

Abstract
Pancreatic cancer is one of the most challenging cancers. Whole genome sequencing studies have been conducted to elucidate the underlying fundamentals underscoring disease behavior. Studies have identified a subgroup of pancreatic cancer patients with distinct molecular and clinical features. Genetic fingerprinting of these tumors is consistent with an unstable genome and defective DNA repair pathways, which creates unique susceptibility to agents inducing DNA damage. BRCA1/2 mutations, both germline and somatic, which lead to impaired DNA repair, are found to be important biomarkers of genomic instability as well as of response to DNA damaging agents. Recent studies have elucidated that PARP inhibitors and platinum agents may be effective to induce tumor regression in solid tumors bearing an unstable genome including pancreatic cancer. In this review we discuss the characteristics of genomic instability in pancreatic cancer along with its clinical implications and the utility of DNA targeting agents particularly PARP inhibitors as a novel treatment approach.
Keywords: Pancreatic cancer, genomic instability, DNA repair, BRCA1, BRCA2, PALB2, ATM, PARP1, ARID1, MSH, PARP inhibitors, cisplatin, synthetic lethality
Introduction
Pancreatic cancer continues to be one of the most challenging malignancies despite vigorous research endeavors and even accepting a now in-depth understanding of the molecular pathogenesis of this disease. Although various genes have been identified to be critical in the development of pancreatic adenocarcinoma, targeted treatment has not changed the course of the cancer to date, in part as the key mutated genes are tumor suppressor genes which cannot be effectively drugged. Newer cytotoxic regimens, such as FOLFIRINOX[1] and nab-paclitaxel combined with gemcitabine [2] have significantly but incrementally improved outcomes in metastatic pancreatic adenocarcinoma. A recent analysis suggests that pancreatic cancer will remain one of most lethal malignancies in the proximate future given current incidence and mortality trends[3]. Therefore, further investigation is warranted to better understand the disease biology and develop new therapeutic approaches to alter the outcomes of this disease. In several studies, diverse genetic alterations have been identified to be involved in pancreatic carcinogenesis including activations of proto-oncogenes such as Kirsten rat sarcoma viral oncogenes homolog(KRAS), and Transforming growth factor beta (TGF-β) signaling and as well as loss of tumor suppressor gene activity such as p53, Cyclin-Dependent Kinase Inhibitor 2A (CDKN2A) and Breast Cancer Susceptibility Genes (BRCAs)[4]. Familial pancreatic cancer accounts for about 5% to 10% of all cases and is typically associated with mutations in tumor suppressor genes[5]. Some examples include CDKN2A which is related to familial atypical multiple mole melanoma (FAMMM) syndrome and BRCA1/BRCA2 which are related to hereditary breast-ovarian cancer syndrome[4]. Although the main drivers of pancreatic carcinogenesis are oncogenes, recent studies have uncovered the impact of tumor suppressor genes particularly genes related to DNA damage response/repair on disease behavior and treatment outcomes. Increasing availability of genome sequencing technology has led the research community to conduct genome wide studies and consequent identification of a distinct subpopulation of pancreatic cancers with unstable genomic properties due to mutations in DNA repair genes which creates a unique vulnerability to DNA targeting agents such as DNA-damaging cytotoxic agents and Poly-ADP ribose polymerase (PARP) inhibitors. Herein, we discuss the characteristics of genomic instability in pancreatic adenocarcinoma and related therapeutic opportunities.
Genomic Instability and Pancreatic Cancer
Recent genome wide studies have elucidated diverse signaling pathways either activated or silenced in multi-stage pancreatic carcinogenesis[6–12] (Table 1). Although these discoveries indicate the complexity of pancreatic cancer development along with the diversity of genetic alterations potentially accounting for varied clinical behavior, they also have enhanced our understanding of the genetic fingerprint of pancreatic cancer as well as the targetability of molecular pathways that may influence clinical outcomes.
Genomic instability refers to a high frequency of deleterious changes within the genomic structure as a consequence of impaired DNA repair response[13]. Silencing or deleterious mutations in tumor suppressor genes, particularly in checkpoint and DNA repair genes such as p53, p16 and Ataxia Telangiectasia Mutated (ATM), have been associated with the development of many malignancies. Many of these genes have also been associated with genomic instability. BRCA1/BRCA2 mutations[14], make DNA susceptible to deleterious changes due to lack of DNA repair response[15] which is further discussed below. Mutations in the ATM gene, another key gene in DNA damage response and repair, are also related to increased genetic alterations such as deletion or insertion of new nucleotides and even interchromosomal translocations [16,17]. A key tumor suppressor gene, p53, is another important signaling gene for maintaining genomic stability via checkpoint functions as well as a direct activator of DNA damage response[18]. Consistent with its physiological function, loss of p53 activity has been implicated in settings of high genomic instability[13,19]. Replication protein A1 (RPA1), Partner and Localizer of BRCA2 (PALB2), and RAD51 are other important mediators of DNA maintenance and were recently found to be other potential biomarkers of genomic instability[10].
Genomic sequencing of 13 pancreatic cancer patients has shed some light on an important relationship between genomic instability and disease progression[8]. In this study, Campbell et al. observed continuous genetic rearrangements throughout the development and progression of pancreatic cancer as a consequence of genomic instability. For example, metastatic samples of different sites from the same patient showed diverse genetic alterations in different loci suggesting an unstable genome provides additional characteristics to metastatic lesions originating from the same parental clone[8]. This genetic diversity explains a challenge observed by clinicians; heterogeneous response to therapy and subsequent progression of some of lesions with maintained disease control in others after initial treatment response. Disease heterogeneity driven by an unstable genome may also show distinct properties in different tumors. For example, while pancreatic cancer typically bears intrachromosomal/interchromosomal rearrangements and deletions, breast cancer has more tandem duplications along with genomic amplifications in oncogenes[8]. These data indicate that genomic instability promotes distinct mechanisms of carcinogenesis throughout the development of different cancers potentially correlating with diverse disease behaviors in different cancers.
Genomic instability in cancer cells is also an important determinant of disease outcome. For example, impaired Mismatch Repair (MMR) activity and related high microsatellite instability (MSI), which is observed in Lynch syndrome typically with right sided colon tumors with mucinous and lymphocytic infiltrates, and causes high genomic instability with thousands of mutations and is related to a more favorable prognosis compared to MSI low colorectal cancer[20–22]. More recent data has provided proof of concept that immune therapy targeting in this subgroup with a high mutational load may lead to significant therapeutic benefit[23]. Chromosomal instability (CIN), on the other hand, seen more often in sporadic colorectal cancer and associated with left sided colon cancers, is correlated with more aggressive tumors with poor prognostic markers[24]. High genomic instability has also led to improved survival outcomes in gastric tumors patients compared to patients with a relatively stable genome[25]. Consistent with findings from other gastrointestinal malignancies, whole genome analysis of pancreatic adenocarcinoma patients elucidated the co-segregation of impaired DNA repair genes (including BRCA1, BRCA2) and genomic instability along with an association between favorable outcome and high genomic instability [10].
DNA Repair Pathways, Cell Cycle, and the Fingerprint of Defective DNA Repair in Pancreas Cancer: “BRCAness” and Beyond
Recent studies interrogating the genome have elucidated multiple defects in DNA repair pathways and their consequences on the molecular behavior of pancreatic cancer cells. ATM, which has been found to be mutated in sporadic and hereditary pancreatic cancer[6,26], is one of the important mediators of DNA damage response and repair pathway along with ATR. ATM initiates a cascade of reactions which triggers various signal mediators and downstream proteins to initiate check point control via p53[27]. Furthermore, double-strand DNA breaks directly activate the cascade of ATM and BRCA1/BRCA2 to launch DNA-repair[17] (Figure 1). BRCA1/BRCA2 are two crucial proteins in activation and operation of homologous recombination (HR) in double-strand DNA break repair. BRCAs, particularly BRCA2, interacts with homologous repair (HR) initiating proteins such as RAD51 and recruit DNA repair assembly[28,29]. PALB2 performs a critical role in recruitment of DNA repair machinery to activate HR and its mutations have been found to be related to an increased risk of pancreatic cancer[30]. HR, unlike other double-stranded DNA break repair pathways such as non-homologous end-joining pathway (NHEJ) or single strand annealing (SSA) uses homologous DNA to repair the breaks which yields error-free and high quality outcomes[31]. A switch from HR to other DNA repair pathways confers an increased risk of alterations in DNA sequence which creates frequent deleterious changes in genetic material due to low-fidelity DNA repair and genetic rearrangements[32].
Figure 1. Cell cycle and check points.
