillation without typical ECG findings of Brugada syndrome. FEBS Lett 2000; 479:29-34; PMID:10940383; http://dx.doi.org/ 10.1016/S0014-5793(00)01875-5 [DOI] [PubMed] [Google Scholar]
- [12].Benson DW, Wang DW, Dyment M, Knilans TK, Fish FA, Strieper MJ, Rhodes TH, George AL Jr. Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J Clin Invest 2003; 112:1019-28; PMID:14523039; http://dx.doi.org/ 10.1172/JCI200318062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Musa H, Kline CF, Sturm AC, Murphy N, Adelman S, Wang C, Yan H, Johnson BL, Csepe TA, Kilic A, et al.. SCN5A variant that blocks fibroblast growth factor homologous factor regulation causes human arrhythmia. Proc Natl Acad Sci U S A 2015; 112:12528-33; PMID:26392562; http://dx.doi.org/ 10.1073/pnas.1516430112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wei EQ, Barnett AS, Pitt GS, Hennessey JA. Fibroblast growth factor homologous factors in the heart: a potential locus for cardiac arrhythmias. Trends Cardiovasc Med 2011; 21:199-203; PMID:22867699; http://dx.doi.org/ 10.1016/j.tcm.2012.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Goldfarb M. Fibroblast growth factor homologous factors: evolution, structure, and function. Cytokine Growth Factor Rev 2005; 16:215-20; PMID:15863036; http://dx.doi.org/ 10.1016/j.cytogfr.2005.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Goldfarb M. Voltage-gated sodium channel-associated proteins and alternative mechanisms of inactivation and block. Cell Mol Life Sci 2012; 69:1067-76; PMID:21947499; http://dx.doi.org/ 10.1007/s00018-011-0832-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Liu CJ, Dib-Hajj SD, Renganathan M, Cummins TR, Waxman SG. Modulation of the cardiac sodium channel Nav1.5 by fibroblast growth factor homologous factor 1B. J Biol Chem 2003; 278:1029-36; PMID:12401812; http://dx.doi.org/ 10.1074/jbc.M207074200 [DOI] [PubMed] [Google Scholar]
- [18].Rush AM, Wittmack EK, Tyrrell L, Black JA, Dib-Hajj SD, Waxman SG. Differential modulation of sodium channel Na(v)1.6 by two members of the fibroblast growth factor homologous factor 2 subfamily. Eur J Neurosci 2006; 23:2551-62; PMID:16817858; http://dx.doi.org/ 10.1111/j.1460-9568.2006.04789.x [DOI] [PubMed] [Google Scholar]
- [19].Lou JY, Laezza F, Gerber BR, Xiao M, Yamada KA, Hartmann H, Craig AM, Nerbonne JM, Ornitz DM. Fibroblast growth factor 14 is an intracellular modulator of voltage-gated sodium channels. J Physiol 2005; 569:179-93; PMID:16166153; http://dx.doi.org/ 10.1113/jphysiol.2005.097220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].van Swieten JC, Brusse E, de Graaf BM, Krieger E, van de Graaf R, de Koning I, Maat-Kievit A, Leegwater P, Dooijes D, Oostra BA, et al.. A mutation in the fibroblast growth factor 14 gene is associated with autosomal dominant cerebellar ataxia [corrected]. Am J Hum Genet 2003; 72:191-9; PMID:12489043; http://dx.doi.org/ 10.1086/345488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wang Q, Bardgett ME, Wong M, Wozniak DF, Lou J, McNeil BD, Chen C, Nardi A, Reid DC, Yamada K, et al.. Ataxia and paroxysmal dyskinesia in mice lacking axonally transported FGF14. Neuron 2002; 35:25-38; PMID:12123606; http://dx.doi.org/ 10.1016/S0896-6273(02)00744-4 [DOI] [PubMed] [Google Scholar]
- [22].Wu QF, Yang L, Li S, Wang Q, Yuan XB, Gao X, Bao L, Zhang X. Fibroblast growth factor 13 is a microtubule-stabilizing protein regulating neuronal polarization and migration. Cell 2012; 149:1549-64; PMID:22726441; http://dx.doi.org/ 10.1016/j.cell.2012.04.046 [DOI] [PubMed] [Google Scholar]