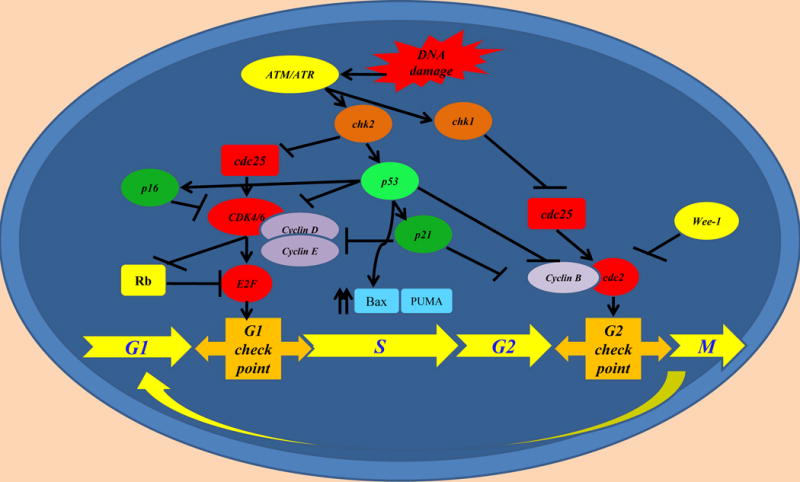
Upon DNA damage, many pathways regulating the cell cycle are activated. Mutations in genes negatively controlling the cell cycle have been demonstrated in carcinogenesis in various tumors.
Aforementioned DNA repair with non-homologous pathways and related errors in DNA repair and replication create certain genotypic and phenotypic features called “BRCAness”[10,32]. This phenotype was first described in breast cancer and it is characterized by invasive behavior, necrosis and lymphocytic infiltration[32,33]. The BRCAness genotype in pancreatic cancer typically harbors extensive intrachromosomal rearrangements (>200 structural changes) compared to tumors with a relatively stable genome (<50 structural changes) as a result of defective DNA maintenance[10]. Although this BRCAness pattern in cancer cells genome may lend a genetic plasticity and possibly diverse molecular behavior, it also confers a unique vulnerability to DNA targeting agents. Therefore, BRCAness genotype is consistent with hyperdynamic genomic rearrangement process with many deleterious changes in genes that directly alter genomic structure and molecular characteristics of those effected cancer cells. BRCAness signature also determines clinical features and behavior of this disease. For example, those affected patients, particularly patients with germlineBRCA1 mutations, tend to develop pancreatic cancer at relatively earlier ages (mean age of 60.3) versus 71 years in average age onset and 30% of those patients have resectable disease at the time of diagnosis[34,35]. Their overall survival also appears to be better particularly for individuals exposed to platinum based treatments [34].
The Role of PARP1 in DNA Repair Pathways and Evidence from Preclinical Studies Pertaining to Antitumor Effects of DNA Targeting Agents and Synthetic Lethality
PARP1, poly (ADP-ribose) polymerase 1, is a crucial nuclear enzyme of cellular homeostasis that modifies many nuclear proteins by poly ADP-ribosylation[36]. One of the important functions of PARP1 is activation of DNA damage response particularly in single-stranded DNA break repair (SSBR). Spontaneous single-stranded DNA breaks are recognized by PARP1 and recruitment of XRCC1 (X-ray Repair Cross-Complementing Protein 1), which functions as a scaffold protein in SSBR, is mediated by PARP1[37]. This process is followed by recruitment of base exchange complex (BER) which repairs single stranded breaks[38]. There is also evidence suggesting that PARP1 is involved in NHEJ, another pathway that functions in double-strand DNA break repair[39]. Moreover, PARP1 appears to be an enhancer of HR pathways. One study suggested that decreased HR activity is due to suppression by NEHJ pathway in PARP1 mutant cells lines[40]. A study in breast cancer, including BRCA mutant tumors, observed up-regulation of PARP1[41].
The upregulation of PARP1 along with its pivotal role in DNA break repair has led to investigation of the targetability of this critical enzyme. Bryant et al. first explored the role of PARP inhibitors in cancer cells and identified a collapsed replication fork in BRCA2 mutant cancer clones which induced apoptosis proceeded by cell cycle arrest[42]. This study followed by other preclinical work demonstrates that both BRCA1 and BRCA2 mutant cancers cells are very sensitive to PARP inhibition leading to further chromosomal instability and apoptosis in the absence of HR mediated DNA repair[43]. Both studies reported that impaired PARP1 activity in the setting of BRCA1/BRCA2 mutant cancer cells created multiple single and double stranded DNA breaks and that these DNA breaks were associated with irreparable DNA injuries which subsequently induced cell cycle arrest particularly in G2 phase and apoptosis (Figure 2). This concept, dysfunction of at least two or more genes related to a certain pathway leading to irreversible cellular damage and apoptosis while one of them is compatible with viability, named as synthetic lethality has created a new approach in cancer therapeutics. There is also evidence that PARP inhibitors lead to increased Wee-1 expression, a cell cycle check point regulator [44]. This suggests that cancer cell induced physiologic cell cycle arrest at G2 phase before proceeding mitosis avoids further DNA damage and occurrence of ultimate DNA injury. Consistent with this observation, combined models of PARP inhibitors with check point inhibitors enhance radiosensitization in combined model systems[45–47]. Another recent study also observed that ARID1A mutations may be related to impaired DNA repair pathway and PARP inhibitor sensitivity in cell lines bearing this mutation[48].
Figure 2. Inducing DNA damages and apoptosis in HR-impaired pancreatic cancer cells.
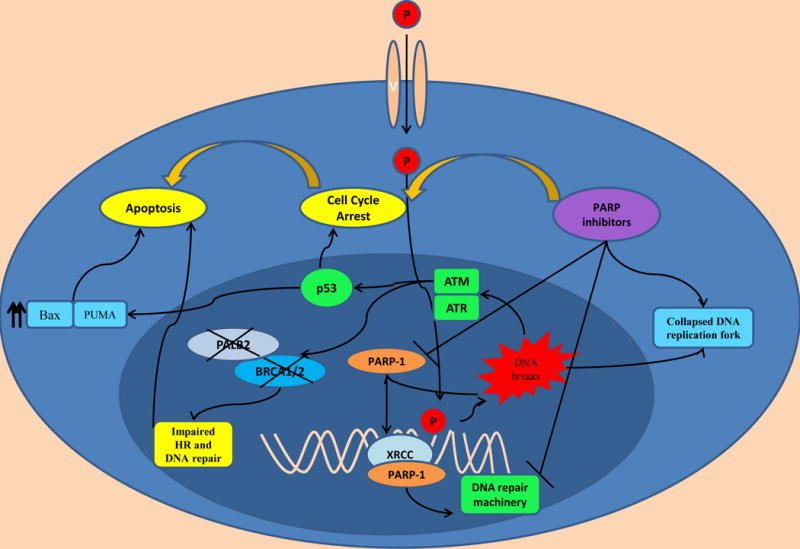
Figure 1. PARP inhibitors and platinum agents (P) induce cell cycle arrest via leading to irreparable DNA damages and collapsed DNA replication fork. HR: homologous repair. XRCC: X-Ray Repair Cross-Complementing Protein 1. PALB2: Partner And Localizer Of BRCA. P: platinum based agents
Screening for Biomarkers of Genomic Instability
BRCA1 and BRCA2 as described above are key important genes of HR-mediated DNA repair and germline mutations in these genes are associated with pancreatic cancer development [49,50]. Screening for mutations in these two genes is well established in breast and ovarian cancer particularly in patients deemed as high risk based on personal or family history. However, criteria for genetic screening of BRCA mutations in pancreatic cancer are evolving. Although their prevalence is considerably lower in pancreatic cancer, approximately 5–7% in an unselected population, as stated above, BRCA mutant pancreatic cancers bear distinct molecular and clinical features which necessitate identification of these mutations to optimize treatment based on this genetic biomarker. Moreover, detection of these mutations may also provide a better stratification of pancreatic cancer patients for enrollment in clinical trials given that BRCA mutations may have prognostic value.
Several studies have investigated the prevalence of BRCA mutations in pancreatic cancer populations. One study suggests, approximately 19% of familial pancreatic cancers (families with two first degrees relatives with pancreatic cancer) are attributable to BRCA2 mutations[51]. Germline BRCA mutations particularly BRCA2 mutations are more prevalent in Ashkenazi Jews and may be seen in more than 2% of this population[52]. One study showed that up to 14.2% of Ashkenazi Jewish patients with a family history of pancreatic cancer harbor a BRCA mutation[53]. A recent cohort indicated this prevalence may be seen in about 4.6% of unselected pancreatic cancer patients[54] suggesting germline BRCA mutations are a more significant contributor to pancreatic carcinogenesis in Ashkenazi Jews population. In a recent study from Memorial Sloan Kettering Cancer Center, 175 pancreatic carcinoma patients referred for genetic testing on the basis of personal/family history were evaluated in a cohort study and of those, 159 (91%) patients underwent germline genetic testing which revealed 24 deleterious mutations (15.1%) of which BRCA2 mutation was the commonest and seen at a rate of 54% (13/24). Other observed mutations were in 4 other genes, including BRCA1 (16.6%), 1 PALB2(4,1%), 2 CDK4N2(8.3%) and 4 MMRs genes (16.6%)[35]. In the same study 96 of the tested patients were Ashkenazi Jewish and 15 (15.6%) patients had one of these deleterious germline mutations of which 11 were BRCA2 mutations (73%). In non-Ashkenazi Jewish a similar rate was observed (14.3%). Interestingly, when mutations were limited to BRCA1/BRCA2, mutations rates in Ashkenazi and non-Ashkenazi Jewish population were 13.5%and 6.3% respectively. Moreover, the authors found a mutation rate of 7.4% in predisposing genes (2 of 27 patients) in Ashkenazi Jewish patients even without any family of history of a BRCA-associated cancer. These collective data provide support that germline screening for a BRCA mutation in pancreatic cancer patients with Ashkenazi Jewish heritage should be strongly considered. Another study by Holter et al. investigated the prevalence of BRCA mutations in pancreatic cancers patients in a large cohort[54]. In this study the authors reported germline BRCA1 and BRCA2 mutational frequency as 1% and 3.6% respectively. Most striking was the finding that more than 50% of patients with a BRCA mutation did not match BRCA screening criteria of the National Comprehensive Cancer Network (NCCN) or Ontario Ministry of Health and Long Term Care (Ontario MHLTC) guidelines pointing that germline penetrance features of BRCA mutations in pancreatic cancer population could be different and modification of both guidelines for BRCA screening is needed in the pancreatic cancer field. In the same study, the authors recommended to screen for a BRCA mutation in pancreatic cancer patients who has a history of at least one or more first or second degree family member affected by a breast or ovarian cancer. Routine screening for germline mutations in other genes including PALB2, RPA1 and mismatch repair genes needs to be further investigated given their relatively low prevalence in pancreatic cancer.
The impact of somatic BRCA mutations on response to DNA targeting agents is currently being investigated. A recent whole genome sequencing study by Waddell et al[10] reported overlapping features in both somatic and germline mutations such as unstable genome along with possible DNA targeting agent sensitivity in both groups. This suggests that gained mutation of DNA repair genes throughout the carcinogenesis process may also create synthetic lethality and a larger subset of patients may benefit from these evolving treatment approaches. However, whether a defect in BRCA as a result of a germline/founder mutation may create distinct disease behavior compared to progressor mutations (clonal expansion related mutations) needs to be further investigated. For example, progressor mutations may occur in subclones of tumors-whereas germline mutations globally involve all tumors clones and progressor mutations creates subclones with multiple compensatory pathways with distinct characteristics.
Evidence from Clinical Studies of DNA targeting agents PARP Inhibitors
Given promising results observed in preclinical work, the utility of PARP inhibitors has been investigated in pancreatic cancer. Fong et al. investigated the safety and efficacy of olaparib, a potent oral PARP inhibitor, as a single agent in 60 advanced stage solid tumor patients (22 patients had a germline BRCA1 or BRCA2 mutation) in a phase I study[55]. Patients received various doses and dose limiting toxicities were observed at 400 mg and 600 mg twice daily. PARP inhibition (>90%) has been demonstrated starting from 60 mg twice daily dosing. In the same study, the authors further investigated olaparib in germline BRCA mutant patients with a dose of 200 mg twice daily continuously. Antitumor activity assessed in 19 patients of which 12 of the 19 (63%) had either biomarker or radiological response. Of those 12 patients, 9 (75%) patients had a RECIST radiologic response. None of the enrolled pancreatic cancer patients were a BRCA mutation carrier. Another early study evaluated olaparib in a phase I study in combination with topotecan for safety and clinical effect. Fatigue and gastrointestinal symptoms were the most common adverse effects along with dose limiting neutropenia[56] (Table 2). The combination of olaparib and topotecan was not recommended to be further studied due to dose limiting myelosuppression and concern for subtherapeutic maximum tolerated dosing. Further, olaparib was studied as a single agent in a phase II study which showed significant clinical activity in BRCA1/BRCA2 mutant pancreatic cancer patients who were previously treated with gemcitabine[57]. In this study, a total of 298 patients were enrolled and of these, 23 patients had pancreatic cancer bearing a germline BRCA mutation. The dose of olaparib administered in the trial was 400 twice daily continuously. Sixty-five percent of pancreatic cancer patients enrolled in the trial were previously treated witha platinum based therapy. Twenty-two percent of pancreatic cancer patients responded to treatment and 35% of patients had stable disease for more than 8 weeks. The response rate to olaparib was similar in both patients who were previously exposed to platinum therapy, however, the status of platinum resistance of these patients was not reported, making the signal of efficacy in the platinum-pre-treated population hard to interpret. Most common side effects observed were fatigue, nausea and vomiting along with grade ≥3 anemia observed in 17.4% of the participants. Of note, two of the 298 patients developed leukemia and one patient developed myelodysplastic syndrome (MDS) which were attributed to olaparib and or related to prior therapy. Most recently, this drug has been approved by the FDA with a recommended dose of 400 twice daily for germline BRCAmutant advanced ovarian cancer in a third-line setting after significant progression free survival (PFS) improvement was observed in a phase III trial [58], although no overall survival benefit was reported. Currently a phase III randomized clinical trial of olaparib is being conducted in pancreatic cancer patients with a germline BRCA mutation following induction treatment of at least 4 months of platinum based therapy for front-line treatment of metastatic pancreatic adenocarcinoma (NCT02184195) (Table 3).
Table 2.
Completed Clinical Trials Evaluating the Role ofPARP Inhibitors in Pancreatic Cancer and Other Solid Tumors
| Study | Study design | Targeting agent/Dose | Adverse Effects (All grades)/Dose | Outcomes |
|---|---|---|---|---|
| Fong et al [55] | Phase I clinical trial in multiple advanced cancers including 22 with a germline BRCA mutation cancer | Olaparib 200 mg twice daily | Anemia, thrombocytopenia, diarrhea, dyspepsia, leukopenia | N= 22 BRCA mutant patients, 19 patients were assessable and 12 of 19 (63%) patients responded to treatment. One patient had sustained response for 76 weeks. One of 3 BRCA mutant breast cancer patients had complete response for 60 weeks. No BRCA mutant pancreatic cancer patient was enrolled in clinical trial |
| Samol et al [56] | Phase I clinical trial in multiple advanced cancers in combination with topotecan | Olaparib 100 mg twice daily | Fatigue and gastrointestinal symptoms. Dose limiting neutropenia and thrombocytopenia | Combination of topotecan and olaparib was not feasible due to dose limiting toxicity. Further combined studies with topotecan not recommended |
| Kauffman et al [57] | Phase II randomized trial in advance stage cancers (germline BRCA mutant tumors) | Olaparib 400 mg twice daily | Fatigue and gastrointestinal symptoms. Anemia (17%). N= 2 developed leukemia and 1 patient had MDS attributed to olaparib | Overall response rate was 26.2%. Tumor response among pancreatic cancer patients with germline BRCA mutation was 21.7%, stable disease (>8 weeks):35%. Response rates were similar in platinum naive and previously exposed patients (25% vs 20%): unclear whether platinum refractory. Similar response rates were observed both in germline BRCA1 and BRCA2 subgroups |
| Sandhu et al [59] | Phase I dose escalation trial | Niraparib 300mg daily | Anemia (48%), nausea (42%), thrombocytopenia (35%), anorexia (26%), neutropenia (24%) | Up to 40% partial response rate in BRCA mutant ovarian cancer (RECIST); 50% in breast cancer patients. Partial response in N= 3 of 9 (33%) platinum-resistant BRCA mutant ovarian cancer. Only one pancreatic cancer was enrolled and this patient did not respond. |
| Berlin et al [65] | Phase I dose escalation and safety trial in combination with FOLFIRI | Veliparib (escalated doses) | Diarrhea (61%), nausea (60%), vomiting (48%), neutropenia(59%), fatigue (47%), anemia(41%), alopecia (41%) | N= 96 patients with varied solid tumors. N= 12 patients (12.5%) had a partial response. Response rates in pancreatic, ovarian andbreast cancer; 14%, 33% and 22% respectively. One ovarian cancer patient had also complete response. Phase II dose veliparib determined as 200 mg twice daily orally on days 1–5 and 15–19 every 28 days. |
| Pishvaian et al [65] | Phase I/II study of veliparib in combination with 5-FU and oxalipatin in metastatic pancreatic cancer patients (BRCA status was not assessed) | Veliparin 300 mg twice daily | Myelosuppresion; primarily thrombocytopenia, | Response rate was 14 %. PFS and OS were 2.9 and 5.4 months respectively. PFS and OS were 4.3 vs 7.7 months respectively in treatment naive patients. Response rate was 18% in previously untreated patients. |
| O’Reilly et al [66] | Phase IB trial in combination with gemcitabine and cisplatin (patients with both mutant and wild type BRCA) | Veliparib 80 mg Twice daily | Anemia, thrombocytopenia, neutropenia and fatigue. One fatal bowel perforation | Partial response and stable disease were 56% and 44% respectively in BRCA mutant patients. Five patients with BRCA mutations (56%) continued treatment at the time of report. No partial response in non-BRCA patients only short-limited stable disease. Recommended phase 2 dose was determined 80 mg twice daily days 1–12 with cisplatin 25 mg/m2 and gemcitabine 600 mg/m2 day 3, 10, q 3 weeks |
| Lowery et al [67] | Phase II trial of veliparib as a single agent in patients with previously treated BRCA or PALB2-mutated pancreas adenocarcinoma | Veliparip 400 mg twice daily | Anemia, thrombocytopenia, neutropenia and fatigue | Four patients (25%) had stable disease on treatment > 4 months(4,6,6,9 months). One patient had unconfirmed partial response (4 months). Ten patients (62.5%) had progression. Median progression free survival was 52 days. Nearly all patients had prior platinum therapy/platinum refractory |
Table 3.