- [23].Wang C, Hennessey JA, Kirkton RD, Graham V, Puranam RS, Rosenberg PB, Bursac N, Pitt GS. Fibroblast growth factor homologous factor 13 regulates Na+ channels and conduction velocity in murine hearts. Circ Res 2011; 109:775-82; PMID:21817159; http://dx.doi.org/ 10.1161/CIRCRESAHA.111.247957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Munoz-Sanjuan I, Smallwood PM, Nathans J. Isoform diversity among fibroblast growth factor homologous factors is generated by alternative promoter usage and differential splicing. J Biol Chem 2000; 275:2589-97; PMID:10644718; http://dx.doi.org/ 10.1074/jbc.275.4.2589 [DOI] [PubMed] [Google Scholar]
- [25].Dover K, Solinas S, D'Angelo E, Goldfarb M. Long-term inactivation particle for voltage-gated sodium channels. J Physiol 2011; 588:3695-711; http://dx.doi.org/ 10.1113/jphysiol.2010.192559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang C, Hoch EG, Pitt GS. Identification of novel interaction sites that determine specificity between fibroblast growth factor homologous factors and voltage-gated sodium channels. J Biol Chem 2011; 286:24253-63; PMID:21566136; http://dx.doi.org/ 10.1074/jbc.M111.245803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Laezza F, Gerber BR, Lou JY, Kozel MA, Hartman H, Craig AM, Ornitz DM, Nerbonne JM. The FGF14(F145S) mutation disrupts the interaction of FGF14 with voltage-gated Na+ channels and impairs neuronal excitability. J Neurosci 2007; 27:12033-44; PMID:17978045; http://dx.doi.org/ 10.1523/JNEUROSCI.2282-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Pablo JL, Wang C, Presby MM, Pitt GS. Polarized localization of voltage-gated Na+ channels is regulated by concerted FGF13 and FGF14 action. Proc Natl Acad Sci U S A 2016; 113(19):E2665-74, Epub ahead of print; PMID:27044086; http://dx.doi.org/ 10.1073/pnas.1521194113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Laezza F, Lampert A, Kozel MA, Gerber BR, Rush AM, Nerbonne JM, Waxman SG, Dib-Hajj SD, Ornitz DM. FGF14 N-terminal splice variants differentially modulate Nav1.2 and Nav1.6-encoded sodium channels. Mol Cell Neurosci 2009; 42:90-101; PMID:19465131; http://dx.doi.org/ 10.1016/j.mcn.2009.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Vassilev PM, Scheuer T, Catterall WA. Identification of an intracellular peptide segment involved in sodium channel inactivation. Science 1988; 241:4873; http://dx.doi.org/ 10.1126/science.2458625 [DOI] [PubMed] [Google Scholar]
- [31].West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, Catterall WA. A cluster of hydrophobic amino acid residues required for fast Na(+)-channel inactivation. Proc Natl Acad Sci U S A 1992; 89:10910-4; PMID:1332060; http://dx.doi.org/ 10.1073/pnas.89.22.10910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kambouris NG, Nuss HB, Johns DC, Marban E, Tomaselli GF, Balser JR. A revised view of cardiac sodium channel “blockade” in the long-QT syndrome. J Clin Invest 2000; 105:1133-40; PMID:10772658; http://dx.doi.org/ 10.1172/JCI9212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hennessey JA, Marcou CA, Wang C, Wei EQ, Tester DJ, Torchio M, Dagradi F, Crotti L, Schwartz PJ, Ackerman MJ, et al.. FGF12 is a candidate Brugada syndrome locus. Heart Rhythm 2013; 10:1886-94; PMID:24096171; http://dx.doi.org/ 10.1016/j.hrthm.2013.09.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wang C, Chung BC, Yan H, Lee SY, Pitt GS. Crystal structure of the ternary complex of a NaV C-terminal domain, a fibroblast growth factor homologous factor, and calmodulin. Structure 2012; 20:1167-76; PMID:22705208; http://dx.doi.org/ 10.1016/j.str.2012.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.