Ongoing/recently completed trials of PARP inhibitors in pancreatic cancer patients
| Study/Sponsor | Study design/Primary outcomes | Targeting agent/Dose | Study group | Current status |
|---|---|---|---|---|
| NCT0218419 AstraZeneca |
A Phase III, randomized, double blind, placebo controlled, multicentre study Primary: PFS |
Olaparib 300 mg twice daily |
Germline BRCA mutated metastatic pancreatic cancer patients whose disease has not progressed on first line platinum based chemotherapy eligible for enrolment following 4 months of platinum-based therapy | Recruiting patients |
|
NCT01123876 AbbVie |
A phase I, open labeled dose escalation study, dose determination and safety | Veliparib | Advance stage cancers (including pancreatic cancer) with a confirmed BRCA1/BRCA2/PALB2 mutation | Completed. Pending final results |
|
NCT01585805 National Cancer Institute Memorial Sloan Kettering Cancer Center |
A Phase II, randomized multicenter study of veliparib in commbination with cisplatin and gemcitabine Primary: Tumor response rate |
Veliparib | Advanced stage pancreatic adenocarcinoma patients with known BRCA1/BRCA2 or PALB2 mutation | Recruiting |
|
NCT02042378 Clovis Oncology, Inc. |
A Phase II, open label, non- randomized, non-comparative, multicenter study Primary: Safety and tumor response rate |
Rucaparib | Locally advanced or metastatic pancreatic adenocarcinoma patients with a known BRCA mutation (germline or somatic) | Not recruiting / Pending final results |
|
NCT01489865 AbbVie |
A Phase I/ II study of ABT-888 in combination of FOLFOX6 Primary: Safety and tumor response rate |
Veliparib | Metastatic pancreatic cancer patients with a history either wildtype or with BRCA1/BRCA2 or PALB2 mutations | Recruiting |
| NCT01098946 National Cancer Institute |
A Phase I/ II study of BMN-673 Primary: Safety and tumor response rate |
BMN-673 | Advance stage solid tumors with a BRCA mutation | Recruiting |
| NCT02184195 | A phase III randomized, double blind, placebo controlled multicenter study of maintenance olaparib monotherapy Primary: PFS |
Olaparib | Patients with gBRCA mutated metastatic pancreatic cancer following front-line platinum based therapy | Recruiting |
| NCT 01296763 | A Randomized multi-center phase I/II Trial of irinotecan, cisplatin, mitomycin C with or without olaparib | Olaparib | Advanced stage pancreatic cancer patients with or without a BRCA mutation | Not recruiting / Pending final results |
Niraparib, another PARP inhibitor has been evaluated in a phase I study in which the recommended treatment dose was established as 300 mg twice daily[59]. In this study one hundred patients were enrolled and a partial response rate in BCRA mutant ovarian and breast of 40% and 50% respectively, was observed. Interestingly a partial response rate of 33% (3 of 9) in platinum resistant BRCA mutant ovarian cancer was also reported. This rate was only 5% in non-BRCA carrier platinum resistant ovarian cancer patients. The most common side effects were related to bone marrow suppression including anemia (48%), thrombocytopenia (35%), neutropenia (24%) along with gastrointestinal symptoms such as nausea and vomiting. Currently, phase II/phase III studies of this agent are being conducted in breast and ovarian cancer (NCT01847274,NCT01905592, NCT02354586). Iniparib, another putated member of the PARP inhibitor family, showed a complete pathologic response in a patient with pancreatic cancer and a germline BRCA2 mutation[60]. Combination of this agent with chemotherapeutic agents showed initial promising activity in breast cancer particularly in BRCA mutant settings[61]. A phase II study of iniparib in combination with chemotherapy improved PFS and overall survival in triple negative breast cancer[62]. However this signal was not confirmed in a randomized phase III trial[63]. A back to bench study of iniparib on HR defected cells reported lack of specific PARP inhibition which further raised questions about the use of iniparib as a PARP targeting agent[64].
Preliminary results of a phase I study of another PARP inhibitor, veliparib showed similar toxicity profile including bone marrow toxicity along and was well tolerated as a single agent and in combination with FOLFIRI(5-FU, folinic acid, Irinotecan)[65]. Veliparib was administered twice daily on days 1–5 and 15–19 every 28 days orally in combination with FOLFIRI to advanced stage solid tumor patients with unknown BRCA mutation status. Best objective responses were observed in ovarian and breast cancer patients and rates were reported 33% and 22% respectively. Two of 14 (14%) pancreatic cancer patients enrolled in this trial had a partial response. Recommended phase II dose of veliparib was determined 200mg twice daily in combination with FOLFIRI. Veliparib in combination with FOLFOX is currently being investigated in a single arm non-randomized phase I/II study as chemotherapy sensitizer(NCT01489865). Patients received veliparib in escalating doses twice a day for days 1–7 of each 14-day cycle of FOLFOX regimen. Metastatic pancreatic cancer patients were enrolled and BRCA mutation status was not included in an early analysis which showed 14% response rate [66]. Reported PFS and OS were 2.9 and 5.4 respectively and myelosuppression was the main dose limiting toxicity. These rates were 4.3 and 7.7 months in untreated patients respectively. Recommended phase II dose was determined as 300 mg twice daily. O’Reilly et al. reported preliminary results of a phase IB study of veliparib in combination with cisplatin and gemcitabine in BRCAmutant and BRCAwild type pancreatic cancer patients and observed promising activity in the former [67]. Veliparib was administered on days 1–12 or 1–21 orally along with gemcitabine and cisplatin on days 3 and 10. Authors reported 66% partial response and 44% stable disease in BRCA mutant patients whereas no partial response and short-term disease stability only was observed in non-BRCA patients. The recommended Phase II dose was determined as 80 mg twice daily for days 1–12 in conjunction with cisplatin and gemcitabine administered on days 3 and 10 every 3 weeks. Phase II of single agent study of veliparib in 16 previously treated advanced stage pancreatic cancer patient showed 4(25%) stable disease more than 4 months, however, most of these patients had progressed on prior platinum therapy. Median PFS was 52 days with a range of 12 to 173 days. Common side effects observed in this study included fatigue and hematologic toxicities[68]. A randomized phase II study of cisplatin and gemcitabine with/without veliparib is currently being conducted in a germline BRCA and PALB2 mutated untreated stage III and IV population (NCT01585805).
Rucaparib is another member of PARP inhibitor family and has been evaluated in pancreatic cancer as a single agent in a phase II study (NCT02042378). In this study pancreatic cancer patients with a germline or somatic BRCA mutation were recruited to receive rucaparib 600 mg twice daily. This study has temporarily closed to recruitment and data are awaited. Other PARP inhibiting agents(Table 4) including BMN 673 (talazoparib) are also in development (NCT01989546). A further study by the Southwest Oncology Group (SWOG) is planned to evaluate the addition of FOLFIRI +/− veliparib in gemcitabine-pre-treated pancreas adenocarcinoma in a second-line setting. Archival tissue will be collected on all patients and markers of homologous repair deficiency will be retrospectively interrogated.
Table 4.
PARP Inhibitors in Development/Approved
| PARP inhibitor | Company | Dosing in trials | Adverse effects/Drug toxicity | Route |
|---|---|---|---|---|
| Olaparib* (AZD2281) | AstraZeneca | 400 mg twice daily | Fatigue, nausea, vomiting, diarrhea, headache, anemia, thrombocytopenia | Oral |
| Veliparib (ABT-888) | AbbVie (Abbott) | 400 mg twice daily | Anemia, thrombocytopenia, fatigue, nausea, vomiting | Oral |
| Rucaparib (AG014699) | Pfizer | 600 mg twice daily (oral) | Fatigue, nausea, vomiting, diarrhea, anorexia, anemia, thrombocytopenia | Oral/IV |
| Niraparib (MK4827) | Tesaro/Merck | 300 mg daily | Thrombocytopenia, neutropenia, anemia, nausea, vomiting, anorexia, pneumonitis | Oral/IV |
| Talazoparib (BMN673) | Medivation | 1 mg daily | Thrombocytopenia, neutropenia, anemia, fatigue, nausea, alopecia | Oral |
| INO-1001 | Inotek | 200 mg twice daily | Thrombocytopenia, neutropenia, nausea, hepatotoxicity, hyponatremia | IV |
| CEP-9722 | Cephalon/Teva | 750 mg daily | Lymphopenia, anemia, astenia, weight loss, nausea, vomiting, diarrhea | Oral |
FDA-approved for germline BRCA mutant ovarian cancer as 3rd line treatment
Platinum Based Treatment in Pancreatic Cancer Patients with Genomic Instability
Platinum-based agents directly target DNA and create cross-links between the strands and subsequently induce DNA breaks[69]. Significant anti-tumor effect induced by platinum based-agents has been elucidated in BRCAmutant ovarian cancer in the early 2000s. [70] Therefore, platinum-induced synthetic lethality has been also explored in pancreatic cancer patients with BRCA mutations. One retrospective study found a response rate of 83%(5 of 6 of patients) by RECIST criteria in metastatic pancreatic cancer patients who received a platinum-based agent as a first line treatment[71]. Authors also reported one patient who received FOLFIRINOX treatment who also had a complete response. A retrospective cohort of pancreatic cancer patients with a BRCA mutation reported that locally advanced and metastatic patients who received platinum based treatment had better survival outcomes compared in a non-randomized fashion to patients who were not treated with platinum agent(P=0.036)[34]. The majority of the patients were treated with gemcitabine and cisplatin and three patients received FOLFIRINOX and one patient gemcitabine and oxaliplatin. Overall survivals in BRCA1 and BRCA2 mutations subgroups were 15 and 13 months respectively with no statistical significant difference observed(P=0.77). A recent whole exome study also elucidated sensitivity of HR deficient pancreatic cancer to platinum based agents[6]. Authors reported 80% (4 of 5 patients) response to cisplatin based therapy in pancreatic cancer patients with BRCA signature. Similar platinum sensitivity has also been observed in mouse models bearing tumors with high genomic instability induced by defective DNA damage response and repair.
Although platinum agents appears to be effective in pancreatic cancer with genomic instability, evolving evidence suggests that cancer cells may ultimately develop resistance. An initial cell-line study identified secondary somatic mutations in BRCA2 gene of a cisplatin resistant BRCA2mutant ovarian cancer cell line derived from ascites of a relapsed patient[72]. This study was followed by a mechanistic and translational study in BRCA2 mutant breast cancer which uncovered the role of secondary sporadic mutations that restored BRCA2 function and induced consequent platinum-resistance[73]. In this study acquired mutations were shown to induce base deletions in germline-mutated BRCA2 gene causing restoration in expression of DNA binding site of this protein which is sufficient for its activation. Importantly, the authors also reported PARP inhibitor resistance in the same clones which developed secondary mutations. Consistent with this data, a study evaluating BRCA1 and BRCA2 mutant ovarian and breast cancer observed secondary somatic mutations leading to cisplatin and PARP inhibitor resistant cancers particularly in patients who were previously treated with platinum-based agents[74]. A study interrogating acquired resistance in germline-mutated BRCA1 tumors elucidated stabilization of mutant BRCA1 by heat shock protein 90 (HSP90) which provided an ability to bind DNA repair pathway proteins[75]. The authors reported concurrent acquired PARP inhibitor and cisplatin resistance conferred by HSP90 mediated restoration in BRCA1 function. A preclinical model system of BRCA mutant breast cancer suggested cisplatin resistance may be arising from cancer stem cells expressing high CD29 along with medium CD24 expression pointing that tumor heterogeneity is one of the possible mechanism for platinum-resistance[76]. Another study in pancreatic adenocarcinoma cells demonstrated resistance to DNA damaging agent including PARP inhibitors and platinum-based agents mediated by Wee-1 induced cell cycle arrest[44]. Although these studies suggested concurrent PARP resistance in platinum-resistant BRCA mutant cancer cells, there is also clinical evidence that PARP inhibitors may be active in platinum-resistance tumors. Fong et al observed a 45% response rate by RECIST criteria in platinum-resistant ovarian cancer as well as platinum refractory disease (23%) [77] suggesting utility of using PARP inhibitors in patients progressing on a platinum-based treatment. There are direct implications for trial design pertaining to the outcome of these latter issues. For example, data to date in pancreatic cancer suggests that PARP inhibitors may have most utility in a platinum sensitive setting. It remains to be seen whether concurrent or sequential administration of PARP inhibitors with platinum-based therapy is a preferred strategy in this disease.
Expert Commentary
In the light of the collective scientific evidence, we can confidently state that there is a subgroup of pancreatic patients with a distinct genomic profile predicting a favorable response to platinum-based agents. Genomic instability, a result of deleterious mutations of genes functioning in HR mediated DNA repair pathways, yields an exceptional vulnerability in cancer cells named synthetic lethality. Inhibition of other DNA repair pathways compensating for this genomic instability, such as PARP1, or directly DNA targeting agents causes overwhelming DNA damage, with platinum based drugs, inducing apoptotic cell death and tumor regression. Thus far, genomic studies have identified many genes that might underscore genomic instability including, BRCA1, BRCA2, PALB2, ATM, ATR, ARID1A, CHEK1/2, RPA1,MMR genes and with additional genes likely to be identified going forward. Of these genes, BRCA1/BRCA2 and PALB2 have been the most commonly studied genes and the signature of BRCA mutation, ‘BRCAness’, has been found to be an important predictor of treatment response[10].
Discoveries in the genomic signature of pancreatic cancer could lead to further characterization of this disease and open new paths for new treatment approaches. For example, there is significant evidence implicating other genes controlling the cell cycle as well as the fact that DNA repair response may be related to genomic instability in genes such ATM, ATR, MMR genes and even p53 which is a frequently mutated gene in pancreatic cancer. Currently, mutations in these genes have not been rigorously studied with regard to DNA-damaging agents and PARP inhibitors. Combined strategies of PARP inhibitors to induce cell cycle progression with injury in DNA may cause further DNA damage and subsequent apoptosis. At this juncture, targeting genes controlling the G2 check point could be a future approach in the setting of genomic instability lacking PARP inhibitor response. Furthermore, combining platinum based agents with PARP inhibitors, an approach currently being studied in clinical trials (NCT01585805), may provide a more pronounced therapeutic impact. Early evidence from clinical studies[57,77]suggests the potential use of PARP inhibitors and platinum-agents as an alternative of each other in patients who develop treatment resistance to one. Although data is conflicting in other aforementioned translational studies for a possible dual-resistance in patients who gain secondary BRCA mutations or other acquired mechanisms of resistance[73,74], further studies will enhance our understanding and elucidate the utility of this approach in pancreatic cancer patients.
While PARP inhibitors may improve the outcomes in pancreatic cancer, their long-term toxicity should be carefully evaluated given the potential for generating mutations in healthy DNA which could lead to further oncogenic activation. For example, observed secondary malignancies in clinical trials of PARP inhibitors such as leukemia and myelodyplastic syndrome [56] suggest genomic instability in a setting of germline BRCA mutation may lead to further malignancies due to inhibited compensatory DNA repair pathways. However a contribution from platinum agents is also likely as these drugs have a small but known risk of myelodysplasia/acute leukemia[78]. These observations suggest that PARP inhibitors may yield advanced genomic instability and subsequently major genetic rearrangements and structural changes in chromosomes. To diminish these adverse effects, cancer specific drug delivery methods can be studied along with optimal targeting combinations. Moreover, the impact of PARP inhibitors on cancer stem cells (CSC) may need to be investigated to be able to obtain more sustained treatment response given many studies indicate treatment resistance particularly in chemotherapeutic agents arise from CSCs[79]. For example, the expression profile of PARP1 and frequency of secondary mutations of BRCA genes may differ in cancer stem clones which may impact the apoptotic effect DNA targeting agents.
Five Year View
Based on the aforementioned evidence, pancreatic cancer patients with genomic instability may clearly benefit from DNA targeting agents, and PARP inhibitors. We strongly believe that vulnerability of genomic structure in patients with defective DNA repair pathway leads to significant tumor suppression and clinical response will bring PARP inhibitors and platinum based agents as standard treatment options for this patient population. Progress of science may also elucidate whether anoptimal strategy is combining PARP inhibitors and platinum agents or using them in a sequential manner toachieve more sustained clinical response, minimize emergence of resistance and mitigate toxicity. In the near future, we may also better understand the clinical utility ofthese agents in patients with resectable /borderline resectablepancreatic cancer in the setting ofadjuvant and/or neoadjuvant treatment. Although genomic signature models of pancreatic cancer may elucidate further clue for precision medicine, the applicability to daily clinical practice remains to be addressed. A recent cohort of pancreatic cancer patients for individualized treatment reported many challenges including quality of tumor specimen, time frame between genomic profiling and clinical practice, and identifying sufficient number of patients[80] However, we believe that as science progresses and significance of genomic models and actionable genes in daily clinical practice becomes more substantial, quality improvement in tissue processing and time frame will likely advance. For example, the cost of whole genome sequencing, which has been another drawback, has substantially diminished along with increased number genomic sequencing modalities. Therefore, we believe that scientific discoveries will bring innovations in daily practice.
Genomic instability and related hyperdynamic mutational status perhaps continues following development of tumors due to advanced metabolic stress in the tumor microenvironment. The protein expression profile of this subgroup may present distinct characteristics compared to tumors with stable genomes. Mutational load in pancreatic cancer with defective DNA repair may be higher compared to a stable genome setting, an observation that as yet is speculative. The impact of this antigenic diversity as a result of varied mutational changes on disease behavior such as the cancer cell tumor microenvironment interaction or even more importantly cancer cells and immune system interactions, warrant further investigation to elucidate fingerprints of this pancreatic cancer subgroup. Evidence from other solid tumor studies with similar genomic repair defects indicate a potential clinical benefit of immune-modulation in genomically unstable pancreatic cancers. Considering tumor infiltrating lymphocytes observed in MSI-high colorectal cancer bears high genomic instability and may predict check point inhibitor response in colorectal cancer patients[23], utility of immune-modulation in pancreatic cancer patients with unstable genome remains to be investigated. Moreover, whether an unstable genome in pancreatic cancer impacts metastatic behavior of the disease warrants further evaluation to better characterize this unique subgroup of patients. It has also yet to be identified whether BRCA1/BRCA2 mutant tumors have prognostic as well as predictive implications.
Lastly, to date, BRCA mutations have been widely used as a biomarker of genomic instability particularly in the context of germline mutations. The question at this time point is of who to screen for a BRCA mutation. Recent studies suggest that many patients who have an underlying germline BRCA mutation are beyond the BRCA screening guidelines (NCCN and Ontario). Until updates in screening guidelines come to pass, opportunities for an enhanced precision medicine approach are being missed. In light of this, while continuing our efforts to better define the at-risk patient population, we must consider expansion of the guidelines for germline screening for pancreatic adenocarcinoma patients. Somatic mutations and epigenetic silencing of BRCA genes currently are not routinely evaluated for, however with the increased availability of somatic profiling, we expect that additional data will emerge in the near future.
In summary, PARP inhibitors and platinum-based treatments promise a significant future for pancreatic cancer particularly in BRCA mutant patients and potentially in other genetic alteration settings related to genomic instability. Further investigation will shed insight on the aforementioned areas to better understand applicability of these new approaches and tailor to the relevant patient population.
Table 1.
Whole genome/exome studies in pancreatic cancer
| Study | Study samples | Genomic discoveries | Conclusions and implications |
|---|---|---|---|
| Biankin et al [6] | N= 99 pancreatic cancer patients with resected stage I or stage II disease | Novel mutated genes were identified in different signaling pathways: chromatin modification (EPC1 and ARID2), DNA damage repair (ATM) and other pathways (ZIM2, MAP2K4, NALCN, SLC16A4 and MAGEA6) Somatic alterations were revealed in axon guidance pathway (SLIT/ROBO signaling). SLIT receptor ROBO2 was found to be associated with better survival outcomes. |
Involvement of aberrant axon guidance pathway was first demonstrated in pancreatic cancer A potentially targetable pathway was elucidated ROBO expression may be used as biomarker of prognosis. |
| Alexandrov et al [7] | N= 7,042 primary of 30 different types tumors (including pancreatic cancer) | More than 20 signature models were identified with distinct features An overlapping mutational pattern in breast, ovarian and pancreatic cancer was identified and classified as signature 3. This signature was associated with BRCA mutations Signature 6 was present in various tumors including colorectal, endometrial, prostate and pancreatic cancers and this signature was related to DNA mismatch repair deficiency |
There are mutational patterns and signatures related to genetic diversity and complex disease behavior Signature models can be used to guide the treatment based on altered pathways and related vulnerability Genetic biomarkers can be identified for prognostic and treatment purposes using the signature models |
| Campbell et al [8] | N= 13 metastatic pancreatic cancer patients | G1-S transition phase is dysregulated in pancreatic cancer with conserved G2-M phase Genomic instability persists following dissemination of disease and yields subclones with distinct behaviors Genetic heterogeneity is present among metastasis initiating cancer cells Tandem forces of genomic instability and clonal selection lead to driver mutations and genetic rearrangements resulting in genetic heterogeneity among disseminated cancer cells |
Cancer cells preserve G2-M phase likely to evade apoptosis related to DNA injury Targeting G2-M transition phase may yield anti-tumor effect Genomic heterogeneity brought by genomic instability could be an underlying reason for observed diverse disease behavior in clinical settings |
| Yachida et al [9] | N= 7 patients with metastatic pancreatic cancer. | Clones representing metastatic foci are present in primary tumor. Genetic heterogeneity is observed before dissemination of cancer cells At least a decade is required between occurrence of the first founder mutation and acquisition of malignant cell properties Five or more years are needed for acquisition of metastatic capacity for a non-metastatic founder cancer cell |
Genetic heterogeneity proceeds acquisition of metastatic features and could be related Extended time window is required for progression of non-metastatic clone to metastatic clone This long time interval may be used to detect non-metastatic disease by screening methods |
| Waddell et al [10] | N= 100 pancreatic cancer patients | Four genomic subtypes of pancreatic adenocarcinoma were identified; stable, locally rearranged, scattered and unstable Genomic instability and BRCA mutations were found to be directly related Genomic instability may occur in the setting of mutations involving other DNA repair genes such as PALB2 and RPA Genomic instability with BRCA signature predicts platinum based treatment response |
Genomic instability in pancreatic cancers creates a critical susceptibility to platinum-based agents Other genes of the DNA repair pathways such as PALB2 may also create a similar vulnerability Patients with BRCA and other DNA repair pathway gene mutations can benefit from DNA targeting agents such as PARP inhibitors |
| Witkiewicz et al. [12] | N= 109 pancreatic cancer patients | Genes related to Fanconi Anemia as well as ATM, CHEK2, BCLAF1 (BCL2-associated Transcription Factor), BRCA1 and BRCA2 were observed to be mutated or deleted at relatively high frequency indicating frequent alterations DNA repair pathways (>35%) Genetic alterations in chromatin remodelling SWI/ SNF pathway observed in 42% of cases Loss of RNF43 or AXIN1 may be biomarkers of porcupine and tankyrase inhibitors targeting the beta-catenin pathway Loss of CDKN2A or amplification of CDK4/ CCND1 may confer sensitivity to CDK4/6 inhibitors RBM10 (RNA Binding Motif Protein 10) mutations were associated with improved survival. ARID1A is associated with poor prognosis |
DNA repair pathway genes are frequently affected throughout pancreatic carcinogenesis Mutations in DNA repair pathways may carry prognostic value in pancreatic cancer and could be potential targets for future treatments Frequent alterations in chromatin remodeling could be biomarker of genomic plasticity in pancreatic cancer Targeted therapy can be tailored based on mutational signature of disease. |
Key Issues.
Genome-wide studies have revealed evidence suggesting that genomic instability in pancreatic cancer has a unique genetic signature with relatively increased deleterious changes in DNA and an unstable genome status which significantly impacts tumor response to treatment.
Defective HR-mediated DNA repair as a result of mutations in BRCA1/BRCA2 genes is directly related with genomic instability and creates a unique vulnerability in cancer cells to DNA targeting agents which induce irreversible DNA damages, further unstable genome and ultimately apoptosis.
Many other genes active in DNA repair and stabilization pathways such as PALB2, RPA1, MMRs, ATM, and ARID1A may also be related to a similar genomic pattern. Whether tumors with somatic mutations in DNA repair genesbearsimilar genetic signatures to those with germline mutations requires further study.
Preclinical studies suggest that PARP inhibitors abrogate DNA repair in HR defected cancer cells and inhibit tumor growth in BRCA mutant tumors including pancreatic cancers. Inhibition of DNA repair in cancer cells induces further genomic instability which alters cell cycle process.
Clinical studies indicate PARP inhibitors and platinum-based agents may have significant anti-tumor effect in pancreatic cancer patients with BRCA mutant tumors. Based on preliminary data, PARP inhibitor may become part of standard care in pancreatic cancer treatment in the next 5 years.
To date, there is limited evidence for this benefit in tumors with other genes related to genomic instability such as PALB2, RPA1, and MMR genes. Further investigation is warranted.
Prior platinum resistance may not exclude benefit to PARP inhibitor treatment, however, mechanisms of resistance appear to be similar and this area needs detailed study in pancreas cancer.
The exact prevalence of BRCA mutations in unselected pancreatic cancer patients is unknown. Studies suggest that approximately 5–7% of pancreatic cancer patients could be related to germline BRCA mutations.
Expansion of screening guidelines for germline BRCA1/BRCA2 and other genetic biomarkers of defective DNA repair should be strongly considered to better identifypancreatic adenocarcinoma patients with genomic instability. Typical indicators including Ashkenazi heritage and a personal or family history of malignancy, clearly miss at least a third to half of patients who have an underlying germline BRCA mutation.
Acknowledgments
Financial
EM O’Reilly has received research funding from Celgene, Myriad Genetics, Abbvie and Astra Zeneca and consulting fees from Celgene and Pfizer.
Footnotes
Competing interests disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
Reference annotations
* Of interest
** Of considerable interest
- 1.Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. New England Journal of Medicine. 2011;364(19):1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 2.Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. New England Journal of Medicine. 2013;369(18):1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74(11):2913–2921. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 4.Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. The Lancet. 2004;363(9414):1049–1057. doi: 10.1016/S0140-6736(04)15841-8. [DOI] [PubMed] [Google Scholar]
- 5.Hruban RH. Familial pancreatic cancer. Archives of pathology & laboratory medicine. 2009;133(3):365. doi: 10.5858/133.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491(7424):399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7*.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. doi: 10.1038/nature12477. This study elucidated an overlapping signature model for breast, ovarian and pancreatic cancer which was associated with germline BRCA mutations. This model was related to microhomology at rearrangements junctions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8*.Campbell PJ, Yachida S, Mudie LJ, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467(7319):1109–1113. doi: 10.1038/nature09460. This paper first demonstrated dynamic genomic alterations causing driver mutations in different metastatic foci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yachida S, Jones S, Bozic I, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467(7319):1114–1117. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10**.Waddell N, Pajic M, Patch A-M, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518(7540):495–501. doi: 10.1038/nature14169. This study identified that an unstable genomic pattern was associated with impaired DNA repair pathways in pancreatic cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Witkiewicz AK, McMillan EA, Balaji U, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nature communications. 2015;6 doi: 10.1038/ncomms7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability—an evolving hallmark of cancer. Nature reviews Molecular cell biology. 2010;11(3):220–228. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- 14.Futreal PA, Liu Q, Shattuck-Eidens D, et al. BRCA1 mutations in primary breast and ovarian carcinomas. Science. 1994;266(5182):120–122. doi: 10.1126/science.7939630. [DOI] [PubMed] [Google Scholar]
- 15.Gudmundsdottir K, Ashworth A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene. 2006;25(43):5864–5874. doi: 10.1038/sj.onc.1209874. [DOI] [PubMed] [Google Scholar]
- 16.Song H, Hollstein M, Xu Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nature cell biology. 2007;9(5):573–580. doi: 10.1038/ncb1571. [DOI] [PubMed] [Google Scholar]
- 17.Cortez D, Wang Y, Qin J, Elledge SJ. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science. 1999;286(5442):1162–1166. doi: 10.1126/science.286.5442.1162. [DOI] [PubMed] [Google Scholar]
- 18.Marión RM, Strati K, Li H, et al. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature. 2009;460(7259):1149–1153. doi: 10.1038/nature08287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fukasawa K, Wiener F, Vande WG, Mai S. Genomic instability and apoptosis are frequent in p53 deficient young mice. Oncogene. 1997;15(11):1295–1302. doi: 10.1038/sj.onc.1201482. [DOI] [PubMed] [Google Scholar]
- 20.Popat S, Hubner R, Houlston R. Systematic review of microsatellite instability and colorectal cancer prognosis. Journal of Clinical Oncology. 2005;23(3):609–618. doi: 10.1200/JCO.2005.01.086. [DOI] [PubMed] [Google Scholar]
- 21.Hemminki A, Mecklin JP, Järvinen H, Aaltonen LA, Joensuu H. Microsatellite instability is a favorable prognostic indicator in patients with colorectal cancer receiving chemotherapy. Gastroenterology. 2000;119(4):921–928. doi: 10.1053/gast.2000.18161. [DOI] [PubMed] [Google Scholar]
- 22.Gryfe R, Kim H, Hsieh ET, et al. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. New England Journal of Medicine. 2000;342(2):69–77. doi: 10.1056/NEJM200001133420201. [DOI] [PubMed] [Google Scholar]
- 23**.Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. The New England journal of medicine. 2015;372(26):2509–2520. doi: 10.1056/NEJMoa1500596. This paper reported a benefit to checkpoint inhibitor therapy in defective DNA repair caused by mismatch repair deficiency in cancer patients. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walther A, Houlston R, Tomlinson I. Association between chromosomal instability and prognosis in colorectal cancer: a meta-analysis. Gut. 2008 doi: 10.1136/gut.2007.135004. [DOI] [PubMed] [Google Scholar]
- 25.dos Santos NR, Seruca R, Constancia M, Seixas M, Sobrinho-Simoes M. Microsatellite instability at multiple loci in gastric carcinoma: clinicopathologic implications and prognosis. Gastroenterology. 1996;110(1):38–44. doi: 10.1053/gast.1996.v110.pm8536886. [DOI] [PubMed] [Google Scholar]
- 26.Roberts NJ, Jiao Y, Yu J, et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer discovery. 2012;2(1):41–46. doi: 10.1158/2159-8290.CD-11-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banin S, Moyal L, Shieh S-Y, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281(5383):1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- 28.Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Molecular cell. 1999;4(4):511–518. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- 29.Powell SN, Kachnic LA. Roles of BRCA1 and BRCA2 in homologous recombination, DNA replication fidelity and the cellular response to ionizing radiation. Oncogene. 2003;22(37):5784–5791. doi: 10.1038/sj.onc.1206678. [DOI] [PubMed] [Google Scholar]
- 30.Jones S, Hruban RH, Kamiyama M, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324(5924):217–217. doi: 10.1126/science.1171202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li X, Heyer W-D. Homologous recombination in DNA repair and DNA damage tolerance. Cell research. 2008;18(1):99–113. doi: 10.1038/cr.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nature Reviews Cancer. 2004;4(10):814–819. doi: 10.1038/nrc1457. [DOI] [PubMed] [Google Scholar]
- 33*.Hedenfalk I, Duggan D, Chen Y, et al. Gene-expression profiles in hereditary breast cancer. N Engl J Med. 2001;344(8):539–548. doi: 10.1056/NEJM200102223440801. This article first described “BRCAness” pattern in BRCA mutant breast cancer patients. [DOI] [PubMed] [Google Scholar]
- 34*.Golan T, Kanji Z, Epelbaum R, et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. British journal of cancer. 2014;111(6):1132–1138. doi: 10.1038/bjc.2014.418. This study suggested a clinical benefit with use of platinum based agents in BRCA mutant pancreatic cancer patients. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salo-Mullen EE, O’Reilly EM, Kelsen DP, et al. Identification of germline genetic mutations in patients with pancreatic cancer. Cancer. 2015 doi: 10.1002/cncr.29664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim MY, Zhang T, Kraus WL. Poly (ADP-ribosyl) ation by PARP-1:PAR-laying’NAD+ into a nuclear signal. Genes & development. 2005;19(17):1951–1967. doi: 10.1101/gad.1331805. [DOI] [PubMed] [Google Scholar]
- 37.Caldecott KW. Single-strand break repair and genetic disease. Nature Reviews Genetics. 2008;9(8):619–631. doi: 10.1038/nrg2380. [DOI] [PubMed] [Google Scholar]
- 38.Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell research. 2008;18(1):27–47. doi: 10.1038/cr.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang M, Wu W, Wu W, et al. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic acids research. 2006;34(21):6170–6182. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hochegger H, Dejsuphong D, Fukushima T, et al. Parp-1 protects homologous recombination from interference by Ku and Ligase IV in vertebrate cells. The EMBO journal. 2006;25(6):1305–1314. doi: 10.1038/sj.emboj.7601015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Domagala P, Huzarski T, Lubinski J, Gugala K, Domagala W. PARP-1 expression in breast cancer including BRCA1-associated, triple negative and basal-like tumors: possible implications for PARP-1 inhibitor therapy. Breast cancer research and treatment. 2011;127(3):861–869. doi: 10.1007/s10549-011-1441-2. [DOI] [PubMed] [Google Scholar]
- 42**.Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly (ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. doi: 10.1038/nature03443. This article demonstrated the rationale of using PARP inhibitors in cancer cells with defective DNA. [DOI] [PubMed] [Google Scholar]
- 43.Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 44.Lal S, Burkhart RA, Beeharry N, et al. HuR posttranscriptionally regulates WEE1: implications for the DNA damage response in pancreatic cancer cells. Cancer research. 2014;74(4):1128–1140. doi: 10.1158/0008-5472.CAN-13-1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karnak D, Engelke CG, Parsels LA, et al. Combined inhibition of Wee1 and PARP1/2 for radiosensitization in pancreatic cancer. Clinical Cancer Research. 2014;20(19):5085–5096. doi: 10.1158/1078-0432.CCR-14-1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vance S, Liu E, Zhao L, et al. Selective radiosensitization of p53 mutant pancreatic cancer cells by combined inhibition of Chk1 and PARP1. Cell Cycle. 2011;10(24):4321–4329. doi: 10.4161/cc.10.24.18661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morgan MA, Parsels LA, Zhao L, et al. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer research. 2010;70(12):4972–4981. doi: 10.1158/0008-5472.CAN-09-3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shen J, Peng Y, Wei L, et al. ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Inhibitors. Cancer Discov. 2015;5(7):752–767. doi: 10.1158/2159-8290.CD-14-0849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Iqbal J, Ragone A, Lubinski J, et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. British journal of cancer. 2012;107(12):2005–2009. doi: 10.1038/bjc.2012.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thompson D, Easton DF, Consortium BCL Cancer incidence in BRCA1 mutation carriers. Journal of the National Cancer Institute. 2002;94(18):1358–1365. doi: 10.1093/jnci/94.18.1358. [DOI] [PubMed] [Google Scholar]
- 51.Hahn SA, Greenhalf B, Ellis I, et al. BRCA2 germline mutations in familial pancreatic carcinoma. Journal of the National Cancer Institute. 2003;95(3):214–221. doi: 10.1093/jnci/95.3.214. [DOI] [PubMed] [Google Scholar]
- 52.Struewing JP, Hartge P, Wacholder S, et al. The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. New England Journal of Medicine. 1997;336(20):1401–1408. doi: 10.1056/NEJM199705153362001. [DOI] [PubMed] [Google Scholar]
- 53.Stadler ZK, Salo-Mullen E, Patil SM, et al. Prevalence of brca1 and brca2 mutations in ashkenazi jewish families with breast and pancreatic cancer. Cancer. 2012;118(2):493–499. doi: 10.1002/cncr.26191. [DOI] [PubMed] [Google Scholar]
- 54*.Holter S, Borgida A, Dodd A, et al. Germline BRCA mutations in a large clinic-based cohort of patients with pancreatic adenocarcinoma. Journal of Clinical Oncology, JCO. 2015;20147401:2059. doi: 10.1200/JCO.2014.59.7401. This report provides a detailed study of germline BRCA in a cohort of patients with pancreas cancer. [DOI] [PubMed] [Google Scholar]
- 55.Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 56.Samol J, Ranson M, Scott E, et al. Safety and tolerability of the poly (ADP-ribose) polymerase (PARP) inhibitor, olaparib (AZD2281) in combination with topotecan for the treatment of patients with advanced solid tumors: a phase I study. Investigational new drugs. 2012;30(4):1493–1500. doi: 10.1007/s10637-011-9682-9. [DOI] [PubMed] [Google Scholar]
- 57.Kaufman B, Shapira-Frommer R, Schmutzler RK, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. Journal of Clinical Oncology, JCO. 2014;20142728:2056. doi: 10.1200/JCO.2014.56.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. New England Journal of Medicine. 2012;366(15):1382–1392. doi: 10.1056/NEJMoa1105535. [DOI] [PubMed] [Google Scholar]
- 59.Sandhu SK, Schelman WR, Wilding G, et al. The poly (ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. The lancet oncology. 2013;14(9):882–892. doi: 10.1016/S1470-2045(13)70240-7. [DOI] [PubMed] [Google Scholar]
- 60.Fogelman DR, Wolff RA, Kopetz S, et al. Evidence for the Efficacy of Iniparib, a PARP-1 Inhibitor, in BRCA2-associated Pancreatic Cancer. Anticancer research. 2011;31(4):1417–1420. [PubMed] [Google Scholar]
- 61.Telli ML, Jensen KC, Vinayak S, et al. Phase II Study of Gemcitabine, Carboplatin, and Iniparib As Neoadjuvant Therapy for Triple-Negative and BRCA1/2 Mutation–Associated Breast Cancer With Assessment of a Tumor-Based Measure of Genomic Instability: PrECOG 0105. Journal of Clinical Oncology, JCO, 2014.2057. 2015;0085 doi: 10.1200/JCO.2014.57.0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.O’Shaughnessy J, Osborne C, Pippen JE, et al. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N Engl J Med. 2011;364(3):205–214. doi: 10.1056/NEJMoa1011418. [DOI] [PubMed] [Google Scholar]
- 63.O’Shaughnessy J, Schwartzberg L, Danso MA, et al. Phase III study of iniparib plus gemcitabine and carboplatin versus gemcitabine and carboplatin in patients with metastatic triple-negative breast cancer. J Clin Oncol. 2014;32(34):3840–3847. doi: 10.1200/JCO.2014.55.2984. [DOI] [PubMed] [Google Scholar]
- 64.Patel AG, De Lorenzo SB, Flatten KS, Poirier GG, Kaufmann SH. Failure of iniparib to inhibit poly (ADP-Ribose) polymerase in vitro. Clinical Cancer Research. 2012;18(6):1655–1662. doi: 10.1158/1078-0432.CCR-11-2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Berlin J, Ramanathan RK, Strickler JH, et al. JOURNAL OF CLINICAL ONCOLOGY. AMER SOC CLINICAL ONCOLOGY; 2318 MILL ROAD, STE 800, ALEXANDRIA, VA 22314 USA: 2014. A phase 1 dose-escalation study of veliparib with bimonthly FOLFIRI in patients with advanced solid tumors. Ed.ˆ(Eds) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pishvaian MJ, Wang H, Zhuang T, et al. JOURNAL OF CLINICAL ONCOLOGY. AMER SOC CLINICAL ONCOLOGY; 2318 MILL ROAD, STE 800, ALEXANDRIA, VA 22314 USA: 2013. A phase I/II study of ABT-888 in combination with 5-fluorouracil (5-FU) and oxaliplatin (Ox) in patients with metastatic pancreatic cancer (MPC) (Ed.ˆ(Eds) [Google Scholar]
- 67.O’Reilly E, Lowery M, Segal M, Smith S. Phase IB trial of cisplatin (C), gemcitabine (G), and veliparib (V) in patients with known or potential BRCA or PALB2-mutated pancreas adenocarcinoma (PC) J Clin Oncol. 2014;32:5s. [Google Scholar]
- 68.Lowery MA, Kelsen DP, Smith SC, et al. JOURNAL OF CLINICAL ONCOLOGY. AMER SOC CLINICAL ONCOLOGY; 2318 MILL ROAD, STE 800, ALEXANDRIA, VA 22314 USA: 2015. Phase II trial of veliparib (V) in patients (pts) with previously treated BRCA or PALB2-mutated (mut) pancreas adenocarcinoma (PC) (Ed.ˆ(Eds) [Google Scholar]
- 69.Carey LA. Targeted chemotherapy? Platinum in BRCA1-dysfunctional breast cancer. Journal of Clinical Oncology. 2010;28(3):361–363. doi: 10.1200/JCO.2009.24.0838. [DOI] [PubMed] [Google Scholar]
- 70.Cass I, Baldwin RL, Varkey T, Moslehi R, Narod SA, Karlan BY. Improved survival in women with BRCA-associated ovarian carcinoma. Cancer. 2003;97(9):2187–2195. doi: 10.1002/cncr.11310. [DOI] [PubMed] [Google Scholar]
- 71.Lowery MA, Kelsen DP, Stadler ZK, et al. An emerging entity: pancreatic adenocarcinoma associated with a known BRCA mutation: clinical descriptors, treatment implications, and future directions. The oncologist. 2011;16(10):1397–1402. doi: 10.1634/theoncologist.2011-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sakai W, Swisher EM, Jacquemont C, et al. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer research. 2009;69(16):6381–6386. doi: 10.1158/0008-5472.CAN-09-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sakai W, Swisher EM, Karlan BY, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451(7182):1116–1120. doi: 10.1038/nature06633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Norquist B, Wurz KA, Pennil CC, et al. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. Journal of Clinical Oncology. 2011;29(22):3008–3015. doi: 10.1200/JCO.2010.34.2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Johnson N, Johnson SF, Yao W, et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc Natl Acad Sci U S A. 2013;110(42):17041–17046. doi: 10.1073/pnas.1305170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shafee N, Smith CR, Wei S, et al. Cancer stem cells contribute to cisplatin resistance in Brca1/p53–mediated mouse mammary tumors. Cancer research. 2008;68(9):3243–3250. doi: 10.1158/0008-5472.CAN-07-5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fong PC, Yap TA, Boss DS, et al. Poly (ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. Journal of Clinical Oncology. 2010;28(15):2512–2519. doi: 10.1200/JCO.2009.26.9589. [DOI] [PubMed] [Google Scholar]
- 78.Travis LB, Holowaty EJ, Bergfeldt K, et al. Risk of leukemia after platinum-based chemotherapy for ovarian cancer. New England Journal of Medicine. 1999;340(5):351–357. doi: 10.1056/NEJM199902043400504. [DOI] [PubMed] [Google Scholar]
- 79.Lee CJ, Dosch J, Simeone DM. Pancreatic cancer stem cells. Journal of Clinical Oncology. 2008;26(17):2806–2812. doi: 10.1200/JCO.2008.16.6702. [DOI] [PubMed] [Google Scholar]
- 80.Chantrill LA, et al. Precision Medicine for Advanced Pancreas Cancer: The Individualized Molecular Pancreatic Cancer Therapy (IMPaCT) Trial. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21:2029–2037. doi: 10.1158/1078-0432.CCR-15-0426. [DOI] [PubMed] [Google Scholar]